
Abstract
Glucose-6-phosphate (G6P) is a common alternative carbon source for various bacteria, and its uptake usually relies on the hexose phosphate antiporter UhpT. In the human pathogenic bacterium Staphylococcus aureus, the ability to utilize different nutrients, particularly alternative carbon source uptake in glucose-limiting conditions, is essential for its fitness in the host environment during the infectious process. It has been reported that G6P uptake in S. aureus is regulated by the three-component system HptRSA. When G6P is provided as the only carbon source, HptRSA could sense extracellular G6P and activate uhpT expression to facilitate G6P utilization. However, the regulatory mechanism of HptRSA is still unclear. In this study, we further investigated the HptRSA system in S. aureus. First, we confirmed that HptRSA is necessary for the normal growth of this pathogen in chemically defined medium with G6P supplementation, and we discovered that HptRSA could exclusively sense extracellular G6P compared to the other organophosphates we tested. Next, using isothermal titration calorimetry, we found that HptA could bind to G6P, suggesting that it may be the G6P sensor. After that experiment, using an electrophoresis mobility shift assay, we verified that the response regulator HptR could directly bind to the uhpT promoter and identified a putative binding site from −67 to −96-bp. Subsequently, we created different point mutations in the putative binding site and revealed that the entire 30-bp sequence is essential for HptR regulation. In summary, we unveiled the regulatory mechanism of the HptRSA system in S. aureus, HptA most likely functions as the G6P sensor, and HptR could implement its regulatory function by directly binding to a conserved, approximately 30-bp sequence in the uhpT promoter.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Carbon sources are required during the lifecycle of microorganisms and can be utilized to generate energy and serve as fundamental substrate for metabolism. Glucose is the primary and the most preferred carbon source [1]. However, in some special cases, this primary carbon source is seriously limited. For example, when pathogens intrude into the cells of the host tissue, the milieu usually lacks glucose and is not suitable for optimal survival. Therefore, in addition to the glucose transporter, bacteria have developed multiple transporters for the uptake of alternative carbon sources [2, 3]. To avoid wasting energy, the uptake of alternative carbon source is often strictly controlled by a variety of regulators [4–6]. It has recently been reported that in the major human pathogen Staphylococcus aureus, the uptake of the alternative carbon source glucose-6-phosphate (G6P) can be facilitated by a two-component system (TCS) HptRS [7].
The TCS is a type of signal transduction system which has been extensively found in nearly all prokaryotes and a few eukaryotes, and it mainly functions as basic stimulus–response mechanism that assists organisms in their response to various stimuli from the surrounding milieu [8]. S. aureus has been demonstrated to possess 16 known TCSs on the bacterial chromosome [9], and their functions are important in many pathways. These TCSs include AgrAC, which is responsible for autoinduced peptide (AIP) quorum sensation and phenol-soluble modulin expression [10, 11], LytRS, which is responsible for autolysis [12], and VraSR, which is involved in antimicrobial resistance [13]. HptRS is the first identified TCS which is responsible for carbon source metabolism in S. aureus, which expands the function of the TCS in this pandemic pathogen. HptS is a membrane-bound histidine kinase, and HptR is a cytoplasmic response regulator. The modulation of G6P uptake by HptRS is dependent on a hexose phosphate antiporter UhpT, which functions through exchanging internal inorganic phosphate for external G6P [1]. A previous study showed that UhpT expression was modulated by the TCS HptRS and an additional regulatory protein HptA. S. aureus lacking HptRSA exhibited deficient growth when G6P was the only available carbon source [7]. However, the molecular mechanism of this signal transduction system remains elusive.
In this study, we first compared the homology between the Hpt operon in S. aureus and the operon of the well-studied hexose phosphate uptake regulator Uhp in Escherichia coli, and found that there is low sequence similarity between the proteins. Next, we discovered that the Hpt system exclusively senses G6P compared to a series of other organophosphates. In addition, we found that the accessory protein HptA, an extracellular protein which showed low homology to its counterpart UhpC in E. coli, could directly bind to exogenous G6P and was predicted to play an important role in G6P perception and signal initiation. Therefore, besides HptRS, HptA is also required for the Hpt system, and we preferred to denote Hpt as a three-component system. We also proved that HptR could modulate uhpT expression by directly binding to its promoter region and further identified a conserved binding site which differs from that in E. coli. In summary, our study provides some insight details on the molecular mechanism of the three-component regulatory system HptRSA and complemented the results from previous research.
Materials and methods
Bacterial strains, plasmids, and growth conditions
The bacterial strains and plasmids used in this study are listed in Table 1. E. coli DH5α and BL21 cells were cultured in Luria-Bertani broth (LB) medium (BD Biosciences, Franklin Lakes, NJ, USA) containing the appropriate antibiotics (100 μg/ml ampicillin sodium salt or 50 μg/ml kanamycin sulfate). S. aureus and its derivative strains were cultured in tryptic soy broth (TSB) medium (BD Biosciences, Franklin Lakes, NJ, USA) containing erythromycin (2.5 μg/ml) or chloramphenicol (15 μg/ml) as necessary.
Construction of the hptRSA mutant and the complementary strain
The hptRS mutant strain was constructed using the pBT2 temperature-sensitive shuttle vector, as previously described [14]. The ermB resistance gene was used to replace the hptRS gene. To create the hptRSA mutant strain, we used a pBTs plasmid, which is a derivative of the pBT2 plasmid [15, 16], thus producing a marker-less mutation. The strains with allelic replacements of hptRS with ermB as well as those with the hptRSA deletion were verified by polymerase chain reaction (PCR) and sequencing.
For complementation of the hptRSA mutation, a 4047-bp fragment of the hptRSA gene containing the promoter region was amplified and cloned into pLI50 to generate pLIhptRSA, which was then introduced into the ΔhptRSA mutant to generate the ChptRSA strain. For complementation of hptRS, a 522-bp fragment of the hptA ORF was deleted from pLIhptRSA to generate pLIhptRS, which was subsequently transferred into the ΔhptRS and ΔhptRSA mutants to generate ChptRS and ChptRSt, respectively. All of the primers used in this study are listed in Table 2.
Growth of S. aureus in chemically defined medium
To analyze the utilization of G6P as the primary carbon source, PN medium was used as the chemically defined medium, as previously described [17], with modifications. The original formulation is described in Table S1. For the medium containing G6P as the carbon source, the glucose in the PN medium was replaced with G6P (PN-G6P). For the growth analysis, the cultures that had been incubated overnight in TSB medium were collected, washed twice with PBS, and then diluted 1:100 in 100 ml of PN or PN-G6P medium. The cultures were incubated at 37 °C with constant shaking (220 rpm), and the growth of the cultures was monitored each hour by measuring the OD600 using an ELx800™ Microplate Reader (Bio-tek, Winooski, VT, USA). Each data point represents the mean and standard derivation from three independent experiments. For the G6P-induced expression analysis, each strain was initially inoculated in PN medium, incubated for 6 h at 37 °C, and then treated with 19 mM G6P. The cells were collected just before G6P was added or at various time points after the addition of G6P (1, 5, and 30 min). The collected cells were used for RNA isolation and real-time RT-PCR analysis.
Total RNA isolation and real-time RT-PCR
The S. aureus cells were collected at the indicated time points to isolate the total RNA. Then, the cells were treated with 1 ml of RNAiso Plus (TaKaRa, Kyoto, Japan) and 0.1-mm diameter silica beads in a FastPrep-24 Automated system (MP Biomedicals, Solon, OH, USA), and the residual DNA was removed using RNase-free DNase I (TaKaRa, Kyoto, Japan). For reverse-transcription, the cDNAs were synthesized using the PrimeScript first-strand cDNA synthesis kit (TaKaRa, Kyoto, Japan). Real-time PCR was performed using the SYBR Premix Ex Taq (TaKaRa, Kyoto, Japan) and the StepOne real-time PCR system (Applied Biosystems, Carlsbad, CA, USA). The quantity of the cDNA measured by real-time PCR was normalized to the quantity of the pta cDNA [18]. The results represent the means of three independent experiments.
Construction of the lacZ reporter for uhpT promoter activity
A 146-bp fragment of the uhpT promoter region was amplified and cloned into pOS1-lacZ [19] to generate pOSuhpT. The recombinant plasmid contained an in-frame fusion of lacZ with the uhpT promoter and its first 6 codons. For the 6-bp linker substitution in the promoter, site-directed mutagenesis was performed via PCR, as previously described [20], with some modifications. For example, a DNA fragment containing the entire length of the pOSuhpT plasmid, with the exception of the promoter region of uhpT from −128 to −107, was amplified using pOSuhpT as the template and mp-uhpT-1f and mp-uhpT-1r as the primers. The PCR product was digested with DpnI and SalI and ligated to generate pOSuhpTmp1, in which the uhpT promoter region from −128 to −107 was substituted by a 6-bp SalI recognition site. Then, pOSuhpT was introduced into the hptRS mutant and the RN4220 strain and were designated as PTM and PTR, respectively. The pOSuhpTmp1–pOSuhpTmp5 plasmids were introduced into the RN4220 strain to generate PTM1R–PTM5R, respectively (Table 1). For the point mutation in the pOSuhpT plasmid, the entire plasmid was first amplified using the primers listed in Table 2. The PCR products were treated with DpnI for 3 h at 37 °C to degrade the plasmid template, and then the recycled fragments were treated with polynucleotide kinase (New England Biolabs, MA, USA) for 30 min at 37 °C to initiate self-ligation and form puhpTr1, puhpTr2 and puhpTr3. Subsequently, the plasmids were transferred into the S. aureus RN4220 strain to generate PTR1R, PTR2R, and PTR3R, respectively (Table 1).
β-Galactosidase assay
S. aureus cells containing pOSuhpT or its derivative plasmid were incubated in PN medium for 6 h. The different inducers were added as indicated, and the cells were incubated for an additional hour. The cells were collected from 100 μl cultures by centrifugation and washed twice with PBS. Then, the cells were resuspended in 100 μl of ABT buffer (60 mM K2HPO4, 40 mM KH2PO4, 100 mM NaCl, and 0.1 % Triton X-100) [21] containing 30 μg/ml lysostaphin and incubated for 15 min at 37 °C. The supernatant was treated with 100 μl each of the ABT buffer and substrate (4 mg/ml o-nitrophenyl-β-d-galactoside (ONPG), 60 mM K2HPO4, 40 mM KH2PO4, and 100 mM NaCl) and incubated for 10 min at 37 °C. The reaction was stopped by adding 1 ml of 1 M Na2CO3. The colorimetric changes were quantified at 420 nm using a spectrophotometer (DU730, Beckman coulter, Brea, CA, USA). The β-galactosidase activity was expressed as Miller units [22]. The results represent the means of three independent experiments.
Purification of HptR and HptA in vitro
The 6-His-tagged HptR and HptA proteins were cloned and purified using standard procedures. Briefly, the full-length hptR and hptA fragments were cloned into the pET28a (+) expression vector (Novagen, Merck, Darmstadt, Germany) and transformed into E. coli BL21 (DE3) cells. The transformants were incubated in LB medium until the cells reached an OD600 of 0.5, followed by induction with 0.05 mM isopropyl-β-d-1-thiogalactopyranoside (IPTG) overnight at 16 °C. The cells were harvested and lysed via sonication in lysis buffer (20 mM Tris–HCl, pH 8.0, and 200 mM NaCl). The proteins were purified using a nickel-nitrilotriacetic acid agarose solution (QIAGEN, Valencia, CA, USA) according to the manufacturer’s recommendations. The bound proteins were eluted using elution buffer (200 mM imidazole, 20 mM Tris–HCl, pH 8.0, and 200 mM NaCl). The imidazole in the eluents was removed using a Centrifuge Biomax-5 column (Millipore, Billerica, MA, USA). The HptR solution was supplemented with 30 % glycerol and stored at −80 °C until further use. For HptA, which was used for the isothermal titration calorimetry measurements, the collected eluent was further applied to size exclusion chromatography using a Superdex 200 column (GE Healthcare, Piscataway, NJ, USA) and cation exchange chromatography using an SP Sepharose Fast Flow column. The purity of the proteins was analyzed via SDS-PAGE, and the protein concentrations were determined using the Bradford assay and bovine serum albumin as the standard.
For the in vitro HptR phosphorylation, we used lithium potassium acetyl phosphate as the phosphoryl group donor [23]. Briefly, 10 μM HptR was equilibrated in buffer containing 50 mM Tris–HCl at pH 8.0, 50 mM KCl, 5 mM MgCl2, and 10 % glycerol (phosphorylation buffer). Lithium potassium acetyl phosphate (Sigma-Aldrich, St. Louis, MO, USA) was added to a final concentration of 50 mM, and this mixture was incubated for 60 min at 37 °C.
Isothermal titration calorimetry
The isothermal titration calorimetry (ITC) measurements were performed using an ITC-200 titration calorimeter (MicroCal, Northampton, MA, USA). The measurements were performed in lysis buffer at 25 °C. Approximately 1 mM G6P was titrated into the HptA protein (50 μM). The control experiments were performed under identical conditions by titrating the lysis buffer into the HptA protein. The ITC data were subsequently analyzed using the MicroCal Origin 7.0 software.
Electrophoretic mobility shift assay (EMSA)
The DNA fragments containing the uhpT promoter region were amplified from the S. aureus NCTC8325 genomic DNA. The PCR products were labeled using the digoxigenin (DIG) gel shift kit (Roche, Indianapolis, IN, USA) according to the manufacturer’s instructions. The labeled fragment was incubated with various amounts of phosphorylated HptR in 10 μl of incubation buffer (10 mM Tris–HCl, pH 8.0, 100 mM NaCl, and 1 mM EDTA) for 15 min at 25 °C. After incubation, the mixtures were electrophoresed using a 3.5 % native polyacrylamide gel in 0.5 × Tris–borate–EDTA (TBE) buffer. The band shifts were detected and analyzed according to the manufacturer’s instructions. The images were captured using an ImageQuant LAS 4000 mini imaging system (GE Healthcare, Piscataway, NJ, USA). The unlabeled uhpT promoter was added at an approximately 50-fold concentration of the labeled promoter as a specific competitor. The unlabeled fragments of the pta ORF region (50-fold amount) were added as non-specific competitors.
Statistical analysis
All experimental results represent the means of three independent experiments. We utilized two-tailed unpaired t tests and analysis of variance to determine the statistical significance of the data. Differences with a P value of <0.05 (one asterisk) were considered statistically significant, P < 0.01 (two asterisks).
Results
Comparison of the S. aureus Hpt operon with the E. coli Uhp operon
In S. aureus, HptRSA is an atypical TCS. In addition to the gene hptS encoding the histidine kinase and the gene hptR encoding the response regulator, a third gene, hptA, is also present in the operon, and all three genes function together to accomplish the signal transduction process. Therefore, it should be called a three-component system. The gene located next to hptRSA encodes a transporter UhpT, which is highly conserved in numerous bacterial species. UhpT is a type of hexose phosphate antiporter and belongs to the major facilitator superfamily, which is the largest and most diverse superfamily of secondary active transporters [1]. In E. coli, it has been well documented that the expression of uhpT is strictly modulated by the hexose phosphate utilization regulatory system UhpABC [24]. The UhpT transporter in the S. aureus NCTC8325 strain shares relatively high homology with the G6P antiporter UhpT in the E. coli MG1655 strain (48.3 % identity and 63.8 % similarity). Here, we compared the Uhp operon and the Hpt operon, and found that uhpT and uhpABC in the MG1655 strain are in the same direction, while in the NCTC8325 strain, uhpT and hptRSA are in the opposite direction. In addition, the Uhp operon begins with the regulatory protein UhpA, while the Hpt operon begins with the additional protein HptA. Moreover, sequence analysis revealed that HptRSA exhibits low similarity to UhpABC (Fig. 1).

Upper panel—E. coli MG1655 Uhp locus; lower panel—S. aureus NCTC8325 Hpt locus
HptRSA system has high substrate sensing specificity
We first investigated the growth status of S. aureus in chemically defined PN medium and confirmed a previous result which showed that the HptRSA mutant of S. aureus displayed deficient growth when G6P was supplied as the only carbon source (Fig. S1). Next, we used a lacZ reporter system to monitor uhpT translation. We found that when G6P was added, the UhpT level in the wild-type strain increased significantly, while the hptRS mutant strain showed no obvious change upon G6P addition (Fig. 2a). In addition, we examined the uhpT transcriptional levels after G6P addition in the wild-type strain, the hptRS mutant strain and the complementary strain, and similar results were obtained (Fig. 2b). Although the uhpT level changed significantly, the hptS level changed very little after G6P addition (Fig. 2c, d), indicating that the hpt operon was not autoinduced.

Expression levels of uhpT and hptS upon G6P addition. a β-Galactosidase activity of the uhpT promoter in the PTR and PTM strains with or without induction with 19 mM G6P. PTR: RN4220 strain with pOSuhpT; PTM: ΔhptRS strain with pOSuhpT. b Expression level of the uhpT transcript in the wild-type strain, the hptRS mutant, and the complementary strain at different time points after the addition of 19 mM G6P. c Expression level of the hptS transcript in the wild-type strain at different time points after the addition of 19 mM G6P. d Expression level of the hptS transcript in the ChptRS strain at different time points after the addition of 19 mM G6P. The results represented the means of three independent experiments. *P < 0.05, **P < 0.01, according to a two-tailed unpaired t test
It has been reported that in addition to G6P, the HptRSA system could sense glycerol-3-phosphate as well [7]. Therefore, to explore whether other organophosphates could also activate the HptRSA system, we tested glycerol-2-phosphate, creatine phosphate, 6-aminopurine phosphate, glucose-1-phosphate, fructose-1,6-diphosphate, and pyridoxal-5-phosphate as potential inducers, but found that none of them were able to activate uhpT expression (Fig. 3). This suggested that the Hpt system senses relatively specific substrates.

β-Galactosidase activity of the uhpT promoter in the wild-type strain following stimulation with various inducers (G6P, glucose-1-phosphate, fructose-1,6-diphosphate, glycerol-2-phosphate, creatine phosphate, 6-aminopurine phosphate, and pyridoxal-5-phosphate; all at 19 mM). The miller units are shown as mean ± SD from three independent experiments. **P < 0.01, according to a two-tailed unpaired t test
HptA can directly bind to G6P in vitro
In the measurement of the bacterial growth, we found that when we expressed the hptRS genes in the hptRSA mutant strain, the ability of bacterial cells to utilize G6P was still not completely restored (Fig. S1F), and a previous study also showed that hptA mutant strain has defect in utilizing extracellular G6P [7]; therefore, HptA likely plays an important role in sensing G6P and activating the entire signal transduction pathway. To investigate the G6P sensing ability of HptA, we purified the full-length HptA protein with a C-terminal His tag (Fig. 4a). Next, we conducted an ITC assay to explore the interaction between HptA and the G6P molecule. We used 50 μM HptA, and G6P was dissolved in the same protein lysis buffer to a final concentration of 1 mM. The K d of G6P for HptA was determined to be 7.58 μM, and the change in the binding free energy change ΔG was calculated to be −6.99 kcal/mol (Fig. 4b), indicating that HptA could bind to the G6P molecule. Therefore, based on these results, we concluded that HptA is responsible for sensing extracellular G6P.

Ability of HptA to bind G6P. a SDS-PAGE electrophoresis of purified HptA (36.9 KD) b The ITC titration fitting curve of HptA with G6P. The HptA concentration was 50 μM, and the G6P concentration was 1 mM
HptR regulates uhpT by directly binding to its promoter
It has been well recognized that many TCSs regulate their targets by directly binding to the promoter regions of their target genes [25–27]. As the lack of hptRS could severely inhibit uhpT expression, we were interested in whether the regulatory protein HptR could directly bind to the target gene uhpT. We used purified HptR containing a His tag and a 143-bp DNA probe containing the uhpT promoter region to perform an EMSA. The results clearly indicated that the DNA bands were shifted after phosphorylated HptR was incubated with the uhpT probe (Fig. 5). Moreover, the intensity of the shifted bands increased as the amount of HptR increased. After an approximately 50-fold excess concentration of the unlabeled uhpT probe was added, the shifted band disappeared. Alternatively, the addition of a non-specific probe did not change the intensity of the shifted band, indicating that HptR specifically binds to the uhpT promoter region.

EMSA of the uhpT probe using phosphorylated HptR. The first lane corresponds to the free uhpT probe (2 nM), the second through fourth lanes correspond to the uhpT probe in the presence of increasing amounts of UhpA (0.5, 1, and 2 μM), the fifth lane contains the same components as the fourth lane and a 50-fold excess of the unlabeled probe as a specific competitor, and the sixth lane contains the same components as the fourth lane and a 50-fold excess of the unlabeled pta ORF region fragments as non-specific competitors
Part of the uhpT promoter sequence (−67 to −96) is required for HptR binding
To identify the HptR-binding region on the uhpT promoter, we used a 6-bp linker substitution to delete the uhpT promoter region in the lacZ reporter system. A 103-bp fragment (from −25 to −128) was divided into five regions of approximately 20-bp each, which were individually replaced by the 6-bp linker sequence (“GTCGAC,” SalI linker) to generate different reporter plasmids. After the addition of G6P, only the deletion of the region from −106 to −128 did not significantly affect the expression of β-galactosidase (Fig. 6a, b), and a large HptR-binding region still remained after the region involved in RNA polymerase binding (−10 region, −35 region) was excluded. To explore the HptR-binding sequence, we performed phylogenetic footprinting using promoter sequences from orthologous uhpT in staphylococci (Staphylococcus epidermidis, Staphylococcus massilienses, Staphylococcus pseudintermedius, and Staphylococcus simiae) and identified several conserved motifs. After excluding the RNA polymerase-binding region, a 30-bp HptR-binding sequence (GTTCAGTATTTTGGATAATTTAATAATTTT) was identified, and this sequence is located from −67 to −96 of uhpT (Fig. 6c).

Determination of the HptR-binding sites on the uhpT promoter. a Schematic representation of the sequences of the uhpT promoter-lacZ reporter plasmids. The blank boxes indicate the regions of the sequence that were replaced with the 6-bp linker, the numbers denote the distance from the start codon “ATG,” and the boxes on the right side indicate the first 6 amino acids of UhpT that were fused to the lacZ sequence. b β-Galactosidase activity of the uhpT promoter in the S. aureus RN4220 strain using the reporter plasmids shown in a. c The orthologous uhpT gene sequences were analyzed using a CLUSTAL Multiple Sequence alignment. The putative elements were underlined; the arrow shows the UhpT start codon, and the numbers on the top match those of the substituted region shown in a. PTR: RN4220 strain with pOSuhpT; PTM1R: RN4220 strain with puhpTmp1; PTM2R: RN4220 strain with puhpTmp2; PTM3R: RN4220 strain with puhpTmp3; PTM4R: RN4220 strain with puhpTmp4; PTM5R: RN4220 strain with puhpTmp5. The miller units are shown as mean ± SD from three independent experiments. **P < 0.01, according to a two-tailed unpaired t test
To further examine the nucleotides that are essential for HptR binding, we created a series of uhpT promoter variants carrying different point mutations in the 30-bp conserved binding sequence. We noticed that the sequence can be divided into two parts: one part “GTTCAGTATTTTGGA” with no obvious characteristics, and another part “TAATTTAATAATTTT” that contains the direct repeat “TAATTTNNTAATTT,” which frequently serves as protein-binding site. To investigate whether the two parts are both necessary for HptR recognition, the two parts were each mutated by random substitutions of different nucleotides, and then the mutated sequences were cloned into pOS1 plasmid (Fig. 7a). Regardless of the sequence that was mutated, the results showed that the β-galactosidase activity was significantly affected, suggesting that the two parts are both required for HptR binding (Fig. 7b). In addition, to determine whether the direct repeat structure is required for binding and not the nucleotide sequence, we substituted the “TAATTTNNTAATTT” repeat with another direct repeat “CTTCCCNNCTTCCC”; however, the β-galactosidase activity was still largely impaired (Fig. 7a, b). In summary, the nucleotide sequence itself and not the structure is required for HptR to recognize the uhpT promoter, and all 30 nucleotides are essential for binding.

Conserved 30-bp nucleotide sequence is essential for UhpA binding to uhpT. a, The 30-bp conserved sequence from the uhpT promoter in the pOSuhpT plasmid was mutated as follows: random nucleotide substitutions in the initial 15-bp to generate puhpTr1, random nucleotide substitutions in the last 15-bp to generate puhpTr2, and other direct repeat nucleotide substitutes in the last 15-bp to generate puhpTr3. The underlined sequence shows the conserved HptR-binding region. The boxed sequences indicate the substituted nucleotides. b β-Galactosidase activity of the uhpT promoter in S. aureus expressing the original reporter plasmid and those with different nucleotide substitutions. PTR: RN4220 strain with pOSuhpT; PTR1R: RN4220 strain with puhpTr1; PTR2R: RN4220 strain with puhpTr2; PTR3R: RN4220 strain with puhpTr3. The miller units are shown as mean ± SD from three independent experiments. **P < 0.01, according to a two-tailed unpaired t test
Discussion
G6P is a major alternative carbon source for the growth of many bacteria, such as E. coli and Salmonella typhimurium. Thus, UhpT, which is responsible for the utilization of extracellular G6P, is widely conserved among bacteria [28]. UhpT is shown to be regulated by the UhpABC operon, and this hexose phosphate utilization system is found in a wide range of gram-negative bacteria such as Escherichia coli [29, 30] and Vibrio cholerae [31]. Nevertheless, studies of the G6P utilization regulatory system in gram-positive bacteria are still rare and are not systematic. In a recent study, a signal transduction system called HPT (hexose phosphate transport) was shown to regulate G6P uptake by activating uhpT in the prevalent gram-positive pathogen S. aureus [7]. This Hpt operon does not share high homology with the Uhp operon and encodes three proteins: HptS, HptR and HptA. HptRS is classified as a standard two-component system and functions in signal transduction and genetic regulation; the additional protein HptA is also essential for the system. Therefore, this system should be called a three-component signal system, HptRSA. In addition, bioinformatics analysis on another gram-positive bacterium, Clostridium botulinum, which harbors the uhpT gene and an adjacent putative three-component system, shows that the three-component system with unknown function shares structure homology with HptRSA in S. aureus. The findings in S. aureus may provide a new model for a G6P uptake regulatory system in many gram-positive bacteria.
In E. coli, UhpC is recognized as an auxiliary protein that assists UhpB in sensing G6P [32]. Upon recognition of extracellular G6P, UhpC induces autophosphorylation of UhpB [33]. UhpC is a structural analog that shares approximately 32 % identity with UhpT and possesses the ability to sense and transport G6P [34, 35]. Compared to UhpC, the S. aureus homologous protein HptA has a quite different structure. The sequence structure analysis indicates that UhpC contains 12 transmembrane domains, while HptA does not contain any transmembrane domains. The difference between these proteins may be due to the differences in their cell wall composition, because E. coli is a gram-negative bacterium that has a thin cell wall and large periplasmic space, while S. aureus is a gram-positive bacterium that has a thicker and more complex cell wall, and thus, evolutionary divergence may have occurred. Using a prediction from an online tool, an N-terminal signal peptide was identified in HptA, suggesting that it may be a membrane-anchored protein or secreted protein. Therefore, we inferred that HptA may not have the ability to transport G6P, unlike UhpC in E. coli. However, we confirmed that HptA can directly bind to G6P, indicating that it retains the ability to sense G6P. Moreover, we found that the hptA mutant strain grew better than the hptRSA mutant strain in chemically defined medium where G6P was provided as the only carbon source, and this implies that the membrane-bound HptS protein likely has the ability to sense G6P in the absence of HptA. We speculated that in the absence of HptA, the HptRS system may be less effective at G6P uptake, and the interaction between HptA and HptS would increase the ability of the Hpt system to sense G6P. However, these hypotheses require further study.
It has been reported that E. coli UhpA binds to two sites in the uhpT promoter, a strong-binding site and a weak-binding site, which comprise an approximately 50-bp region [36]. In S. aureus, we found that HptR, the homologous protein of UhpA, binds to an approximately 30-bp region of the DNA in the uhpT promoter, suggesting that HptR possesses a binding mechanism which is different from that of UhpA. In addition, it has been reported that the HptRSA system also controls the resistance to phosphomycin in S. aureus [7]. Therefore, it is not clear whether the HptRSA system is able to modulate other genes, which requires additional research.
As the widespread pathogenic bacterium, S. aureus frequently accumulates in host cells where could protect it from the host defense mechanism and antibiotics [37]. Since G6P serves as a key component in energy and carbon metabolism and constitutes one of the major intermediate products of the glycolysis pathway, the molecule is always present in the cellular cytosol [34]. In the cytoplasmic G6P-dominant environment, Hpt system could enhance G6P uptake to help S. aureus maintain the carbon source required for cellular basal metabolism. It is reported that S. aureus with an hptRSA mutation showed decreased survival and replication rates in macrophages cell such as THP-1 cells [7], indicating that the Hpt system plays an important role during the infection process.
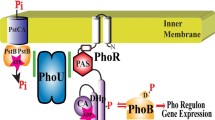
Our results indicate that although the HptRSA system in S. aureus functions in a similar manner to the UhpABC system in E. coli, the signal sensing and regulatory mechanism differs from each other. It seems that the putative secreted protein HptA in S. aureus only possesses the G6P sensing ability, and the interaction mechanism between HptA and HptS needs to be further investigated. The activated regulatory protein HptR could directly bind to a conserved, approximately 30-bp sequence in the uhpT promoter, and further enhance its expression (Fig. 8). In conclusion, this study has revealed a distinct regulatory mechanism on hexose phosphate utilization in S. aureus, which differs from that in E. coli.

Schematic diagram of the Hpt regulatory system-mediated G6P uptake process in S. aureus. a In the absence of glucose, G6P is the sole carbon source in the milieu, and the extracellular G6P is perceived by the sensor protein HptA. Then, an unknown mechanism transfers the signal to the histidine kinase HptS, and HptS itself may also have a reduced ability to sense G6P. Upon stimulation, HptS becomes autophosphorylated. b Activated HptS phosphorylates the response regulator HptR in a single phosphotransfer step. c Phosphorylated HptR binds to a conserved sequence in the uhpT promoter and subsequently facilitates G6P transport. d Following stimulation with exogenous G6P, more UhpT protein is produced, resulting in increased G6P uptake
References
Law CJ, Maloney PC, Wang DN (2008) Ins and outs of major facilitator superfamily antiporters. Annu Rev Microbiol 62:289–305. doi:10.1146/annurev.micro.61.080706.093329
Chen J (2013) Molecular mechanism of the Escherichia coli maltose transporter. Curr Opin Struct Biol 23(4):492–498. doi:10.1016/j.sbi.2013.03.011
Buckwalter CM, King SJ (2012) Pneumococcal carbohydrate transport: food for thought. Trends Microbiol 20(11):517–522. doi:10.1016/j.tim.2012.08.008
Schumacher MA, Allen GS, Diel M, Seidel G, Hillen W, Brennan RG (2004) Structural basis for allosteric control of the transcription regulator CcpA by the phosphoprotein HPr-Ser46-P. Cell 118(6):731–741. doi:10.1016/j.cell.2004.08.027
Nam TW, Park YH, Jeong HJ, Ryu S, Seok YJ (2005) Glucose repression of the Escherichia coli sdhCDAB operon, revisited: regulation by the CRP*cAMP complex. Nucleic Acids Res 33(21):6712–6722. doi:10.1093/nar/gki978
Vastermark A, Saier MH Jr (2014) The involvement of transport proteins in transcriptional and metabolic regulation. Curr Opin Microbiol 18:8–15. doi:10.1016/j.mib.2014.01.002
Park JY, Kim JW, Moon BY, Lee J, Fortin YJ, Austin FW, Yang SJ, Seo KS (2015) Characterization of a novel two-component regulatory system, HptRS, the regulator for the hexose phosphate transport system in Staphylococcus aureus. Infect Immun 83(4):1620–1628. doi:10.1128/IAI.03109-14
Bourret RB, Silversmith RE (2010) Two-component signal transduction. Curr Opin Microbiol 13(2):113–115. doi:10.1016/j.mib.2010.02.003
Kuroda M, Ohta T, Uchiyama I, Baba T, Yuzawa H, Kobayashi I, Cui LZ, Oguchi A, Aoki K, Nagai Y, Lian JQ, Ito T, Kanamori M, Matsumaru H, Maruyama A, Murakami H, Hosoyama A, Mizutani-Ui Y, Takahashi NK, Sawano T, Inoue R, Kaito C, Sekimizu K, Hirakawa H, Kuhara S, Goto S, Yabuzaki J, Kanehisa M, Yamashita A, Oshima K, Furuya K, Yoshino C, Shiba T, Hattori M, Ogasawara N, Hayashi H, Hiramatsu K (2001) Whole genome sequencing of meticillin-resistant Staphylococcus aureus. Lancet 357(9264):1225–1240. doi:10.1016/S0140-6736(00)04403-2
Novick RP, Geisinger E (2008) Quorum sensing in staphylococci. Annu Rev Genet 42:541–564. doi:10.1146/annurev.genet.42.110807.091640
Queck SY, Jameson-Lee M, Villaruz AE, Bach TH, Khan BA, Sturdevant DE, Ricklefs SM, Li M, Otto M (2008) RNAIII-independent target gene control by the agr quorum-sensing system: insight into the evolution of virulence regulation in Staphylococcus aureus. Mol Cell 32(1):150–158. doi:10.1016/j.molcel.2008.08.005
Brunskill EW, Bayles KW (1996) Identification and molecular characterization of a putative regulatory locus that affects autolysis in Staphylococcus aureus. J Bacteriol 178(3):611–618
Yin S, Daum RS, Boyle-Vavra S (2006) VraSR two-component regulatory system and its role in induction of pbp2 and vraSR expression by cell wall antimicrobials in Staphylococcus aureus. Antimicrob Agents Chemother 50(1):336–343. doi:10.1128/AAC.50.1.336-343.2006
Sun H, Yang Y, Xue T, Sun B (2013) Modulation of cell wall synthesis and susceptibility to vancomycin by the two-component system AirSR in Staphylococcus aureus NCTC8325. BMC Microbiol 13:286. doi:10.1186/1471-2180-13-286
Hu J, Zhang X, Liu X, Chen C, Sun B (2015) Mechanism of reduced vancomycin susceptibility conferred by walK mutation in community-acquired methicillin-resistant Staphylococcus aureus strain MW2. Antimicrob Agents Chemother 59(2):1352–1355. doi:10.1128/AAC.04290-14
Bae T, Schneewind O (2006) Allelic replacement in Staphylococcus aureus with inducible counter-selection. Plasmid 55(1):58–63. doi:10.1016/j.plasmid.2005.05.005
Pattee PA, Neveln DS (1975) Transformation analysis of three linkage groups in Staphylococcus aureus. J Bacteriol 124(1):201–211
Valihrach L, Demnerova K (2012) Impact of normalization method on experimental outcome using RT-qPCR in Staphylococcus aureus. J Microbiol Methods 90(3):214–216. doi:10.1016/j.mimet.2012.05.008
Liu Y, Mu C, Ying X, Li W, Wu N, Dong J, Gao Y, Shao N, Fan M, Yang G (2011) RNAIII activates map expression by forming an RNA–RNA complex in Staphylococcus aureus. FEBS Lett 585(6):899–905. doi:10.1016/j.febslet.2011.02.021
Xue T, Zhao L, Sun H, Zhou X, Sun B (2009) LsrR-binding site recognition and regulatory characteristics in Escherichia coli AI-2 quorum sensing. Cell Res 19(11):1258–1268. doi:10.1038/cr.2009.91
Horsburgh MJ, Clements MO, Crossley H, Ingham E, Foster SJ (2001) PerR controls oxidative stress resistance and iron storage proteins and is required for virulence in Staphylococcus aureus. Infect Immun 69(6):3744–3754. doi:10.1128/IAI.69.6.3744-3754.2001
Zhang X, Bremer H (1995) Control of the Escherichia coli rrnB P1 promoter strength by ppGpp. J Biol Chem 270(19):11181–11189. doi:10.1074/jbc.270.19.11181
Belcheva A, Verma V, Korenevsky A, Fridman M, Kumar K, Golemi-Kotra D (2012) Roles of DNA sequence and sigma A factor in transcription of the vraSR operon. J Bacteriol 194(1):61–71. doi:10.1128/JB.06143-11
Weston LA, Kadner RJ (1988) Role of uhp genes in expression of the Escherichia coli sugar-phosphate transport system. J Bacteriol 170(8):3375–3383
Koenig RL, Ray JL, Maleki SJ, Smeltzer MS, Hurlburt BK (2004) Staphylococcus aureus AgrA binding to the RNAIII-agr regulatory region. J Bacteriol 186(22):7549–7555. doi:10.1128/JB.186.22.7549-7555.2004
Stauff DL, Torres VJ, Skaar EP (2007) Signaling and DNA-binding activities of the Staphylococcus aureus HssR-HssS two-component system required for heme sensing. J Biol Chem 282(36):26111–26121. doi:10.1074/jbc.M703797200
Nygaard TK, Pallister KB, Ruzevich P, Griffith S, Vuong C, Voyich JM (2010) SaeR binds a consensus sequence within virulence gene promoters to advance USA300 pathogenesis. J Infect Dis 201(2):241–254. doi:10.1086/649570
Island MD, Wei BY, Kadner RJ (1992) Structure and function of the uhp genes for the sugar phosphate transport system in Escherichia coli and Salmonella typhimurium. J Bacteriol 174(9):2754–2762
Kornberg HL, Smith J (1969) Genetic control of hexose phosphate uptake by Escherichia coli. Nature 224(5226):1261–1262. doi:10.1038/2241261a0
Webber CA, Kadner RJ (1995) Action of receiver and activator modules of UhpA in transcriptional control of the Escherichia coli sugar phosphate transport system. Mol Microbiol 15(5):883–893. doi:10.1111/j.1365-2958.1995.tb02358.x
Moisi M, Lichtenegger S, Tutz S, Seper A, Schild S, Reidl J (2013) Characterizing the hexose-6-phosphate transport system of Vibrio cholerae, a utilization system for carbon and phosphate sources. J Bacteriol 195(8):1800–1808. doi:10.1128/JB.01952-12
Island MD, Kadner RJ (1993) Interplay between the membrane-associated UhpB and UhpC regulatory proteins. J Bacteriol 175(16):5028–5034
Schwoppe C, Winkler HH, Neuhaus HE (2002) Properties of the glucose-6-phosphate transporter from Chlamydia pneumoniae (HPTcp) and the glucose-6-phosphate sensor from Escherichia coli (UhpC). J Bacteriol 184(8):2108–2115. doi:10.1128/Jb.184.8.2108-2115.2002
Schwoppe C, Winkler HH, Neuhaus HE (2003) Connection of transport and sensing by UhpC, the sensor for external glucose-6-phosphate in Escherichia coli. Eur J Biochem 270(7):1450–1457. doi:10.1046/j.1432-1033.2003.03507.x
Kadner RJ, Island MD, Dahl JL, Webber CA (1994) A transmembrane signaling complex controls transcription of the Uhp sugar-phosphate transport-system. Res Microbiol 145(5–6):381–387. doi:10.1016/0923-2508(94)90085-X
Chen Q, Kadner RJ (2000) Effect of altered spacing between uhpT promoter elements on transcription activation. J Bacteriol 182(16):4430–4436. doi:10.1128/Jb.182.16.4430-4436.2000
Lowy FD (1998) Staphylococcus aureus infections. N Engl J Med 339(8):520–532. doi:10.1056/NEJM199808203390806
Bruckner R (1997) Gene replacement in Staphylococcus carnosus and Staphylococcus xylosus. FEMS Microbiol Lett 151(1):1–8
Lee CY, Buranen SL, Ye ZH (1991) Construction of single-copy integration vectors for Staphylococcus aureus. Gene 103(1):101–105
Acknowledgments
The authors thank the Network on Antimicrobial Resistance in Staphylococcus aureus (NARSA) for providing the bacterial strains. This study was supported by the National Natural Science Foundation of China (Grant 31070116).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no financial or commercial conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Yang, Y., Sun, H., Liu, X. et al. Regulatory mechanism of the three-component system HptRSA in glucose-6-phosphate uptake in Staphylococcus aureus . Med Microbiol Immunol 205, 241–253 (2016). https://doi.org/10.1007/s00430-015-0446-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00430-015-0446-6