
Abstract
Leishmaniasis is one of the most important infectious diseases worldwide; a vaccine is still not available. Infected dendritic cells (DC) are critical for the initiation of protective Th1 immunity against Leishmania major. Phagocytosis of L. major by DC leads to cell activation, IL-12 release and (cross-) presentation of Leishmania antigens by DC. Here, we review the role of Fcγ receptor- and B cell-mediated processes for parasite internalization by DC. In addition, the early events after parasite inoculation that consist of mast cell activation, parasite uptake by skin-resident macrophages (MΦ), followed by neutrophil and monocyte immigration and DC activation are described. All these events contribute significantly to antigen processing in infected DC and influence resulting T cell priming in vivo. A detailed understanding of the role of DC for the development of efficient anti-Leishmania immunity will aid the development of potent anti-parasite drugs and/or vaccines.
Similar content being viewed by others

Introduction
Cutaneous leishmaniasis as model disease
Infections with Leishmania spp. represent a serious health problem in large parts of the world. Leishmaniasis is endemic in 88 countries of southern Europe, Central and South America, Africa, the Middle East and the Indian subcontinent [1, 2]. Currently, 12 million people are suffering from leishmaniasis, and an estimated 1–1.5 million new cases every year and a death toll of around 70,000 annually are observed [3]. Due to these numbers, the development of new drugs or vaccines against leishmaniasis has recently received more attention. In addition to being a disabling and socioeconomically important disease, leishmaniasis has emerged as a model system of parasitic skin infections and studies in leishmaniasis have greatly improved our understanding of skin/parasite interactions.
Leishmaniasis is caused by protozoan parasites of the genus Leishmania. The disease is transmitted to the host via sand flies. Dermatotropic species of Leishmania—the main focus of this review—induce granulomatous skin reactions, while viscerotropic species induce hepatosplenomegaly.
The early events that take place in skin and lymph node (LN) after infection have been studied extensively. After inoculation of infectious stage metacyclic promastigotes into the upper dermis by the bite of a sand fly, Leishmania primarily locate to the phagolysosomes of skin-resident macrophages (MΦ) (Fig. 1) [2]. Here, the parasite transforms to obligate intracellular amastigotes and replicates. Release of free amastigotes into the tissue leads to infection of other phagocytes. Finally, the parasite’s life cycle is complete upon re-infection of a sand fly during another blood meal.

Role of MΦ and DC in cutaneous leishmaniasis. The parasite L. major is inoculated into skin by the bite of a sand fly. The infectious stage metacyclic promastigotes adhere to MΦ via complement receptor (CR) 3 which mediates phagocytosis. Within MΦ, parasites transform into amastigotes and replicate, the infection remains ‘silent’. Promastigote parasites adhere to surfaces of DC without entering the cell. Later, when free amastigotes are released from lysed MΦ, DC internalize this parasite life form via FcγRI or FcγRIII requiring parasite opsonization with IgG produced by B cells. In contrast to MΦ, infection of DC with L. major leads to cell activation, upregulation of MHC class I and II as well as co-stimulatory molecules. Infected DC migrate to draining LN, where they present parasite antigens to both CD4+ and CD8+ T cells. In parallel, cytokine production by DC contributes to T helper cell (Th) differentiation towards protective Th1/Tc1-dependent immunity
Defence mechanisms against Leishmania major
After several weeks post-infection, an influx of inflammatory neutrophils (PMN), monocyte-derived MΦ and (later) also dendritic cells (DC) into skin lesions is observed, after which adaptive immune responses are elicited with priming and activation of antigen-specific T cells (Fig. 1) [4–6]. At this stage, clinically apparent skin lesions are observed. Finally, IFNγ-producing antigen-specific CD4+ Th1 and CD8+ Tc1 cells induce healing of the lesions by activating infected MΦ to eliminate the parasite via nitric oxide (NO).
Prior studies showed that infected DC are the critical APC responsible for T cell priming in Leishmania infections [7–10]. Uptake of L. major by DC results in activation and interleukin (IL)-12 release. Infected DC efficiently stimulate CD4+ and CD8+ T cells and vaccinate against leishmaniasis. In contrast, complement receptor (CR) 3–dependent phagocytosis of L. major by MΦ leads exclusively to MHC class II–restricted antigen presentation to primed, but not naive, T cells, and no IL-12 production [4, 11].
Due to the fact that an effective Leishmania vaccine does not exist and (skin) DC are critical regulators of the anti-Leishmania immune response, DC are attractive targets for immuno-therapeutic approaches. Thus, it is essential to understand the precise role of DC in leishmaniasis.
Phagocytosis of Leishmania parasites by antigen-presenting cells
As described above, both MΦ and DC play important roles in the defence mechanisms against Leishmania infections. Even though both cell types are antigen-presenting cells (APC) and of myeloid lineage, L. major uptake by these cells leads to major differences in the functional consequences. Additionally, we documented strong differences in parasite internalization [12]: (1) Skin DC preferentially take up L. major amastigotes, the obligate intracellular life form of the parasite, rather than promastigotes transmitted by sand flies, whereas MΦ efficiently phagocytose both life forms [7, 13, 14], (2) the phagocytotic capacity of DCs is limited as compared with that of MΦ [7], (3) L. major-infected DC, unlike infected MΦ, release IL-12 and efficiently induce Th1/Tc1 differentiation of naive cells [7, 8, 15, 16], and (4) although both cell types present Leishmania antigen via the MHC class II pathway, only DC prime and restimulate L. major-specific CD8+ T cells [17]. Prior studies showed that MΦ internalize L. major parasites via CR3 [18] and that CR3 signalling in MΦ is responsible for their inability to release IL-12 after infection with the parasites [18].
Parasite recognition by DC
Receptors involved in DC phagocytosis
Because of the differences in cellular behaviour of MΦ and DC after infection, we speculated that differences in the receptors utilized for parasite internalization exist. Confirmingly, we showed that parasite uptake by DC was independent of CR3. To this aim, DC deficient in CD18—not expressing CR3 or CR4—were used for in vitro infections. Here, no difference in the percentage of infected DC and in the number of parasite per cell was observed [12]. However, a previous report suggested that uptake of L. major amastigotes by cell suspensions containing 30 % primary Langerhans cells (LC) was decreased in the presence of anti-CR3 [14]. Interestingly, even though we were unable to demonstrate an effect of CR3/4 involvement in parasite uptake by DC, we consistently observed tight adherence of even serum-opsonized promastigote life forms to the surface of DC after 18 h co-cultures (Fig. 1). Thus, it is possible that the initial steps leading to parasite/DC contact are mediated by complement-associated mechanisms. In line, in other inflammatory processes, co-engagement of CR3 with other receptors such as FcγR has been described [19].
We also assessed the involvement of other candidate phagocytosis receptors, for example, the mannose receptor or CD205 [12]. None of the tested inhibitors affected the uptake of L. major by DC. Thus, similar to CR3 and CR4, C-type lectins appeared to be dispensable for phagocytosis of the parasites by DCs.
As described above, DC preferentially internalize L. major amastigotes as compared to promastigotes. Experimentally, amastigotes need to be prepared from lesional tissue, whereas infectious stage metacyclic promastigotes can be isolated and enriched from parasite cultures. While assessing potential differences between these life forms, we studied the isotypes of parasite-specific antibodies that are bound to parasite surfaces. Using flow cytometry, we determined that complement-opsonized parasite preparations displayed membrane-bound natural IgM (found on both promastigotes and amastigotes) [20]. Thus, IgM was not involved in parasite uptake by DC. However, only amastigotes additionally displayed IgG (IgG1 as well as IgG2a/b) on their surface. This suggested that immunoglobulins are involved in L. major internalization by DC. To confirm this, we next used amastigotes isolated from B cell-deficient JHT or μMT mice. These parasites are devoid of IgG on their surface and were not phagocytosed by DC in vitro, and, in line, in subsequent experiments, promastigotes opsonized with antibodies derived from serum of immunized mice were taken up rapidly [12]. Finally, we determined that parasite uptake was dramatically decreased in vitro and in vivo only if DC from FcγR−/− or FcγRI × FcγRIII−/− were used. In summary, we identified FcγRI and FcγRIII as important mediators for DC infection in leishmaniasis. Interestingly, both receptors were capable of efficiently compensating for each other.
Disease outcome in B cell- or Fcγ receptor-deficient mice
In vivo, DC infiltration of L. major-infected skin lesions coincides with the appearance of lesional B cells and parasite-specific antibodies in sera [12, 17]. Consequently, skin of infected B cell-deficient μMT and JHT mice and FcγR−/− mice contained fewer parasite-infected DC in vivo [12]. Infected B cell-deficient mice as well as FcγR−/− or FcγRI−/− × FcγRIII−/− mice (all on C57BL/6 background) showed similarly increased disease susceptibility as assessed by lesion volumes and parasite burdens. The B cell-deficient mice also displayed impaired T cell priming and dramatically reduced IFNγ production, and all these deficits were normalized by infection with IgG-opsonized parasites. As expected, since FcγRI and FcγRIII compensated for each other, FcγRI−/−, FcγRII−/−, as well as FcγRIII−/− mice all exhibited no phenotype.
Genetical differences matter
All data described above were relevant especially in physiological low dose infections with 1,000 infectious stage metacyclic promastigotes inoculated into dermal ear skin [17]. Interestingly, our findings in C57BL/6 mice were not in line with those of prior reports using BALB/c mice. It is well accepted that genetically determined disease outcome in leishmaniasis is determined on the level of various immune cells [2]: Whereas C57BL/6 mice—similar to infected humans with no immune deficits—develop skin lesions that ultimately heal due to parasite-eradicating IFNγ release from antigen-specific Th1/Tc1 cells, BALB/c mice succumb to infection with L. major because of enhanced Th2/Th17 development [2, 21]. In BALB/c mice, the strong pathological Th2-dominated immune response is associated with excessive immunoglobulin levels of all classes. In addition, a strongly improved disease outcome of BALB/c FcγR−/− and B cell-deficient BALB/c JHT mice was explained by decreased IL-10 release from MΦ [22].
Summary and open questions
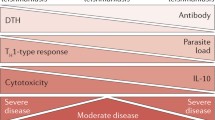
Altogether, these data demonstrated that DC and MΦ use different receptors to recognize and ingest L. major with different outcomes, and indicate that B cell-derived, parasite-reactive IgG and DC FcγRI and FcγRIII are essential for optimal development of protective immunity. It appears that at later stages of infection, the balance between (1) FcγR-mediated induction of IL-12 from DC and (2) FcγR-associated IL-10 release from infected MΦ is ultimately responsible for disease outcome.
This pivotal role for antibodies to parasites in the priming of T cell immunity by DC raises the question of how the initial B cell response to the parasite itself develops [23]. Does it evolve in the absence of infected DC? Alternatively, early on after parasite inoculation, natural IgG may substitute for the antigen-specific IgG that are found at later stages post-infection. Former work has suggested that parasites can enter host cells via a process called ‘apoptotic mimicry’ and it is also known that L. major parasites are covered with phospholipids on their surface [24, 25]. Thus, natural IgG recognizing, for example, antiphospholipid antibodies may play a role in early parasite uptake by DC.
Proper DC activation requires skin mast cells
Role of mast cells for disease outcome in cutaneous leishmaniasis
Mast cells (MC) are key effector cells in type I hypersensitivity reactions or in response to parasites and play an important role in the regulation of protective adaptive immune responses against pathogens. Strategically located in the skin, they are known to contribute to the control of parasitic skin infections by L. major [26, 27]. First, MC degranulation and activation were demonstrated after interaction with L. major both in vitro and in vivo (Fig. 2) [26–29]. Next, L. major-infected MC-deficient Kit W /Kit W-v mice (on a Leishmania-resistant C57BL/6 background) developed markedly larger skin lesions than normal Kit +/+ mice did, and cutaneous reconstitution with MC resulted in normalization of lesion development. Kit W /Kit W-v lesions contained significantly more parasites, and infections resulted in enhanced spreading of parasites to the spleens as compared to controls. Antigen-specific T cell priming was delayed in Kit W /Kit W-v mice, and cytokine responses were skewed towards Th2. Notably, local skin MC reconstitution at sites of infection was sufficient for the induction of systemic protection.

Skin MC regulate the development of protective anti-parasite immunity. Parasite inoculation into skin leads to activation of MC as assessed by degranulation and TNFα release. MC-derived TNFα leads to recruitment of neutrophils (PMN) into the tissue, which in turn release MIP-1α/β that is chemotactic for immigrating inflammatory MΦ. In parallel, improper activation of DC was observed in the absence of MC, which leads to decreased levels of IL-12 and defects in the development of efficient anti-parasite immunity. Finally, it is unclear to date whether MC directly contribute to antigen presentation and T cell priming in this T cell-dependent disease
The c-kit mutation in Kit W /Kit W-v mice impairs melanogenesis and results in anaemia, sterility, and markedly reduced levels of tissue MC. In contrast, Kit W-sh /Kit W-sh mice, bearing the W-sash (W sh) inversion mutation, also show MC deficiency, but normal levels of major classes of other differentiated hematopoietic and lymphoid cells [30]. As a result, intradermal inoculation of high doses and physiologically relevant low doses of L. major resulted in significantly worsened disease with larger lesions in MC-deficient Kit W-sh /Kit W-sh compared to infected C57BL/6 control mice ([31], and unpublished data). This was correlated to enhanced parasite burdens in ears and spleens in Kit W-sh /Kit W-sh mice. Additionally, skin draining LN cells from infected Kit W-sh /Kit W-sh and C57BL/6 mice were isolated and pulsed with soluble Leishmania antigen to analyse antigen-specific cytokine production. Here, the IL-10 production was significantly increased in MC-deficient mice, which strongly supports a shift towards a Th2 response. Thus, using two independent mouse models for MC deficiency, we showed that MC are critically involved in regulating disease outcome after L. major infection into skin.
Involvement of MC in inflammatory cell recruitment
The early inflammatory response after inoculation of L. major is dominated by an influx of polymorphonuclear neutrophils (PMN) and MΦ [4, 6]. MΦ are the major constituents of skin granulomas, and parasite containment is the result of efficient granuloma formation in cutaneous leishmaniasis. In the end, MΦ are the main effector cells for parasite elimination, since IFNγ release from T cells induces NO-mediated parasite killing by MΦ [2]. PMN, in contrast, are recruited to the inoculation site within minutes post-infection [32]. Interestingly, Leishmania possess some means to survive their early encounter with recruited PMN at the bite site [6]. In addition, PMN serve as Trojan horse that mediates parasite survival and entry into tissue MΦ [33, 34] and DC [35].
In some models, MC are the main cells initiating cell recruitment to effector organs. Thus, using a model of biogel-induced skin granuloma formation, we assessed the role of MC for PMN followed by MΦ recruitment [36]. Kit W /Kit W-v mice as well as mice deficient in the MC product TNFα exhibited markedly reduced MΦ numbers in cutaneous granulomas. MΦ recruitment was restored in Kit W /Kit W-v mice reconstituted with MC from Kit+/+ or TNFα+/+, but not from TNFα−/− mice. MC-TNFα-dependent MΦ influx required prior recruitment of MIP-1α/β-producing PMN, as PMN depletion before biogel injection completely inhibited MΦ influx, which was restored after reconstitution with PMN supernatants. These findings showed that MΦ recruitment to skin granulomas is the result of a sequence of inflammatory processes initiated by MC-derived TNFα followed by PMN influx and MIP-1α/β release.
As a conclusion, we hypothesized that MC could be crucial for recruiting MΦ to sites of infection as well. And indeed, in infections with L. major, we and others detected significantly fewer 7/4+ PMN, F4/80+ MΦ, and—interestingly—CD11c+/MHC class II+ DC in leishmaniasis skin lesions in Kit W /Kit W-v mice (Fig. 2) ([26] and Nathan Peters/Bethesda, personal communication). The latter was associated with decreased levels of IL-12 production found in draining LN. Thus, MC-dependent recruitment of the most important APC to skin, MΦ, PMN and DC, contributes to control of disease in leishmaniasis and facilitates proper T cell priming against L. major infection.
DC activation and T cell priming is regulated by MC
Prior work suggested that MC can promote T cell activation indirectly through the stimulation of APC in vivo; for example, MC induced the migration of DC [37] and LC [38, 39] to draining LN, where antigen presentation occurs. To further identify the underlying mechanism of the immunoregulatory capacity of MC, we recently investigated the impact of MC on DC maturation and function in more detail [31]. Peritoneal cultured MC directly bound to immature DC and this cell-to-cell crosstalk resulted in an increased expression of the DC maturation markers CD86, CD80 and CD40 [31]. The supernatants of MC/DC co-cultures contained several T cell modulating cytokines, for example, IL-6 and TGFβ. Interestingly, in these in vitro experiments, the presence of MC further increased the amount of IL12p70 release of lipopolysaccharide (LPS)-matured DC (Fig. 2). Our results also demonstrated that the crosstalk of DC with MC strongly impacts the subsequent capacity of DC to activate CD4+ T cells and to polarize the T cells towards a Th1 or Th17 response. In line, using Kit W-sh /Kit W-sh mice, we showed that after L. major infection, decreased production of Th1 and Th17 cytokines is observed also in vivo.
MC are a potential source of many mediators that can influence the development, recruitment, phenotype, proliferation and activation of T cells in vitro and in vivo. Thus, during the initial phase of infection MC could also directly influence T cell priming and subsequent immune responses. Here, MC may act as APC themselves [40]. MC are most often associated with Th2-type inflammatory responses, but, more recently, MC were shown to be an important source of IL-12 during peritonitis, which promoted PMN and survival of infected mice [41].
Antigen processing in DC
Role of T cells for disease outcome
It is well accepted that protection against Leishmania infections is T-cell-dependent. For example, T cell-deficient nude or severe combined immunodeficient (SCID) mice failed to control fatal dissemination of L. major mice [42, 43]. An inability to control disease was also observed in MHC class II−/− and CD4-deficient mice. Interestingly, despite the absence of CD4+ T cells, a population of functional helper T cells (CD8− αβTCR+ T cells) developed after infection of CD4-deficient C57BL/6 mice with L. major [44]. These cells were MHC II-restricted and produced IFNγ after challenge with parasite antigens. These data indicated that T cell lineage commitment and peripheral function may not stringently depend on the function of the CD4 molecule [44]. Disease susceptibility also resulted from sustained depletion of CD4+ cells by monoclonal anti-CD4 antibodies, although transient depletion at the time of infection allows the side-effect of protective Th1 cells in otherwise susceptible BALB/c mice [45–47]. The latter effect is the result of transient depletion of so-called pre-primed, Leishmania homolog of receptors for activated c kinase (LACK)-reactive Vβ4 Vα8+ CD4+ T cells, with most LACK-specific T cells producing IL-4, but not IFNγ [48]. LACK-induced preferential expansion of parasite-specific Th2 cells and a low level of IFNγ production resulted in progressive infection of BALB/c mice and fatal outcome [49]. Interestingly, similar LACK-reactive T cells with preferential IL-4 release also exist in resistant C57BL/6 cells, but in smaller numbers [2].
Several studies have indicated that IFNγ release by CD8+ L. major-specific T cells (Tc1) also promotes the development of protective immunity [17, 50–52]. Thus, CD8 as well as MHC class I knockout mice were unable to control infection [17]. In line, antigen-specific IFNγ release from total LN cells results from equal production on this cytokine by CD4+ and CD8+ T cells in vivo, [21]. In addition, using (DC-based) vaccinations with fusion proteins consisting of HIV-1 TAT and the Leishmania-specific protein LACK leading to preferential induction of LACK-specific CD8 responses resulted in significantly smaller lesion volumes compared to mice immunized with LACK alone [53].
Evidence for T cell priming by DC
In experimental cutaneous leishmaniasis, infected DC appear to be the most important inducers of protective immunity. Once activated by parasite uptake, they process the antigen both in the MHC class I and in the MHC class II pathway, thereby efficiently stimulating both CD4+ and CD8+ T cells [12, 17]. In line, other groups have previously shown that murine skin-derived DC (e.g. epidermal LC) internalize L. major and transport the parasites from the skin via the lymphatics into the T cell areas of draining LN to stimulate parasite-specific T cell responses [54–56]. In addition, using low dose infections mimicking physiological sand fly bite transmission, DC immigration into the infection site and local IL-12 production is observed between week 5 and 6 post-infection shortly before the first T cells are recruited [5], whereas infected MΦ can already be found within days after parasite inoculation. Others have previously shown that impaired DC activation/migration (e.g. in CCR2−/− mice) results in worsened disease outcome due to defects in T cell priming [57]. Finally, Baldwin et al. [10] systematically examined the parasite load in different APC subsets isolated from draining LN of Leishmania-susceptible BALB/c and resistant C57BL/6 mice. Although parasites were already detectable in LN a few hours after infection with 103 metacyclic promastigotes, parasites were not found in DC until week 3, indicating that at this point of time the main infected cell type may be MΦ. Interestingly, when using this physiologically relevant low dose model, T cell priming does not occur before 4–5 weeks post-infection [10].
The contribution of MΦ to T cell activation in L. major infections is less clear. Comparative migration assays with infected epidermal LC and MΦ from BALB/c and athymic BALB/c nude mice as well as mixed labelling immunohistology of draining LN early in infection provided evidence that infected MΦ were, in contrast to LC, not able to migrate to dLN and therefore may not induce antigen-specific T cell responses [58]. In the mammalian body, IL-12 is the main and most important cytokine inducing anti-Leishmania immunity, which requires IFNγ release by primed T cells. Host MΦ from susceptible and resistant mice were, in contrast to DC, not efficiently activated by uptake of parasites via CR3-dependent phagocytosis, and IL-12 synthesis is selectively inhibited. This leads to the production of a broad range of cytokines (e.g. IL-2, IL-10) from CD4+ T cells, which may favour survival of the host organism by MHC class II-restricted antigen presentation by MΦ to primed, but not naïve, T cells [59, 60]. Overall, infected MΦ may thus be less important for T cell priming against L. major antigens. However, they may contribute to restimulation of antigen-specific CD4+ T cells that have immigrated to the site of infection at later stages post-infection.
PMN and DC co-localize at sites of acute inflammation in the skin after parasite inoculation. Interestingly, a recent study indicated that infected PMN in skin expressed elevated apoptotic markers, resulting in enhanced capturing by skin DC [35]. Thus, after 1 day, the majority of infected DC recovered from skin stained positive for neutrophil markers, indicating that they acquired their parasites via uptake of infected PMN. When infected, these DC were actively downmodulated. Consequently, PMN depletion led to enhanced activation marker expression on DC and improved DC-mediated Leishmania antigen presentation of CD4+ T cells. The findings suggest that during the acute PMN response early after infection, the parasites actively promote immunosuppression by entering DC via PMN shuttles, whereas at later stages, ‘direct’ DC uptake of L. major supports the development of protective T cell responses.
Cross-presentation of exogenous parasite antigens by DC
It is long believed that exogenous antigens, like parasite proteins, are mainly presented via the MHC class II pathway (a vesicular system) for priming of CD4+ T cells. Interestingly, however, only infected DC, but not MΦ, are also capable of priming and restimulation of CD8+ T cells [17, 61, 62]. The presentation of exogenous antigen as endogenous antigen such as self proteins was named ‘cross-presentation’ [63, 64]. In general, APC present endogenous cytosolic proteins via MHC class I molecules towards CD8+ T cells. Degradation of the proteins is performed by the proteasome [65] and resulting peptides are then transported to the endoplasmatic reticulum by the transporter associated with antigen presentation (TAP), where they bind to MHC class I molecules which are then presented on the surface to CD8+ T cells [65].
How the parasite antigens enter the cytoplasm is unknown so far. Interestingly, prior work using immune complexes and tumour models has already demonstrated that the development of efficient CD8 T-cell-dependent anti-tumour immunity was dependent on Fcγ signalling [64, 66]. Thus, the utilization of FcγR by DC for parasite internalization (as compared to CR3 by MΦ) may explain their ability to prime CD8+ T cells for parasite antigens. To further understand CD8+ T cell priming by infected DC, L. major promastigotes were transfected with a plasmid to express ovalbumin (OVA) fragments [62]. Here, it was shown that parasite-secreted OVA was more immunogenic compared to nonsecreted OVA, suggesting that secreted parasite proteins are more efficiently processed as endogenous antigens than others [62]. Interestingly, studies by Bertholet et al. and our group have furthermore revealed that DC-mediated antigen presentation of L. major peptides to CD8+ T cells is (immuno-)proteasome/TAP-independent [67, 68], thus independent of these two major nonlysosomal protein degradation machineries. The involvement of alternative cytosolic pathways of cross-presentation encompassing protease candidates such as tripeptidyl peptidase II and nardilysin will have to be investigated.
Summary and outlook
In summary, these data indicate that infected DC are crucial for T cell priming in cutaneous leishmaniasis, whereas other infected APC such as MΦ mainly contribute to MHC class II-restricted CD4+ T cell restimulation. However, until now, at least five different DC subsets have been described in skin. As described below, all of these may have different functions for disease outcome, whereby epidermal LC may be the most important cells best equipped to prime CD8+ T cells against L. major [69]. However, using novel tools such as cell-specific ablation of, for example, MHC class I or II on CD11c+ or LysM+ cells will allow for a detailed analysis of the respective role of these APC for T cell activation.
How important is DC infection for disease outcome?
DC as major source of Th1/Tc1-promoting IL-12
C57BL/6 mice lacking IL-12 showed progressive disease. Similarly, continuous treatment of BALB/c mice with recombinant IL-12 redirected the early Th2 response to Th1 and promoted resistance [70–72]. Interestingly, a strong, more sustained Th2 response in anti-IL-12-treated resistant mice led to disease exacerbation. Of note, the anti-IL-12 antibody treatment has the greatest effect when delayed until seven days after infection [73]. This indicated that production of IL-12 is delayed normally even in resistant mice. Indirectly, this coincides with the delayed appearance of infected DC both at the site of infection and in the draining LN [17]. As described above, DC appear to be the major and only cellular source for bioactive IL-12 after infection [7, 8].
DC subsets in skin: it’s not that easy…
Overall, however, only indirect evidence is present so far that points to an important role of DC for disease outcome in leishmaniasis. Only recently, new experimental approaches such as constitutive or conditional lineage ablation allow for an investigation into specific cell functions in vivo. The situation is further complicated by the fact that DC in skin can be subdivided into at least 6 subtypes. In addition, all of these subsets may have different functions for resulting immune responses [74]. Evidence exists that suggest that the different DC subsets express different activation markers, such as Toll-like receptors (TLR) [74], supporting the hypothesis that they are triggered by different signals in vivo resulting in divergent immune responses.
DC are divided in type-1 interferon-producing plasmacytoid DC (pDC) [75] and conventional DCs (cDC) [76] in nonlymphoid tissues, in circulation, and in lymphoid tissues. The cDC can be further subdivided into lymphoid tissue-resident DC that are present in the thymus, spleen and LN, and into migratory DC that act as sentinels in the periphery. These latter migratory DC are, according to their tissue localization, LC in the epidermis or dermal DC (dDC) in the dermis. The migratory DC have a mature phenotype upon reaching the LN, whereas the lymphoid-tissue-resident DC have an immature phenotype and are active in antigen uptake and processing [77]. Finally, DC that are not found in the steady state, but develop after inflammation or infection, include the monocyte-derived DCs (mo-DC) [78]. These mo-DC are found in particularly large numbers in lesions after L. major infection. Even though it is well accepted that APC are crucial for T cell priming during infection and that skin-derived LC and dDC are the main APC to activate T cells during infections with L. major, it is still unclear which of the mentioned DC subtypes is the key player [69].
Langerin (CD207) is a C-type lectin predominantly expressed by LC [79, 80], but Langerin expression was also found on some murine CD8α+ LN DC [81, 82]. Recently, a new subset of Langerin+ dDC that is independent from epidermal LC was identified [83–85]. In addition to LC in transit from the epidermis, the dermis appears to contain two more subsets of Langerin+ dDC (distinguished by differential CD103 expression) and two subsets of Langerinneg dDC that differ in CD11b expression [86].
Experimental approaches such as inducible cell lineage ablation enable the investigation into specific cell functions in vivo. Langerin-DTR knock-in mice express the human DTR under control of the langerin promoter and allow for the inducible ablation of Langerin+ cells [87]. We recently demonstrated that after physiological low dose infection with metacyclic promastigotes of L. major, mice lacking Langerin+ DC developed significantly smaller ear lesions, reduced parasite burdens, and an increased Th1 response as compared to their control littermates. Depending on the timing of the DT treatment protocol used, it was possible to completely deplete LC, while Langerin+ dDC are largely restored [88]. Alternatively, bone marrow chimera with Langerin-DTR and wild-type mice allow for depletion of LC or dDC only. Interestingly, selective depletion of LC alone led to significantly reduced lesion sizes, enhanced IFNγ/IL-4 and IFNγ/IL-10 ratios, and reduced numbers of regulatory T cells (Treg), indicating that LC and not Langerin+ dDC were responsible for the suppressive effect (Fig. 3) [89]. Interestingly, in contrast, bone marrow chimeras, in which only Langerin+ dDC can be depleted, showed no phenotype.

Various skin dendritic cell subsets with different roles for disease outcome. L. major-infected skin contains at least 6 different subsets of DC: epidermal LC, dermal Langerin+ DC, dermal Langerinneg DC and monocyte-derived inflammatory DC. All subsets have been shown to be infected with L. major in vivo and migrate to the draining LN. Recently, we have shown that—in contrast to all other DC subsets—epidermal LC regulate anti-Leishmania immunity by inducing regulatory T cells (Treg). These Treg promote parasite persistence and counterbalance the parasite-eliminating, IFNγ-producing effector T cells (Teff). Whether the various dermal DC (dDC) perform differing tasks during the initiation or maintenance of a protective T cell response in L. major infections is under investigation. The identification of “the protective” DC subset is important for vaccine development
Summary and conclusion
To induce protective adaptive immunity against Leishmania spp., IL-12 release by DC (and that of other cytokines such as IL-1α [90, 91], IL-27 [92], IL-23 [21]) is a crucial step and known to be an important promoter of Th1 differentiation. FcγR-mediated phagocytosis appears to be one important trigger of this type of response by DC after parasite internalization. In addition, MHC class I- and II-restricted antigen presentation towards both CD4+ and CD8+ T cells by DC is essential for appropriate T cell priming against parasite antigens. Some of the underlying mechanisms, for example, such as how cross-presentation of exogeneous parasite epitopes occurs, need to be studied in detail in the future. Overall, it is still unclear which skin DC subtype is the definitive key population to induce protective immunity and there is only indirect evidence present so far to answer the question whether this DC population must be infected (with live parasites) or not.
References
The references marked with an asterisk result from the work within project part A1 of the collaborative research center (SFB) 490
Herwaldt BL (1999) Leishmaniasis. Lancet 354:1191–1199
Sacks D, Noben-Trauth N (2002) The immunology of susceptibility and resistance to Leishmania major in mice. Nat Rev Immunol 2:845–858
Bogdan C (2012) Leishmaniasis in rheumatology, haematology and oncology: epidemiological, immunological and clinical aspects and caveats. Ann Rheum Dis 71:60–66
*von Stebut E (2007) Immunology of cutaneous leishmaniasis: the role of mast cells, phagocytes and dendritic cells for protective immunity. Eur J Dermatol 17:115–122
Belkaid Y, Mendez S, Lira R, Kadambi N, Milon G, Sacks D (2000) A natural model of Leishmania major infection reveals a prolonged “silent” phase of parasite amplification in the skin before the onset of lesion formation and immunity. J Immunol 165:969–977
Peters NC, Sacks DL (2009) The impact of vector-mediated neutrophil recruitment on cutaneous leishmaniasis. Cell Microbiol 11:1290–1296
*von Stebut E, Belkaid Y, Jakob T, Sacks DL, Udey MC (1998) Uptake of Leishmania major amastigotes results in activation and interleukin 12 release from murine skin-derived dendritic cells: implications for the initiation of anti-Leishmania immunity. J Exp Med 188:1547–1552
*von Stebut E, Belkaid Y, Nguyen BV, Cushing M, Sacks DL, Udey MC (2000) Leishmania major-infected murine Langerhans cell-like dendritic cells from susceptible mice release IL-12 after infection and vaccinate against experimental cutaneous Leishmaniasis. Eur J Immunol 30:3498–3506
Misslitz AC, Bonhagen K, Harbecke D, Lippuner C, Kamradt T, Aebischer T (2004) Two waves of antigen-containing dendritic cells in vivo in experimental Leishmania major infection. Eur J Immunol 34:715–725
Baldwin T, Henri S, Curtis J, O’Keeffe M, Vremec D, Shortman K, Handman E (2004) Dendritic cell populations in Leishmania major-infected skin and draining lymph nodes. Infect Immun 72:1991–2001
*von Stebut E (2007) Cutaneous Leishmania infection: progress in pathogenesis research and experimental therapy. Exp Dermatol 16:340–346
*Woelbing F, Kostka SL, Moelle K, Belkaid Y, Sunderkoetter C, Verbeek S, Waisman A, Nigg AP, Knop J, Udey MC, von Stebut E (2006) Uptake of Leishmania major by dendritic cells is mediated by Fcγ receptors and facilitates acquisition of protective immunity. J Exp Med 203:177–188
Locksley RM, Fowell DJ, Shinkai K, Wakil AE, Lacy D, Bix M (1998) Development of CD4+ effector T cells and susceptibility to infectious diseases. Adv Exp Med Biol 452:45–52
Blank C, Fuchs H, Rappersberger K, Röllinghoff M, Moll H (1993) Parasitism of epidermal Langerhans cells in experimental cutaneous leishmaniasis with Leishmania major. J Infect Dis 167:418–425
Flohé SB, Bauer C, Flohé S, Moll H (1998) Antigen-pulsed epidermal Langerhans cells protect susceptible mice from infection with the intracellular parasite Leishmania major. Eur J Immunol 28:3800–3811
Ahuja SS, Mummidi S, Malech HL, Ahuja SK (1998) Human dendritic cell (DC)-based anti-infective therapy: engineering DCs to secrete functional IFN-γ and IL-12. J Immunol 161:868–876
*Belkaid Y, von Stebut E, Mendez S, Lira R, Caler E, Bertholet S, Udey MC, Sacks D (2002) CD8+ T cells are required for primary immunity in C57BL/6 mice following low-dose, intradermal challenge with Leishmania major. J Immunol 168:3992–4000
Schönlau F, Scharffetter-Kochanek K, Grabbe S, Pietz B, Sorg C, Sunderkötter C (2000) In experimental leishmaniasis deficiency of CD18 results in parasite dissemination associated with altered macrophage functions and incomplete Th1 cell response. Eur J Immunol 30:2729–2740
Köhl J (2006) Self, non-self, and danger: a complementary view. Adv Exp Med Biol 586:71–94
Domínguez M, Toraño A (1999) Immune adherence-mediated opsonophagocytosis: the mechanism of Leishmania infection. J Exp Med 189:25–35
*Lopez Kostka S, Dinges S, Griewank K, Iwakura Y, Udey MC, von Stebut E (2009) IL-17 promotes progression of cutaneous leishmaniasis in susceptible mice. J Immunol 182:3039–3046
Miles SA, Conrad SM, Alves RG, Jeronimo SM, Mosser DM (2005) A role for IgG immune complexes during infection with the intracellular pathogen Leishmania. J Exp Med 201:747–754
Editor’s Choice (2006) Primed by parasites. Science 311:579–581
Wanderley JL, Barcinski MA (2010) Apoptosis and apoptotic mimicry: the Leishmania connection. Cell Mol Life Sci 67:1653–1659
Wanderley JL, Moreira ME, Benjamin A, Bonomo AC, Barcinski MA (2006) Mimicry of apoptotic cells by exposing phosphatidylserine participates in the establishment of amastigotes of Leishmania (L) amazonensis in mammalian hosts. J Immunol 176:1834–1839
*Maurer M, Lopez Kostka S, Siebenhaar F, Moelle K, Metz M, Knop J, von Stebut E (2006) Skin mast cells control T cell-dependent host defense in Leishmania major infections. FASEB J 20:2460–2467
Wershil BK, Theodos CM, Galli SJ, Titus RG (1994) Mast cells augment lesion size and persistence during experimental Leishmania major infection in the mouse. J Immunol 152:4563–4571
Bidri M, Vouldoukis I, Mossalayi MD, Debre P, Guillosson JJ, Mazier D, Arock M (1997) Evidence for direct interaction between mast cells and Leishmania parasites. Parasite Immunol 19:475–483
Grimaldi G Jr, Soares MJ, Moriearty PL (1984) Tissue eosinophilia and Leishmania mexicana eosinophil interactions in murine cutaneous leishmaniasis. Parasite Immunol 6:397–408
Grimbaldeston MA, Nakae S, Kalesnikoff J, Tsai M, Galli SJ (2007) Mast cell-derived interleukin 10 limits skin pathology in contact dermatitis and chronic irradiation with ultraviolet B. Nat Immunol 8:1095–1104
*Dudeck A, Suender CA, Kostka SL, von Stebut E, Maurer M (2011) Mast cells promote Th1 and Th17 responses by modulating dendritic cell maturation and function. Eur J Immunol 41:1883–1893
Peters NC, Egen JG, Secundino N, Debrabant A, Kimblin N, Kamhawi S, Lawyer P, Fay MP, Germain RN, Sacks D (2008) In vivo imaging reveals an essential role for neutrophils in leishmaniasis transmitted by sand flies. Science 321:970–974
Laskay T, van Zandbergen G, Solbach W (2008) Neutrophil granulocytes as host cells and transport vehicles for intracellular pathogens: apoptosis as infection-promoting factor. Immunobiology 213:183–191
John B, Hunter CA (2008) Immunology. Neutrophil soldiers or Trojan Horses? Science 321:917–918
Ribeiro-Gomes FL, Peters NC, Debrabant A, Sacks DL (2012) Efficient capture of infected neutrophils by dendritic cells in the skin inhibits the early anti-Leishmania response. PLoS Pathog 8:e1002536
*von Stebut E, Metz M, Milon G, Knop J, Maurer M (2003) Early macrophage influx to sites of cutaneous granuloma formation is dependent on MIP-1α/β released from neutrophils recruited by mast cell-derived TNFα. Blood 101:210–215
Suto H, Nakae S, Kakurai M, Sedgwick JD, Tsai M, Galli SJ (2006) Mast cell-associated TNF promotes dendritic cell migration. J Immunol 176:4102–4112
Bryce PJ, Miller ML, Miyajima I, Tsai M, Galli SJ, Oettgen HC (2004) Immune sensitization in the skin is enhanced by antigen-independent effects of IgE. Immunity 20:381–392
Jawdat DM, Albert EJ, Rowden G, Haidl ID, Marshall JS (2004) IgE-mediated mast cell activation induces Langerhans cell migration in vivo. J Immunol 173:5275–5282
Mekori YA, Metcalfe DD (1999) Mast cell-T cell interactions. J Allergy Clin Immunol 104:517–523
Nakano N, Nishiyama C, Kanada S, Niwa Y, Shimokawa N, Ushio H, Nishiyama M, Okumura K, Ogawa H (2007) Involvement of mast cells in IL-12/23 p40 production is essential for survival from polymicrobial infections. Blood 109:4846–4855
Mitchell GF (1983) Murine cutaneous leishmaniasis: resistance in reconstituted nude mice and several F1 hybrids infected with Leishmania tropica major. J Immunogenet 10:395–412
Holaday BJ, Sadick MD, Wang Z-E, Reiner SL, Heinzel FP, Parslow TG, Locksley RM (1991) Reconstitution of Leishmania immunity in severe combined immunodeficient mice using Th1- and Th2-like lines. J Immunol 147:1653–1658
Locksley RM, Reiner SL, Hatam F, Littman DR, Killeen N (1993) Helper T cells without CD4: control of leishmaniasis in CD4-deficient mice. Science 261:1448–1451
Titus RG, Ceredig R, Cerottini JC, Louis JA (1985) Therapeutic effect of anti-L3T4 monoclonal antibody GK1.5 on cutaneous leishmaniasis in genetically-susceptible BALB/c mice. J Immunol 135:2108–2114
Titus RG, Milon G, Marchal G, Vassalli P, Cerottini JC, Louis JA (1987) Involvement of specific Lyt-2+ T cells in the immunological control of experimentally induced murine cutaneous leishmaniasis. Eur J Immunol 17:1429–1433
Heinzel FP, Sadick MD, Mutha SS, Locksley RM (1991) Production of interferon gamma, interleukin 2, interleukin 4, and interleukin 10 by CD4+ lymphocytes in vivo during healing and progressive murine leishmaniasis. Proc Natl Acad Sci USA 88:7011–7015
Mougneau E, Altare F, Wakil AE, Zheng S, Coppola T, Wang ZE, Waldmann R, Locksley RM, Glaichenhaus N (1995) Expression cloning of a protective Leishmania antigen. Science 268:563–566
Heinzel FP, Sadick MD, Holaday BJ, Coffman RL, Locksley RM (1989) Reciprocal expression of interferon gamma or interleukin 4 during the resolution or progression of murine leishmaniasis. Evidence for expansion of distinct helper T cell subsets. J Exp Med 169:59–72
Müller I, Kropf P, Louis JA, Milon G (1994) Expansion of gamma interferon-producing CD8+ T cells following secondary infection of mice immune to Leishmania major. Infect Immun 62:2575–2581
Erb K, Blank C, Ritter U, Bluethmann H, Moll H (1996) Leishmania major infection in major histocompatibility complex class II-deficient mice: CD8+ T cells do not mediate a protective immune response. Immunobiology 195:243–260
Uzonna JE, Joyce KL, Scott P (2004) Low dose Leishmania major promotes a transient T helper cell type 2 response that is down-regulated by interferon gamma-producing CD8+ T cells. J Exp Med 199:1559–1566
*Kronenberg K, Brosch S, Butsch F, Tada Y, Shibagaki N, Udey MC, von Stebut E (2010) Vaccination with TAT-antigen fusion protein induces protective, CD8+ T cell-mediated immunity against Leishmania major. J Invest Dermatol 130:2602–2610
Macatonia SE, Knight SC, Edwards AJ, Griffiths S, Fryer P (1987) Localization of antigen on lymph node dendritic cells after exposure to the contact sensitizer fluorescein isothiocyanate. Functional and morphological studies. J Exp Med 166:1654–1667
Larsen CP, Steinman RM, Witmer-Pack M, Hankins DF, Morris PJ, Austyn JM (1990) Migration and maturation of Langerhans cells in skin transplants and explants. J Exp Med 172:1483
Kripke ML, Munn CG, Jeevan A, Tang JM, Bucana CJ (1990) Evidence that cutaneous antigen-presenting cells migrate to regional lymph nodes during contact sensitization. J Immunol 145:2833–2838
Sato N, Ahuja SK, Quinones M, Kostecki V, Reddick RL, Melby PC, Kuziel WA, Ahuja SS (2000) CC chemokine receptor (CCR) 2 is required for Langerhans cell migration and localization of T helper cell type 1 (Th1)-inducing dendritic cells. Absence of CCR2 shifts the Leishmania major-resistant phenotype to a susceptible state dominated by Th2 cytokines, b cell outgrowth, and sustained neutrophilic inflammation. J Exp Med 192:205–218
Moll H, Fuchs H, Blank C, Röllinghoff M (1993) Langerhans cells transport Leishmania major from the infected skin to the draining lymph node for presentation to antigen-specific T cells. Eur J Immunol 23:1595–1601
Reiner SL, Zheng S, Wang Z-E, Stowring L, Locksley RM (1994) Leishmania promastigotes evade interleukin 12 (IL-12) induction by macrophages and stimulate a broad range of cytokines from CD4+ T cells during initiation of infection. J Exp Med 179:447–456
Carrera L, Gazzinelli RT, Badolato R, Hieny S, Müller W, Kühn R, Sacks DL (1996) Leishmania promastigotes selectively inhibit interleukin 12 induction in bone marrow-derived macrophages from susceptible and resistant mice. J Exp Med 183:515–526
Ramachandra L, Boom WH, Harding CV (2008) Class II MHC antigen processing in phagosomes. Methods Mol Biol 445:353–377
Bertholet S, Debrabant A, Afrin F, Caler E, Mendez S, Tabbara KS, Belkaid Y, Sacks DL (2005) Antigen requirements for efficient priming of CD8+ T cells by Leishmania major-infected dendritic cells. Infect Immun 73:6620–6628
Miller JF, Kurts C, Allison J, Kosaka H, Carbone F, Heath WR (1998) Induction of peripheral CD8+ T-cell tolerance by cross-presentation of self antigens. Immunol Rev 165:267–277
den Haan JM, Lehar SM, Bevan MJ (2000) CD8+ but not CD8− dendritic cells cross-prime cytotoxic T cells in vivo. J Exp Med 192:1685–1696
Kloetzel PM (2001) Antigen processing by the proteasome. Nat Rev Mol Cell Biol 2:179–187
Kalergis AM, Ravetch JV (2002) Inducing tumor immunity through the selective engagement of activating Fcgamma receptors on dendritic cells. J Exp Med 195:1653–1659
Bertholet S, Goldszmid R, Morrot A, Debrabant A, Afrin F, Collazo-Custodio C, Houde M, Desjardins M, Sher A, Sacks D (2006) Leishmania antigens are presented to CD8+ T cells by a transporter associated with antigen processing-independent pathway in vitro and in vivo. J Immunol 177:3525–3533
*Brosch S, Tenzer S, Akkad N, Lorenz B, Schild H, von Stebut E (2012) Priming of Leishmania-reactive CD8+ T cells in vivo does not require LMP7-containing immunoproteasomes. J Invest Dermatol 132:1302–1305
Brewig N, Kissenpfennig A, Malissen B, Veit A, Bickert T, Fleischer B, Mostböck S, Ritter U (2009) Priming of CD8+ and CD4+ T cells in experimental leishmaniasis is initiated by different dendritic cell subtypes. J Immunol 182:774–783
Sypek JP, Chung CL, Mayor SE, Subramanyam JM, Goldman SJ, Sieburth DS, Wolf SF, Schaub RG (1993) Resolution of cutaneous leishmaniasis: interleukin-12 initiates a protective T helper type 1 immune response. J Exp Med 177:1797–1802
Heinzel FP, Schoenhaut DS, Rerko RM, Rosser LE, Gately MK (1993) Recombinant interleukin-12 cures mice infected with Leishmania major. J Exp Med 177:1505–1509
Mattner F, Magram J, Ferrante J, Launois P, Di Padova K, Behin R, Gately MK, Louis JA, Alber G (1996) Genetically resistant mice lacking interleukin-12 are susceptible to infection with Leishmania major and mount a polarized Th2 cell response. Eur J Immunol 26:1553–1559
Heinzel FP, Rerko RM, Ahmed F, Pearlman E (1995) Endogenous IL-12 is required for control of Th2 cytokine responses capable of exacerbating leishmaniasis in normally resistant mice. J Immunol 155:730–739
*von Stebut E (2011) Research in practice: different dendritic cell types in skin with various functions—important implications for intradermal vaccines. J Dtsch Dermatol Ges 9:506–509
Asselin-Paturel C, Boonstra A, Dalod M, Durand I, Yessaad N, Dezutter-Dambuyant C, Vicari A, O’Garra A, Biron C, Briere F, Trinchieri G (2001) Mouse type I IFN-producing cells are immature APCs with plasmacytoid morphology. Nat Immunol 2:1144–1150
Wu L, Dakic A (2004) Development of dendritic cell system. Cell Mol Immunol 1:112–118
Wilson NS, El Sukkari D, Belz GT, Smith CM, Steptoe RJ, Heath WR, Shortman K, Villadangos JA (2003) Most lymphoid organ dendritic cell types are phenotypically and functionally immature. Blood 102:2187–2194
Leon B, Lopez-Bravo M, Ardavin C (2007) Monocyte-derived dendritic cells formed at the infection site control the induction of protective T helper 1 responses against Leishmania. Immunity 26:519–531
Valladeau J, Clair-Moninot V, Dezutter-Dambuyant C, Pin JJ, Kissenpfennig A, Mattei MG, Ait-Yahia S, Bates EE, Malissen B, Koch F, Fossiez F, Romani N, Lebecque S, Saeland S (2002) Identification of mouse langerin/CD207 in Langerhans cells and some dendritic cells of lymphoid tissues. J Immunol 168:782–792
Takahara K, Omatsu Y, Yashima Y, Maeda Y, Tanaka S, Iyoda T, Clausen BE, Matsubara K, Letterio J, Steinman RM, Matsuda Y, Inaba K (2002) Identification and expression of mouse Langerin (CD207) in dendritic cells. Int Immunol 14:433–444
Kissenpfennig A, Henri S, Dubois B, Laplace-Builhe C, Perrin P, Romani N, Tripp CH, Douillard P, Leserman L, Kaiserlian D, Saeland S, Davoust J, Malissen B (2005) Dynamics and function of Langerhans cells in vivo: dermal dendritic cells colonize lymph node areas distinct from slower migrating Langerhans cells. Immunity 22:643–654
Douillard P, Stoitzner P, Tripp CH, Clair-Moninot V, Ait-Yahia S, McLellan AD, Eggert A, Romani N, Saeland S (2005) Mouse lymphoid tissue contains distinct subsets of langerin/CD207 dendritic cells, only one of which represents epidermal-derived Langerhans cells. J Invest Dermatol 125:983–994
Poulin LF, Henri S, de Bovis B, Devilard E, Kissenpfennig A, Malissen B (2007) The dermis contains langerin+ dendritic cells that develop and function independently of epidermal Langerhans cells. J Exp Med 204:3119–3131
Ginhoux F, Collin MP, Bogunovic M, Abel M, Leboeuf M, Helft J, Ochando J, Kissenpfennig A, Malissen B, Grisotto M, Snoeck H, Randolph G, Merad M (2007) Blood-derived dermal langerin+ dendritic cells survey the skin in the steady state. J Exp Med 204:3133–3146
Bursch LS, Wang L, Igyarto B, Kissenpfennig A, Malissen B, Kaplan DH, Hogquist KA (2007) Identification of a novel population of Langerin+ dendritic cells. J Exp Med 204:3147–3156
Henri S, Poulin LF, Tamoutounour S, Ardouin L, Guilliams M, de Bovis B, Devilard E, Viret C, Azukizawa H, Kissenpfennig A, Malissen B (2010) CD207+ CD103+ dermal dendritic cells cross-present keratinocyte-derived antigens irrespective of the presence of Langerhans cells. J Exp Med 207:189–206
Bennett CL, van Rijn E, Jung S, Inaba K, Steinman RM, Kapsenberg ML, Clausen BE (2005) Inducible ablation of mouse Langerhans cells diminishes but fails to abrogate contact hypersensitivity. J Cell Biol 169:569–576
Noordegraaf M, Flacher V, Stoitzner P, Clausen BE (2010) Functional redundancy of Langerhans cells and Langerin+ dermal dendritic cells in contact hypersensitivity. J Invest Dermatol 130:2752–2759
*Kautz-Neu K, Noordegraaf M, Dinges S, Bennett CL, John D, Clausen BE, von Stebut E (2011) Langerhans cells are negative regulators of the anti-Leishmania response. J Exp Med 208:885–891
*von Stebut E, Ehrchen JM, Belkaid Y, Kostka SL, Molle K, Knop J, Sunderkotter C, Udey MC (2003) Interleukin 1α promotes Th1 differentiation and inhibits disease progression in Leishmania major-susceptible BALB/c mice. J Exp Med 198:191–199
Filippi C, Hugues S, Cazareth J, Julia V, Glaichenhaus N, Ugolini S (2003) CD4+ T cell polarization in mice is modulated by strain-specific major histocompatibility complex-independent differences within dendritic cells. J Exp Med 198:201–209
*Zahn S, Wirtz S, Birkenbach M, Blumberg RS, Neurath MF, von Stebut E (2005) Impaired Th1 responses in mice deficient in Epstein-Barr virus-induced gene 3 and challenged with physiological doses of Leishmania major. Eur J Immunol 35:1106–1112
Acknowledgments
The authors would like to thank the German Research Foundation for their support. Work of the authors was supported by the Deutsche Forschungsgemeinschaft (Sonderforschungsbereich 490; STE 1208/6-2, 11-1 and 12-1).
Author information
Authors and Affiliations
Corresponding author
Additional information
Kordula Kautz-Neu, Kirsten Schwonberg, Michael R. Fischer and Anja I. Schermann contributed equally to this work.
Rights and permissions
About this article
Cite this article
Kautz-Neu, K., Schwonberg, K., Fischer, M.R. et al. Dendritic cells in Leishmania major infections: mechanisms of parasite uptake, cell activation and evidence for physiological relevance. Med Microbiol Immunol 201, 581–592 (2012). https://doi.org/10.1007/s00430-012-0261-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00430-012-0261-2