
Abstract
The TSC1 and TSC2 tumor suppressor genes control the activity of mechanistic target of rapamycin (mTOR) pathway. Elevated activity of this pathway in Tsc2+/- mouse model leads to reduction of postsynaptic GABAB receptor–mediated inhibition and hyperexcitability in the medial prefrontal cortex (mPFC). In this study, we asked whether presynaptic GABAB receptors (GABABRs) can compensate this shift of hyperexcitability. Experiments were performed in brain slices from adolescent wild-type (WT) and Tsc2+/- mice. Miniature and spontaneous postsynaptic currents (m/sPSCs) were recorded from layer 2/3 pyramidal neurons in mPFC using patch-clamp technique using a Cs+-based intrapipette solution. Presynaptic GABABRs were activated by baclofen (10 µM) or blocked by CGP55845 (1 µM). Independent on genotype, GABABR modulators bidirectionally change miniature excitatory postsynaptic current (mEPSC) frequency by about 10%, indicating presynaptic GABABR-mediated effects on glutamatergic transmission are comparable in both genotypes. In contrast, frequencies of both mIPSCs and sIPCSs were suppressed by baclofen stronger in Tsc2+/- neurons than in WT ones, whereas CGP55845 significantly increased (m/s)IPSC frequencies only in WT cells. Effects of baclofen and CGP55845 on the amplitudes of evoked (e)IPSCs confirmed these observations. These data indicate (1) that GABAergic synapses are inhibited by ambient GABA in WT but not in Tsc2+/- slices, and (2) that baclofen shifts the E/I ratio, determined as the ratio of (m/s)EPSC frequency to (m/s)IPSC frequency, towards excitation only in Tsc2+/- cells. This excitatory presynaptic GABABR-mediated action has to be taken into account for a possible medication of mental disorders using baclofen.
Graphical abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Tuberous sclerosis complex (TSC) is an autosomal dominant disorder that affects many organs, including the brain. Clinical features of TSC vary between individuals and can include brain tumors, seizures, and neurodevelopmental disorders like autism spectrum disorder (ASD) (for recent review [11, 14]). A loss-of-function mutation in one of the tumor suppressor genes, TSC1 (hamartin) or TSC2 (tuberin) underlies TSC [7]. A mutation of either Tsc1 or Tsc2 leads to an enhanced mTOR pathway activity and ultimately to TSC symptoms [9, 13]. The mTOR pathway plays a pivotal role in the regulation of a wide array of cellular functions, including both excitatory and inhibitory synaptic transmission [26]. However, the specific cascades of cellular changes that alter and/or destabilize the neuronal networks in the brain remain elusive. In particular, the sequence of events that could lead to the appearance of ASD symptoms independently of epileptic ones, i.e., in the conditions observed in Tsc2+/- mice, is of particular interest.
It is generally believed that a physiological balance between excitatory and inhibitory synaptic drives in the central nervous system (CNS) is of crucial importance for normal brain development (but see [1]). Even a transient imbalance between excitatory and inhibitory function (E/I ratio) during early development might lead to an immediate or later manifestation of patho(physio)logical symptoms. Conditional deletion of Tsc1 increases the amplitude of miniature excitatory postsynaptic currents (mEPSCs) in hippocampal neurons, suggesting a postsynaptic strengthening of glutamatergic inputs [4, 27]. In contrast to the Tsc1 model, spontaneous Tsc2 mutation (Tsc2+/-) appears not to affect basal glutamatergic transmission in rat hippocampus [2, 8, 30]. Similar to the strengthening of excitatory synaptic transmission, the weakening of GABAergic inputs disturbs the E/I balance, and this can contribute to development of neurologic deficits in ASD (for review [12, 22]). Reduced frequency of miniature inhibitory postsynaptic currents (mIPSCs) in hippocampus has been reported in the pan-neuronal Tsc1 knockout mouse model [34]. Stimulation of GABAergic transmission with benzodiazepines, allosteric modulators of GABAA receptors, improves deficits in social interaction in BTBR T + Itpr3tf/J (BTBR) mice [10]. In addition to phasic GABAergic transmission, postsynaptic tonic inhibition can influence neuronal network stability [28]. In our recent study, we have shown that GABAAR-mediated postsynaptic inhibition develops by the end of the first postnatal month in the medial prefrontal cortex (mPFC) of both WT and Tsc2+/- cortices. However, its strength has been found to be not significantly different in both genotypes. In contrast, shunting GABABR-mediated postsynaptic inhibition in the mPSC protects neuronal networks against overexcitation induced by a destabilizing factor, for instance kainate, in WT cortices but not in Tsc2+/- ones [3].
Expression of functional GABABRs in the mPFC has been previously reported [31]. Activation or inhibition of GABABRs influences the frequency of spontaneous EPSCs and modulates the duration of cortical UP-states [31]. Modulatory action of GABABRs on the UP-states has been reported in various species and cortex regions [6, 21]. The reported inhibitory action of baclofen, a GABABR agonist, on spontaneous glutamatergic transmission in the mPFC suggests that functional GABABRs are located on glutamatergic boutons. However, the question of an involvement of GABABRs on presynaptic GABAergic terminals in mPFC in the modulation of synaptic transmission and in particular in the framework of ASD models has not been extensively investigated yet. Interestingly, higher expression level of presynaptic GABABRs has been shown immunohistochemically in the mPFC in a rat model of schizophrenia (APO-SUS). Moreover, reduced paired-pulse ratio of evoked IPSCs observed in this model could be corrected by CGP55845, a GABABR antagonist, indicating a contribution of presynaptic GABABRs located on GABAergic terminals [23]. Changes in GABABR functioning have been also reported in Fmr1-knockout mice, a model of fragile X syndrome. In this model, a selective weakening of GABABR-mediated presynaptic inhibition of glutamatergic synaptic transmission has been reported, while both postsynaptic and presynaptic GABABR-mediated suppression of GABAergic inputs were not modified [15]. As baclofen alleviates behavior deficits only in some, for instance 16p11.2 and BTBP ASD mouse models [24, 25], but not in Fmr1-knockout mice [33], we aimed to investigate the physiological role of presynaptic GABABRs in the mPFC of Tsc2+/- mouse model.
In this work, we aimed to address the following questions: (1) Are functional presynaptic GABABRs expressed in both glutamatergic and GABAergic synaptic terminals? (2) Is their functionality dependent on the genotype? (3) How does activation or blockade of GABABRs affects the E/I balance in the mPFC at the single cell and network level? In order to minimize a potential contribution of postsynaptic GABABRs, E(I)PSCs were recorded using a Cs+-based intrapipette solution. Here, we report that both glutamatergic and GABAergic terminals express functional GABABRs in both genotypes. In case of glutamatergic contacts, an activation/inhibition of presynaptic GABABRs suppresses/potentiates glutamatergic synaptic transmission to the same extent in both genotypes. In contrast to glutamate, the GABAergic system demonstrates genotype-dependent sensitivity to GABABR modulators. Baclofen demonstrates a more robust inhibitory effect in Tsc2+/- neurons compared with WT cells, while CGP55845 enhances GABAergic transmission only in WT neurons. These results demonstrate that GABAergic terminals in both genotypes express functional presynaptic GABABRs. However, the latter are tonically activated by ambient GABA only in WT cortices. Baclofen suppresses the GABAergic system more robustly in the Tsc2+/- mPFC. As a consequence, in Tsc2+/- mice, baclofen treatment shifts the E/I balance towards excitation both at the single cell and at the network level. As baclofen has been suggested as a potential pharmacological treatment of ASD, this excitatory action of baclofen in the mPFC in case of Tsc2 mutation should be taken into account.
Materials and methods
Experimental subjects
Animals (B6;129S4-Tsc2tm1Djk/J, [18]) were provided by Jackson Laboratory. Mice were bred and genotyped by the Translation Animal Center (Mainz). Augmented activity of mTOR in Tsc2+/- animals was confirmed by western blot analysis [3]. All experiments were performed in line with the EU directive 86/609/EEC and approved by Landesuntersuchungsanstalt Rheinland-Pfalz (23.177–07/G 10–1-010). All experiments were designed to minimize the number of animals used.
Brain slice preparation
Wild-type (WT) and Tsc2+/- littermates of postnatal day (P) 25–40 were used for experiments. Brain slice preparation procedure was described elsewhere [3]. Briefly, animals were deeply anesthetized using isoflurane and decapitated. The brain was taken out and submerged in protective artificial cerebrospinal fluid (ACSF), which contained (in mM): 110 choline chloride, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 20 glucose, 11.6 sodium l-ascorbate, 3.1 sodium pyruvate, 0.2 CaCl2, and 5 MgCl2. ACSF was continuously bubbled with a 95% O2 and 5% CO2 gas mixture, to set the pH value. For brain extraction, ACSF was cooled down to zero degrees (°C) using ice. Coronal slices were sectioned at 300 µm using a vibratome (Campden Instruments Ltd., Loughborough, UK). Slices containing the medial prefrontal cortex (mPFC) were selected using the mouse brain atlas [20]. Before recording, slices were allowed to recover in ACSF at room temperature for at least 1 h. For recovery and recording, the ACSF contained (in mM) 125 NaCl, 2.5 KCl, 10 glucose, 1.25 NaH2PO4, 25 NaHCO3, 2CaCl2, and 1 MgCl2, and was continuously oxygenated with a 95% O2 and 5% CO2 gas mixture.
Whole-cell patch-clamp recordings
For electrophysiological recordings, brain slices were transferred into a recording chamber on the microscope stage (Axioscope FS, Zeiss, Germany). Slices were kept continuously perfused with ACSF via a gravity-driven system. The flow rate was set to about 1.5 ml/min. The recording chamber volume was about 0.7 ml. The temperature was kept at 31–32 °C using a temperature-controller (Temperaturcontroller III, Luigs & Neumann, Ratingen, Germany).
Firstly, slices were visually inspected using a × 5 objective prior to any experiment. After this, a × 40 objective (Zeiss, Oberkochen, Germany) was used to identify and select layer 2/3 pyramidal neurons according to morphological criteria. Pipettes were pulled from borosilicate glass capillaries using a P-87 horizontal puller (Sutter Instrument Co., Novato, USA). Their resistance amounted to 3–6 MOhm when filled with intrapipette solution. The latter contained (in mM) 125 CsOH, 125 gluconic acid, 5 CsCl, 10 EGTA, 10 HEPES, 2 MgCl2, 2 Na-ATP, and 0.4 Na-GTP. pH was adjusted to 7.3 using CsOH. Osmolarity was measured and set to 320 mOsm using sucrose.
Acquisition of postsynaptic currents (PSCs) was performed using an EPC-10 amplifier using the TIDA 5.24 software (HEKA Elektronik, Lambrecht, Germany). The liquid junction potential was not corrected. Recorded signals were filtered at 3 kHz and digitally sampled at 10 kHz. After successful establishment of whole-cell configuration, hyperpolarizing pulses of 10 mV were applied to enable an estimation of passive properties of the cell such as cell capacity, access resistance, and series resistance. Series resistance compensation was not applied. Recordings with series resistance higher than 30 MOhm were discarded.
Synaptic currents were recorded in voltage clamp mode. In particular, we recorded spontaneous postsynaptic currents (sPSCs) and miniature postsynaptic currents (mPSCs) in either standard ACSF (sPSCs) or in the presence of 0.5 µM tetrodotoxin (TTX), a blocker of voltage-gated sodium channels (mPSCs). Excitatory postsynaptic currents (s/m)EPSCs were recorded at a holding potential of − 70 mV, the reversal potential for inhibitory postsynaptic currents (IPSCs). In order to acquire (s/m)IPSCs, the holding potential was set to 0 mV, the reversal potential for EPSCs.
Evoked inhibitory postsynaptic currents (eIPSCs) were recorded in the presence of DNQX and APV, the antagonists of AMPA and NMDA receptors, respectively. eIPSCs were elicited using focal electrical stimulation in the vicinity of the patched cell (about 100 µm) via a glass pipette filled with ACSF. A custom-made stimulation unit was used to provide rectangular current pulses. The intensity level was tuned to activate a unitary synaptic input (minimal stimulation, as previously described in [29]). Briefly, inputs were located by slowly moving the stimulation pipette in the vicinity of the cell patched, while a train of four pulses was applied every 4 s. After the input was revealed, stimulation intensity was set about 20% higher than the threshold strength for the identified input. Only traces with stable latency between the stimulation artifact and the evoked response were kept for further analysis. The applied stimulus intensity amounted to 1–2 µA. At least 20 responses were recorded in each cell.
Chemicals
DL-APV, DNQX, and CGP55845 were obtained from Biotrend (Cologne, Germany). All other chemicals were provided from Tocris (Bio-Techne, Wiesbaden, Germany). Aliquots of DNQX and CGP55845 were prepared using dimethylsufoxide (DMSO). All final solutions were prepared shortly before the experiments. Final concentration of DMSO in ACSF was always less than 0.2%.
Data evaluation and statistics
Evoked IPSCs were analyzed using TIDA 5.24 (HEKA Elektronik, Lambrecht, Germany). mPSCs and sPSCs were evaluated using the PeakCount software. The latter utilizes a derivative threshold-crossing algorithm to detect individual PSCs. All detected PSCs were displayed for visual inspection. Data in text and figures are expressed in mean ± SEM. Statistical analysis was performed using GraphPad Prism 5 (GraphPad Software, Inc., San Diego, USA). Differences between means were tested for significance using one-way ANOVA. To test the difference from a hypothetical mean, one sample Student’s t-test was used. Paired values were compared using paired Student’s t-test. Statistical significance was presented using the following rules: *p < 0.05, **p < 0.005, ***p < 0.001, ns—not significant.
Results
Functional GABABRs are located on both glutamatergic and GABAergic presynaptic terminals
In the mPFC, functional GABABRs have been reported to be expressed postsynaptically and presynaptically on glutamatergic boutons [31]. As postsynaptic GABABRs are tonically activated in WT pyramidal neurons, but not in Tsc2+/- cells, the membrane resistance of WT pyramidal neurons around the end of the first postnatal month is significantly lower [3]. To reduce the effect of persistent postsynaptic GABABR-mediated shunting, whole-cell patch-clamp recordings were performed using a Cs2+-based intrapipette solution to block K+ conductance (see the “Methods” section). In this case membrane resistances in WT and Tsc2+/- cells were not significantly different (92 ± 3 and 91 ± 4 MOhm, n = 27 and 26 neurons for WT and Tsc2+/-, respectively, p = 0.94, F = 0.005). Neither baclofen (10 µM), a GABABR agonist (− 1.5 ± 1.4 and − 1.7 ± 1.5 pA, n = 17 and 15 for WT and Tsc2+/-, p = 0.97, F = 0.001), nor CGP55845 (1 µM), a GABABR antagonist (− 0.2 ± 1.5 and − 0.4 ± 2.1 pA, n = 12 and 14 for WT and Tsc2+/-, p = 0.99, F = 0.0001, data not shown), could significantly affect the holding current in both genotypes. Furthermore, neither baclofen nor CGP55845 influenced the mean amplitude of either mEPSCs or mIPSCs (Table 1), confirming that postsynaptic GABABRs mediate their effects mostly via postsynaptic K+ channels (GIRKs) which are blocked by intracellular Cs+ ions in this study.
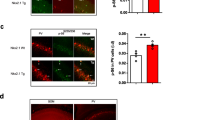
Having confirmed that the target cell is not sensitive to GABABR modulators, we investigated excitatory/inhibitory postsynaptic currents and their sensitivity to baclofen and CGP55845. In the first set of experiments, we recorded mPSCs in the presence of TTX (0.5 µM), an antagonist of voltage-gated Na+ channels. In line with our previous work [3], Tsc2+/- neurons at this age displayed significantly increased frequencies of both mIPSCs (4.3 ± 0.4, n = 14, and 5.8 ± 0.3 Hz, n = 14, in WT and Tsc2+/- neurons, respectively, p = 0.004) and mEPSCs (4.1 ± 0.3, n = 14, and 5.3 ± 0.3 Hz, n = 14, in WT and Tsc2+/- neurons, respectively, p = 0.011). Baclofen (10 µM) reduced mEPSC frequencies both in WT (0.87 ± 0.02, n = 7) and in Tsc2+/- (0.86 ± 0.03, n = 7) cells without any significant difference between the genotypes (p = 0.92, F = 0.01, Fig. 1a, b). Blockade of GABABRs with CGP55845 (1 µM) induced a slight increase in mEPSC frequency in WT (1.11 ± 0.04, n = 8) and Tsc2+/- (1.09 ± 0.03, n = 7) neurons. Also, these values did not differ in a statistically significant way (F = 0.05, p = 0.94, Fig. 1c, d).

GABABR agonist-induced and antagonist-induced effects on mEPSC frequency do not depend on genotype. a Representative mEPSC traces recorded from a WT neuron demonstrate the baclofen-induced effect. b Baclofen (10 µM) significantly reduces mEPSC frequency both in WT (left) and in Tsc2+/- (middle) neurons. Relative baclofen-induced reductions of mEPSC frequency are similar in WT and Tsc2+/- cells (right panel). c Representative mEPSC traces recorded from Tsc2+/- cells demonstrate the CGP55845-induced effect. d CGP55845 (1 µM) significantly increases mEPSC frequency both in WT (left) and in Tsc2+/- (middle) cells. Relative CGP55845-induced elevations of mEPSC frequency are comparable in WT and Tsc2+/- neurons (right panel)
In contrast to mEPSCs, GABAergic mIPSCs demonstrated a genotype-dependent sensitivity to GABABR modulators. Baclofen decreased mIPSC frequency in WT cells (0.86 ± 0.03, n = 7) and in Tsc2+/- neurons (0.61 ± 0.03, n = 7). This difference was statistically significant (p < 0.0001, F = 47.1, Fig. 2a, b). CGP55845 strongly increased mIPSC frequency in WT neurons (1.59 ± 0.12, n = 8) but only slightly elevated mIPSC frequency in Tsc2+/- cells (1.07 ± 0.05, n = 7). The difference between genotypes was statistically significant (F = 14.4, p = 0.0022, Fig. 2c, d), indicating stronger tonic GABABR-mediated inhibition of synaptic GABA release in WT neurons.

GABABR agonist-induced and antagonist-induced effects on mIPSC frequency differ in WT and Tscs2 ± cells. a Representative mIPSC traces recorded from a Tsc2+/- neuron demonstrate the baclofen-induced effect. b Baclofen (10 µM) significantly reduces mIPSC frequency both in WT (left) and in Tsc2+/- (middle) neurons. Relative baclofen-induced reduction of mIPSC frequency is significantly larger in Tsc2+/- cells compared with that in WT ones (right panel). c Representative mIPSC traces recorded from a WT cell demonstrate the CGP55845-induced effect. d CGP55845 (1 µM) significantly increases mIPSC frequency in WT (left) but not in Tsc2+/- (middle) neurons. Relative CGP55845-induced elevation of mEPSC frequency is significantly stronger in WT neurons compared with that in Tsc2+/- cells (right panel)
Evoked IPSCs confirm presynaptic location of GABABRs
The above data obtained using CGP55845 suggests that presynaptic GABABRs on GABAergic terminals in WT cortices are tonically activated under control condition. To corroborate this result, we recorded evoked (e)IPSCs using a minimal stimulation approach (see the “Methods” section). Presumably due to the variability of inhibitory inputs (with amplitude ranging from about 80 to 400 pA), the mean eIPSC amplitudes did not differ in the two genotypes (192 ± 37 vs 188 ± 26 pA, n = 14 and 12 for WT and Tsc2+/-, respectively, p = 0.94, F = 0.007, Fig. 3a, b). However, GABAergic inputs demonstrated a different sensitivity to GABABR modulators in WT and Tsc2+/- slices. Baclofen reduced the mean eIPSC amplitude stronger in Tsc2+/- cells (0.49 ± 0.06, n = 12) compared with WT (0.66 ± 0.05, n = 9, p = 0.027, F = 5.66) neurons. CGP55845 increased the mean eIPSC amplitude in WT cells (1.38 ± 0.12, n = 7) in a more pronounced way than in Tsc2+/- neurons (1.03 ± 0.06, n = 8, p = 0.02, F = 7.01, Fig. 3a, c). Next, we applied two consecutive pulses separated by 500 ms interstimulus interval (ISI) and calculated the paired-pulse ratio (PPR500). The latter was defined as the ratio of the mean amplitude of the second eIPSCs to the mean amplitude of the first eIPSCs. If presynaptic GABABRs in WT cortices are tonically activated, their blockade with CGP55845 should decrease the PPR500. Indeed, in the presence of CGP55845, PPR500 was significantly smaller compared with control conditions (0.83 ± 0.04 and 0.72 ± 0.04 in control and in the presence of CGP55845, respectively, n = 7, t = 4.22, p = 0.006, paired Student’s test), thus confirming our hypothesis. In contrast to WT neurons, in Tsc2+/- cells CGP55845 increased PPR500 to 0.91 ± 0.04 from 0.86 ± 0.03 in control (n = 8, t = − 2.9, p = 0.022, paired Student’s test, Fig. 3d). As CGP55845 failed to affect the mean amplitude of the first eIPSCs in Tsc2+/- neurons (Fig. 3c), this data suggests that GABA released by the first stimulus inhibits the second release in an autocrine manner.

GABABR agonist-induced and antagonist-induced effects on eIPSCs. a Representative eIPSC traces recorded from a WT (top) and Tsc2+/- (bottom) cell in control and in the presence of either baclofen (10 µM) or CGP55845 (1 µM). Traces are an average of 20 consecutive recordings. b Mean eIPSC amplitudes are not significantly different in WT and Tsc2+/- neurons. c Baclofen reduces the mean eIPSC amplitude stronger in Tcs2 ± cells compared with WT neurons (left), whereas CGP55845 increases the mean eIPSC amplitude in WT cells but not in Tsc2+/- neurons (right panel). d In the presence of CGP55845 (CGP), the paired-pulse ratio at 500 ms interstimulus interval (PPR500) is reduced in WT neurons (left) but increased in Tsc2+/- ones (right panel)
Presynaptic GABABRs’ effects on the E/I ratio at a single cell level depend on genotype
The above data show that glutamatergic terminals demonstrate comparable sensitivity to GABABR modulators in both genotypes, while this is not the case at GABAergic synapses. Next, we defined the E/I ratio as the ratio of mEPSC frequency to the mIPSC frequency and ask whether GABABR modulators affect this ratio in a genotype-dependent manner. Figure 4 shows that the E/I ratios under control conditions were comparable in WT (1.1 ± 0.1, n = 14) and in Tsc2+/- neurons (1.09 ± 0.03, n = 14, p = 0.71, F = 0.13). Baclofen increased the E/I ratio only slightly in WT cells (1.19 ± 0.04, n = 7), but caused a significantly larger shift towards excitation in Tsc2+/- neurons (1.63 ± 0.13, n = 7). This difference between the two genotypes was statistically significant (n = 17 in both cases, p = 0.009, F = 9.68, Fig. 4a, c). CGP55845 failed to influence the E/I ratio in Tsc2+/- neurons (1.1 ± 0.06, n = 7), but moved the E/I ratio towards inhibition in WT cells (0.71 ± 0.7, n = 7). The difference between the genotypes was statistically significant (p = 0.0008, F = 18.5, Fig. 4b, c). Thus, activation of presynaptic GABABRs results in a shift towards excitation in Tsc2+/- slices. In contrast, in WT slices, the baclofen-induced effect on the E/I ratio is relatively small, whereas CGP55845 causes a robust change in the opposite direction (i.e., towards inhibition).

GABABR agonist-induced and antagonist-induced effects on the E/I ratio at a single cell level. a Representative mEPSC (top) and mIPSC (bottom) traces recorded from the same Tsc2+/- neuron in control (left) and in the presence of baclofen (10 µM, right panel). b Representative mEPSC (top) and mIPSC (bottom) traces of recorded from the same WT cell in control (left) and in the presence of CGP55845 (1 µM, CGP, right panel). c Statistical data shows that the E/I ratios are comparable in WT and Tsc2+/- neurons in control (left). The baclofen-induced increase in the E/I ratio is more pronounced in Tsc2+/- cells than in WT cells (middle). CGP55845 decreases the E/I ratio significantly stronger in WT neurons compared with Tsc2+/- cells (right panel)
GABABR-mediated effects on the network E/I ratio depend on genotype
As GABABRs in mPFC are expressed both presynaptically and postsynaptically, we asked what could be the effect of GABABR modulation on the network (net-)E/I ratio. Using Cs2+-based intrapipette solution and standard ASCF, i.e., without TTX added, we recorded alternatively spontaneous (s)EPSCs and (s)IPSCs from the same cell (Fig. 5). In this configuration, both presynaptic and postsynaptic GABABR will be activated/blocked by baclofen/CGP55845 in the whole slice with exception of the patched cell. In this case, we defined the net-E/I ratio as a ratio of sEPSC frequency to sIPSC frequency. Under control conditions the net-E/I ratios were comparable in WT (0.93 ± 0.03, n = 11) and in Tsc2 (0.85 ± 0.04, n = 11, p = 0.12, F = 2.63). Baclofen failed to affect the net-E/I ratio in WT cells (0.91 ± 0.05, n = 7), but shifted it towards excitation in Tsc2+/- neurons (1.29 ± 0.05, n = 7). This difference was statistically significant (p = 0.001, F = 22.6). CGP55845 reduced the net-E/I ratio in WT neurons (0.77 ± 0.04, n = 6), but only slightly influenced the net-E/I ratio in Tsc2+/- cells (0.96 ± 0.04, n = 7). This difference between the genotypes was statistically significant (p = 0.008, F = 11.1). Thus, on the network level, neither activation nor inhibition of GABABRs shifts the net-E/I ratio towards excitation in WT animals. In contrast to WT slices, GABABR activation strongly potentiates neuronal activity in Tsc2+/- cortices.

GABABR agonist-induced and antagonist-induced effects on network (net-)E/I ratio. a Representative sEPSC (top) and sIPSC (bottom) traces recorded from the same Tsc2+/- neuron in control (left) and in the presence of baclofen (10 µM, right panel). b Representative sEPSC (top) and sIPSC (bottom) traces of recorded from the same WT neuron in control (left) and in the presence of CGP55845 (1 µM, right panel). c Statistical data shows that the net-E/I ratios are comparable in WT and Tsc2+/- neurons in control (left). The baclofen-induced increase in the net-E/I ratio is more pronounced in Tsc2+/- cells than that in WT cells (middle). CGP55845 decreases the net-E/I ratio significantly stronger in WT neurons compared with Tsc2+/- cells (right panel)
Discussion
In this study, we used whole-cell patch-clamp recordings to investigate synaptic function in layer 2/3 pyramidal neurons in the mPFC in acute brain slices obtained from adolescent WT and Tsc2+/- mice. In our previous work, we have shown that by the end of the first postnatal month, tonically activated GABABRs mediate postsynaptic shunting inhibition in WT neurons, but not in Tsc2+/- cells, leading to hyperexcitability of mPFC in Ts2 ± animals [3].
In this study, we show that (1) functional presynaptic GABABRs are present in both excitatory and inhibitory terminals in the mPFC of both WT and Tsc2+/- neurons; (2) in the case of glutamatergic synaptic transmission, GABABR modulations produce comparable effects in the two genotypes; (3) modulation of GABABRs exerts different effects on GABAergic transmission in the two genotypes. In fact, blockade of GABABRs disinhibited GABAergic transmission in WT neurons, but not in Tsc2+/- ones, indicating a tonic GABABR-mediated inhibition of GABA release by ambient GABA in WT slices. On the other hand, activation of GABABRs suppressed GABAergic activity in Tsc2+/- cells much stronger than that in WT neurons, showing that the decreased tonic activation of presynaptic GABABRs in Tsc2+/- neurons is not necessarily mediated by their decreased functionality. Furthermore, (4) the GABABR modulation effects on the network activity differ between the genotypes. Unexpectedly, activation of GABABRs shifts the E/I balance towards excitation in Tsc2+/- mice. Since GABABRs can be activated by ambient GABA, both elevation and decrease of extracellular GABA concentration hardly affect the E/I balance in WT mPFC, while this balancing effect of post-/presynaptic GABABR-mediated inhibition is compromised in Tsc2+/- mPFC.
Presynaptic GABABRs in the mPFC
Expression of functional presynaptic GABABRs at glutamatergic terminals and postsynaptically in pyramidal neurons has been reported in rat mPFC [31]. Baclofen and CGP55432 can decrease and increase sEPSC frequency in pyramidal neurons, respectively. However, as the experiments were performed using a K+-based intrapipette solution, the involvement of postsynaptic GABABRs cannot be completely excluded. In addition, a previous study reported that GABABR modulators affect the duration of UP-states and DOWN-states in the mPFC [31]. Using a genetic approach, an involvement of presynaptic GABABRs in regulation of UP-states and DOWN-states has been directly shown in mouse enthorinal cortex [6]. Instead of genetic ablation of presynaptic GABABRs, we used a Cs+-based intrapipette solution to block postsynaptic K+ conductance and in turn, the effects of postsynaptic GABABRs. As both baclofen and CGP55845 failed to influence the holding currents, membrane resistance and the mean amplitudes of postsynaptic currents, we assume a minimal influence of postsynaptic GABABRs on the parameters measured in this work.
Glutamatergic synaptic transmission
In the first set of experiments, we investigated the effect of modulation of GABABRs activity on glutamatergic synaptic transmission. The presynaptic location of GABABRs to glutamatergic synapses in the mPFC has been previously reported in mice [5] and rats [31]. In the rat mPFC, both baclofen and CGP55845 produced significant effects on sEPSCs. This is in line with the data obtained in this study. Both baclofen and CGP55845 significantly affected the frequency of mEPSCs both in WT and in Tsc2+/- cells (Fig. 1), confirming that functional GABABRs are located on glutamatergic synaptic terminals. The magnitude of GABABR modulator-induced changes of mEPSC frequencies in WT neurons amounted to about 10% in all cases, which is comparable with data reported in [31]. We failed to observe any significant difference between Tsc2+/- and WT cells. As the CGP55845-induced elevation of mEPSC frequency is significant in both genotypes, presynaptic GABABRs on glutamatergic boutons are tonically activated by ambient GABA in both cases. We conclude that glutamatergic terminals both in WT and in Tsc2+/- mPFC express functional GABABRs, and the GABABR-mediated effects are comparable in the two genotypes.
GABAergic synaptic transmission
Although the (patho-)physiological role of presynaptic GABABRs located on GABAergic terminals is still elusive, their possible involvement in the manifestation of neuropsychological symptoms has been postulated. In this work, we report that GABAergic synaptic terminals are sensitive to GABABR modulators both in WT and in Tsc2+/- cortices, confirming that functional GABABRs are located on presynaptic GABAergic boutons in the mPFC. However, the strength of a modulator-induced change depends on the genotype. Stimulation of GABABRs with baclofen decreased mIPSC frequency and the mean eIPCS amplitude significantly stronger in Tsc2+/- neurons compared to WT ones, while the blockade of GABABRs increased mIPSC frequency and the mean eIPSC amplitude in WT cells, but not in Tsc2+/- ones (Figs. 2 and 3). The latter result shows that GABA release from GABAergic terminals is tonically inhibited by ambient GABA in WT cells, but not in Tsc2+/- neurons. Interestingly, GABABR modulators differently affected PPR of eIPSCs in WT and Tsc2+/- neurons. In the WT cells, CGP55845 strongly increased the mean amplitude of the first eIPSCs and decreased PPR, indicating that presynaptic GABA release probability at WT terminals is tonically suppressed by ambient GABA. In contrast, in the Tsc2+/- neurons, CGP55845 did not affect the mean amplitude of the first eIPSCs, but significantly increased PPR at 500 ms ISI, indicating an autocrine GABABR-mediated inhibition of presynaptic GABA release.
Our results indicate that the tonic activation of inhibitory and excitatory synapses via GABABRs is dependent on the type of synapse. A potential explanation could lie in a different local extracellular GABA concentration at excitatory versus inhibitory synapses. We suggest an involvement of GABA transporters (GATs), which level was found to be increased in the amygdala of mice exposed to valproic acid, in conjunction with a reduction in GAD67 [17].
E/I ratio at a single cell level
The observed differences in GABABR-mediated effects on excitatory and inhibitory transmission raise the question of whether presynaptic GABABRs can influence the E/I balance in the mPFC. As we recorded mEPSCs and mIPSCs from the same cell, it is possible to calculate both the E/I ratio and its sensitivity to GABABR modulators in individual neurons. Since both mEPSC and mIPSC frequencies were increased in Tsc2+/- cells compared with that in WT neurons, the E/I ratio was not significantly different in the investigated age group (Fig. 4 and [3]). As CGP55845 hardly affected mIPSC frequency in Tsc2+/- cells, it only slightly shifted the E/I ratio. In contrast, the GABABR blockade strongly reduced the E/I ratio in WT neurons, i.e., shifted it towards inhibition. Given the activation of GABABRs had a strong inhibitory effect on mIPSC frequency in Tsc2+/- neurons, baclofen also caused a marked increase in E/I ratio (Fig. 4c). Surprisingly, in WT cells, baclofen also significantly elevated the E/I ratio, although to a smaller extent (~ 20% versus ~ 60% in WT and Tsc2+/- neurons, respectively). Activation of GABABRs, a Gi/o coupled receptor [19], is traditionally believed to mediate inhibition [16, 32] and a deficit of GABABR-mediated inhibition of glutamatergic inputs can lead to hyperactivity in the cortex [15]. However, in the Tsc2+/- mouse model, presynaptic GABABRs on GABAergic synaptic terminals appear to be not activated by ambient GABA at resting conditions, resulting in higher GABA release probability which is in line with the increased mIPSC frequency recorded. Baclofen or physiologically elevated extracellular GABA concentration can activate them and reduce the phasic inhibitory drive, leading to increased neuronal activity in the mPFC.
E/I ratio on the network level
The above conclusion is valid at a single cell level. As mEPSCs/mIPSCs were recorded in the presence of TTX, a blocker of voltage-gated Na+ channels, and using a Cs+-based intrapipette solution, the influence of postsynaptic GABABRs on cell excitability is probably negligible. On the other hand, usage of Cs+-based intrapipette solution but without TTX added to ACSF would block postsynaptic GABABRs only in the patched neuron, but not in rest of the slice. Recordings of sEPSCs and sIPSCs would then report the effects of GABABR modulators on the complete neuronal network. As both sEPSCs and sIPSCs were acquired from the same cell, the net-E/I ratio can be calculated. Figure 5 and [3] show that the control net-E/I ratios in WT and Tsc2+/- slices were not significantly different at P25–40. As effects of CGP55845 on both postsynaptic and presynaptic GABABRs on Tsc2+/- neurons were rather minimal, GABABR blockade produced minimal effects on the net-E/I ratio in Tsc2+/- genotype. Although CGP55845 blocked tonically activated postsynaptic GABABRs in WT slices, disinhibition of phasic GABAergic transmission shifted the net-E/I ratio towards inhibition (Fig. 5). And vice versa, although baclofen via presynaptic GABABRs inhibited phasic GABA release, in parallel, it activated postsynaptic GABABRs shifting the net-E/I ratio towards inhibition as well. Thus presynaptic and postsynaptic GABABRs operate in concert to prevent hyperactivity of mPFC in WT cortex independently on the extracellular GABA level. This is however not the case in the Tsc2+/- mPFC, where GABABR activation strongly suppressed phasic GABA release, and the lack of postsynaptic GABABR-mediated inhibition ultimately resulted in an overall excitatory baclofen action on the network (Fig. 5). These results obtain particular relevance if we consider that modulation of GABABRs has been contemplated as a potential treatment for ASD symptoms in both mice and humans. The overall excitatory effect of baclofen on the network activity observed in this study should be taken into consideration in case of therapeutic application of drugs acting on GABABRs.
References
Antoine MW, Langberg T, Schnepel P, Feldman DE (2019) Increased excitation-inhibition ratio stabilizes synapse and circuit excitability in four autism mouse models. Neuron 101:648–661. https://doi.org/10.1016/j.neuron.2018.12.026
Auerbach BD, Osterweil EK, Bear MF (2011) Mutations causing syndromic autism define an axis of synaptic pathophysiology. Nature 480:63–68. https://doi.org/10.1038/nature10658
Bassetti D, Lombardi A, Kirischuk S, Luhmann HJ (2020) Haploinsufficiency of Tsc2 leads to hyperexcitability of medial prefrontal cortex via weakening of tonic GABAB receptor-mediated inhibition. Cereb Cortex 30:6313–6324. https://doi.org/10.1093/cercor/bhaa187
Bateup HS, Johnson CA, Denefrio CL, Saulnier JL, Kornacker K, Sabatini BL (2013) Excitatory/inhibitory synaptic imbalance leads to hippocampal hyperexcitability in mouse models of tuberous sclerosis. Neuron 78:510–522. https://doi.org/10.3389/fnmol.2013.00028
Burke KJ Jr, Keeshen CM, Bender KJ (2018) Two forms of synaptic depression produced by differential neuromodulation of presynaptic calcium channels. Neuron 99:969–984. https://doi.org/10.1016/j.neuron.2018.07.030
Craig MT, Mayne EW, Bettler B, Paulsen O, McBain CJ (2013) Distinct roles of GABAB1a- and GABAB1b-containing GABAB receptors in spontaneous and evoked termination of persistent cortical activity. J Physiol 591:835–843. https://doi.org/10.1113/jphysiol.2012.248088
Dabora SL, Jozwiak S, Franz DN, Roberts PS, Nieto A, Chung J, Choy YS, Reeve MP, Thiele E, Egelhoff JC, Kasprzyk-Obara J, Domanska-Pakiela D, Kwiatkowski DJ (2001) Mutational analysis in a cohort of 224 tuberous sclerosis patients indicates increased everity of TSC2, compared with TSC1, disease in multiple organs. Am J Hum Genet 68:64–80
Ehninger D, Han S, Shilyansky C, Zhou Y, Li W, Kwiatkowski DJ, Ramesh V, Silva AJ (2008) Reversal of learning deficits in a Tsc2+/- mouse model of tuberous sclerosis. Nat Med 14:843–848. https://doi.org/10.1086/316951
Gao X, Zhang Y, Arrazola P, Hino O, Kobayashi T, Yeung RS, Ru B, Pan D (2002) Tsc tumour suppressor proteins antagonize amino-acid-TOR signalling. Nat Cell Biol 4:699–704. https://doi.org/10.1038/ncb847
Han S, Tai C, Jones CJ, Scheuer T, Catterall WA (2014) Enhancement of inhibitory neurotransmission by GABAA receptors having alpha2,3-subunits ameliorates behavioral deficits in a mouse model of autism. Neuron 81:1282–1289. https://doi.org/10.1016/j.neuron.2014.01.016
Hasbani DM, Crino PB (2018) Tuberous sclerosis complex. Handb Clin Neurol 148:813–822. https://doi.org/10.1016/B978-0-444-64076-5.00052-1
Heaney CF, Kinney JW (2016) Role of GABA(B) receptors in learning and memory and neurological disorders. Neurosci Biobehav Rev 63:1–28. https://doi.org/10.1016/j.neubiorev.2016.01.007
Inoki K, Li Y, Zhu T, Wu J, Guan KL (2002) TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol 4:648–657. https://doi.org/10.1038/ncb839
Islam MP, Roach ES (2015) Tuberous sclerosis complex. Handb Clin Neurol 132:97–109. https://doi.org/10.1016/B978-0-444-62702-5.00006-8
Kang JY, Chadchankar J, Vien TN, Mighdoll MI, Hyde TM, Mather RJ, Deeb TZ, Pangalos MN, Brandon NJ, Dunlop J, Moss SJ (2017) Deficits in the activity of presynaptic gamma-aminobutyric acid type B receptors contribute to altered neuronal excitability in fragile X syndrome. J Biol Chem 292:6621–6632. https://doi.org/10.1074/jbc.M116.772541
Kohl MM, Paulsen O (2010) The roles of GABAB receptors in cortical network activity. Adv Pharmacol 58:205–229. https://doi.org/10.1016/s1054-3589(10)58009-8
Olexová L, Štefánik P, Kršková L (2016) Increased anxiety-like behaviour and altered GABAergic system in the amygdala and cerebellum of VPA rats - an animal model of autism. Neurosci Lett 629:9–14. https://doi.org/10.1016/j.neulet.2016.06.035
Onda H, Lueck A, Marks PW, Warren HB, Kwiatkowski DJ (1999) Tsc2(+/-) mice develop tumors in multiple sites that express gelsolin and are influenced by genetic background. J Clin Invest 104:687–695. https://doi.org/10.1172/JCI7319
Padgett CL, Slesinger PA (2010) GABAB receptor coupling to G-proteins and ion channels. Adv Pharmacol 58:123–147. https://doi.org/10.1016/s1054-3589(10)58006-2
Paxinos G, Franklin K (2012) The mouse brain. Elsevier Academic Press.
Perez-Zabalza M, Reig R, Manrique J, Jercog D, Winograd M, Parga N, Sanchez-Vives MV (2020) Modulation of cortical slow oscillatory rhythm by GABA(B) receptors: an in vitro experimental and computational study. J Physiol 598:3439–3457. https://doi.org/10.1113/jp279476
Robertson CE, Ratai EM, Kanwisher N (2016) Reduced GABAergic action in the autistic brain. Curr Biol 26:80–85. https://doi.org/10.1016/j.cub.2015.11.019
Selten MM, Meyer F, Ba W, Valles A, Maas DA, Negwer M, Eijsink VD, van Vugt RW, van Hulten JA, van Bakel NH, Roosen J, van der Linden RJ, Schubert D, Verheij MM, Kasri NN, Martens GJ (2016) Increased GABAB receptor signaling in a rat model for schizophrenia. Sci Rep 6:34240. https://doi.org/10.1038/srep34240
Silverman JL, Pride MC, Hayes JE, Puhger KR, Butler-Struben HM, Baker S, Crawley JN (2015) GABAB receptor agonist R-baclofen reverses social deficits and reduces repetitive behavior in two mouse models of autism. Neuropsychopharmacology 40:2228–2239. https://doi.org/10.1038/npp.2015.66
Stoppel LJ, Kazdoba TM, Schaffler MD, Preza AR, Heynen A, Crawley JN, Bear MF (2018) R-Baclofen reverses cognitive deficits and improves social interactions in two lines of 16p11.2 deletion mice. Neuropsychopharmacology 43:513–524. https://doi.org/10.1038/npp.2017.236
Tang SJ, Reis G, Kang H, Gingras AC, Sonenberg N, Schuman EM (2002) A rapamycin-sensitive signaling pathway contributes to long-term synaptic plasticity in the hippocampus. Proc Natl Acad Sci U S A 99:467–472. https://doi.org/10.1073/pnas.012605299
Tavazoie SF, Alvarez VA, Ridenour DA, Kwiatkowski DJ, Sabatini BL (2005) Regulation of neuronal morphology and function by the tumor suppressors Tsc1 and Tsc2. Nat Neurosci 8:1727–1734. https://doi.org/10.1038/nn1566
Tong XJ, Hu Z, Liu Y, Anderson D, Kaplan JM (2015) A network of autism linked genes stabilizes two pools of synaptic GABA(A) receptors. Elife 4:e09648. https://doi.org/10.7554/eLife.09648
Unichenko P, Dvorzhak A, Kirischuk S (2013) Transporter-mediated replacement of extracellular glutamate for GABA in the developing murine neocortex. Eur J Neurosci 38:3580–3588. https://doi.org/10.1111/ejn.12380
von der Brelie C, Waltereit R, Zhang L, Beck H, Kirschstein T (2006) Impaired synaptic plasticity in a rat model of tuberous sclerosis. Eur J Neurosci 23:686–692. https://doi.org/10.1111/j.1460-9568.2006.04594.x
Wang Y, Neubauer FB, Luscher HR, Thurley K (2010) GABAB receptor-dependent modulation of network activity in the rat prefrontal cortex in vitro. Eur J Neurosci 31:1582–1594. https://doi.org/10.1111/j.1460-9568.2010.07191.x
Yudin Y, Rohacs T (2018) Inhibitory G(i/O)-coupled receptors in somatosensory neurons: potential therapeutic targets for novel analgesics. Mol Pain 14:1–16. https://doi.org/10.1177/1744806918763646
Zeidler S, Pop AS, Jaafar IA, de Boer H, Buijsen RAM, de Esch CEF, Nieuwenhuizen-Bakker I, Hukema RK, Willemsen R (2018) Paradoxical effect of baclofen on social behavior in the fragile X syndrome mouse model. Brain Behav 8:e00991. https://doi.org/10.1002/brb3.991
Zhao JP, Yoshii A (2019) Hyperexcitability of the local cortical circuit in mouse models of tuberous sclerosis complex. Mol Brain 12:6–0427. https://doi.org/10.1186/s13041-019-0427-6
Acknowledgements
The technical assistance of Beate Krumm and Nicole Knauer is highly appreciated.
Funding
Open Access funding enabled and organized by Projekt DEAL. This work was supported by the Deutsche Forschungsgemeinschaft (DFG, grant CRC1080-B10 to HJL).
Author information
Authors and Affiliations
Contributions
Conceptualization, SK and HJL; data acquisition and analysis, DB and SK; and writing and editing, DB, SK and HJL.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bassetti, D., Luhmann, H.J. & Kirischuk, S. Presynaptic GABAB receptor–mediated network excitation in the medial prefrontal cortex of Tsc2+/- mice. Pflugers Arch - Eur J Physiol 473, 1261–1271 (2021). https://doi.org/10.1007/s00424-021-02576-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-021-02576-5