
Abstract
Macrophages reside in virtually every organ. First arising during embryogenesis, macrophages replenish themselves in the adult through a combination of self-renewal and influx of bone marrow-derived monocytes. As large phagocytic cells, macrophages participate in innate immunity while contributing to tissue-specific homeostatic functions. Among the key metabolic tasks are senescent red blood cell recycling, free heme detoxification, and provision of iron for de novo hemoglobin synthesis. While this systemic mechanism involves the shuttling of iron between spleen, liver, and bone marrow through the concerted function of defined macrophage populations, similar circuits appear to exist within the microenvironment of other organs. The high turnover of iron is the prerequisite for continuous erythropoiesis and tissue integrity but challenges macrophages’ ability to maintain cellular iron homeostasis and immune function.
This review provides a brief overview of systemic, microenvironmental, and cellular aspects of macrophage iron handling with a focus on exciting and unresolved questions in the field.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Systemic aspects of iron recycling
The mononuclear phagocyte system (MPS) encompasses monocytes and macrophages residing throughout the body [9, 82, 85]. A central role of this multifunctional system beyond immunity and tissue repair is to control the body’s metabolic needs for iron. The maintenance of iron homeostasis at the systemic level requires iron recycling (Fig. 1), much of which is contained in senescent red blood cells (RBC). This is a prerequisite for sufficient de novo synthesis of hemoglobin (Hb) in the bone marrow (BM), a process which consumes as much as 20–25 mg of iron per day [78, 179]. The capacity of the small intestine to absorb dietary iron, in comparison to the daily needs of iron, is rather low; in humans under steady-state conditions, 1–2 mg of iron is absorbed from the diet in the duodenum and upper jejunum. Thus, dietary iron absorption only compensates for obligatory losses of the metal, mainly through desquamation of the epidermis and the intestinal epithelium, and menstrual bleeding [168].

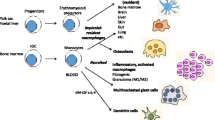
a In steady state, 20–25 mg/day of iron is needed to generate new red blood cells (RBC) in the bone marrow (depicted as vertebral body). The RBC of an adult human form the body’s largest iron pool (1500–2500 mg contained in hemoglobin [Hb]). After an average lifetime of 120 days, aged RBC (aRBC) are trapped in the spleen, recognized by red pulp macrophages (RPM), and eliminated. Their Hb is recycled, and ferroportin (Fpn)-1 exports iron into sinusoidal capillaries where it is loaded onto transferrin (TF). TF-bound iron is utilized by developing erythroblasts (EB) via TF receptor (Tfr)-1. Erythroid island macrophages assist in iron uptake and differentiation. Plasma iron content is sensed by hepatocytes, presumably via Tfr1 and other iron-sensitive molecules. They affect the transcriptional activation of hepcidin, the major iron hormone, in hepatocytes. Hepcidin acts as negative feedback regulator of iron influx by targeting Fpn1 resulting in degradation of the latter and thus reduction of iron transfer from the diet and macrophages to the plasma. Kupffer cells (KC) inhibit hepcidin expression by hepatocytes and also engage in erythrophagocytosis. b The lifespan of RBC can be reduced as a consequence of autoinflammation and subsequent hemolysis, radical formation and toxin activity or based on intrinsic structural or metabolic defects. The rapid accumulation of damaged RBC (dRBC) may overwhelm the spleen’s clearance capacity. In such a scenario, KC engulf dRBC which may result in cell death. As back-up system, monocytes are recruited from the bone marrow and possibly spleen via chemokines CCL2 and CCL3 and their receptor CCR2. These monocytes encounter a niche in the liver and differentiate into KC-like cells that express Fpn1 which is induced by several mechanisms including the growth factor macrophage colony-stimulating factor (M-CSF). Fpn1-mediated iron export sustains increased erythropoiesis in the bone marrow to compensate for losses by RBC damage
Intestinal iron absorption is regulated, to a large extent, at the basolateral membrane of enterocytes [59, 248]. It is at this location that ferroportin-1 (Fpn1; AKA solute carrier family 40 member 1) fulfills its function as the sole cellular efflux protein for ionic iron, shuttling apically absorbed iron to the circulation, where it is oxidized and loaded onto transferrin (TF) [173]. The presence of Fpn1 in the absorptive epithelium is under negative control of hepcidin, which is mainly secreted by hepatocytes in response to high circulating and tissue amounts of iron or upon stimulation by inflammatory mediators [171, 229]. This negative feedback loop exists because of the Janus-faced nature of iron. Iron forms the central cation of hemoglobin, other heme-containing proteins (such as myoglobin and cytochrome P450 enzymes including the mitochondrial electron transport chain), and iron-sulfur cluster-containing enzymes found in most cells [179, 180]. Free labile forms of iron and heme, however, are potentially toxic and threaten tissue integrity due to their pro-oxidative properties which are largely based on the capacity of iron to non-enzymatically boost reactive oxygen species (ROS) production via Fenton chemistry [89, 119].
Given its essential role in metabolism and cellular processes such as mitochondrial respiration, an efficient and tightly controlled pathway for maintaining iron in the body must exist and likely depends on factors controlling the recognition, uptake, and degradation of erythrocytes by macrophages [113]. In steady-state conditions, RBC recycling mainly takes place in the spleen [48, 213]. In conditions of excessive RBC damage, liver macrophages (Kupffer cells, henceforward abbreviated as KC) dominate RBC clearance (as detailed in “Liver—the iron regulatory organ” section) [231].
During massive hemolysis, circulating hemoglobin (Hb) and heme are bound by hemopexin (HPX) and haptoglobin, respectively, to avoid intravasal radical formation via the catalytic action of iron [203, 235, 244]. However, when the capacity of red pulp macrophages (the iron-recycling macrophage population in the spleen, henceforward abbreviated as RPM) and KC is overwhelmed, the cells die, presumably by a specific form of cell death called ferroptosis that is induced by iron-mediated oxidative stress and lipid peroxidation [54]. To compensate, blood-derived monocytes fill the partial niche after KC loss and differentiate into a transient macrophage population that has a unique phenotype including the high expression of the CC-chemokine receptor 2 (CCR2) [231]. Indeed, monocyte recruitment from the BM to the liver is mediated by a liver-specific, KC-derived CC-chemokine gradient of CCL2 and CCL3 that attracts CCR2+ CCR5+ monocytes to the organ. As a consequence, the BM enhances myelopoiesis and releases additional monocytes to the circulation to meet the increased demand (Fig. 1b). This chemokine-mediated monocyte recruitment is essential because if disrupted pharmacologically, damaged RBC accumulate in glomerular capillaries resulting in renal failure [231]. Comparable phenomena of KC death, although necroptotic by nature, and replacement of KCs from BM-derived monocytes have been reported in Listeria monocytogenes infection [21] raising the possibility that the mechanism is a general stress response that can be fine tuned according to whether the initial stressor is endogenous (e.g., RBC, heme) or exogenous (e.g., a pathogen).
In theory, additional mechanisms by which macrophages may influence systemic iron homeostasis (other than its recycling) include control of dietary iron absorption or excretion. It is generally thought that no regulated pathway of iron excretion exists in the mammalian organism. Duodenal iron absorption, however, appears to rely on macrophages, albeit indirectly via modulating hepcidin production by hepatocytes (see “Liver—the iron regulatory organ” section). To the best of our knowledge, no data exist on whether lamina propria macrophages in the small intestine affect absorptive enterocyte iron transporter expression or function, although this is possible given that macrophages produce small amounts of hepcidin in response to inflammatory stimuli [185, 227]. However, one might speculate that intestinal infection might alter absorption by shifting intestinal stem cell differentiation towards non-absorptive progeny such as Paneth or Tuft cells and by promoting recruitment of inflammatory macrophages [39, 64]. Although hypothetical, such a mechanism might be initiated by myeloid cells since lamina propria macrophages and dendritic cells (DCs) will be one of the first cell types to engage in host response to invasive pathogens [125].
Erythrophagocytosis
The physiologic senescence of RBC is associated with mechanical and biochemical alterations of their membrane such as reduced flexibility, exposure of phosphatidylserine on their surface, and clustering of band 3 followed by binding of antibodies and complement [97, 237]. The microanatomy of splenic sinusoidal capillaries prevents the unhindered passage of these senescent RBC. Macrophages, however, recognize cell surface markers of senescence via scavenger receptors such as Tim4, SR-AI, and CD36 [114, 155, 156, 202, 251]. Damaged and aged RBC are engulfed by RPM (flow cytometrically phenotyped as F4/80low CD11blow Fpn1+) and phagocytosed. The molecular machinery of RBC recycling is relatively well described, and its function is essential to tissue integrity given the pro-oxidative capacity of free labile heme. This capacity is potentiated by the extraordinarily high number of about 1.2 × 109 heme moieties contained within a single RBC and the fact that RBC are the body’s most abundant cell type comprising approximately 84% of our cells [118, 208]. It thus appears conceivable that a stand-by mechanism for heme detoxification exists: The molecular switch to turn on the protective pathways within cells is the transcriptional repressor Btb and Cnc homology 1 (Bach1). Bach1 is a basic leucine zipper (bZip) transcriptional factor which has a high affinity for heme, enabling it to act as a sensor of cellular heme levels. Rising heme levels induce Bach1 proteosomal degradation, releasing the brake on two types of down-stream pathways. First, the competitive inhibition of nuclear factor (erythroid-derived 2)-like 2 (Nrf2) binding to antioxidant-response elements (ARE) ceases for genes containing AREs [51, 145, 221], such as ferritin h chain (FTH), FPN1, heme-regulated gene-1 (HRG1), and heme oxygenase (Hmox1) [102, 190]. Second, the degradation of Bach1 induces expression of the transcription factor (an E26 transformation-specific transcription factor of the Spi subfamily) SpiC independently of Nrf2 [116, 211]. SpiC is a transcription factor that is predominately expressed by RPM and erythroid island macrophages (EIM) in the BM [88]. It is essential to the development of these cell types as Spic −/− mice have drastically reduced numbers of RPM and EIM which lead to reduced proerythroblast numbers in the steady state, supporting the idea that iron recycling is essential to sustain iron-sufficient erythropoiesis [58].
Hypoxia-inducible factors (HIFs) provide an alternative mechanism to adjust heme iron recycling by macrophages to the degree of anemia and the need for de novo Hb synthesis. Anemia inevitably results in low oxygen concentrations in the tissue microenvironment which counteract HIF degradation in the proteasome so that these transcription factors become available to trans-activate genes containing hypoxia-responsive elements (HREs). HIF-HRE interaction in response to hypoxia thus stimulates iron release from macrophages by inducing Hmox1 and Fpn1 as well as one of its associated ferroxidases, ceruloplasmin [37, 63, 200]. Although many genes involved in heme iron recycling are thus HIF-inducible, HIF-1α and HIF-2α in macrophages are dispensable for heme iron recycling under hemolytic stress [147]. This suggests that HIF isoforms fulfill redundant functions in macrophages or that they are not essential because Bach1-controlled pathways are dominant.
Bach1 is not the only cellular sensor for heme. Intracellular free heme can also induce signaling via nuclear receptors Rev-Erbα and Rev-Erbß [29, 132, 187]. Furthermore, the inflammasome activation via cryopyrin (AKA NLRP3) is sensitive to surplus intracellular heme while extracellular free heme can activate Toll-like receptor 4 (TLR4), supporting the idea that free heme is an endogenous danger signal [60, 65, 66, 131]. Given its inherent potential toxicity, the majority of free intracellular heme is promptly degraded by Hmox. Hmox1, which itself is induced via Nrf2 in response to heme or other oxidative stressors, is widely expressed in the MPS. Hmox2, by contrast, is a constitutive isoform carrying important functions in the nervous and reproductive systems [89]. Both Hmox isoenzymes cleave heme’s protoporphyrin ring which yields bilirubin, carbon monoxide, and iron in equimolar amounts. The iron released during this process is either stored within ferritin (FT), incorporated into iron-containing proteins, or shuttled to the extracellular space via Fpn1. Outside the cell, ferrous iron needs to be oxidized to its ferric form by cell membrane-anchored hephaestin or plasmatic ceruloplasmin. Thereafter, ferric iron can be bound to transferrin (TF) and the bulk of it will re-enter the BM and be utilized in RBC production. Therefore, the incorporation of iron into protoporphyrin IX to form a new heme moiety closes the circle of heme iron recycling.
Local circuits
Liver—the iron regulatory organ
The liver acts as central orchestrator of iron metabolism through various mechanisms. First, among many other carrier proteins, hepatocytes synthesize TF to cargo ferric iron in the circulation and keep free ionic iron at minimal levels. Second, even small fluctuations in plasma iron such as those originating from a single dose of an iron supplement are sensed by the liver and counterbalanced by an accordant change in hepcidin expression [158]. An increase in plasma iron stimulates hepcidin secretion which dampens macrophage iron egress and to a lesser extent duodenal iron absorption following its interaction with Fpn1. Conversely, KC appear to dampen hepatocytes’ hepcidin expression since KC depletion with clodronate liposomes augments hepcidin and leads to a dramatic drop in plasma iron levels according to one study [228]. However, other data suggest that KC are dispensable for the induction of hepcidin in response to iron or inflammation. Studies in Il-6 −/− mice confirmed that IL-6 is an important mediator to induce hepcidin in response to TLR4 ligation but KC depletion did not alter hepcidin induction in this model [137]. While the physiological role of these observations therefore remains controversial [137, 157], it is tempting to speculate that KC may fine-tune hepcidin production or that a liver-intrinsic circuit reports on the amount of iron acquired by KC through erythrophagocytosis. A diphtheria toxin-mediated depletion of C-type lectin domain family 4 member F (Clec4F), a marker specific to KC, might help to test this hypothesis [207].
Hepatocytes are also central effectors of the acute phase reaction that is induced during extracellular hemolysis and thereby produce haptoglobin and HPX which are rapidly consumed. Haptoglobin and HPX scavenge free Hb and heme, respectively, and are ligands to CD163 and CD91 which are primarily present on macrophages including KC [100, 120]. The destruction of large amounts of RBC in the circulation further activates F4/80+ CD11blow KC to respond with erythrophagocytosis. KCs have a limited tolerance to iron toxicity and die in the setting of massive hemolysis as discussed above [231].
Erythrophagocytosis augments Fpn1 and FT expression by F4/80+ CD11b+ liver-infiltrating monocytes that differentiate to macrophages which transiently replace dying KC. In the liver, M-CSF stimulates Fpn1 expression while GM-CSF, which is produced in the spleen, represses Fpn1 [231]. Upon differentiation, these F4/80+ CD11b+ Fpn1+ monocytes also express SpiC and bone morphogenetic protein 6 (Bmp6). While SpiC may program them to recycle iron efficiently, the iron-inducible factor, Bmp6, may have paracrine effects on adjacent hepatocytes by instructing them to increase hepcidin expression, thereby lowering circulating iron levels. However, the major production of Bmp6 in the liver is by yet another liver-resident cell type, sinusoidal endothelial cells [28]. Since Bmp6 stimulates both hepcidin production by hepatocytes [6] and transcriptionally regulates macrophage effector functions [123], it may broadly orchestrate the interplay between various liver-resident cell types. Intriguingly, there might be direct transfer of iron from macrophages to hepatocytes after erythrophagocytosis because iron contained in damaged RBC first accumulates in myeloid cells but is distributed to hepatocytes within 1 week. The underlying mechanism remains elusive but may employ Fpn1 as well as alternative routes of iron trafficking via FT secretion and uptake via FT receptors [34, 90, 130]. In fact, it has been shown that macrophages are the major source of plasma FT [40].
The importance of Fpn1 for liver cell functions is evident from the clinical manifestations of ferroportin disease (type 4 hemochromatosis) which is caused by various mutations in the SLC40A1 (encoding FPN1) gene [148, 189]. Classical loss-of-function mutations of SLC40A1 result in reduced iron transport capacity that are attributable to an amino acid exchange in the iron pore or to protein misfolding of FPN1. The mutations mainly manifest in the MPS (i.e., by iron retention in KC) because the high iron turnover of macrophages cannot be met by 50% of functional FPN1 molecules transcribed from the intact SLC40A1 allele [58, 149]. However, non-classical Fpn1 mutations SLC40A1 are gain-of-function mutations that cause hepcidin resistance of the FPN1 protein. In this case, hepcidin cannot bind to FPN1 and does not cause its internalization and lysosomal degradation. Consequently, a phenotype of functional hepcidin deficiency that is similar to classical type I hemochromatosis with hepatocellular iron overload occurs [2, 188, 246]. In summary, hepatocytes, KC, and endothelial cells cooperate to maintain the body’s iron homeostasis and we are beginning to decipher the signals that coordinate the function of these and other liver-resident cell types.
Spleen—iron recycler in steady state
Similar to the liver, several cell populations co-exist in the spleen. RPM ingest erythrocytes in steady state, eliminating more than 2 × 106 of these cells per second in a healthy adult (as deduced from the RBC production rate assuming equilibrium). This enormous task has implications for the spleen’s immune function. For instance, CD47 is a “do not eat me” signal expressed by many cell types including RBC [178]. CD47 is recognized by signal regulatory protein alpha (SIRPα) on macrophages which then ignore the intact RBC. In contrast, CD47-deficient RBC are rapidly cleared from the circulation. Aging RBC partially lose surface expression of CD47 [109]. Given that CD47 specifically and substantially inhibits FcγR-mediated phagocytosis of IgG-carrying RBC, the CD47 reduction with senescence is thought to enable the elimination of aged RBC. RPM are not the only ones that recognize CD47, though. The absence of CD47 on RBC is also sensed by splenic CD4+ DCs. Lacking the inhibitory input of CD47-SIRPα interaction, they are licensed to augment MHC-II, CD86, and CCR7 and start their migration to the T cell zone to stimulate T cells [253]. However, it remains unclear whether or not erythrophagocytosis by RPM and CD4+ DC activation affect each other.
In response to increased demand for erythrophagocytosis, the spleen also produces abundant amount of GM-CSF, which inhibits Fpn1 expression in macrophages [231]. It has thus been proposed that the emergency mechanism of iron recycling is less efficient under stress conditions in the spleen. This fundamental difference between spleen and liver may be based on the fact that iron alters the binding affinity of antibodies: Both ferrous iron and free heme interact with IgG to enlarge the antibody repertoire. Specifically, heme-IgG complexes have a broader ability to recognize bacterial antigens and kill intact bacteria. In addition, Fe-IgG complexes protect from E. coli sepsis suggesting another possible link between erythrophagocytosis and adaptive immunity [52, 53]. Another aspect underlying reduced erythrophagocytosis in the spleen under stress conditions including infections can be attributed to the fact that iron loading of macrophages dampens their innate anti-microbial effector function directed against invading pathogens [212]. While the function of RPM in continuous erythrophagocytosis is clear, we are lacking a comprehensive understanding of how iron recycling in the splenic red pulp and the organ’s immune functions affect each other.
Muscle—venue for a closed circuit for iron recycling following myolysis?
A mechanism comparable to hemolysis-induced RBC recycling in the liver has recently been described in the skeletal muscle in the context of rhabdomyolysis. Rhabdomyolysis, the death of the skeletal muscle, poses a fundamental risk for the body’s integrity because myoglobin, the oxygen transfer protein, is a heme-containing protein that can contribute to iron-mediated radical damage. Additionally, myoglobin’s molecular weight of 17 kDa allows it to penetrate the glomerular filtration apparatus of the kidney which operates at a cutoff of 30–50 kDa so that in rhabdomyolysis large quantities of myoglobin end up in the renal tubuli where they can cause acute kidney injury (crush kidney). Therefore, the skeletal muscle is equipped with a machinery that protects from free myoglobin/heme-mediated damage. This includes HPX formation and myeloid cell recruitment. Macrophages infiltrating the injured skeletal muscle induce the expression of Hmox1, CD163, and FT to contain and detoxify heme [41]. It has further been suggested that the induction of Hmox1 and Fpn1 by the infiltrating monocytes allows for local heme iron recycling so that the regenerating myoblasts have enough iron for de novo myoglobin synthesis. It will be interesting to see whether myeloid cells also affect iron metabolism of skeletal muscle during more physiological conditions such as growth and whether there are perturbations in myopathies other than rhabdomyolysis.
Heart—an organ exquisitely sensitive to iron toxicity
The presence of a local mechanism of iron recycling by monocytes in skeletal muscle raises the question whether similar circuits exist throughout the body. Given that myocardial ischemia induces substantial recruitment of monocytes from blood, BM, and spleen to the site of damage and its vicinity, these cells may also encounter myoglobin molecules. However, the adult myocardium lacks regenerative potential suggesting that iron handling by monocytes infiltrating the injured heart muscle may be different from that of monocytes infiltrating damaged skeletal muscle [92]. Alternatively, it is possible that cardiac fibroblasts use iron recycled by monocytes for proliferation and collagen synthesis [80].
Resident cardiac macrophages are one of the major non-parenchymal cells of the heart [223]. They have well-established roles in the pathogenesis of ischemic myocardial injury as well as in tissue repair thereafter [93, 96]. In addition, their roles in the metabolic activity of the adjacent myocardium and in chronic cardiac diseases such as congestive heart failure are increasingly appreciated [199], and macrophages may either tailor the delivery of iron to the myocardium or accumulate and detoxify heme which may mainly arise from tissue injury [161, 214].
The susceptibility of the heart to pathological iron loading is well reflected in Friedreich’s ataxia, an autosomal recessive disease with a complex neurological phenotype that inevitably affects the heart; congestive heart failure is the most frequent cause of death in these patients. Frataxin localizes to mitochondria where it fulfills functions in the assembly of iron-sulfur clusters acting as the mitochondrial iron chaperone [24].
Given the extraordinary energy generation by mitochondria in cardiomyocytes, it is likely that the cardiac phenotype of Friedreich’s ataxia is primarily a parenchymal one that occurs as a consequence of mitochondrial heme accumulation due to defective iron utilization [98]. However, it is interesting to note that the number of CD68+ macrophages is increased in the myocardium of Friedreich ataxia patients and that these cells contain higher amounts of FT. It has been proposed that Fpn1-mediated iron export from cardiac macrophages may be impaired in Friedreich’s disease [115].
Since cardiomyocytes are exquisitely sensitive to iron overload and iron overload disorders commonly affect the heart, a better understanding of iron handling by resident or recruited myeloid cells in the cardiac microenvironment may open new therapeutic options. However, cardiac macrophages are already an established diagnostic target of contrast material in magnetic resonance imaging (MRI). Specifically, nanoscale iron-containing compounds (IONs, for iron oxide nanoparticles) have been designed to being selectively taken up by macrophages [183]. After exposure to magnetic fields, these compounds cause a signal alteration which is affected by processes such as ischemia or inflammation secondary to macrophage activation.
Blood vessels—arterial macrophage iron overload as hallmark of atherosclerosis
Iron accumulation in arterial macrophages has long been suggested to contribute to the pathogenesis of atherosclerosis [220]. A comprehensive pathophysiological understanding of this observation is still pending. However, it is well documented that arterial macrophages, typically identified by CD68 histochemistry on histologic sections within atherosclerotic lesions, contain larger amounts of FT than those outside plaques [129, 256]. Accordingly, high levels of plasma FT are an independent risk factor for the severity of carotid artery disease [110, 111], and high circulating iron levels have been linked to impaired endothelial function and intima media thickening, two early predictors of atherosclerosis [76]. Published evidence suggests that microhemorrhages occur in the vasa vasora of the arterial wall and that macrophages remove RBC debris by phagocytosis, which increases their iron levels without overt activation [22, 87, 201]. In addition, atherosclerosis is a lipid-driven chronic inflammatory disorder closely linked to the metabolic syndrome, and higher plasma hepcidin levels are associated with vascular damage as documented in a small but well-conducted ultrasonographic pilot study [238]. That said, hepcidin levels do not predict an increased risk for myocardial infarction or stroke in a population-based study [182].
The specific role of macrophage iron efflux in the progression of atherosclerosis is controversial. While the presence of the hypofunctional flatiron mutation of Slc40a1 in apolipoprotein E-deficient (Apoe −/−) mice did not affect atherosclerosis [107], systemic administration of the hepcidin-antagonist LDN-193189 (LDN), which inhibits Bmp-induced hepcidin transcription [10, 27, 230], blocked the differentiation of macrophages into foam cells. This latter effect was based on enhanced expression of ATB-binding cassette transporters ABCA1 and ABCG1, two major cholesterol efflux proteins, and on increased cholesterol transfer to apolipoprotein A1 (ApoA1) [198]. Importantly, LDN treatment also reduced atherosclerotic lesion size, suggesting that modulating systemic iron metabolism affects atherosclerosis progression. A more selective approach that only targets arterial wall macrophages may be warranted as the metabolic syndrome can be associated not only with functional iron deficiency (predicted to be ameliorated by hepcidin antagonism) but also with iron overload (predicted to be aggravated by systemic hepcidin inhibition).
The dysregulation of iron metabolism in arterial macrophages is not limited to the hepcidin-Fpn1 axis. In humans, expression of the ferroxidases hephaestin and ceruloplasmin was reduced in atherosclerosis [105]. Accordingly, there is in vitro evidence that iron-induced ROS contribute to LDL oxidation and that oxidized LDL particles induce FTH and hepcidin transcription in macrophages which may reduce Fpn1 levels and thus retain cellular iron [144].
In conclusion, evidence is accumulating for a cross-talk between the regulators of iron and cholesterol metabolism both systemically and locally in the microenvironment of the atherosclerotic plaque. However, due to a lack of mechanistic insight, we are not able yet to therapeutically target one metabolite or metabolic pathway in the attempt to beneficially affect the other.
Brain—cerebral iron overload is implicated in neurodegeneration
In the central nervous system (CNS), macrophages appear as microglia and are in close proximity to neurons, astrocytes, and oligodendrocytes. The microglia depend on the transcription factors PU.1 (encoded by SPI1) and “Spalt-like”-1 (Sall1) [25] and are important for maintaining CNS homeostasis in physiological conditions and for restoring it after injury [84]. This paradigm has also been addressed in the context of iron metabolism. For instance, after experimental intracranial hemorrhage (ICH), microglia clear RBC but may also sustain inflammation and cause secondary damage. It is thus conceivable that the molecular machinery for RBC degradation and heme iron recycling including Hmox1 and Nramp1 [213] is essential for microglial function [19]. Indeed, long-term induction of Hmox1 promotes the resolution of ICH [260]. However, this may not be a feasible therapeutic approach to be readily translated into clinical practice because various cerebral cell types differ in their cytoprotective enzyme repertoire and thus in their sensitivity to iron-induced damage [255]. Cell type-specific overexpression of Hmox1 in astrocytes, although neuroprotective in some in vitro models [240] and in ICH in young mice, is deleterious in other settings [133, 215, 216] and results in spontaneous cerebral iron deposition and consecutive movement and psychiatric abnormalities with aging [36, 218]. When the iron chelator deferoxamine (DFO), however, is administered early after experimental ICH, microglial activation is blunted and reduced local concentrations of tumor necrosis factor (TNF) and interleukin (IL)-1ß are associated with protection from neuronal death. Therefore, early DFO treatment in experimental ICH translates into improved neurologic outcome [222].
Local iron accumulation in the brain (particularly in neurons and astrocytes) is linked to several neurodegenerative disorders, collectively referred to as neurodegeneration with brain iron accumulation (NBIA) [127]. Although iron-mediated generation of ROS and injury to mitochondria are likely mechanisms [196, 204], the interplay between microglia, neurons, and other cell types in cerebral iron homeostasis is incompletely characterized. A paradigmatic yet rare NBIA condition is the neuroferritinopathy that is caused by a mutation in the coding region of the FTL gene [44, 45]. Although FTH carries the ferroxidase activity that is required to oxidize ferrous iron and to contain it within the FT shell, FTL facilitates the mineralization of ferric iron thus supporting FTH activity. The composition of FT is cell type-specific, and central neuronal FT are L chain-rich [236]. In addition, mutated FTL proteins may have a dominant negative effect on FT’s stability and resistance to oxidative damage [12, 140, 160]. It would appear that neuronal iron efflux is insufficient to remove surplus and potentially harmful iron from the cytoplasm. Whether or not a transfer of iron between neurons and microglial cells exists is unclear. However, it has been proposed that the siderophore-binding peptide lipocalin-2 (NGAL, Lcn2) is secreted by activated microglia and other cell types including neurons to transfer iron across cellular membranes. Notably, Lcn2 also affects a variety of brain functions from cognition to emotional stress [104]. Interestingly, the hyperferritinemia-cataract syndrome that is caused by a mutation in a specific non-coding region of the FTL gene, the iron-responsive element (IRE), is not associated with iron accumulation in the brain but with the formation of ferritin crystals in the ocular lenses, resulting in cataracts in early adulthood [23, 83, 195].
Several common neurodegenerative disorders such as Alzheimer’s and Parkinson’s diseases are also characterized by iron deposition, but they are not considered as classical NBIA. Whether or not the latter is causatively involved in the pathophysiology of the disease or a consequence of neurodegeneration is currently not known, as the influx of iron-rich inflammatory cells and their subsequent death may contribute to neuronal iron overload [233]. In addition, an intrinsic defect of neurons in cellular iron handling may exist. For instance, in neuronal cells exposed to amyloid beta peptide fragments, iron chelation with DFO or deferiprone (DFP) reduces ROS generation via the NADPH oxidase [181]. Apart from neurons, activated microglia can accumulate iron in Alzheimer’s disease, e.g., in the hippocampus as detected my ultra-high-resolution MR [258]. A similar observation has been made in the setting of the HFE H63D polymorphism, which is otherwise associated with type I hereditary hemochromatosis and has been linked to neurodegeneration. Mice with the homologous mutation in Hfe have increased FTL chain expression in microglia, suggesting iron overload [169]. This phenotype is discrepant from the iron-poor one of other Hfe-deficient macrophage populations, but the underlying mechanism remains unknown.
Similar to cardiac imaging, IONs are selectively taken up by microglia as a function of its activation state. Conceivably, microglial activation due to ischemia or inflammation results in MR signal alterations that can be used for diagnostics. However, microglia are also exquisitely sensitive to the potential toxicity of IONs because they are rapidly degraded within lysosomes and their iron catalyzes ROS generation [184]. Although hepcidin production has been observed in the brain, co-culture models suggest that microglia enhance hepcidin expression by neurons via IL-6 in a paracrine fashion [191]. These findings thus contrast to what has been shown for co-cultured primary Kupffer cells and hepatocytes inasmuch as culture supernatants from primary Kupffer cells blunted hepcidin production [228].
The published studies provide growing insight into how microglia affect brain function, local iron homeostasis, and iron-mediated neurodegeneration. The human brain is probably the most complex system known, and we need to better understand how its different cell types interact both under physiological conditions and challenges such as cognition or memory, how iron affects the function of the various cerebral cell types, and how we can potentially interfere to prevent iron from contributing to CNS disease.
Retina—the location in which iron aggravates light damage
From an evolutionary standpoint, the retina is part of the CNS. It is thus not surprising that it handles iron similarly to the cerebrum. Since photoreceptors are sensitive to iron toxicity, local iron regulatory circuits including the hepcidin-Fpn1 axis operate in the retina [261]. Müller glial cells and endothelial cells of the retina both express Fpn1, but only the latter cell type is sensitive to hepcidin regulation. This suggests that endothelial Fpn1 is the gatekeeper for iron transport into the retina [232]. In addition, experimental light damage to the retina results in microglial activation, its migration to the outer retina, and in induction of FT H and L chains. These effects along with oxidative stress to photoreceptors can be prevented by the oral iron chelator DFP [217]. Therefore, the retina is another organ that is sensitive to iron toxicity but has evidence of endogenous mechanisms to maintain local iron homeostasis in the microenvironment.
Bone—osteoclasts are iron-dependent
Osteoclasts are bone-resident cells of the MPS whose primary function is to absorb bone tissue in order to maintain, repair, and remodel the skeleton. Osteoclast generation requires the fusion of macrophages, the presence of the cytokines RANKL and M-CSF, and iron. Osteoclast precursors take up iron via Tfr1 and utilize six-transmembrane epithelial antigen of prostate (Steap4) as ferrireductase [262]. In addition, the transcription factor peroxisome proliferator-activated receptor-gamma coactivator 1 beta (PGC-1ß) is essential for osteoclast differentiation [101]. Apparently, both Tfr1-mediated iron uptake and PGC-1ß activation ensure mitochondrial respiration in osteoclasts as prerequisite for energy generation and proton secretion. While PGC-1ß transactivates genes of the mitochondrial respiratory chain, iron acquired via Tfr1 is incorporated into heme moieties and iron-sulfur clusters in mitochondria ensuring activity of their enzyme repertoire. Conceivably, iron overload is often associated with skeletal disease. For instance, surplus iron in thalassemic patients may promote osteoclast differentiation and function and thus contribute to bone resorption [101].
Arthropathy of the second and third metacarpophalangeal joints is a typical manifestation of type I hemochromatosis [94]. Elevated vascular cell adhesion molecule 1 (VCAM1) levels are an accurate biomarker to diagnose this condition, but whether the adhesion molecule is also involved in its pathogenesis remains unclear [170]. Of interest, circulating VCAM1 is shed by the metalloproteinase ADAM metallopeptidase domain 17 (ADAM17, AKA TNF alpha-converting enzyme (TACE)) which is activated by inflammatory stimuli [81]. In addition, membrane VCAM1 is expressed on CD169+ macrophages in the spleen and provides a molecular anchor to retain hematopoietic stem cells in the murine spleen, although it is unclear whether this is also relevant to arthropathy [61]. Taken together, despite the essential role of iron for osteoclast development and the clear association of skeletal disease and iron overload, we have little information about underlying mechanisms.
Bone marrow—are erythroblasts “nursed” with iron?
Embedded in cavities and the spongiosa of bones lies the BM as organ of hematopoiesis. Erythroid island macrophages (EIM) are a population of BM-resident macrophages that support erythropoiesis as “nurse cells” [17, 192]. The smallest erythropoietic unit thus consists of a central EIM surrounded by several erythroblasts (and stromal cells). This microarchitecture is maintained by adhesive interactions because EIM depend on a set of adhesion molecules. For instance, EIM express the αV integrin which binds ICAM4 on erythroid cells, and ICAM4 KO mice have reduced numbers of erythropoietic islands [126]. Similarly, macrophage VCAM1 interacts with the integrin-α4ß1 on erythroblasts. It is well characterized that erythroblasts expel their nuclei during differentiation and that these are taken up by EIM after phosphatidylserine exposure via Tim4 and MER proto-oncogene, tyrosine kinase (MerTK) [234, 254].
The transcription factor SpiC and its down-stream effector Hmox1 are both essential for EIM differentiation, since ablation of either gene reduces EIM number [73, 88]. Less clear however remains the role of EIM and Hmox1 in local iron management in the BM and mechanisms by which EIM may assist erythroblasts in iron acquisition. One possibility is that erythroblasts acquire ionic iron via their proximity to Fpn1 on EIM. However, since Fpn1 exports only divalent iron and erythroblasts largely rely on Tfr1 (AKA CD71) to satisfy their iron demand, one can speculate that either abundant amounts of ceruloplasmin and TF (to oxidize divalent iron) are present in erythroid islands or that direct cell-to-cell transfer of iron from EIM to erythroblasts exists. However, the deletion of Fpn1 by Cre recombinase expressed under the lysozyme M promoter results in iron accumulation in macrophages, including BM macrophages, and mild anemia. This suggests that EIM utilize Fpn1 to export iron and thus function as iron-rich nurse cells [259]. Such a mechanism may be particularly relevant in contexts when erythropoiesis is hyperactive yet inefficient and when local hemolysis occurs in the BM (i.e., in thalassemia syndromes). Another hypothesis is that EIM act as “iron buffers” that continuously releases adequate amounts of iron even when plasma iron levels fluctuate. Yet, another idea is that despite their other essential functions EIM contribute little to the iron acquisition by erythroblasts and that the latter import any TF-bound iron the blood stream delivers. Data in support of this concept has been generated using the CD169-DTR model: The depletion of EIM (and other CD169+ macrophages) had only little impact on the restoration of erythropoiesis after BM transplantation. Concretely, a moderate decrease of iron supply to erythroblasts as deduced from the reduced Hb content in reticulocytes was observed [38]. In conclusion, much needs to be learned about local mechanisms and their regulation of iron shuttling from EIM to erythroblasts at various stages of their differentiation.
The complexity of macrophage iron handling at the cellular level
Macrophages express a range of factors mediating iron uptake (summarized in the Table 1) which essentially face only two pathways for iron efflux, Fpn1 for ionic iron and feline leukemia virus subtype C receptor (Flvcr) for heme iron (Fig. 2), although alternatives routes may exist [49, 136]. One such mechanism may involve Lcn2 and one of its receptors, LcnR (AKA SLC22A17). Lcn2 binds distinct types of so-called siderophores, low molecular mass compounds with remarkably high affinity and specificity for ferric iron. These are best characterized in bacteria, but eukaryotes including fungi and mammals produce siderophores, too [16, 42, 67, 86, 106]. While some of these compounds such as citrate are ubiquitous substrates, other mammalian siderophores such as catechols are probably synthesized from commensal metabolites or dietary precursors [11, 50, 252]. Much needs to be learned about the biology of mammalian siderophores whose important functions may extend beyond mitochondrial iron homeostasis, erythropoiesis, and host defense [47, 134, 135].

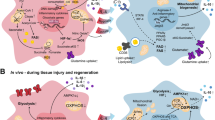
a In homeostatic conditions, iron uptake and release pathways are coordinated ensuring efficient iron recycling from aged red blood cells (aRBC) and iron delivery to sites of erythropoiesis. Apart from erythrophagocytosis, iron uptake mechanisms include (left to right) ferritin (FT) receptors such as scavenger receptor class A member (Scara)-5 and T-cell immunoglobulin and mucin domain-containing molecule (Tim)-2, transferrin (TF; depicted as monomer for simplicity) and its receptor Tfr1, the ferric reductase duodenal cytochrome B (DcytB) and divalent metal transporter (Dmt)-1, lactoferrin (LF) and its receptor LfR, lipocalin (Lcn)-2 and its receptor LcnR, haptoglobin-hemoglobin (Hb) complexes and their receptor CD163, and hemopexin (HPX)-heme complexes and their receptor CD91. Endosomal iron transporters Dmt1, natural resistance-associated macrophage protein-1 (Nramp1), and heme-regulated gene-1 (HRG1) shift ferrous iron or heme to the cytoplasm, from where the former can be exported via ferroportin (Fpn)-1 and the latter metabolized by heme oxygenase (Hmox)-1 or exported via feline leukemia virus subgroup C receptor (Flvcr). Macrophage iron homeostasis ensures proper function including the activity of pivotal transcription factors nuclear factor (NF)-IL6, NF-κB, and hypoxia-inducible factor (HIF)-1α, all of which are iron regulated. b Macrophage iron overload may result from an increase in circulating or local hepcidin concentrations, an elevated TF saturation, free iron excess, or hemolysis with subsequent accumulation of damaged RBC (dRBC), free Hb, or free heme. If severe, all these mechanisms will overwhelm the macrophage’s capacity to contain, store, and detoxify iron resulting in an increase of free iron, heme, and/or FT in cells. On the one hand, surplus intracellular iron may serve as nutrient for intraphagosomal or cytoplasmatic pathogens (e.g. for Gram-negative rods in the Tfr1-endosome and the cytoplasm). On the other hand, iron overload differentially affects distinct innate immune pathways. Surplus iron blocks the binding of transcription factors NF-IL6 and HIF-1α to their respective target promoter sequences. In parallel, iron facilitates the generation of ROS and thus NF-κB activation. Therefore, iron overload may cause a dysbalance in the transcriptional response to macrophage activation
As discussed above, scavenger receptors for the uptake of senescent or damaged RBC and for the clearance of complexed and free forms of Hb and heme are expressed at high levels in liver and spleen yet may be induced at other locations in response to inflammatory stimuli. These scavenger receptors are coupled to a down-stream machinery of heme detoxification. Its two major functional components are HRG1 which shifts heme from the endosome/lysosome to the cytoplasm and Hmox1 which then metabolizes heme [117]. In addition to the phagocyte-specific scavenger receptors, Tfr1 and divalent metal transporter 1 (Dmt1), two ubiquitous iron import proteins, are also expressed by macrophages.
A broad spectrum of iron uptake pathways
Macrophages express a range of factors mediating iron uptake, although the relative importance of these factors to different macrophage populations has not been systematically studied. It is well established that RPM and KC express the complete machinery for RBC clearance and heme iron recycling [72, 88, 231]. At either of these locations (spleen and liver), CD163 and CD91, two members of the scavenger receptor family, are expressed at high levels. Whereas CD163 binds both free and haptoglobin-bound Hb, CD91 accepts heme-HPX complexes. However, many other molecules participate in the overall process. Among these, heme-regulated gene 1 (HRG1) shifts heme from the endosome/lysosome to the cytoplasm, which is then metabolized by Hmox1 [117]. At the cell surface membrane, macrophages possess the heme exporter Flvcr and its genetic deletion results in RPM iron overload [108]. Tfr1, which is also known as CD71, is widely expressed by isolated macrophages, and it may form a major route of iron uptake in the absence of RBC (an indirect iron source), though it also appears to be important in adaptive immunity [103, 138]. Dmt1 shifts iron from the extracellular space to the cytoplasm and, in its alternately spliced form, from the Tfr1 endosome to the cytoplasm [69, 99, 139]. At either location, Dmt1 only accepts ferrous iron along with other divalent cations, such as copper or manganese, and co-localizes with an iron reductase [152, 177].
The control of cytosolic iron levels
FT is the central mechanism of iron storage in macrophages and other cell types (25, 179, (236). However, LysM-Cre FTH mice have no apparent phenotype suggesting that iron storage and trafficking are well controlled in myeloid cells by other mechanisms [46, 239]. One such mechanism to maintain cytosolic iron levels are the iron regulatory proteins (IRPs) 1 and 2 [5, 179, 195]. IRPs sense free labile iron and control cellular iron homeostasis at the post-transcriptional level: Due to their ability to interact with iron-responsive elements (IREs) that are present in the non-coding mRNA sequences of Tfr1, Dmt1, Fpn1, FTH, and FTL chains, fluctuations in intracellular iron are promptly counterbalanced [179, 195]. Interestingly, the functions of IRPs1 and 2 are redundant in macrophages and only targeted deletion of both isoforms via the LysM-Cre system results in increased FT levels and intracellular iron retention [193]. The deletion of both IRPs in macrophages does not affect RBC recycling but impairs their nutritional immune functions against intracellular bacteria, such as Salmonella Typhimurium [166]. This pathogen preferentially infects murine macrophages and has a proliferative advantage in the absence of both IRPs resulting in increased bacterial burden and reduced survival in a systemic infection model. Therefore, macrophage IRPs are required to regulate their effector functions upon infection with an iron-dependent pathogen.
Interplay of macrophage iron metabolism and immune functions
Macrophage iron metabolism and immune functions are interconnected [57, 79, 165, 247]. Clinical data as well as many studies in infection models show that macrophage iron overload, often resulting from hemolysis or iron supplementation, interferes with their antimicrobial activity (Fig. 2b). Several mechanisms explain this negative effect that surplus iron has on macrophage effector functions and the outcome of infectious diseases. First, a range of intracellular microbes can resist killing by macrophages by gaining access to macrophage iron pools [30, 245]. Second, iron alters gene transcription, translation, and mitochondrial respiration in macrophages [165, 175]. Specifically, macrophage functions driven by IFN-γ are impaired when macrophages are exposed to surplus iron [138, 162, 176] largely because iron directly inhibits the binding activities of the transcription factors nuclear factor interleukin-6 (NF-IL6) and hypoxia-inducible factor (HIF)-1α [55, 153] [176]. Because of its inhibitory effects on NF-IL6 and HIF-1α, iron excess blocks Nos2 transcription and the high output formation of NO which worsens bacterial killing of iron-laden macrophages [91, 164]. In addition, IFN-γ in conjunction with TLR4 ligation results in NF-κB-dependent activation of HIF-1α and increased transcription of TfR1 thus aggravating macrophage iron overload [224]. Moreover, hypoxia impairs NO production by Nos2 and thus blunts the antimicrobial activity of macrophages towards Leishmania major [142]. Another layer of complexity is added by the fact that TLR4 signaling stabilizes HIF-1α activation via induction of FTH and hypoxia stimulates arginase-1 expression in myeloid cells thus undermining Leishmania killing [142, 209]. Mechanistically, these two modes of HIF activation are distinct. While TLR4 signaling results in depletion of iron as co-substrate which prolyl hydroxylases (PHDs) require to tag HIF for degradation, hypoxia, per definition, impairs PHD activity due to a lack of their substrate oxygen. Nevertheless, both hypoxia (secondary to anemia or impaired microcirculation) and the presence of TLR ligands may be present at sites of infection in close spatial or temporal association. In addition, monocytes may be exposed to fluctuating levels of both stimuli (hypoxia and inflammation) as they travel to sites of injury and infiltrate the tissue to differentiate and become sessile. Therefore, due to its sensitivity to both oxygen and iron, the transcription factor HIF-1α appears to hold a central position in the interplay of macrophage iron homeostasis and immunity.
In contrast to its inhibitory effect on NO, iron stimulates the non-enzymatic generation of ROS which may promote their antimicrobial activity but contribute to tissue damage and the pathogenesis of many inflammatory disorders. In this context, iron present in heme can stimulate the production of ROS and of pro-inflammatory cytokines such as TNF, IL-1ß, and IL-6 [241]. In contrast, arginase-1, which depletes the Nos2 substrate arginine by converting it to ornithine [62, 205], is reduced by heme. These effects may contribute to the inflammatory characteristics of sickle cell disease and can be ameliorated by the heme scavenger HPX. Accordingly, free heme, which is predicted to accumulate when Hmox1 is inhibited, activates NF-κB to induce TNF and Nos2 transcription resulting in improved control of Salmonella infection [154]. Intriguingly, the accumulation of heme during the course of sepsis-induced hemolysis also impairs phagocyte functions due to interference with actin cytoskeleton rearrangements resulting in poor outcome [146].
A phagolysosomal transporter with homology to Dmt1 (Nramp2) is Nramp1 [8, 20, 71, 75]. Its cation transport function is directly linked to the antimicrobial activity of macrophages and DCs [74, 163, 219]. In addition, the functional form of Nramp1 in myeloid cells shifts the balance in T helper cell activity towards Th1, which may increase resistance to infections while increasing the susceptibility to autoimmune diseases [167, 226]. However, the directionality of iron transport is still under some debate [32, 71, 225, 249]: Most studies have shown that Nramp1 shifts iron and other divalent cations from the phagolysosome to the cytosol. This pathway is important for iron recycling after erythrophagocytosis [213] and serves as a mechanism of nutrient withdrawal that is particularly efficient in host defense against Mycobacterium, Salmonella, and Leishmania species [31]. However, some data suggest that Nramp1 in fact pumps iron into the phagolysosome where it may boost ROS generation [121, 122].
As discussed earlier, nanoparticles are increasingly used in clinical routine for imaging studies. Although intended to be otherwise inert, it has recently been uncovered in tumor models that nanoparticles may alter the biology of both tumor cells and tumor-associated macrophages, albeit with favorable results. Two elegant studies have shown that the application of nanoparticles to tumor-bearing mice is sufficient to induce clinical remission. One of these papers implicated a direct tumoricidal effect of iron delivered by ultrasmall polyethylene glycol particles and attributed tumor regression to the induction of ferroptosis in malignant cells [112]. The other study suggested that IONs stimulate TNF expression while inhibiting arginase-1 which results in improved killing of tumor cells [257]. These novel and exciting data support the general idea that the modulation of macrophage iron homeostasis is a promising way to control their immune functions and alter myeloid-driven disease processes in infection, auto-immunity, atherosclerosis, and possibly also cancer [143, 167]
Conclusion
The MPS comprises a heterogeneous population of tissue-specific leukocytes. It is a cornerstone of innate immunity, regulates the adaptive immune system, and is indispensable for tissue development, homeostasis, and repair. One of macrophages’ central metabolic functions is to eliminate senescent and damaged RBCs and to recycle their heme-bound iron to maintain systemic and cellular iron homeostasis. The role of macrophages in iron handling preserves iron homeostasis and tissue integrity. Under physiologic conditions, macrophages recycle the iron for erythropoiesis, while under hemolytic stress, macrophages detoxify heme to prevent iron toxicity towards parenchymal cells and tissues.
Only recently, we have begun to appreciate the specific functions of various macrophage populations in the body and to study their potential roles iron metabolism. New technologies are entering the field including systems biology approaches, in situ LASER capture, single cell RNA sequencing, cell type-specific genomic editing with CRISPR/Cas9, and cell type-specific vectors to silence genes of interest [43]. Such methods will be important to critically interrogate results obtained by more drastic and unspecific approaches (such as global macrophage depletion or MAC sorting) and to understand the spatiotemporal orchestration of iron metabolism at the single cell level and in the context of the tissue microenvironment characterized by cell-to-cell contact and paracrine signals. Most of all, we face the challenge to translate findings obtained in small animal models to human subjects to improve patient care.
Abbreviations
- ABCA1, ABCG1:
-
ATP binding cassette transporter A1 or G1, respectively
- ADAM17:
-
ADAM metallopeptidase domain 17
- ApoA1, ApoE:
-
Apolipoprotein A1 or E, respectively
- ARE:
-
Antioxidant response elements
- Bach1:
-
Btb And and Cnc Homology homology 1
- BM:
-
Bone marrow
- Bmp6:
-
Bone morphogenetic protein 6
- CCL:
-
CC-chemokine ligand
- CCR:
-
CC-chemokine receptor
- CD:
-
Cluster of differentiation
- Clec4F:
-
C-type Type lectin domain family 4 member F
- Cas9:
-
CRISPR- associated protein 9
- CNS:
-
Central nervous system
- CRISPR:
-
Clustered regulatory interspaced short palindromic repeats
- DC:
-
Dendritic cell
- DcytB:
-
Duodenal cytochrome B
- DFO:
-
Deferoxamine
- Dmt1:
-
Divalent metal transporter 1
- DFP:
-
Deferiprone
- EB:
-
Erythroblast
- EIM:
-
Erythroid island macrophages
- FcγR:
-
Fragment, crystallizable, gamma receptor
- Fpn1:
-
Ferroportin 1
- Flvcr:
-
Feline leukemia virus subtype C receptor
- FT:
-
Ferritin
- FTH, FTL:
-
Ferritin heavy chain or light chain, respectively
- F4/80:
-
(Murine macrophage marker) EGF-like module-containing mucin-like hormone receptor-like 1
- GM-CSF:
-
Granulocyte-macrophage colony-stimulating factor
- Hb:
-
Hemoglobin
- HFE:
-
Type I hemochromatosis gene
- HIF:
-
Hypoxia-inducible factor
- Hmox:
-
Heme oxygenase
- HPX:
-
Hemopexin
- HRE:
-
Hypoxia- responsive element
- HRG1:
-
Heme-regulated gene 1
- ICH:
-
Intracranial hemorrhage
- IFN:
-
Interferone
- Ig:
-
Immunoglobulin
- IL:
-
Interleukin
- ION:
-
Iron oxide nanoparticle
- IRE:
-
Iron responsive element
- IRP:
-
Iron regulatory protein
- KC:
-
Kupffer cell
- Lcn2:
-
Lipocalin 2
- LcnR:
-
Lcn2 receptor
- LDN:
-
LDN-193189 a BMP inhibitor that decreases hepcidin expression
- LF:
-
Lactoferrin
- LfR:
-
Lactoferrin receptor
- MAC:
-
Magnetic activated cell (sorting)
- M-CSF:
-
Macrophage colony-stimulating factor
- MerTK:
-
MER proto-oncogene, tyrosine kinase
- MHC-II:
-
Major histocompatibiliy complex class II
- MPS:
-
Mononuclear phagocyte system
- MRI:
-
Magnetic resonance imaging
- NADPH oxidase:
-
Nicotinamide adenine dinucleotide phosphate-oxidase
- NBIA:
-
Neurodegeneration with brain iron accumulation
- Nrf2:
-
Nuclear factor (erythroid-derived 2)-like 2
- NF-IL6:
-
Nuclear factor interleukin 6
- NF-κB:
-
Nuclear factor kappa B
- NLPR3:
-
NACHT, LRR, and PYD domains-containing protein 3 AKA cryopyrin
- Nramp1:
-
Natural resistance-associated macrophage protein 1
- PHD:
-
Prolyl hydroxylase domain proteins
- PGC-1ß:
-
Peroxisome proliferator-activated receptor-gamma coactivator 1 beta
- PU.1:
-
An E26 transformation-specific transcription factor encoded by the SPI1 gene
- RBC:
-
Red blood cell
- Rev-ErbAα, Rev-ErbBα:
-
(Nuclear receptors) NR1D1 and NR1D2, for nuclear receptor subfamily 1, group D, member 1 or member 2, respectively
- RPM:
-
Red pulp macrophage
- ROS:
-
Reactive oxygen species
- Sall1:
-
“Spalt-like”-1
- Scara5:
-
Scavenger receptor class A member 5
- SIRPα:
-
Signal regulatory protein alpha
- SpiC:
-
An E26 transformation-specific transcription factor of the Spi subfamily
- SR-A1:
-
Scavenger receptor class A type 1
- Steap:
-
Six-transmembrane epithelial antigen of prostate
- TF:
-
Transferrin
- Tfr1:
-
Transferrin receptor 1
- Tim:
-
T-cell immunoglobulin- and mucin- domain-containing molecule
- TLR4:
-
Toll-like receptor 4
- VCAM1:
-
Vascular cell adhesion molecule 1
References
Abboud S, Haile DJ (2000) A novel mammalian iron-regulated protein involved in intracellular iron metabolism. J Biol Chem 275:19906–19912
Adams PC, Barton JC (2007) Haemochromatosis. Lancet 370:1855–1860
Aguilar-Martinez P, Biron C, Masmejean C, Jeanjean P, Schved JF (1996) A novel mutation in the iron responsive element of ferritin L-subunit gene as a cause for hereditary hyperferritinemia-cataract syndrome. Blood 88:1895
Anderson GJ, Frazer DM, McKie AT, Vulpe CD (2002) The ceruloplasmin homolog hephaestin and the control of intestinal iron absorption. Blood Cells Mol Dis 29:367–375
Anderson CP, Shen M, Eisenstein RS, Leibold EA (2012) Mammalian iron metabolism and its control by iron regulatory proteins. Biochim Biophys Acta 1823:1468–1483
Andriopoulos B Jr, Corradini E, Xia Y, Faasse SA, Chen S, Grgurevic L, Knutson MD, Pietrangelo A, Vukicevic S, Lin HY, Babitt JL (2009) BMP6 is a key endogenous regulator of hepcidin expression and iron metabolism. Nat Genet 41:482–487
Armitage AE, Eddowes LA, Gileadi U, Cole S, Spottiswoode N, Selvakumar TA, Ho LP, Townsend AR, Drakesmith H (2011) Hepcidin regulation by innate immune and infectious stimuli. Blood 118:4129–4139
Atkinson PG, Blackwell JM, Barton CH (1997) Nramp1 locus encodes a 65 kDa interferon-gamma-inducible protein in murine macrophages. Biochem J 325(Pt 3):779–786
Auffray C, Sieweke MH, Geissmann F (2009) Blood monocytes: development, heterogeneity, and relationship with dendritic cells. Annu Rev Immunol 27:669–692
Babitt JL, Huang FW, Wrighting DM, Xia Y, Sidis Y, Samad TA, Campagna JA, Chung RT, Schneyer AL, Woolf CJ, Andrews NC, Lin HY (2006) Bone morphogenetic protein signaling by hemojuvelin regulates hepcidin expression. Nat Genet 38:531–539
Bao G, Clifton M, Hoette TM, Mori K, Deng SX, Qiu A, Viltard M, Williams D, Paragas N, Leete T, Kulkarni R, Li X, Lee B, Kalandadze A, Ratner AJ, Pizarro JC, Schmidt-Ott KM, Landry DW, Raymond KN, Strong RK, Barasch J (2010) Iron traffics in circulation bound to a siderocalin (Ngal)-catechol complex. Nat Chem Biol 6:602–609
Baraibar MA, Barbeito AG, Muhoberac BB, Vidal R (2012) A mutant light-chain ferritin that causes neurodegeneration has enhanced propensity toward oxidative damage. Free Radic Biol Med 52:1692–1697
Barton JC, Acton RT (2009) Hemochromatosis and Vibrio vulnificus wound infections. J Clin Gastroenterol 43:890–893
Barton CH, Biggs TE, Baker ST, Bowen H, Atkinson PG (1999) Nramp1: a link between intracellular iron transport and innate resistance to intracellular pathogens. J Leukoc Biol 66:757–762
Beaumont C, Leneuve P, Devaux I, Scoazec JY, Berthier M, Loiseau MN, Grandchamp B, Bonneau D (1995) Mutation in the iron responsive element of the L ferritin mRNA in a family with dominant hyperferritinaemia and cataract. Nat Genet 11:444–446
Behnsen J, Raffatellu M (2016) Siderophores: more than stealing iron. MBio 7:e01397–16
Bessis M, Breton-Gorius J (1959) New observations on the erythroblast islet and rhopheocytosis of ferritin. Rev Hematol 14:165–197
Beutler E (2006) Hemochromatosis: genetics and pathophysiology. Annu Rev Med 57:331–347
Biggs TE, Baker ST, Botham MS, Dhital A, Barton CH, Perry VH (2001) Nramp1 modulates iron homoeostasis in vivo and in vitro: evidence for a role in cellular iron release involving de-acidification of intracellular vesicles. Eur J Immunol 31:2060–2070
Blackwell JM (1996) Structure and function of the natural-resistance-associated macrophage protein (Nramp1), a candidate protein for infectious and autoimmune disease susceptibility. Mol Med Today 2:205–211
Bleriot C, Dupuis T, Jouvion G, Eberl G, Disson O, Lecuit M (2015) Liver-resident macrophage necroptosis orchestrates type 1 microbicidal inflammation and type-2-mediated tissue repair during bacterial infection. Immunity 42:145–158
Boyle JJ, Harrington HA, Piper E, Elderfield K, Stark J, Landis RC, Haskard DO (2009) Coronary intraplaque hemorrhage evokes a novel atheroprotective macrophage phenotype. Am J Pathol 174:1097–1108
Brooks DG, Manova-Todorova K, Farmer J, Lobmayr L, Wilson RB, Eagle RC Jr, St Pierre TG, Stambolian D (2002) Ferritin crystal cataracts in hereditary hyperferritinemia cataract syndrome. Invest Ophthalmol Vis Sci 43:1121–1126
Bulteau AL, O'Neill HA, Kennedy MC, Ikeda-Saito M, Isaya G, Szweda LI (2004) Frataxin acts as an iron chaperone protein to modulate mitochondrial aconitase activity. Science 305:242–245
Buttgereit A, Lelios I, Yu X, Vrohlings M, Krakoski NR, Gautier EL, Nishinakamura R, Becher B, Greter M (2016) Sall1 is a transcriptional regulator defining microglia identity and function. Nat Immunol
Camaschella C, Zecchina G, Lockitch G, Roetto A, Campanella A, Arosio P, Levi S (2000) A new mutation (G51C) in the iron-responsive element (IRE) of L-ferritin associated with hyperferritinaemia-cataract syndrome decreases the binding affinity of the mutated IRE for iron-regulatory proteins. Br J Haematol 108:480–482
Canali S, Vecchi C, Garuti C, Montosi G, Babitt JL, Pietrangelo A (2016a) The SMAD pathway is required for hepcidin response during endoplasmic reticulum stress. Endocrinology 157:3935–3945
Canali S, Zumbrennen-Bullough KB, Core AB, Wang CY, Nairz M, Bouley R, Swirski FK, Babitt JL (2016b) Endothelial cells produce bone morphogenetic protein 6 required for iron homeostasis in mice. Blood 129:405–414
Carter EL, Gupta N, Ragsdale SW (2016) High affinity heme binding to a heme regulatory motif on the nuclear receptor rev-erbbeta leads to its degradation and indirectly regulates its interaction with nuclear receptor Corepressor. J Biol Chem 291:2196–2222
Cassat JE, Skaar EP (2013) Iron in infection and immunity. Cell Host Microbe 13:509–519
Cellier MF (2012) Nramp: from sequence to structure and mechanism of divalent metal import. Curr Top Membr 69:249–293
Cellier MF, Courville P, Campion C (2007) Nramp1 phagocyte intracellular metal withdrawal defense. Microbes Infect 9:1662–1670
Chen H, Su T, Attieh ZK, Fox TC, McKie AT, Anderson GJ, Vulpe CD (2003) Systemic regulation of Hephaestin and Ireg1 revealed in studies of genetic and nutritional iron deficiency. Blood 102:1893–1899
Chen TT, Li L, Chung DH, Allen CD, Torti SV, Torti FM, Cyster JG, Chen CY, Brodsky FM, Niemi EC, Nakamura MC, Seaman WE, Daws MR (2005) TIM-2 is expressed on B cells and in liver and kidney and is a receptor for H-ferritin endocytosis. J Exp Med 202:955–965
Chen C, Samuel TK, Sinclair J, Dailey HA, Hamza I (2011) An intercellular heme-trafficking protein delivers maternal heme to the embryo during development in C. elegans. Cell 145:720–731
Chen-Roetling J, Song W, Schipper HM, Regan CS, Regan RF (2015) Astrocyte overexpression of heme oxygenase-1 improves outcome after intracerebral hemorrhage. Stroke 46:1093–1098
Chiabrando D, Fiorito V, Marro S, Silengo L, Altruda F, Tolosano E (2012) Cell-specific regulation of Ferroportin transcription following experimentally-induced acute anemia in mice. Blood Cells Mol Dis 50:25–30
Chow A, Huggins M, Ahmed J, Hashimoto D, Lucas D, Kunisaki Y, Pinho S, Leboeuf M, Noizat C, van Rooijen N, Tanaka M, Zhao ZJ, Bergman A, Merad M, Frenette PS (2013) CD169(+) macrophages provide a niche promoting erythropoiesis under homeostasis and stress. Nat Med 19:429–436
Clevers H (2013) The intestinal crypt, a prototype stem cell compartment. Cell 154:274–284
Cohen LA, Gutierrez L, Weiss A, Leichtmann-Bardoogo Y, Zhang DL, Crooks DR, Sougrat R, Morgenstern A, Galy B, Hentze MW, Lazaro FJ, Rouault TA, Meyron-Holtz EG (2010) Serum ferritin is derived primarily from macrophages through a nonclassical secretory pathway. Blood 116:1574–1584
Corna G, Caserta I, Monno A, Apostoli P, Manfredi AA, Camaschella C, Rovere-Querini P (2016) The repair of skeletal muscle requires iron recycling through macrophage ferroportin. J Immunol 197:1914–1925
Correnti C, Strong RK (2012) Mammalian siderophores, siderophore-binding lipocalins, and the labile iron pool. J Biol Chem 287:13524–13531
Courties G, Heidt T, Sebas M, Iwamoto Y, Jeon D, Truelove J, Tricot B, Wojtkiewicz G, Dutta P, Sager HB, Borodovsky A, Novobrantseva T, Klebanov B, Fitzgerald K, Anderson DG, Libby P, Swirski FK, Weissleder R, Nahrendorf M (2013) In vivo silencing of the transcription factor IRF5 reprograms the macrophage phenotype and improves infarct healing. J Am Coll Cardiol 63:1556–1566
Cozzi A, Santambrogio P, Privitera D, Broccoli V, Rotundo LI, Garavaglia B, Benz R, Altamura S, Goede JS, Muckenthaler MU, Levi S (2013) Human L-ferritin deficiency is characterized by idiopathic generalized seizures and atypical restless leg syndrome. J Exp Med 210:1779–1791
Curtis AR, Fey C, Morris CM, Bindoff LA, Ince PG, Chinnery PF, Coulthard A, Jackson MJ, Jackson AP, McHale DP, Hay D, Barker WA, Markham AF, Bates D, Curtis A, Burn J (2001) Mutation in the gene encoding ferritin light polypeptide causes dominant adult-onset basal ganglia disease. Nat Genet 28:350–354
Darshan D, Vanoaica L, Richman L, Beermann F, Kuhn LC (2009) Conditional deletion of ferritin H in mice induces loss of iron storage and liver damage. Hepatology 50:852–860
Davuluri G, Song P, Liu Z, Wald D, Sakaguchi TF, Green MR, Devireddy L (2016) Inactivation of 3-hydroxybutyrate dehydrogenase 2 delays zebrafish erythroid maturation by conferring premature mitophagy. Proc Natl Acad Sci U S A 113:E1460–E1469
Delaby C, Pilard N, Hetet G, Driss F, Grandchamp B, Beaumont C, Canonne-Hergaux F (2005) A physiological model to study iron recycling in macrophages. Exp Cell Res 310:43–53
Devireddy LR, Gazin C, Zhu X, Green MR (2005) A cell-surface receptor for lipocalin 24p3 selectively mediates apoptosis and iron uptake. Cell 123:1293–1305
Devireddy LR, Hart DO, Goetz DH, Green MR (2010) A mammalian siderophore synthesized by an enzyme with a bacterial homolog involved in enterobactin production. Cell 141:1006–1017
Dhakshinamoorthy S, Jain AK, Bloom DA, Jaiswal AK (2005) Bach1 competes with Nrf2 leading to negative regulation of the antioxidant response element (ARE)-mediated NAD(P)H:quinone oxidoreductase 1 gene expression and induction in response to antioxidants. J Biol Chem 280:16891–16900
Dimitrov JD, Ivanovska ND, Lacroix-Desmazes S, Doltchinkova VR, Kaveri SV, Vassilev TL (2006) Ferrous ions and reactive oxygen species increase antigen-binding and anti-inflammatory activities of immunoglobulin G. J Biol Chem 281:439–446
Dimitrov JD, Roumenina LT, Doltchinkova VR, Mihaylova NM, Lacroix-Desmazes S, Kaveri SV, Vassilev TL (2007) Antibodies use heme as a cofactor to extend their pathogen elimination activity and to acquire new effector functions. J Biol Chem 282:26696–26706
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, Morrison B 3rd, Stockwell BR (2012) Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149:1060–1072
Dlaska M, Weiss G (1999) Central role of transcription factor NF-IL6 for cytokine and iron-mediated regulation of murine inducible nitric oxide synthase expression. J Immunol 162:6171–6177
Donovan A, Brownlie A, Zhou Y, Shepard J, Pratt SJ, Moynihan J, Paw BH, Drejer A, Barut B, Zapata A, Law TC, Brugnara C, Lux SE, Pinkus GS, Pinkus JL, Kingsley PD, Palis J, Fleming MD, Andrews NC, Zon LI (2000) Positional cloning of zebrafish ferroportin1 identifies a conserved vertebrate iron exporter. Nature 403:776–781
Drakesmith H, Prentice AM (2012) Hepcidin and the iron-infection axis. Science 338:768–772
Drakesmith H, Schimanski LM, Ormerod E, Merryweather-Clarke AT, Viprakasit V, Edwards JP, Sweetland E, Bastin JM, Cowley D, Chinthammitr Y, Robson KJ, Townsend AR (2005) Resistance to hepcidin is conferred by hemochromatosis-associated mutations of ferroportin. Blood 106:1092–1097
Drakesmith H, Nemeth E, Ganz T (2015) Ironing out ferroportin. Cell Metab 22:777–787
Dutra FF, Alves LS, Rodrigues D, Fernandez PL, de Oliveira RB, Golenbock DT, Zamboni DS, Bozza MT (2014) Hemolysis-induced lethality involves inflammasome activation by heme. Proc Natl Acad Sci U S A 111:E4110–E4118
Dutta P, Hoyer FF, Grigoryeva LS, Sager HB, Leuschner F, Courties G, Borodovsky A, Novobrantseva T, Ruda VM, Fitzgerald K, Iwamoto Y, Wojtkiewicz G, Sun Y, Da Silva N, Libby P, Anderson DG, Swirski FK, Weissleder R, Nahrendorf M (2015) Macrophages retain hematopoietic stem cells in the spleen via VCAM-1. J Exp Med 212:497–512
El Kasmi KC, Qualls JE, Pesce JT, Smith AM, Thompson RW, Henao-Tamayo M, Basaraba RJ, Konig T, Schleicher U, Koo MS, Kaplan G, Fitzgerald KA, Tuomanen EI, Orme IM, Kanneganti TD, Bogdan C, Wynn TA, Murray PJ (2008) Toll-like receptor-induced arginase 1 in macrophages thwarts effective immunity against intracellular pathogens. Nat Immunol 9:1399–1406
Fang HY, Hughes R, Murdoch C, Coffelt SB, Biswas SK, Harris AL, Johnson RS, Imityaz HZ, Simon MC, Fredlund E, Greten FR, Rius J, Lewis CE (2009) Hypoxia-inducible factors 1 and 2 are important transcriptional effectors in primary macrophages experiencing hypoxia. Blood 114:844–859
Farin HF, Karthaus WR, Kujala P, Rakhshandehroo M, Schwank G, Vries RG, Kalkhoven E, Nieuwenhuis EE, Clevers H (2014) Paneth cell extrusion and release of antimicrobial products is directly controlled by immune cell-derived IFN-gamma. J Exp Med 211:1393–1405
Fernandez PL, Dutra FF, Alves L, Figueiredo RT, Mourao-Sa D, Fortes GB, Bergstrand S, Lonn D, Cevallos RR, Pereira RM, Lopes UG, Travassos LH, Paiva CN, Bozza MT (2010) Heme amplifies the innate immune response to microbial molecules through spleen tyrosine kinase (Syk)-dependent reactive oxygen species generation. J Biol Chem 285:32844–32851
Figueiredo RT, Fernandez PL, Mourao-Sa DS, Porto BN, Dutra FF, Alves LS, Oliveira MF, Oliveira PL, Graca-Souza AV, Bozza MT (2007) Characterization of heme as activator of Toll-like receptor 4. J Biol Chem 282:20221–20229
Fischbach MA, Lin H, Liu DR, Walsh CT (2006) How pathogenic bacteria evade mammalian sabotage in the battle for iron. Nat Chem Biol 2:132–138
Fleming MD, Trenor CC 3rd, Su MA, Foernzler D, Beier DR, Dietrich WF, Andrews NC (1997) Microcytic anaemia mice have a mutation in Nramp2, a candidate iron transporter gene. Nat Genet 16:383–386
Fleming MD, Romano MA, Su MA, Garrick LM, Garrick MD, Andrews NC (1998) Nramp2 is mutated in the anemic Belgrade (b) rat: evidence of a role for Nramp2 in endosomal iron transport. Proc Natl Acad Sci U S A 95:1148–1153
Flo TH, Smith KD, Sato S, Rodriguez DJ, Holmes MA, Strong RK, Akira S, Aderem A (2004) Lipocalin 2 mediates an innate immune response to bacterial infection by sequestrating iron. Nature 432:917–921
Forbes JR, Gros P (2001) Divalent-metal transport by NRAMP proteins at the interface of host-pathogen interactions. Trends Microbiol 9:397–403
Franken L, Klein M, Spasova M, Elsukova A, Wiedwald U, Welz M, Knolle P, Farle M, Limmer A, Kurts C (2015) Splenic red pulp macrophages are intrinsically superparamagnetic and contaminate magnetic cell isolates. Sci Rep 5:12940
Fraser ST, Midwinter RG, Coupland LA, Kong S, Berger BS, Yeo JH, Andrade OC, Cromer D, Suarna C, Lam M, Maghzal GJ, Chong BH, Parish CR, Stocker R (2015) Heme oxygenase-1 deficiency alters erythroblastic island formation, steady-state erythropoiesis and red blood cell lifespan in mice. Haematologica 100:601–610
Fritsche G, Dlaska M, Barton H, Theurl I, Garimorth K, Weiss G (2003) Nramp1 functionality increases inducible nitric oxide synthase transcription via stimulation of IFN regulatory factor 1 expression. J Immunol 171:1994–1998
Fritsche G, Nairz M, Theurl I, Mair S, Bellmann-Weiler R, Barton HC, Weiss G (2007) Modulation of macrophage iron transport by Nramp1 (Slc11a1). Immunobiology 212:751–757
Gaenzer H, Marschang P, Sturm W, Neumayr G, Vogel W, Patsch J, Weiss G (2002) Association between increased iron stores and impaired endothelial function in patients with hereditary hemochromatosis. J Am Coll Cardiol 40:2189–2194
Galy B, Ferring-Appel D, Kaden S, Grone HJ, Hentze MW (2008) Iron regulatory proteins are essential for intestinal function and control key iron absorption molecules in the duodenum. Cell Metab 7:79–85
Ganz T (2012) Macrophages and systemic iron homeostasis. J Innate Immun 4:446–453
Ganz T, Nemeth E (2015) Iron homeostasis in host defence and inflammation. Nat Rev Immunol 15:500–510
Gardi C, Arezzini B, Fortino V, Comporti M (2002) Effect of free iron on collagen synthesis, cell proliferation and MMP-2 expression in rat hepatic stellate cells. Biochem Pharmacol 64:1139–1145
Garton KJ, Gough PJ, Philalay J, Wille PT, Blobel CP, Whitehead RH, Dempsey PJ, Raines EW (2003) Stimulated shedding of vascular cell adhesion molecule 1 (VCAM-1) is mediated by tumor necrosis factor-alpha-converting enzyme (ADAM 17). J Biol Chem 278:37459–37464
Ginhoux F, Guilliams M (2016) Tissue-resident macrophage ontogeny and homeostasis. Immunity 44:439–449
Girelli D, Corrocher R, Bisceglia L, Olivieri O, De Franceschi L, Zelante L, Gasparini P (1995) Molecular basis for the recently described hereditary hyperferritinemia-cataract syndrome: a mutation in the iron-responsive element of ferritin L-subunit gene (the “Verona mutation”). Blood 86:4050–4053
Goldmann T, Wieghofer P, Jordao MJ, Prutek F, Hagemeyer N, Frenzel K, Amann L, Staszewski O, Kierdorf K, Krueger M, Locatelli G, Hochgerner H, Zeiser R, Epelman S, Geissmann F, Priller J, Rossi FM, Bechmann I, Kerschensteiner M, Linnarsson S, Jung S, Prinz M (2016) Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat Immunol 17:797–805
Gordon S, Taylor PR (2005) Monocyte and macrophage heterogeneity. Nat Rev Immunol 5:953–964
Haas H (2014) Fungal siderophore metabolism with a focus on Aspergillus fumigatus. Nat Prod Rep 31:1266–1276
Habib A, Polavarapu R, Karmali V, Guo L, Van Dam R, Cheng Q, Akahori H, Saeed O, Nakano M, Pachura K, Hong CC, Shin E, Kolodgie F, Virmani R, Finn AV (2015) Hepcidin-ferroportin axis controls toll-like receptor 4 dependent macrophage inflammatory responses in human atherosclerotic plaques. Atherosclerosis 241:692–700
Haldar M, Kohyama M, So AY, Kc W, Wu X, Briseno CG, Satpathy AT, Kretzer NM, Arase H, Rajasekaran NS, Wang L, Egawa T, Igarashi K, Baltimore D, Murphy TL, Murphy KM (2014) Heme-mediated SPI-C induction promotes monocyte differentiation into iron-recycling macrophages. Cell 156:1223–1234
Hamza I, Dailey HA (2012) One ring to rule them all: trafficking of heme and heme synthesis intermediates in the metazoans. Biochim Biophys Acta 1823:1617–1632
Han J, Seaman WE, Di X, Wang W, Willingham M, Torti FM, Torti SV (2011) Iron uptake mediated by binding of H-ferritin to the TIM-2 receptor in mouse cells. PLoS One 6:e23800
Haschka D, Nairz M, Demetz E, Wienerroither S, Decker T, Weiss G (2015) Contrasting regulation of macrophage iron homeostasis in response to infection with Listeria monocytogenes depending on localization of bacteria. Metallomics 7:1036–1045
Haubner BJ, Schneider J, Schweigmann U, Schuetz T, Dichtl W, Velik-Salchner C, Stein JI, Penninger JM (2015) Functional recovery of a human neonatal heart after severe myocardial infarction. Circ Res 118:216–221
He S, Chousterman BG, Fenn A, Anzai A, Nairz M, Brandt M, Hilgendorf I, Sun Y, Ye YX, Iwamoto Y, Tricot B, Weissleder R, Macphee C, Libby P, Nahrendorf M, Swirski FK (2016) Lp-PLA2 antagonizes left ventricular healing after myocardial infarction by impairing the appearance of reparative macrophages. Circ Heart Fail 8:980–987
Heiland GR, Aigner E, Dallos T, Sahinbegovic E, Krenn V, Thaler C, Weiss G, Distler JH, Datz C, Schett G, Zwerina J (2010) Synovial immunopathology in haemochromatosis arthropathy. Ann Rheum Dis 69:1214–1219
Hentze MW, Muckenthaler MU, Galy B, Camaschella C (2010) Two to tango: regulation of mammalian iron metabolism. Cell 142:24–38
Hilgendorf I, Gerhardt LM, Tan TC, Winter C, Holderried TA, Chousterman BG, Iwamoto Y, Liao R, Zirlik A, Scherer-Crosbie M, Hedrick CC, Libby P, Nahrendorf M, Weissleder R, Swirski FK (2014) Ly-6Chigh monocytes depend on Nr4a1 to balance both inflammatory and reparative phases in the infarcted myocardium. Circ Res 114:1611–1622
Hornig R, Lutz HU (2000) Band 3 protein clustering on human erythrocytes promotes binding of naturally occurring anti-band 3 and anti-spectrin antibodies. Exp Gerontol 35:1025–1044
Huang ML, Becker EM, Whitnall M, Suryo Rahmanto Y, Ponka P, Richardson DR (2009) Elucidation of the mechanism of mitochondrial iron loading in Friedreich’s ataxia by analysis of a mouse mutant. Proc Natl Acad Sci U S A 106:16381–16386
Hubert N, Hentze MW (2002) Previously uncharacterized isoforms of divalent metal transporter (DMT)-1: implications for regulation and cellular function. Proc Natl Acad Sci U S A 99:12345–12350
Hvidberg V, Maniecki MB, Jacobsen C, Hojrup P, Moller HJ, Moestrup SK (2005) Identification of the receptor scavenging hemopexin-heme complexes. Blood 106:2572–2579
Ishii KA, Fumoto T, Iwai K, Takeshita S, Ito M, Shimohata N, Aburatani H, Taketani S, Lelliott CJ, Vidal-Puig A, Ikeda K (2009) Coordination of PGC-1beta and iron uptake in mitochondrial biogenesis and osteoclast activation. Nat Med 15:259–266
Iwasaki K, Mackenzie EL, Hailemariam K, Sakamoto K, Tsuji Y (2006) Hemin-mediated regulation of an antioxidant-responsive element of the human ferritin H gene and role of Ref-1 during erythroid differentiation of K562 cells. Mol Cell Biol 26:2845–2856
Jabara HH, Boyden SE, Chou J, Ramesh N, Massaad MJ, Benson H, Bainter W, Fraulino D, Rahimov F, Sieff C, Liu ZJ, Alshemmari SH, Al-Ramadi BK, Al-Dhekri H, Arnaout R, Abu-Shukair M, Vatsayan A, Silver E, Ahuja S, Davies EG, Sola-Visner M, Ohsumi TK, Andrews NC, Notarangelo LD, Fleming MD, Al-Herz W, Kunkel LM, Geha RS (2016) A missense mutation in TFRC, encoding transferrin receptor 1, causes combined immunodeficiency. Nat Genet 48:74–78
Jha MK, Lee S, Park DH, Kook H, Park KG, Lee IK, Suk K (2014) Diverse functional roles of lipocalin-2 in the central nervous system. Neurosci Biobehav Rev 49:135–156
Ji J, Zhou Y, Hao S, Wang Q, Li K, Qiao T (2015) Low expression of ferroxidases is implicated in the iron retention in human atherosclerotic plaques. Biochem Biophys Res Commun 464:1134–1138
Jones RL, Peterson CM, Grady RW, Cerami A (1980) Low molecular weight iron-binding factor from mammalian tissue that potentiates bacterial growth. J Exp Med 151:418–428
Kautz L, Gabayan V, Wang X, Wu J, Onwuzurike J, Jung G, Qiao B, Lusis AJ, Ganz T, Nemeth E (2013) Testing the iron hypothesis in a mouse model of atherosclerosis. Cell Rep 5:1436–1442
Keel SB, Doty RT, Yang Z, Quigley JG, Chen J, Knoblaugh S, Kingsley PD, De Domenico I, Vaughn MB, Kaplan J, Palis J, Abkowitz JL (2008) A heme export protein is required for red blood cell differentiation and iron homeostasis. Science 319:825–828
Khandelwal S, van Rooijen N, Saxena RK (2007) Reduced expression of CD47 during murine red blood cell (RBC) senescence and its role in RBC clearance from the circulation. Transfusion 47:1725–1732
Kiechl S, Aichner F, Gerstenbrand F, Egger G, Mair A, Rungger G, Spogler F, Jarosch E, Oberhollenzer F, Willeit J (1994) Body iron stores and presence of carotid atherosclerosis. Results from the Bruneck Study. Arterioscler Thromb 14:1625–1630
Kiechl S, Willeit J, Egger G, Poewe W, Oberhollenzer F (1997) Body iron stores and the risk of carotid atherosclerosis: prospective results from the Bruneck study. Circulation 96:3300–3307
Kim SE, Zhang L, Ma K, Riegman M, Chen F, Ingold I, Conrad M, Turker MZ, Gao M, Jiang X, Monette S, Pauliah M, Gonen M, Zanzonico P, Quinn T, Wiesner U, Bradbury MS, Overholtzer M (2016) Ultrasmall nanoparticles induce ferroptosis in nutrient-deprived cancer cells and suppress tumour growth. Nat Nanotechnol 11:977–985
Knutson MD, Oukka M, Koss LM, Aydemir F, Wessling-Resnick M (2005) Iron release from macrophages after erythrophagocytosis is up-regulated by ferroportin 1 overexpression and down-regulated by hepcidin. Proc Natl Acad Sci U S A 102:1324–1328
Kobayashi N, Karisola P, Pena-Cruz V, Dorfman DM, Jinushi M, Umetsu SE, Butte MJ, Nagumo H, Chernova I, Zhu B, Sharpe AH, Ito S, Dranoff G, Kaplan GG, Casasnovas JM, Umetsu DT, Dekruyff RH, Freeman GJ (2007) TIM-1 and TIM-4 glycoproteins bind phosphatidylserine and mediate uptake of apoptotic cells. Immunity 27:927–940
Koeppen AH, Ramirez RL, Becker AB, Bjork ST, Levi S, Santambrogio P, Parsons PJ, Kruger PC, Yang KX, Feustel PJ, Mazurkiewicz JE (2015) The pathogenesis of cardiomyopathy in Friedreich ataxia. PLoS One 10:e0116396
Kohyama M, Ise W, Edelson BT, Wilker PR, Hildner K, Mejia C, Frazier WA, Murphy TL, Murphy KM (2009) Role for Spi-C in the development of red pulp macrophages and splenic iron homeostasis. Nature 457:318–321
Korolnek T, Hamza I (2014) Like iron in the blood of the people: the requirement for heme trafficking in iron metabolism. Front Pharmacol 5:126
Korolnek T, Hamza I (2015) Macrophages and iron trafficking at the birth and death of red cells. Blood 125:2893–2897
Koskenkorva-Frank TS, Weiss G, Koppenol WH, Burckhardt S (2013) The complex interplay of iron metabolism, reactive oxygen species, and reactive nitrogen species: insights into the potential of various iron therapies to induce oxidative and nitrosative stress. Free Radic Biol Med 65:1174–1194
Kristiansen M, Graversen JH, Jacobsen C, Sonne O, Hoffman HJ, Law SK, Moestrup SK (2001) Identification of the haemoglobin scavenger receptor. Nature 409:198–201
Kuhn DE, Baker BD, Lafuse WP, Zwilling BS (1999) Differential iron transport into phagosomes isolated from the RAW264.7 macrophage cell lines transfected with Nramp1Gly169 or Nramp1Asp169. J Leukoc Biol 66:113–119
Kuhn DE, Lafuse WP, Zwilling BS (2001) Iron transport into mycobacterium avium-containing phagosomes from an Nramp1(Gly169)-transfected RAW264.7 macrophage cell line. J Leukoc Biol 69:43–49
Kwon SJ, Lee GT, Lee JH, Kim WJ, Kim IY (2009) Bone morphogenetic protein-6 induces the expression of inducible nitric oxide synthase in macrophages. Immunology 128:e758–e765
Lambe T, Simpson RJ, Dawson S, Bouriez-Jones T, Crockford TL, Lepherd M, Latunde-Dada GO, Robinson H, Raja KB, Campagna DR, Villarreal G Jr, Ellory JC, Goodnow CC, Fleming MD, McKie AT, Cornall RJ (2009) Identification of a Steap3 endosomal targeting motif essential for normal iron metabolism. Blood 113:1805–1808
Lavin Y, Merad M (2014) Macrophages: gatekeepers of tissue integrity. Cancer Immunol Res 1:201–209
Lee G, Lo A, Short SA, Mankelow TJ, Spring F, Parsons SF, Yazdanbakhsh K, Mohandas N, Anstee DJ, Chasis JA (2006) Targeted gene deletion demonstrates that the cell adhesion molecule ICAM-4 is critical for erythroblastic island formation. Blood 108:2064–2071
Levi S, Finazzi D (2014) Neurodegeneration with brain iron accumulation: update on pathogenic mechanisms. Front Pharmacol 5:99
Levy JE, Jin O, Fujiwara Y, Kuo F, Andrews NC (1999) Transferrin receptor is necessary for development of erythrocytes and the nervous system. Nat Genet 21:396–399
Li W, Xu LH, Forssell C, Sullivan JL, Yuan XM (2008) Overexpression of transferrin receptor and ferritin related to clinical symptoms and destabilization of human carotid plaques. Exp Biol Med (Maywood) 233:818–826
Li JY, Paragas N, Ned RM, Qiu A, Viltard M, Leete T, Drexler IR, Chen X, Sanna-Cherchi S, Mohammed F, Williams D, Lin CS, Schmidt-Ott KM, Andrews NC, Barasch J (2009) Scara5 is a ferritin receptor mediating non-transferrin iron delivery. Dev Cell 16:35–46
Li Q, Fu W, Yao J, Ji Z, Wang Y, Zhou Z, Yan J, Li W (2014a) Heme induces IL-1beta secretion through activating NLRP3 in kidney inflammation. Cell Biochem Biophys 69:495–502
Li T, Eheim AL, Klein S, Uschner FE, Smith AC, Brandon-Warner E, Ghosh S, Bonkovsky HL, Trebicka J, Schrum LW (2014b) Novel role of nuclear receptor Rev-erbalpha in hepatic stellate cell activation: potential therapeutic target for liver injury. Hepatology 59:2383–2396
Lin SH, Song W, Cressatti M, Zukor H, Wang E, Schipper HM (2015) Heme oxygenase-1 modulates microRNA expression in cultured astroglia: implications for chronic brain disorders. Glia 63:1270–1284
Liu Z, Ciocea A, Devireddy L (2014a) Endogenous siderophore 2,5-dihydroxybenzoic acid deficiency promotes anemia and splenic iron overload in mice. Mol Cell Biol 34:2533–2546
Liu Z, Reba S, Chen WD, Porwal SK, Boom WH, Petersen RB, Rojas R, Viswanathan R, Devireddy L (2014b) Regulation of mammalian siderophore 2,5-DHBA in the innate immune response to infection. J Exp Med 211:1197–1213
Lok HC, Sahni S, Jansson PJ, Kovacevic Z, Hawkins CL, Richardson DR (2016) A nitric oxide storage and transport system that protects activated macrophages from endogenous nitric oxide cytotoxicity. J Biol Chem 291:27042–27061
Lou DQ, Lesbordes JC, Nicolas G, Viatte L, Bennoun M, Van Rooijen N, Kahn A, Renia L, Vaulont S (2005) Iron- and inflammation-induced hepcidin gene expression in mice is not mediated by Kupffer cells in vivo. Hepatology 41:1056–1064
Ludwiczek S, Aigner E, Theurl I, Weiss G (2003) Cytokine-mediated regulation of iron transport in human monocytic cells. Blood 101:4148–4154
Ludwiczek S, Theurl I, Muckenthaler MU, Jakab M, Mair SM, Theurl M, Kiss J, Paulmichl M, Hentze MW, Ritter M, Weiss G (2007) Ca2+ channel blockers reverse iron overload by a new mechanism via divalent metal transporter-1. Nat Med 13:448–454
Luscieti S, Santambrogio P, Langlois d'Estaintot B, Granier T, Cozzi A, Poli M, Gallois B, Finazzi D, Cattaneo A, Levi S, Arosio P (2010) Mutant ferritin L-chains that cause neurodegeneration act in a dominant-negative manner to reduce ferritin iron incorporation. J Biol Chem 285:11948–11957
Macedo MF, de Sousa M, Ned RM, Mascarenhas C, Andrews NC, Correia-Neves M (2004) Transferrin is required for early T-cell differentiation. Immunology 112:543–549
Mahnke A, Meier RJ, Schatz V, Hofmann J, Castiglione K, Schleicher U, Wolfbeis OS, Bogdan C, Jantsch J (2014) Hypoxia in Leishmania major skin lesions impairs the NO-dependent leishmanicidal activity of macrophages. J Invest Dermatol 134:2339–2346
Mair SM, Nairz M, Bellmann-Weiler R, Muehlbacher T, Schroll A, Theurl I, Moser PL, Talasz H, Fang FC, Weiss G (2011) Nifedipine affects the course of Salmonella enterica serovar Typhimurium infection by modulating macrophage iron homeostasis. J Infect Dis 204:685–694
Marques L, Negre-Salvayre A, Costa L, Canonne-Hergaux F (2016) Iron gene expression profile in atherogenic Mox macrophages. Biochim Biophys Acta 1862:1137–1146
Marro S, Chiabrando D, Messana E, Stolte J, Turco E, Tolosano E, Muckenthaler MU (2010) Heme controls ferroportin1 (FPN1) transcription involving Bach1, Nrf2 and a MARE/ARE sequence motif at position -7007 of the FPN1 promoter. Haematologica 95:1261–1268
Martins R, Maier J, Gorki AD, Huber KV, Sharif O, Starkl P, Saluzzo S, Quattrone F, Gawish R, Lakovits K, Aichinger MC, Radic-Sarikas B, Lardeau CH, Hladik A, Korosec A, Brown M, Vaahtomeri K, Duggan M, Kerjaschki D, Esterbauer H, Colinge J, Eisenbarth SC, Decker T, Bennett KL, Kubicek S, Sixt M, Superti-Furga G, Knapp S (2016) Heme drives hemolysis-induced susceptibility to infection via disruption of phagocyte functions. Nat Immunol
Mathieu JR, Heinis M, Zumerle S, Delga S, Le Bon A, Peyssonnaux C (2014) Investigating the real role of HIF-1 and HIF-2 in iron recycling by macrophages. Haematologica 99:e112–e114
Mayr R, Janecke AR, Schranz M, Griffiths WJ, Vogel W, Pietrangelo A, Zoller H (2010) Ferroportin disease: a systematic meta-analysis of clinical and molecular findings. J Hepatol 53:941–949
Mayr R, Griffiths WJ, Hermann M, McFarlane I, Halsall DJ, Finkenstedt A, Douds A, Davies SE, Janecke AR, Vogel W, Cox TM, Zoller H (2011) Identification of mutations in SLC40A1 that affect ferroportin function and phenotype of human ferroportin iron overload. Gastroenterology 140(2056–2063):e2051
McKie AT, Barlow DJ (2004) The SLC40 basolateral iron transporter family (IREG1/ferroportin/MTP1). Pflugers Arch 447:801–806
McKie AT, Marciani P, Rolfs A, Brennan K, Wehr K, Barrow D, Miret S, Bomford A, Peters TJ, Farzaneh F, Hediger MA, Hentze MW, Simpson RJ (2000) A novel duodenal iron-regulated transporter, IREG1, implicated in the basolateral transfer of iron to the circulation. Mol Cell 5:299–309
McKie AT, Barrow D, Latunde-Dada GO, Rolfs A, Sager G, Mudaly E, Mudaly M, Richardson C, Barlow D, Bomford A, Peters TJ, Raja KB, Shirali S, Hediger MA, Farzaneh F, Simpson RJ (2001) An iron-regulated ferric reductase associated with the absorption of dietary iron. Science 291:1755–1759
Melillo G, Taylor LS, Brooks A, Musso T, Cox GW, Varesio L (1997) Functional requirement of the hypoxia-responsive element in the activation of the inducible nitric oxide synthase promoter by the iron chelator desferrioxamine. J Biol Chem 272:12236–12243
Mitterstiller AM, Haschka D, Dichtl S, Nairz M, Demetz E, Talasz H, Soares MP, Einwallner E, Esterbauer H, Fang FC, Geley S, Weiss G (2016) Heme oxygenase 1 controls early innate immune response of macrophages to Salmonella typhimurium infection. Cell Microbiol 18:1374–1389
Miyanishi M, Tada K, Koike M, Uchiyama Y, Kitamura T, Nagata S (2007) Identification of Tim4 as a phosphatidylserine receptor. Nature 450:435–439
Moestrup SK, Moller HJ (2004) CD163: a regulated hemoglobin scavenger receptor with a role in the anti-inflammatory response. Ann Med 36:347–354
Montosi G, Corradini E, Garuti C, Barelli S, Recalcati S, Cairo G, Valli L, Pignatti E, Vecchi C, Ferrara F, Pietrangelo A (2005) Kupffer cells and macrophages are not required for hepatic hepcidin activation during iron overload. Hepatology 41:545–552
Moretti D, Goede JS, Zeder C, Jiskra M, Chatzinakou V, Tjalsma H, Melse-Boonstra A, Brittenham G, Swinkels DW, Zimmermann MB (2015) Oral iron supplements increase hepcidin and decrease iron absorption from daily or twice-daily doses in iron-depleted young women. Blood 126:1981–1989
Muckenthaler M, Roy CN, Custodio AO, Minana B, deGraaf J, Montross LK, Andrews NC, Hentze MW (2003) Regulatory defects in liver and intestine implicate abnormal hepcidin and Cybrd1 expression in mouse hemochromatosis. Nat Genet 34:102–107
Muhoberac BB, Baraibar MA, Vidal R (2013) Iron loading-induced aggregation and reduction of iron incorporation in heteropolymeric ferritin containing a mutant light chain that causes neurodegeneration. Biochim Biophys Acta 1812:544–548
Nahrendorf M, Swirski FK (2016) Innate immune cells in ischaemic heart disease: does myocardial infarction beget myocardial infarction? Eur Heart J 37:868–872
Nairz M, Fritsche G, Brunner P, Talasz H, Hantke K, Weiss G (2008) Interferon-gamma limits the availability of iron for intramacrophage Salmonella typhimurium. Eur J Immunol 38:1923–1936
Nairz M, Fritsche G, Crouch ML, Barton HC, Fang FC, Weiss G (2009) Slc11a1 limits intracellular growth of Salmonella enterica sv. Typhimurium by promoting macrophage immune effector functions and impairing bacterial iron acquisition. Cell Microbiol 11:1365–1381
Nairz M, Schleicher U, Schroll A, Sonnweber T, Theurl I, Ludwiczek S, Talasz H, Brandacher G, Moser PL, Muckenthaler MU, Fang FC, Bogdan C, Weiss G (2013) Nitric oxide-mediated regulation of ferroportin-1 controls macrophage iron homeostasis and immune function in Salmonella infection. J Exp Med 210:855–873
Nairz M, Haschka D, Demetz E, Weiss G (2014) Iron at the interface of immunity and infection. Front Pharmacol 5:152
Nairz M, Ferring-Appel D, Casarrubea D, Sonnweber T, Viatte L, Schroll A, Haschka D, Fang FC, Hentze MW, Weiss G, Galy B (2015a) Iron regulatory proteins mediate host resistance to salmonella infection. Cell Host Microbe 18:254–261
Nairz M, Schroll A, Demetz E, Tancevski I, Theurl I, Weiss G (2015b) 'Ride on the ferrous wheel'—the cycle of iron in macrophages in health and disease. Immunobiology 220:280–294
Nairz M, Theurl I, Wolf D, Weiss G (2016) Iron deficiency or anemia of inflammation?: differential diagnosis and mechanisms of anemia of inflammation. Wien Med Wochenschr 166:411–423
Nandar W, Neely EB, Unger E, Connor JR (2013) A mutation in the HFE gene is associated with altered brain iron profiles and increased oxidative stress in mice. Biochim Biophys Acta 1832:729–741
Nell-Duxneuner V, Axmann R, Husar-Memmer E, Dallos T, Datz C, Stadlmayr A, Aigner E, Englbrecht M, Schett G, Zwerina J (2013) VCAM-1 serum levels are associated with arthropathy in hereditary haemochromatosis. Ann Rheum Dis 72:2006–2010
Nemeth E, Ganz T (2006) Regulation of iron metabolism by hepcidin. Annu Rev Nutr 26:323–342
Nemeth E, Rivera S, Gabayan V, Keller C, Taudorf S, Pedersen BK, Ganz T (2004a) IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J Clin Invest 113:1271–1276
Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, Ganz T, Kaplan J (2004b) Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 306:2090–2093
Nicolas G, Bennoun M, Devaux I, Beaumont C, Grandchamp B, Kahn A, Vaulont S (2001) Lack of hepcidin gene expression and severe tissue iron overload in upstream stimulatory factor 2 (USF2) knockout mice. Proc Natl Acad Sci U S A 98:8780–8785
Oexle H, Gnaiger E, Weiss G (1999) Iron-dependent changes in cellular energy metabolism: influence on citric acid cycle and oxidative phosphorylation. Biochim Biophys Acta 1413:99–107
Oexle H, Kaser A, Most J, Bellmann-Weiler R, Werner ER, Werner-Felmayer G, Weiss G (2003) Pathways for the regulation of interferon-gamma-inducible genes by iron in human monocytic cells. J Leukoc Biol 74:287–294
Ohgami RS, Campagna DR, Greer EL, Antiochos B, McDonald A, Chen J, Sharp JJ, Fujiwara Y, Barker JE, Fleming MD (2005) Identification of a ferrireductase required for efficient transferrin-dependent iron uptake in erythroid cells. Nat Genet 37:1264–1269
Olsson M, Oldenborg PA (2008) CD47 on experimentally senescent murine RBCs inhibits phagocytosis following Fcgamma receptor-mediated but not scavenger receptor-mediated recognition by macrophages. Blood 112:4259–4267
Pantopoulos K, Weiss G, Hentze MW (1996) Nitric oxide and oxidative stress (H2O2) control mammalian iron metabolism by different pathways. Mol Cell Biol 16:3781–3788
Pantopoulos K, Porwal SK, Tartakoff A, Devireddy L (2012) Mechanisms of mammalian iron homeostasis. Biochemistry 51:5705–5724
Part K, Kunnis-Beres K, Poska H, Land T, Shimmo R, Zetterstrom Fernaeus S (2015) Amyloid beta25-35 induced ROS-burst through NADPH oxidase is sensitive to iron chelation in microglial Bv2 cells. Brain Res 1629:282–290
Pechlaner R, Kiechl S, Mayr M, Santer P, Weger S, Haschka D, Bansal SS, Willeit J, Weiss G (2016) Correlates of serum hepcidin levels and its association with cardiovascular disease in an elderly general population. Clin Chem Lab Med 54:151–161
Penno E, Johnsson C, Johansson L, Ahlstrom H (2006) Macrophage uptake of ultra-small iron oxide particles for magnetic resonance imaging in experimental acute cardiac transplant rejection. Acta Radiol 47:264–271
Petters C, Thiel K, Dringen R (2015) Lysosomal iron liberation is responsible for the vulnerability of brain microglial cells to iron oxide nanoparticles: comparison with neurons and astrocytes. Nanotoxicology 10:332–342
Peyssonnaux C, Zinkernagel AS, Datta V, Lauth X, Johnson RS, Nizet V (2006) TLR4-dependent hepcidin expression by myeloid cells in response to bacterial pathogens. Blood 107:3727–3732
Pham CG, Bubici C, Zazzeroni F, Papa S, Jones J, Alvarez K, Jayawardena S, De Smaele E, Cong R, Beaumont C, Torti FM, Torti SV, Franzoso G (2004) Ferritin heavy chain upregulation by NF-kappaB inhibits TNFalpha-induced apoptosis by suppressing reactive oxygen species. Cell 119:529–542
Phelan CA, Gampe RT Jr, Lambert MH, Parks DJ, Montana V, Bynum J, Broderick TM, Hu X, Williams SP, Nolte RT, Lazar MA (2010) Structure of Rev-erbalpha bound to N-CoR reveals a unique mechanism of nuclear receptor-co-repressor interaction. Nat Struct Mol Biol 17:808–814
Pietrangelo A (2007) Hemochromatosis: an endocrine liver disease. Hepatology 46:1291–1301
Pietrangelo A (2010) Hereditary hemochromatosis: pathogenesis, diagnosis, and treatment. Gastroenterology 139:393–408 408 e391-392
Pietsch EC, Chan JY, Torti FM, Torti SV (2003) Nrf2 mediates the induction of ferritin H in response to xenobiotics and cancer chemopreventive dithiolethiones. J Biol Chem 278:2361–2369
Qian ZM, He X, Liang T, Wu KC, Yan YC, Lu LN, Yang G, Luo QQ, Yung WH, Ke Y (2014) Lipopolysaccharides upregulate hepcidin in neuron via microglia and the IL-6/STAT3 signaling pathway. Mol Neurobiol 50:811–820
Ramos P, Casu C, Gardenghi S, Breda L, Crielaard BJ, Guy E, Marongiu MF, Gupta R, Levine RL, Abdel-Wahab O, Ebert BL, Van Rooijen N, Ghaffari S, Grady RW, Giardina PJ, Rivella S (2013) Macrophages support pathological erythropoiesis in polycythemia vera and beta-thalassemia. Nat Med 19:437–445
Recalcati S, Alberghini A, Campanella A, Gianelli U, De Camilli E, Conte D, Cairo G (2006) Iron regulatory proteins 1 and 2 in human monocytes, macrophages and duodenum: expression and regulation in hereditary hemochromatosis and iron deficiency. Haematologica 91:303–310
Roetto A, Papanikolaou G, Politou M, Alberti F, Girelli D, Christakis J, Loukopoulos D, Camaschella C (2003) Mutant antimicrobial peptide hepcidin is associated with severe juvenile hemochromatosis. Nat Genet 33:21–22
Rouault TA (2006) The role of iron regulatory proteins in mammalian iron homeostasis and disease. Nat Chem Biol 2:406–414
Rouault TA (2013) Iron metabolism in the CNS: implications for neurodegenerative diseases. Nat Rev Neurosci 14:551–564
Rucker P, Torti FM, Torti SV (1996) Role of H and L subunits in mouse ferritin. J Biol Chem 271:33352–33357
Saeed O, Otsuka F, Polavarapu R, Karmali V, Weiss D, Davis T, Rostad B, Pachura K, Adams L, Elliott J, Taylor WR, Narula J, Kolodgie F, Virmani R, Hong CC, Finn AV (2012) Pharmacological suppression of hepcidin increases macrophage cholesterol efflux and reduces foam cell formation and atherosclerosis. Arterioscler Thromb Vasc Biol 32:299–307
Sager HB, Hulsmans M, Lavine KJ, Moreira MB, Heidt T, Courties G, Sun Y, Iwamoto Y, Tricot B, Khan OF, Dahlman JE, Borodovsky A, Fitzgerald K, Anderson DG, Weissleder R, Libby P, Swirski FK, Nahrendorf M (2016) Proliferation and recruitment contribute to myocardial macrophage expansion in chronic heart failure. Circ Res 119:853–864
Sarkar J, Seshadri V, Tripoulas NA, Ketterer ME, Fox PL (2003) Role of ceruloplasmin in macrophage iron efflux during hypoxia. J Biol Chem 278:44018–44024
Schaer CA, Schoedon G, Imhof A, Kurrer MO, Schaer DJ (2006) Constitutive endocytosis of CD163 mediates hemoglobin-heme uptake and determines the noninflammatory and protective transcriptional response of macrophages to hemoglobin. Circ Res 99:943–950
Schaer DJ, Vinchi F, Ingoglia G, Tolosano E, Buehler PW (2014) Haptoglobin, hemopexin, and related defense pathways-basic science, clinical perspectives, and drug development. Front Physiol 5:415
Schaer CA, Deuel JW, Schildknecht D, Mahmoudi L, Garcia-Rubio I, Owczarek C, Schauer S, Kissner R, Banerjee U, Palmer AF, Spahn DR, Irwin DC, Vallelian F, Buehler PW, Schaer DJ (2016) Haptoglobin preserves vascular nitric oxide signaling during hemolysis. Am J Respir Crit Care Med 193:1111–1122
Schipper HM (1999) Glial HO-1 expression, iron deposition and oxidative stress in neurodegenerative diseases. Neurotox Res 1:57–70
Schleicher U, Paduch K, Debus A, Obermeyer S, Konig T, Kling JC, Ribechini E, Dudziak D, Mougiakakos D, Murray PJ, Ostuni R, Korner H, Bogdan C (2016) TNF-mediated restriction of arginase 1 expression in myeloid cells triggers type 2 NO synthase activity at the site of infection. Cell Rep 15:1062–1075
Schroll A, Eller K, Feistritzer C, Nairz M, Sonnweber T, Moser PA, Rosenkranz AR, Theurl I, Weiss G (2012) Lipocalin-2 ameliorates granulocyte functionality. Eur J Immunol 42:3346–3357
Scott CL, Zheng F, De Baetselier P, Martens L, Saeys Y, De Prijck S, Lippens S, Abels C, Schoonooghe S, Raes G, Devoogdt N, Lambrecht BN, Beschin A, Guilliams M (2016) Bone marrow-derived monocytes give rise to self-renewing and fully differentiated Kupffer cells. Nat Commun 7:10321
Sender R, Fuchs S, Milo R (2016) Revised estimates for the number of human and bacteria cells in the body. PLoS Biol 14:e1002533
Siegert I, Schodel J, Nairz M, Schatz V, Dettmer K, Dick C, Kalucka J, Franke K, Ehrenschwender M, Schley G, Beneke A, Sutter J, Moll M, Hellerbrand C, Wielockx B, Katschinski DM, Lang R, Galy B, Hentze MW, Koivunen P, Oefner PJ, Bogdan C, Weiss G, Willam C, Jantsch J (2015) Ferritin-mediated iron sequestration stabilizes hypoxia-inducible factor-1alpha upon LPS activation in the presence of ample oxygen. Cell Rep 13:2048–2055
Smith KD (2007) Iron metabolism at the host pathogen interface: lipocalin 2 and the pathogen-associated iroA gene cluster. Int J Biochem Cell Biol 39:1776–1780
Soares MP, Hamza I (2016) Macrophages and iron metabolism. Immunity 44:492–504
Soares MP, Weiss G (2015) The iron age of host-microbe interactions. EMBO Rep 16:1482–1500
Soe-Lin S, Apte SS, Andriopoulos B Jr, Andrews MC, Schranzhofer M, Kahawita T, Garcia-Santos D, Ponka P (2009) Nramp1 promotes efficient macrophage recycling of iron following erythrophagocytosis in vivo. Proc Natl Acad Sci U S A 106:5960–5965
Sohn YS, Ghoti H, Breuer W, Rachmilewitz E, Attar S, Weiss G, Cabantchik ZI (2012) The role of endocytic pathways in cellular uptake of plasma non-transferrin iron. Haematologica 97:670–678
Song W, Su H, Song S, Paudel HK, Schipper HM (2006) Over-expression of heme oxygenase-1 promotes oxidative mitochondrial damage in rat astroglia. J Cell Physiol 206:655–663
Song L, Song W, Schipper HM (2007) Astroglia overexpressing heme oxygenase-1 predispose co-cultured PC12 cells to oxidative injury. J Neurosci Res 85:2186–2195
Song D, Song Y, Hadziahmetovic M, Zhong Y, Dunaief JL (2012a) Systemic administration of the iron chelator deferiprone protects against light-induced photoreceptor degeneration in the mouse retina. Free Radic Biol Med 53:64–71
Song W, Zukor H, Lin SH, Liberman A, Tavitian A, Mui J, Vali H, Fillebeen C, Pantopoulos K, Wu TD, Guerquin-Kern JL, Schipper HM (2012b) Unregulated brain iron deposition in transgenic mice over-expressing HMOX1 in the astrocytic compartment. J Neurochem 123:325–336
Stober CB, Brode S, White JK, Popoff JF, Blackwell JM (2007) Slc11a1, formerly Nramp1, is expressed in dendritic cells and influences major histocompatibility complex class II expression and antigen-presenting cell function. Infect Immun 75:5059–5067
Sullivan JL (2009) Iron in arterial plaque: modifiable risk factor for atherosclerosis. Biochim Biophys Acta 1790:718–723
Sun J, Hoshino H, Takaku K, Nakajima O, Muto A, Suzuki H, Tashiro S, Takahashi S, Shibahara S, Alam J, Taketo MM, Yamamoto M, Igarashi K (2002) Hemoprotein Bach1 regulates enhancer availability of heme oxygenase-1 gene. EMBO J 21:5216–5224
Sun YM, Wang YT, Jiang L, Xue MZ (2016) The effects of deferoxamine on inhibition for microglia activation and protection of secondary nerve injury after intracerebral hemorrhage in rats. Pak J Pharm Sci 29:1087–1093
Swirski FK, Nahrendorf M (2013) Leukocyte behavior in atherosclerosis, myocardial infarction, and heart failure. Science 339:161–166
Tacchini L, Gammella E, De Ponti C, Recalcati S, Cairo G (2008) Role of HIF-1 and NF-kappaB transcription factors in the modulation of transferrin receptor by inflammatory and anti-inflammatory signals. J Biol Chem 283:20674–20686
Techau ME, Valdez-Taubas J, Popoff JF, Francis R, Seaman M, Blackwell JM (2007) Evolution of differences in transport function in Slc11a family members. J Biol Chem 282:35646–35656
Theurl I, Fritsche G, Ludwiczek S, Garimorth K, Bellmann-Weiler R, Weiss G (2005) The macrophage: a cellular factory at the interphase between iron and immunity for the control of infections. Biometals 18:359–367
Theurl I, Theurl M, Seifert M, Mair S, Nairz M, Rumpold H, Zoller H, Bellmann-Weiler R, Niederegger H, Talasz H, Weiss G (2008a) Autocrine formation of hepcidin induces iron retention in human monocytes. Blood 111:2392–2399
Theurl M, Theurl I, Hochegger K, Obrist P, Subramaniam N, van Rooijen N, Schuemann K, Weiss G (2008b) Kupffer cells modulate iron homeostasis in mice via regulation of hepcidin expression. J Mol Med (Berl) 86:825–835
Theurl I, Aigner E, Theurl M, Nairz M, Seifert M, Schroll A, Sonnweber T, Eberwein L, Witcher DR, Murphy AT, Wroblewski VJ, Wurz E, Datz C, Weiss G (2009) Regulation of iron homeostasis in anemia of chronic disease and iron deficiency anemia: diagnostic and therapeutic implications. Blood 113:5277–5286
Theurl I, Schroll A, Sonnweber T, Nairz M, Theurl M, Willenbacher W, Eller K, Wolf D, Seifert M, Sun CC, Babitt JL, Hong CC, Menhall T, Gearing P, Lin HY, Weiss G (2011) Pharmacologic inhibition of hepcidin expression reverses anemia of chronic inflammation in rats. Blood 118:4977–4984
Theurl I, Hilgendorf I, Nairz M, Tymoszuk P, Haschka D, Asshoff M, He S, Gerhardt LM, Holderried TA, Seifert M, Sopper S, Fenn AM, Anzai A, Rattik S, McAlpine C, Theurl M, Wieghofer P, Iwamoto Y, Weber GF, Harder NK, Chousterman BG, Arvedson TL, McKee M, Wang F, Lutz OM, Rezoagli E, Babitt JL, Berra L, Prinz M, Nahrendorf M, Weiss G, Weissleder R, Lin HY, Swirski FK (2016a) On-demand erythrocyte disposal and iron recycling requires transient macrophages in the liver. Nat Med 22:945–951
Theurl M, Song D, Clark E, Sterling J, Grieco S, Altamura S, Galy B, Hentze M, Muckenthaler MU, Dunaief JL (2016b) Mice with hepcidin-resistant ferroportin accumulate iron in the retina. FASEB J 30:813–823
Thomsen MS, Andersen MV, Christoffersen PR, Jensen MD, Lichota J, Moos T (2015) Neurodegeneration with inflammation is accompanied by accumulation of iron and ferritin in microglia and neurons. Neurobiol Dis 81:108–118
Toda S, Segawa K, Nagata S (2014) MerTK-mediated engulfment of pyrenocytes by central macrophages in erythroblastic islands. Blood 123:3963–3971
Tolosano E, Fagoonee S, Hirsch E, Berger FG, Baumann H, Silengo L, Altruda F (2002) Enhanced splenomegaly and severe liver inflammation in haptoglobin/hemopexin double-null mice after acute hemolysis. Blood 100:4201–4208
Torti FM, Torti SV (2002) Regulation of ferritin genes and protein. Blood 99:3505–3516
Turrini F, Mannu F, Cappadoro M, Ulliers D, Giribaldi G, Arese P (1994) Binding of naturally occurring antibodies to oxidatively and nonoxidatively modified erythrocyte band 3. Biochim Biophys Acta 1190:297–303
Valenti L, Dongiovanni P, Motta BM, Swinkels DW, Bonara P, Rametta R, Burdick L, Frugoni C, Fracanzani AL, Fargion S (2010) Serum hepcidin and macrophage iron correlate with MCP-1 release and vascular damage in patients with metabolic syndrome alterations. Arterioscler Thromb Vasc Biol 31:683–690
Vanoaica L, Darshan D, Richman L, Schumann K, Kuhn LC (2010) Intestinal ferritin H is required for an accurate control of iron absorption. Cell Metab 12:273–282
Vargas MR, Pehar M, Cassina P, Martinez-Palma L, Thompson JA, Beckman JS, Barbeito L (2005) Fibroblast growth factor-1 induces heme oxygenase-1 via nuclear factor erythroid 2-related factor 2 (Nrf2) in spinal cord astrocytes: consequences for motor neuron survival. J Biol Chem 280:25571–25579
Vinchi F, Costa da Silva M, Ingoglia G, Petrillo S, Brinkman N, Zuercher A, Cerwenka A, Tolosano E, Muckenthaler MU (2016) Hemopexin therapy reverts heme-induced proinflammatory phenotypic switching of macrophages in a mouse model of sickle cell disease. Blood 127:473–486
Wang J, Pantopoulos K (2011) Regulation of cellular iron metabolism. Biochem J 434:365–381
Wang J, Chen G, Lee J, Pantopoulos K (2008) Iron-dependent degradation of IRP2 requires its C-terminal region and IRP structural integrity. BMC Mol Biol 9:15
Warkentin DL, Marchand A, Van Lente F (1987) Serum haptoglobin concentrations in concurrent hemolysis and acute-phase reaction. Clin Chem 33:1265–1266
Weinberg ED (2009) Iron availability and infection. Biochim Biophys Acta 1790:600–605
Weiss G (2010) Genetic mechanisms and modifying factors in hereditary hemochromatosis. Nat Rev Gastroenterol Hepatol 7:50–58
Weiss G, Schaible UE (2015) Macrophage defense mechanisms against intracellular bacteria. Immunol Rev 264:182–203
Wessling-Resnick M (2006) Iron imports. III. Transfer of iron from the mucosa into circulation. Am J Physiol Gastrointest Liver Physiol 290:G1–G6
Wessling-Resnick M (2015) Nramp1 and other transporters involved in metal withholding during infection. J Biol Chem 290:18984–18990
White C, Yuan X, Schmidt PJ, Bresciani E, Samuel TK, Campagna D, Hall C, Bishop K, Calicchio ML, Lapierre A, Ward DM, Liu P, Fleming MD, Hamza I (2013) HRG1 is essential for heme transport from the phagolysosome of macrophages during erythrophagocytosis. Cell Metab 17:261–270
Willekens FL, Werre JM, Kruijt JK, Roerdinkholder-Stoelwinder B, Groenen-Dopp YA, van den Bos AG, Bosman GJ, van Berkel TJ (2005) Liver Kupffer cells rapidly remove red blood cell-derived vesicles from the circulation by scavenger receptors. Blood 105:2141–2145
Xiao X, Yeoh BS, Saha P, Tian Y, Singh V, Patterson AD, Vijay-Kumar M (2016) Modulation of urinary siderophores by the diet, gut microbiota and inflammation in mice. J Nutr Biochem 41:25–33
Yi T, Li J, Chen H, Wu J, An J, Xu Y, Hu Y, Lowell CA, Cyster JG (2015) Splenic dendritic cells survey red blood cells for missing self-CD47 to trigger adaptive immune responses. Immunity 43:764–775
Yoshida H, Kawane K, Koike M, Mori Y, Uchiyama Y, Nagata S (2005) Phosphatidylserine-dependent engulfment by macrophages of nuclei from erythroid precursor cells. Nature 437:754–758
Yu X, Song N, Guo X, Jiang H, Zhang H, Xie J (2016) Differences in vulnerability of neurons and astrocytes to heme oxygenase-1 modulation: implications for mitochondrial ferritin. Sci Rep 6:24200
Yuan XM (1999) Apoptotic macrophage-derived foam cells of human atheromas are rich in iron and ferritin, suggesting iron-catalysed reactions to be involved in apoptosis. Free Radic Res 30:221–231
Zanganeh S, Hutter G, Spitler R, Lenkov O, Mahmoudi M, Shaw A, Pajarinen JS, Nejadnik H, Goodman S, Moseley M, Coussens LM, Daldrup-Link HE (2016) Iron oxide nanoparticles inhibit tumour growth by inducing pro-inflammatory macrophage polarization in tumour tissues. Nat Nanotechnol 11:986–994
Zeineh MM, Chen Y, Kitzler HH, Hammond R, Vogel H, Rutt BK (2015) Activated iron-containing microglia in the human hippocampus identified by magnetic resonance imaging in Alzheimer disease. Neurobiol Aging 36:2483–2500
Zhang Z, Zhang F, An P, Guo X, Shen Y, Tao Y, Wu Q, Zhang Y, Yu Y, Ning B, Nie G, Knutson MD, Anderson GJ, Wang F (2011) Ferroportin1 deficiency in mouse macrophages impairs iron homeostasis and inflammatory responses. Blood 118:1912–1922
Zhang Z, Song Y, Li D, Zhu H, Liang R, Gu Y, Pang Y, Qi J, Wu H, Wang J (2017) Distinct role of heme oxygenase-1 in early- and late-stage intracerebral hemorrhage in 12-month-old mice. J Cereb Blood Flow Metab 37:25–38
Zhao L, Li Y, Song D, Song Y, Theurl M, Wang C, Cwanger A, Su G, Dunaief JL (2014) A high serum iron level causes mouse retinal iron accumulation despite an intact blood-retinal barrier. Am J Pathol 184:2862–2867
Zhou J, Ye S, Fujiwara T, Manolagas SC, Zhao H (2013) Steap4 plays a critical role in osteoclastogenesis in vitro by regulating cellular iron/reactive oxygen species (ROS) levels and cAMP response element-binding protein (CREB) activation. J Biol Chem 288:30064–30074
Zohn IE, De Domenico I, Pollock A, Ward DM, Goodman JF, Liang X, Sanchez AJ, Niswander L, Kaplan J (2007) The flatiron mutation in mouse ferroportin acts as a dominant negative to cause ferroportin disease. Blood 109:4174–4180
Zoller H, Theurl I, Koch RO, McKie AT, Vogel W, Weiss G (2003) Duodenal cytochrome b and hephaestin expression in patients with iron deficiency and hemochromatosis. Gastroenterology 125:746–754
Author information
Authors and Affiliations
Corresponding authors
Additional information
This article is part of the special issue on macrophages in tissue homeostasis in Pflügers Archiv – European Journal of Physiology
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Nairz, M., Theurl, I., Swirski, F.K. et al. “Pumping iron”—how macrophages handle iron at the systemic, microenvironmental, and cellular levels. Pflugers Arch - Eur J Physiol 469, 397–418 (2017). https://doi.org/10.1007/s00424-017-1944-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-017-1944-8