
Abstract
Glucose concentrations of normal human airway surface liquid are ~12.5 times lower than blood glucose concentrations indicating that glucose uptake by epithelial cells may play a role in maintaining lung glucose homeostasis. We have therefore investigated potential glucose uptake mechanisms in non-polarised and polarised H441 human airway epithelial cells and bronchial biopsies. We detected mRNA and protein for glucose transporter type 2 (GLUT2) and glucose transporter type 4 (GLUT4) in non-polarised cells but GLUT4 was not detected in the plasma membrane. In polarised cells, GLUT2 protein was detected in both apical and basolateral membranes. Furthermore, GLUT2 protein was localised to epithelial cells of human bronchial mucosa biopsies. In non-polarised H441 cells, uptake of d-glucose and deoxyglucose was similar. Uptake of both was inhibited by phloretin indicating that glucose uptake was via GLUT-mediated transport. Phloretin-sensitive transport remained the predominant route for glucose uptake across apical and basolateral membranes of polarised cells and was maximal at 5–10 mM glucose. We could not conclusively demonstrate sodium/glucose transporter-mediated transport in non-polarised or polarised cells. Our study provides the first evidence that glucose transport in human airway epithelial cells in vitro and in vivo utilises GLUT2 transporters. We speculate that these transporters could contribute to glucose uptake/homeostasis in the human airway.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The luminal surface of the lung from nose to alveoli is lined with a thin layer of fluid (airway surface liquid, ASL). We estimate that glucose concentrations of normal human ASL are 12.5 times lower than blood glucose concentrations [3], similar to observations made in sheep and rat lungs [4, 39]. Low ASL glucose concentrations could contribute to pulmonary defence against infection. Patients admitted to intensive care with high ASL glucose concentrations are more likely to develop lung infections, particularly with methicillin-resistant Staphylococcus aureus (MRSA), than those with low ASL glucose concentrations [34]. Maintenance of low ASL glucose concentrations could form part of a therapeutic strategy against respiratory infection, but mechanisms underlying airway glucose uptake and ASL glucose homeostasis are not fully understood.
ASL glucose concentrations increase as blood glucose is raised and falls as blood glucose falls, but against the transepithelial glucose gradient [3, 42]. This indicates that glucose is cleared from ASL by glucose transporters in the apical membrane of airway epithelial cells.
Glucose transporters can be divided into two groups. The facilitative glucose transporters (GLUTs) transport glucose into or out of the cell dependent on the glucose gradient. Under normal conditions, GLUTs transport glucose into the cell down its concentration gradient which is maintained by the rapid metabolism of glucose to glucose-6-phosphate as soon as it enters the cell. The sodium/glucose co-transporters (SGLT) require the co-transport of Na+ and can utilise the transmembrane Na+ gradient to drive concentrative glucose uptake [45]. Animal studies indicate that both transporter types are functionally expressed in respiratory epithelium. SGLT-1 mRNA, but not protein, was detected in rat and mouse whole lung tissue and rat alveolar type II pneumocytes [18, 10]. Glucose removal from the lumen of fetal sheep and adult rat lungs was inhibited by the SGLT inhibitor phlorizin [4, 38]. Phlorizin also caused a small depolarisation of bovine and sheep tracheal epithelium, implying SGLT activity [40, 21]. GLUT1, GLUT2, GLUT4 and GLUT5 mRNA were detected in rat type II alveolar cells [28]. We have also shown that GLUT1 and GLUT2 mRNA were present in human airway by PCR [41]. Glucose uptake by isolated guinea pig type II pneumocytes was inhibited by the GLUT-blocker phloretin [25]. However, glucose absorption from fluid filled adult rat lungs was not phloretin-sensitive [39], indicating that GLUT transport did not contribute to apical glucose absorption in the distal lung of this species.
The aim of this study was to elucidate mechanisms of glucose transport by human airway epithelial cells. We used immortalised cultured H441 cells, which derive from a papillary adenocarcinoma of the bronchiolar epithelium. When cultured at air interface, these cells form an absorptive epithelial monolayer, exhibit vectorial ion transport processes and have similar morphological and phenotypic characteristics to human bronchiolar epithelium [17]. Observations were then confirmed in intact human bronchial epithelium obtained at bronchoscopy.
Materials and methods
Cell culture
H441 cells obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA) were cultured in RPMI-1640 media + 10% foetal bovine serum (FBS) (Invitrogen, UK); glucose (10 mM); l-glutamine (2 mM); sodium pyruvate (1 mM); insulin (10 μg/ml); transferrin (5 μg/ml); sodium selenite (7 ng/ml); penicillin (100 U/ml); and streptomycin (100 μg/ml). Non-polarised cells were grown in 12-well tissue culture plates. Polarised monolayers were produced by culturing cells for 7 days on permeable 4-µm pore polyester membrane supports (Transwells, Corning, MA, USA). The basolateral membrane was exposed to RPMI media [4% charcoal stripped serum; glucose (10 mM); dexamethasone (200 μM); 3,3′-5-Triiodothyronine (10 nM); l-glutamine (2 mM); sodium pyruvate (1 mM); insulin (10 μg/ml); transferrin (5 μg/ml); sodium selenite (7 ng/ml); penicillin (100 U/ml); and streptomycin (100 μg/ml)] and the apical membrane was at air interface. The day before the experiment, cells were washed with glucose-free RPMI media and incubated in medium with the required glucose concentration.
Glucose uptake experiments
Cultured cells were washed twice with glucose-free transport medium [15 mM HEPES buffer (pH 7.6), 135 mM NaCl, 5 mM KCl, 1.8 mM CaCl2 and 0.8 mM MgCl2] to remove culture medium, then incubated for 15 min at room temperature in the same solution to deplete the cells of intracellular glucose (± inhibitors).
Uptake experiments were performed as described previously [32]. In non-polarised cells, the experiment was initiated by replacing the medium with 0.5 ml transport medium containing 1.0 μCi of radiolabelled glucose or glucose analogue plus 10 mM of non-radiolabelled equivalent glucose or glucose analogue (tracer mix), followed by incubation at room temperature for 10 min. Preliminary experiments indicated that uptake was linear between 0 and 10 min (data not shown). Uptake was terminated by adding 2 ml ice-cold stop solution [15 mM HEPES buffer (pH 7.6); 135 mM choline Cl; 5 mM KCl; 0.8 mM MgSO4; 1.8 mM CaCl2 and 0.2 mM HgCl2]. The cells were then rinsed twice with 2 ml stop solution and lysed in 0.5 ml of 10 mM Tris–HCl (pH 8.0) with 0.2% SDS. Lysed samples were added to 2 ml scintillation cocktail and radio-active emissions determined using a scintillation counter to quantify glucose uptake. Each uptake experiment was performed three times (n = 3) unless otherwise stated. The same basic protocol was used for all experiments with the following modifications: In polarised monolayers, the radioactive tracer mix was added either to the basolateral or apical side of the monolayer.
Most glucose uptake studies were carried out using [3H]-d-glucose, which is transported by all glucose transporters. Radiolabelled glucose analogues [3H]-deoxyglucose (DOG) (Sigma, UK) and [14C]-α-methyl-d-glucopyranoside (AMG) (Amersham, UK) were used to study GLUT- and SGLT-mediated glucose transport, respectively.
The effect of different glucose concentrations on glucose transport was determined by culturing cells at different glucose concentrations (1, 5, 10, and 15 mM) the day before the experiment. Cells from the same passage were used for all uptake experiments at different glucose concentrations to avoid discrepancies in results.
Several inhibitors were used to block glucose transport. Phlorizin (500 μM dissolved in ethanol) is a high affinity inhibitor of Na+–d-glucose co-transport via SGLT in whole tissue [13, 14]. Phloretin (1 mM dissolved in ethanol) and cytochalasin B (10 μM) inhibit glucose transport mediated by GLUTs in whole tissue [24]. Ouabain (1 mM dissolved in ethanol), which inhibits Na+K+ATPase activity, and Na+ substitution with choline were also used in some experiments to determine if glucose transport was coupled to the Na+ gradient (SGLT-mediated). The amount of ethanol used to dissolve inhibitors (5 μl/well, 1% of total volume) was added to cells in control groups (vehicle control, 1% of total volume). Where inhibitors were used in experiments, they were added as soon as cells were transferred from culture to transport medium.
Reverse transcriptase polymerase chain reaction
Total RNA was isolated from H441 cells using the RNeasy kit (Qiagen). cDNA was generated using reverse transcriptase (Superscript II, Invitrogen, Carlsbad, CA, USA). Reverse transcriptase polymerase chain reaction (RT-PCR) was performed using primer sequences derived from regions within GLUT1–GLUT4 and SGLT1 coding regions in a PCR reaction mix [1.5 mM MgCl2, 250 nM of each primer and 0.2 U of AGS gold Taq polymerase (Hybaid, Ashford, Middx, UK)]. PCR products were fractionated on 1.2% agarose gels and visualized by ethidium bromide staining and UV fluorescence. The following primer pairs were used in the reactions. GLUT1 sense: TCCACGGAGCATCTTCGAGA; GLUT1 anti-sense: ATACTGGAAGCACATGCCC (cycling protocol of 94°C for 1 min, 56°C for 1 min and 72°C for 90 s for 40 cycles); GLUT2 sense: CACTGATGGCTGCATGTGGC; GLUT2 anti-sense: ATGTGAACAGGGTAAAGGCC (cycling protocol of 94°C for 1 min, 56°C for 1 min and 72°C for 90 s for 40 cycles); GLUT2 nested sense: CTACTCAACCAGCATTTTTC; GLUT2 nested anti-sense: AACACATAAGGTCCACAGAA (cycling protocol of 94°C for 1 min, 52°C for 1 min and 72°C for 90 s for 40 cycles); GLUT3 sense: AAGGATAACTATAATGG; GLUT3 anti-sense: GGTCTCCTTAGCAGGCT (cycling protocol of 95°C for 45 s, 46°C for 30 s and 72°C for 45 s for 37 cycles); GLUT4 sense: GCCATTGTTATCGGCATTCT; GLUT4 anti-sense: GAGCTGGAGCAGGGACAGT (cycling protocol of 94°C for 1 min, 51°C for 1 min and 72°C for 1 min for 35 cycles). Primers amplifying a region of β-actin were used as an internal control. β-actin sense: CGGGACCTGACTGACTACC; β-actin anti-sense: TGAAGGTAGTTTCGTGGATGC (cycling protocol of 94°C for 3 min, 53°C for 1 min and 72°C for 1 min for 35 cycles). Cycle curves for all sets of PCR primers were performed. The number of cycles performed for each primer set was in the linear range of the curve.
Protein preparation
Total cell protein was prepared from cells scraped from flasks and homogenised in tissue lysis buffer [100 mM Tris pH, 6.8, 1 mM EDTA pH 8.0, 1% NP-40, 10% v/v glycerol and 10 ml/l protease inhibitor cocktail] followed by centrifugation (5 min, 250×g) to remove nuclei. Plasma membranes were isolated as previously described [6]. Briefly, cells were grown in six T75 flasks, lysed and the membranes separated from the intracellular fraction by centrifugation (60,000×g, 4°C, 30 min). Membrane and intracellular fractions were suspended in lysis buffer as above.
Membrane protein biotinylation
Apical or basolateral membrane proteins were biotinylated as previously described [43]. Briefly, after 6 days at air interface, polarised cells were washed with ice-cold PBS. Sulfo-NHS-biotin (0.5 mg/ml) was applied to the apical or basolateral membrane. Cells were then lysed, proteins solubilised and incubated overnight with streptavidin agarose beads. The following day, biotinylated proteins bound to beads were separated from non-biotinylated proteins by centrifugation and samples prepared for immunoblotting.
Western blotting
Membrane protein fractions (50 μg) were separated on 4–12% Bis-Tris acrylamide gels, transferred to polyvinylidine difluoride membranes and incubated overnight at 4oC with anti GLUT1–GLUT4 affinity-purified antisera (1:200; Alpha Diagnostic, San Antonio, TX and Millipore, UK) or mouse monoclonal anti β-actin (1:500, AbCam, Cambridge, UK) or mouse monoclonal anti α1-Na+K+ATPase (1:1,000, Developmental Studies Hybridoma Bank, University of Iowa, USA). Blots were washed three times in PBS + 0.01% TWEEN 20 then incubated with either biotinylated donkey anti-rabbit IgG or rabbit anti-mouse secondary antiserum (1:200) (GE Healthcare, UK) followed by streptavidin horseradish peroxidase (HRP) conjugate (1:200) (GE Healthcare, UK) for 1 h each at room temperature. Immunostained proteins were visualised using enhanced chemiluminescence (ECL) western blot analysis system (NEN Life Science Products, Boston, MA; Western lightning, PerkinElmer, Norwalk, CT, USA) and exposure to X-ray film. All western blots were repeated at least twice. Blots of protein from cells grown at different glucose concentrations were processed simultaneously to allow visual comparisons to be made. Binding specificity of GLUT2 antibodies was confirmed in a repeat experiment where membranes were pre-incubated with antigenic peptides.
Measurement of short circuit current (I sc)
Polarised monolayers were mounted in Ussing chambers with both apical and basolateral surfaces bathed in a physiological salt solution [117 mM NaCl; 25 mM NaHCO3; 4.7 mM KCl; 1.2 mM MgSO4; 1.2 mM KH2PO4; 2.5 mM CaCl2; 11 mM d-glucose, equilibrated with 5% CO2 to pH 7.3–7.4]. The solution was maintained at 37°C, bubbled with premixed gas [21% O2 + 5% CO2] and circulated continuously throughout the experiment. Monolayers were maintained under open circuit conditions whilst transepithelial potential difference (V t) and resistance (R t) were monitored and observed to reach a stable level. The cells were then short circuited by clamping V t at 0 mV using a DVC-4000 voltage/current clamp. The current required to maintain this condition (I sc) was measured and recorded using a PowerLab computer interface. Every 30 s throughout each experiment, the preparations were returned to open circuit conditions for 3 s so that the spontaneous V t could be measured and R t could be calculated as previously described [44].
The contribution of SGLT and epithelial sodium channels (ENaC) to I sc were measured by adding 500 μM phlorizin or vehicle control to the apical or basolateral solution and 10 μM amiloride to the apical bath, respectively. Ion transport across these cells is dependent on Na+K+ATPase activity. Thus, ouabain (1 mM) was added to the basolateral compartment to calculate values for total I sc.
Immunofluorescence staining of human bronchial epithelium
Endobronchial biopsies were obtained at bronchoscopy from two patients with chronic obstructive pulmonary disease and a patient with no airway disease participating in a study approved by King’s College Research Ethics Committee. Participants gave written informed consent for inclusion in the study. Biopsies were frozen immediately at optimal cutting temperature and stored at −80°C. Cryostat sections of 7 μm were mounted on polysine-coated slides. Sections were blocked with 10% chicken serum (ISL) in 0.1% Triton X-100-PBS. Sections were incubated with rabbit polyclonal antibodies raised against the human liver GLUT2 transporter (1:100 dilution in 0.1%Triton-PBS; Chemicon, Hampshire, UK) at room temperature for 1 h. After washing 3 × 10 min in PBS, sections were incubated in FITC-conjugated goat anti-rabbit IgG 1:100 diluted in 0.1% Triton-PBS for 30 min at room temperature. Slides were washed three times for 10 min in PBS, sections were then counterstained with 4′,6-diamidino-2-phenyindole (DAPI; 1:1,000 diluted in 0.1% Triton-PBS) to localise cell nuclei. Sections were also incubated with FITC-conjugated goat anti-rabbit IgG alone or GLUT2 antiserum pre-absorbed with a 10-fold excess of antigenic peptide for 20 min. Images were observed with a fluorescence microscope Zeiss with Axioplan 2 analysis system using the Axiovision 4.5 software. Cell fluorescence was recorded by excitation at 450–490 nm.
Chemicals and reagents
All chemicals and reagents were obtained from Sigma, Poole, UK unless otherwise stated.
Values are reported as mean ± SEM. Statistical analysis was performed using paired Student’s t tests or one-way ANOVA tests (where appropriate). P values of <0.05 were considered statistically significant.
Results
Glucose uptake by non-polarised cells
Non-polarised cells bathed in 10 mM glucose without inhibitors (control) took up 39.7 ± 7.3 nmol labelled d-glucose/mg protein. In the presence of phloretin, d-glucose uptake was lower at 5.6 ± 1.7 nmol/mg protein (p < 0.01, n = 6) compared to control. d-Glucose uptake was not significantly different from control in the presence of phlorizin or ouabain (Fig. 1a).

Glucose uptake in non-polarised H441 cells. Effect of pre-incubation with vehicle (control), 500 μM phlorizin (PZ), 1 mM phloretin (PT), 1 mM ouabain (OU), 10 μM cytochalasin B (Cyt B) or Na+ replacement with choline (choline) on the uptake of 10 mM glucose (traced by [3H]-glucose) (a), [14C]-α-methyl-d-glucopyranoside (AMG) (b) or [3H]-deoxyglucose (DOG) (c). Uptake experiments were performed at 37°C. Data are shown as mean ± SEM. *p < 0.05, significantly different from control. **p < 0.01, significantly different from control
In the absence of inhibitors, AMG uptake was lower than d-glucose uptake at 0.62 ± 0.22 nmol/mg protein (Fig. 1b). None of the inhibitors significantly reduced AMG uptake.
DOG uptake in the absence of inhibitors (control, 44.4 ± 5.1 nmol/mg protein) was similar to d-glucose uptake (Fig. 1c). DOG uptake was significantly lower than in control in the presence of either phloretin (9.7 ± 1.5 nmol/mg protein) or cytochalasin B (3.5 ± 0.7 nmol/mg protein) (p < 0.05, n = 3, respectively), but not significantly lower in the presence of ouabain, phlorizin or choline. Overall, these results demonstrate that non-polarised H441 epithelial cells predominantly take up glucose through GLUT transporters.
Effect of glucose concentration on glucose uptake by non-polarised cells
Glucose uptake by non-polarised cells was measured following incubation in culture medium containing a range of physiological glucose concentrations: 1 mM (hypoglycaemic); 5 mM (normoglycaemic); 10 mM (mildly hyperglycaemic) and 15 mM (hyperglycaemic). Glucose uptake in the absence of inhibitors (control) was lowest at 1 mM glucose (18.4 ± 0.4 nmol/mg protein), increased at 5 mM (25.9 ± 0.3 nmol/mg protein) and was greatest at 10 mM glucose (39.7 ± 7.3 nmol/mg protein) and at 15 mM glucose (34.0 ± 2.7 nmol/mg protein) (n = 3) (Figs. 1a and 2a–c).

Uptake of glucose (traced by [3H]-glucose) by non-polarised H441 cells incubated in culture medium containing 1 mM (a), 5 mM (b) or 15 mM (c) glucose. Cells were pre-incubated with vehicle (control), 500 μM phlorizin (PZ) or 1 mM phloretin (PT) before measurement of glucose uptake. Uptake experiments were performed at 37°C. Data are presented as mean ± SEM. **p < 0.01, significantly different from control
The GLUT inhibitor phloretin significantly reduced glucose uptake at all glucose concentrations (1 mM to 1.2 ± 0.1; 5 mM to 3.6 ± 0.5 and 15 mM to 4.1 ± 0.2 nmol/mg protein, p < 0.01, n = 6, respectively) compared to control studies without inhibitors. Values for 10 mM glucose are given above. Phlorizin did not significantly affect glucose uptake at any glucose concentration (Figs. 1a and 2a–c).
GLUT transporter expression in non-polarised cells
We detected mRNA for GLUT1, GLUT2 and GLUT4 in non-polarised H441 cells (Fig. 3a). In non-polarised cells cultured at 10 mM glucose, a protein of predicted size for GLUT2 (60 kDa) was detected in separated plasma membrane and intracellular protein fractions in approximately equal quantities (Fig. 3b). A 45-kDa product corresponding to GLUT4 protein was detected in total cell protein and the intracellular fraction but was absent from the plasma membrane protein fraction. It is interesting to note that although GLUT1 mRNA was detected, translated protein could not be detected by western blotting in these cells (Fig. 3b). Neither GLUT3 mRNA nor protein was detected (Fig. 3a,b).

a RT-PCR of GLUT1–GLUT4 mRNA sequences from non-polarised H441 cells. Products were resolved on ethidium bromide-stained agarose gels. RT-PCR products corresponding to the correct size for GLUT1 (400 bp), GLUT2 (425 bp) and GLUT4 (299 bp) were amplified. No products corresponding to GLUT3 (411 bp) were detected in H441 cells but were detected in a positive control, Ishikawa cell line (data not shown). Amplification of β-actin was used as a control for the reaction. b Western blot analysis of glucose transporter expression in non-polarised H441 cells. Total protein (Total), intracellular proteins (Intracellular) and plasma membrane protein (Plasma membrane) (50 μg) were resolved on acrylamide gels. Immunostained products corresponding to GLUT2 (∼60 kDa) and GLUT4 (∼45 kDa) were detected. Positive controls (+C) used: GLUT1, skeletal muscle; GLUT2, liver; GLUT3, Ishikawa cells; GLUT4, adipose tissue
Effect of glucose concentration on transporter expression in non-polarised cells
GLUT2 protein was present at 1, 5, 10 and 15 mM glucose (Figs. 3 and 4). There was an apparent increase in the abundance of GLUT2 with increasing glucose concentration. Labelling of GLUT2 was completely inhibited by pre-incubation of the antibody with the corresponding antigenic peptide (Fig. 4). These changes in glucose concentration did not induce expression of GLUT1, GLUT3 or alter the expression of GLUT4 (data not shown).

Effect of 1, 5 and 15 mM glucose on glucose transporter protein expression in non-polarised H441 cells. Total protein (50 μg) was resolved on acrylamide gels. Immunostained products corresponding to GLUT2 (∼60 kDa) and β-actin (∼42 kDa) are shown. Bands corresponding to GLUT2 were completely inhibited by pre-incubating the antibodies with the antigenic peptide against which they were raised
Glucose uptake by polarised cells
Polarised cells bathed in 10 mM glucose without inhibitors (control) took up 24.6 ± 1.4 nmol d-glucose/mg protein and 44.0 ± 1.2 nmol d-glucose/mg protein, respectively, across apical and basolateral membranes (p < 0.05, n = 6) (Fig. 5a). In the presence of phloretin, d-glucose uptake across both membranes was significantly lower than control (apical 2.7 ± 0.1; basolateral 9.8 ± 0.4 nmol/mg protein, p < 0.001, n = 6). In the presence of phlorizin, d-glucose uptake was also significantly lower than control across both membranes (apical 19.6 ± 3.6; basolateral 32.8 ± 0.1 nmol/mg protein, p < 0.05, n = 6). Ouabain had no significant effect on d-glucose uptake across either membrane (Fig. 5a).

Glucose uptake by polarised H441 cells. Effect of pre-incubation with vehicle (C), 500 μM phlorizin (PZ), 1 mM phloretin (PT) and 1 mM ouabain (OU) on the apical or basolateral uptake of 10 mM glucose (traced by [3H]-glucose) (a), [14C]-α-methyl-d-glucopyranoside (AMG) (b) or [3H]-deoxyglucose (DOG) (c). Uptake experiments were performed at 37°C. Data are shown as mean ± SEM. *p < 0.05, significantly different from control. **p < 0.01, significantly different from control. #p < 0.05, significantly different from apical control
In the absence of inhibitors (control), uptake of AMG across apical and basolateral membranes was 2.0 ± 0.9 and 3.5 ± 0.1 nmol/mg protein, respectively (n = 3) (Fig. 5b). In polarised cells, AMG uptake appeared to be slightly greater than in non-polarised cells, but was much lower than d-glucose or DOG uptake and was not inhibited by phlorizin or phloretin.
DOG uptake in the absence of inhibitors (control) was similar to d-glucose uptake (apical 36.6 ± 1.4 nmol/mg protein; basolateral 41.6 ± 0.64 nmol/mg protein) (Fig. 5c). DOG uptake across both membranes was lower than control in the presence of phloretin (apical 4.7 ± 0.5 nmol/mg protein; basolateral 11.36 ± 0.65 nmol/mg protein, p < 0.001, n = 3), but phlorizin had no effect. It is interesting to note that ouabain inhibited uptake of DOG across both membranes. However, taken together, these data indicate that glucose transport via GLUTs remains the predominant route of transport across apical and basolateral membranes when H441 cells are polarised.
Effect of phlorizin on I sc
Apical application of phlorizin induced a small reduction in mean I sc (∼10%) from control 55 ± 3 to 49 ± 3 μA cm−2 (p < 0.05, n = 4). Basolateral application of phlorizin also inhibited a small (∼10%) component of transepithelial I sc from control, 46 ± 5 to 42 ± 5 μA cm−2, p < 0.05, n = 5. Apical application of amiloride inhibited ∼92% of transepithelial I sc, similar to our previous observations in these cells (Fig. 6).

Short circuit current (I sc) measured across polarised H441 cell monolayers before and after application of 500 μM phlorizin (PZ) or 10 μM amiloride (amiloride) to the apical compartment or 500 μM phlorizin (PZ) or 1 mM ouabain (ouabain) to the basolateral compartment. *p < 0.05, significantly different from control. **p < 0.01, significantly different from control
Effect of glucose concentration on glucose uptake by polarised cells
d-Glucose uptake across the apical membrane was lowest at 1 mM glucose, 3.2 ± 0.1 nmol/mg protein (Fig. 7a). It is greater at 5 mM glucose, 28.9 ± 2.3 nmol/mg protein (Fig. 7b) (n = 6, p < 0.05 compared to (cf) 1 mM) and 10 mM glucose, 24.6 ± 1.4 nmol/mg protein (Fig. 5a) (n = 6, p < 0.01), but decreased at 15 mM glucose, 6.5 ± 0.2 nmol/mg protein (Fig. 7c) (n = 6, p < 0.01 cf 5 mM). A similar pattern of d-glucose uptake was observed across the basolateral membrane, being lowest at 1 mM (5.7 ± 0.2 nmol/mg protein), greater at 5 mM (48.0 ± 2.7 nmol/mg protein (n = 6, p < 0.05 cf 1 mM)) and 10 mM (43.9 ± 1.2 nmol/mg protein) (Fig. 5a) and decreasing at 15 mM glucose (18.1 ± 0.6 nmol/mg protein, n = 6, p < 0.05 cf 5 mM) (Fig. 7a–c). These results indicate that H441 cells display maximal glucose uptake under normoglycaemic conditions (5–10 mM) but down-regulate glucose transport under conditions mimicking hyperglycaemia (15 mM glucose). Phloretin significantly inhibited glucose uptake at all glucose concentrations across the apical (p < 0.001, n = 6) and basolateral (p < 0.001, n = 6) membranes (Figs. 7a–c and 5a).

Effect of 1 mM (a), 5 mM (b) and 15 mM (c) glucose on glucose uptake (traced by [3H]-glucose) across apical and basolateral membranes in polarised H441 cells. Cells were pre-incubated with vehicle (C), 500 μM phlorizin (PZ) or 1 mM phloretin (PT) before measurement of glucose uptake across the apical (apical) or basolateral (basolateral) membrane. Uptake experiments were performed at 37°C. Data are presented as mean ± SEM. **p < 0.01, significantly different from corresponding control. #p < 0.05, significantly different from the apical control. †p < 0.001, significantly different from corresponding control values for 1 and 15 mM glucose
Effect of cell polarisation on transporter expression
GLUT2 (but not GLUT1, GLUT3 or GLUT4) proteins were detected in the membranes of polarised cells. GLUT2 was equally distributed in the biotinylated apical and basolateral membrane fractions but was not detected in the intracellular non-bound protein fraction (Fig. 8a). α1-Na+K+ATPase was predominantly located in the basolateral biotinylated fraction and the total protein lysate but not in the non-bound fraction consistent with its distribution in the basolateral membrane of these cells (Fig. 8b). β-actin was present at similar abundance in all total protein preparations but was not detected in apical and basolateral membrane preparations, indicating that these fractions were not contaminated with intracellular proteins (Fig. 8a,b).

Western blot analysis of total protein and non-bound fraction (30 μg) or biotinylated apical and basolateral proteins isolated from 1 mg of total protein extracted from polarised H441 cells. a Immunostained proteins corresponding to GLUT2 (∼60 kDa) were present in the total, biotinylated apical and basolateral protein fractions. b Immunostained proteins corresponding to the α1 subunit of Na+K+ATPase (∼113 kDa) were present predominantly in the total protein and basolateral fractions. a and b Immunostained products corresponding to β-actin (∼42 kDa) were present in the total protein and non-bound fractions. c Effect of 1, 5 and 15 mM glucose on GLUT2 transporter abundance in biotinylated apical and basolateral proteins from polarised H441 cells
Effect of glucose concentration on transporter expression in polarised cells
In polarised cells, basolateral abundance of GLUT2 was apparently greater at 5 mM glucose than at 1 or 15 mM glucose, but we were unable to demonstrate any change in abundance of GLUT2 in the apical membrane (Fig. 8c).
Immunocytochemistry of human bronchial mucosal biopsies
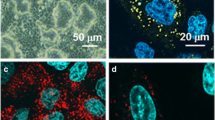
Fluorescence microscopy of three independent biopsies from human bronchial mucosa including one control patient with no airway disease revealed specific staining of epithelial cells with GLUT2 antibody. Representative sequential sections from two biopsies immunostained with GLUT2 antiserum and counterstained with DAPI are shown in Fig. 9. Images a–f and images g–l are from two independent biopsies. GLUT2 immunofluorescence was present only in the epithelial cell layer (Fig. 9a,c,g and i). The epithelial cell membranes were clearly defined and we could not distinguish any differences in intensity between the apical and basolateral membranes indicating that GLUT2 was present in both (Fig. 9a,c). Non-specific binding of anti rabbit FITC was not observed in the absence of GLUT2 antibody (Fig. 9d,f). Furthermore, we could not detect FITC fluorescence in the epithelial cell membrane after pre-absorption with GLUT2 antigenic peptide (Fig. 9j,l).

GLUT2-fluorescent immunohistochemistry of representative sequential frozen sections from two independent human bronchial mucosa biopsies (a–f) and (g–l). GLUT2–FITC fluorescence was localized to the epithelial cells present in the sections (a, c, g and i). DAPI staining of cellular nuclei was observed in all cells visible in the section (b, e, h and k). Merged fluorescence images of GLUT2–FITC and DAPI are shown in (c, f, i and l). No FITC fluorescence was observed in the epithelial cells in the absence of GLUT2 antiserum (d and f). Pre-absorbtion of GLUT 2 antiserum with GLUT2 antigenic peptide also inhibited FITC labelling of epithelial cells (j and l). White arrows indicate the position of epithelial cells. All images were obtained at ×40 magnification. Bar = 10 μm
Discussion
The aim of our study was to characterise glucose transport by human airway epithelial cells, using the H441 cell line as a model. In non-polarised H441 cells, we found that d-glucose uptake was largely inhibited by phloretin, indicating that glucose transport was predominantly via facilitated GLUT transport. In support of this notion, the transport of DOG (which is preferentially transported by GLUTs) was similar to that of d-glucose and inhibited by phloretin and cytochalasin B. We could not determine any significant SGLT mediated glucose transport in non-polarised cells with the use of AMG (which is preferentially transported by SGLT), inhibition of SGLT with phlorizin, by substitution of Na+ with choline to inhibit co-transport of Na+ with glucose or with ouabain to inhibit activity of Na+K+ATPase and the generation of the transmembrane Na+ gradient.
Effect of polarisation on transport
When polarised H441 cell monolayers were grown in 10 mM glucose, phloretin-sensitive transport remained the principal mechanism of glucose uptake across the apical and basolateral membrane. We observed that similar amounts of glucose were transported across the basolateral membrane to non-polarised cells but transport across the apical membrane was significantly less. These findings are consistent with those in cultured renal epithelial layers where phloretin-sensitive uptake was greater across the basolateral membrane than across the apical membrane [33]. In contrast, DOG uptake was similar across both membranes. We currently have no explanation for this difference as both measurements are indicative of glucose uptake via GLUTs [37]. However, it is possible that glucose transporters, additional to those we have investigated, may be present on the basolateral membrane which may have different glucose transport characteristics and inhibitor affinities [27]. We also found that DOG uptake in polarised cell monolayers was inhibited by ouabain. It is interesting to note that Na+-dependent, phloretin-sensitive uptake of DOG has been described in human intestinal Caco-2 cells [5].
We detected a small inhibition of glucose transport across the apical and basolateral membranes of polarised H441 cell monolayers with phlorizin consistent with an inhibition of SGLT transport. The appearance of phlorizin-sensitive glucose and AMG uptake was described in renal LLC-PK cells after polarisation [33, 35, 36]. However, unlike the findings of Rabito et al., we found that uptake of AMG remained low in polarised cells, was not inhibited by phlorizin and did not tally with the phlorizin-inhibitable proportion of d-glucose uptake. Furthermore, inhibition of the transmembrane Na+ gradient with ouabain did not decrease transport of d-glucose. These findings could potentially be explained by an apical SGLT protein acting as a glucosensor, a protein that transports Na+ but not glucose [11, 12] but we could not obtain further evidence to support this notion. The co-transport of glucose with Na+ via SGLT1 is electrogenic [40, 21]. Apical phlorizin inhibited I sc consistent with apical transport of Na+ through SGLT. However, basolateral application of phlorizin also inhibited I sc, which would not be consistent with a functional role for SGLT in the basolateral membrane as it should augment I sc. We also measured phlorizin-sensitive I sc across cells incubated in 1 mM glucose which should promote uptake by low capacity high affinity transporters such as SGLT. However, as we could not determine any difference in the effect of phlorizin on I sc at 1 or 10 mM glucose we did not include this data. Taken together, the inconsistencies in our data raise the possibility that phlorizin elicited non-specific effects in our cells and that there is no functional glucose transport via SGLT. It is interesting to note that we were also unable to show that glucose transport via SGLT made a detectable contribution to electrogenic transport across human nasal epithelium in vivo (E. Baker, personal communication). We have not yet excluded role for SGLT1 or SGLT2 in the human airway but further work will be required to ratify these findings.
Identity of GLUT transporters in H441 lung cells
We detected mRNA and protein for GLUT2 and GLUT4 transporter isoforms in H441 cells. Although we identified GLUT1 mRNA in this cell line we were unable to detect GLUT1 protein. Several studies have reported the presence of GLUT1 mRNA in lung [2, 26]. GLUT1 protein has also been demonstrated in resected lung lesions [29] and lung tumours [8]. Whilst GLUT1 may be at low abundance and beyond the limit of detection using the antiserum described in this study, our results support the findings of Mantych et al. who were also unable to detect GLUT1 protein in mouse alveolar or bronchiolar epithelial cells [30]. We were unable to detect GLUT3 mRNA or protein in H441 cells. It has been detected in non-small cell lung carcinoma [46, 20] but not in normal lung tissue [47].
GLUT2 protein was present in the membrane of both H441 cells and intact human bronchial epithelium obtained at bronchoscopy. GLUT2 protein has previously been described in rat bronchiolar epithelium [10] and in normal airway epithelium [19]. Thus, its presence in H441 cells indicates that these cells could provide a useful model for the study of airway glucose transport in vitro. We found that GLUT2 was predominantly in the membrane of polarised cells with very little in the intracellular compartment. These findings contrast with studies in intestinal epithelium which have indicated that there is an intracellular pool of GLUT2 that can be translocated to the membrane in response to changes in luminal glucose concentration [22, 23]. In polarised H441 cells and bronchial biopsies, we detected GLUT2 expression in both apical and basolateral membranes. This dual distribution is consistent with models for glucose transport in intestinal epithelium where GLUT2 has been localized to the apical and basolateral membrane [1, 23]. The correlation of GLUT-mediated transport and GLUT 2 protein in the membrane of human airway cells indicate for the first time that GLUT2 could make an important contribution to glucose transport and homeostasis in the human airway.
We did not detect GLUT4 protein in the plasma membrane of non-polarised or polarised H441 cells indicating that GLUT4 does not contribute to GLUT-mediated glucose uptake under the conditions we studied. The distribution and function of GLUT4 in human lung therefore remains elusive as it has been detected in human foetal lung and in some lung carcinomas [20, 28]. Its presence in the intracellular protein fraction from H441 cells and its potential ability to translocate to the plasma membrane therefore merits further study. We did not look for other GLUT isoforms in this study, although mRNA for GLUT8 and GLUT10 have been detected in bovine and human lung, respectively [9, 48].
Effect of glucose concentration on GLUT transport
Phloretin-sensitive glucose transport across either membrane was maximal under normoglycaemic conditions (5 mM glucose), was minimal under hypoglycaemic conditions (1 mM glucose) and was also reduced at a high glucose concentration mimicking hyperglycaemia (15 mM glucose). That glucose uptake was lower in 1 mM glucose is consistent with the transport kinetics for GLUT2 where the rate of uptake is proportional to glucose concentration [7, 15, 23]. However, the decrease in glucose uptake at 15 mM would indicate down-regulation of transport via GLUT2. In the gut, GLUT2 protein abundance in the apical membrane of the gut is dynamically regulated by luminal glucose concentration [16, 23, 31] and GLUT2 protein levels were reduced in human foetal hepatocytes by hyperglycaemic conditions [49]. We were unable to demonstrate any obvious changes in GLUT2 protein abundance in the membranes of our cells which would underlie such changes. This could reflect the lack of sensitivity of our assay or could indicate that there are other GLUT transporters contributing to phloretin-sensitive uptake in lung cells. Our data indicate that this is unlikely to be GLUT1, GLUT3 or GLUT4 but more work will be necessary to fully exclude these transporters.
In summary, our data shows that glucose transport across the apical and basolateral membranes of polarised H441 airway epithelial cells utilises GLUT2 transporters. Furthermore, glucose concentration modulated transport through GLUTs. Whilst, we fully recognise that H441 cells are a cancer cell line and may not fully represent glucose transport across airway epithelial cells in vivo, GLUT2 was present also in the epithelial cells of human bronchial mucosa biopsies. Our study provides the first evidence that regulation of glucose transport in human airway could involve GLUT2. The specific role of these transporters in regulating glucose homeostasis across human airway epithelium in health and disease now requires further study.
References
Affleck JA, Helliwell PA, Kellett GL (2003) Immunocytochemical detection of GLUT2 at the rat intestinal brush-border membrane. J Histochem Cytochem 51:1567–1574
Allen CB, Guo XL, White CW (1998) Changes in pulmonary expression of hexokinase and glucose transporter mRNAs in rats adapted to hyperoxia. Am J Physiol 274:L320–L329
Baker EH, Clark N, Brennan AL, Fisher DA, Gyi KM, Hodson ME, Philips BJ, Baines DL, Wood DM (2007) Hyperglycemia and cystic fibrosis alter respiratory fluid glucose concentrations estimated by breath condensate analysis. J Appl Physiol 102:1969–1975
Barker PM, Boyd CAR, Ramsden CA, Strang LB, Walters DV (1989) Pulmonary glucose transport in the fetal sheep. J Physiol 409:15–27
Bissonnette P, Gagne H, Coady MJ, Benabdallah K, Lapointe JY, Berteloot A (1996) Kinetic separation and characterization of three sugar transport modes in Caco-2 cells. Am J Physiol 270:G833–G843
Brot-Laroche E, Dao MT, Alcalde AI, Delhomme B, Triadou N, Alvarado F (1988) Independent modulation by food supply of two distinct sodium-activated d-glucose transport systems in the guinea pig jejunal brush-border membrane. Proc Natl Acad Sci U S A 85:6370–6373
Brown GK (2000) Glucose transporters: structure, function and consequences of deficiency. J Inherit Metab Dis 23:237–246
Chung JK, Lee YJ, Kim SK, Jeong JM, Lee DS, Lee MC (2004) Comparison of [18F]fluorodeoxyglucose uptake with glucose transporter-1 expression and proliferation rate in human glioma and non-small-cell lung cancer. Nucl Med Commun 25:11–17
Dawson PA, Mychaleckyj JC, Fossey SC, Mihic SJ, Craddock AL, Bowden DW (2001) Sequence and functional analysis of GLUT10: a glucose transporter in the Type 2 diabetes-linked region of chromosome 20q12–13.1. Mol Genet Metab 74:186–199
Devaskar SU, deMello DE (1996) Cell-specific localization of glucose transporter proteins in mammalian lung. J Clin Endocrinol Metab 81:4373–4378
Diez-Sampedro A, Eskandari S, Wright EM, Hirayama BA (2001) Na+-to-sugar stoichiometry of SGLT3. Am J Physiol Renal Physiol 280:F278–F282
Diez-Sampedro A, Hirayama BA, Osswald C, Gorboulev V, Baumgarten K, Volk C, Wright EM, Koepsell H (2003) A glucose sensor hiding in a family of transporters. Proc Natl Acad Sci U S A 100:11753–11758
Frasch W, Frohnert PP, Bode F, Baumann K, Kinne R (1970) Competitive inhibition of phlorizin binding by d-glucose and the influence of sodium: a study on isolated brush border membrane of rat kidney. Pflugers Arch 320:265–284
Glossmann H, Neville DM Jr. (1972) Phlorizin receptors in isolated kidney brush border membranes. J Biol Chem 247:7779–7789
Gould GW, Thomas HM, Jess TJ, Bell GI (1991) Expression of human glucose transporters in Xenopus oocytes: kinetic characterization and substrate specificities of the erythrocyte, liver, and brain isoforms. Biochemistry 30:5139–5145
Gouyon F, Caillaud L, Carriere V, Klein C, Dalet V, Citadelle D, Kellett GL, Thorens B, Leturque A, Brot-Laroche E (2003) Simple-sugar meals target GLUT2 at enterocyte apical membranes to improve sugar absorption: a study in GLUT2-null mice. J Physiol 552:823–832
Hermanns MI, Unger RE, Kehe K, Peters K, Kirkpatrick CJ (2004) Lung epithelial cell lines in coculture with human pulmonary microvascular endothelial cells: development of an alveolo-capillary barrier in vitro. Lab Invest 84:736–752
Icard P, Saumon G (1999) Alveolar sodium and liquid transport in mice. Am J Physiol 277:L1232–L1238
Ito T, Noguchi Y, Satoh S, Hayashi H, Inayama Y, Kitamura H (1998) Expression of facilitative glucose transporter isoforms in lung carcinomas: its relation to histologic type, differentiation grade, and tumor stage. Mod Pathol 11:437–443
Ito T, Noguchi Y, Udaka N, Kitamura H, Satoh S (1999) Glucose transporter expression in developing fetal lungs and lung neoplasms. Histol Histopathol 14:895–904
Joris L, Quinton PM (1989) Evidence for electrogenic Na-glucose cotransport in tracheal epithelium. Pflugers Arch 415:118–120
Kellett GL (2001) The facilitated component of intestinal glucose absorption. J Physiol 531:585–595
Kellett GL, Brot-Laroche E (2005) Apical GLUT2: a major pathway of intestinal sugar absorption. Diabetes 54:3056–3062
Kellett GL, Helliwell PA (2000) The diffusive component of intestinal glucose absorption is mediated by the glucose-induced recruitment of GLUT2 to the brush-border membrane. Biochem J 350(Pt 1):155–162
Kemp PJ, Boyd CA (1992) Pathways for glucose transport in type II pneumocytes freshly isolated from adult guinea pig lung. Am J Physiol 263:L612–L616
Kurata T, Oguri T, Isobe T, Ishioka S, Yamakido M (1999) Differential expression of facilitative glucose transporter (GLUT) genes in primary lung cancers and their liver metastases. Jpn J Cancer Res 90:1238–1243
Li J, Hu X, Selvakumar P, Russell RR 3rd, Cushman SW, Holman GD, Young LH (2004) Role of the nitric oxide pathway in AMPK-mediated glucose uptake and GLUT4 translocation in heart muscle. Am J Physiol Endocrinol Metab 287:E834–E841
Mamchaoui K, Makhloufi Y, Saumon G (2002) Glucose transporter gene expression in freshly isolated and cultured rat pneumocytes. Acta Physiol Scand 175:19–24
Mamede M, Higashi T, Kitaichi M, Ishizu K, Ishimori T, Nakamoto Y, Yanagihara K, Li M, Tanaka F, Wada H, Manabe T, Saga T (2005) [18F]FDG uptake and PCNA, Glut-1, and Hexokinase-II expressions in cancers and inflammatory lesions of the lung. Neoplasia 7:369–379
Mantych G, Devaskar U, deMello D, Devaskar S (1991) GLUT 1-glucose transporter protein in adult and fetal mouse lung. Biochem Biophys Res Commun 180:367–373
Marks J, Carvou NJ, Debnam ES, Srai SK, Unwin RJ (2003) Diabetes increases facilitative glucose uptake and GLUT2 expression at the rat proximal tubule brush border membrane. J Physiol 553:137–145
Medina RA, Meneses AM, Vera JC, Guzman C, Nualart F, Astuya A, Garcia MA, Kato S, Carvajal A, Pinto M, Owen GI (2003) Estrogen and progesterone up-regulate glucose transporter expression in ZR-75-1 human breast cancer cells. Endocrinology 144:4527–4535
Miller JH, Mullin JM, McAvoy E, Kleinzeller A (1992) Polarity of transport of 2-deoxy-d-glucose and d-glucose by cultured renal epithelia (LLC-PK1). Biochim Biophys Acta 1110:209–217
Philips BJ, Redman J, Brennan A, Wood D, Holliman R, Baines D, Baker EH (2005) Glucose in bronchial aspirates increases the risk of respiratory MRSA in intubated patients. Thorax 60:761–764
Rabito CA (1981) Localization of the Na+-sugar cotransport system in a kidney epithelial cell line (LLC PK1). Biochim Biophys Acta 649:286–296
Rabito CA (1986) Occluding junctions in a renal cell line (LLC-PK1) with characteristics of proximal tubular cells. Am J Physiol 250:F734–F743
Roman Y, Alfonso A, Louzao MC, Vieytes MR, Botana LM (2001) Confocal microscopy study of the different patterns of 2-NBDG uptake in rabbit enterocytes in the apical and basal zone. Pflugers Arch 443:234–239
Saumon G, Martet G (1996) Effect of changes in paracellular permeability on airspace liquid clearance: role of glucose transport. Am J Physiol 270:L191–L198
Saumon G, Martet G, Loiseau P (1996) Glucose transport and equilibrium across alveolar-airway barrier of rat. Am J Physiol 270:L183–L190
Steel DM, Graham A, Geddes DM, Alton EW (1994) Characterization and comparison of ion transport across sheep and human airway epithelium. Epithel Cell Biol 3:24–31
Wood D, Baines DL, Woollhead AM, Philips BJ, Baker EH (2004) Functional and molecular evidence for glucose transporters in human airway epithelium. Am J Respir Crit Care Med 169:A672
Wood DM, Brennan AL, Philips BJ, Baker EH (2004) Effect of hyperglycaemia on glucose concentration of human nasal secretions. Clin Sci (Lond) 106:527–533
Woollhead AM, Baines DL (2006) Forskolin-induced cell shrinkage and apical translocation of functional enhanced green fluorescent protein-human alphaENaC in H441 lung epithelial cell monolayers. J Biol Chem 281:5158–5168
Woollhead AM, Scott JW, Hardie DG, Baines DL (2005) Phenformin and 5-aminoimidazole-4-carboxamide-1-{beta}-d-ribofuranoside (AICAR) activation of AMP-activated protein kinase inhibits transepithelial Na+ transport across H441 lung cells. J Physiol 566:781–792
Wright EM (2001) Renal Na(+)-glucose cotransporters. Am J Physiol Renal Physiol 280:F10–F18
Younes M, Brown RW, Stephenson M, Gondo M, Cagle PT (1997) Overexpression of Glut1 and Glut3 in stage I nonsmall cell lung carcinoma is associated with poor survival. Cancer 80:1046–1051
Younes M, Lechago LV, Somoano JR, Mosharaf M, Lechago J (1997) Immunohistochemical detection of Glut3 in human tumors and normal tissues. Anticancer Res 17:2747–2750
Zhao FQ, Miller PJ, Wall EH, Zheng YC, Dong B, Neville MC, McFadden TB (2004) Bovine glucose transporter GLUT8: cloning, expression, and developmental regulation in mammary gland. Biochim Biophys Acta 1680:103–113
Zheng Q, Levitsky LL, Mink K, Rhoads DB (1995) Glucose regulation of glucose transporters in cultured adult and fetal hepatocytes. Metabolism 44:1553–1558
Acknowledgements
The α1-Na+K+ATPase monoclonal antibody developed by Dr. D. Fambrough was obtained from the Developmental Studies Hybridoma Bank, developed under the auspices of the NICHD and maintained by the University of Iowa, Department of Biological Sciences, Iowa City, IA55242, USA. This work was supported by the Wellcome Trust (Grant 075049/Z/04/Z).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Kalsi, K.K., Baker, E.H., Medina, R.A. et al. Apical and basolateral localisation of GLUT2 transporters in human lung epithelial cells. Pflugers Arch - Eur J Physiol 456, 991–1003 (2008). https://doi.org/10.1007/s00424-008-0459-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-008-0459-8