
Abstract
Purpose
Carbohydrate (CHO) restriction could be a potent metabolic regulator of endurance exercise-induced muscle adaptations. Here, we determined whether post-exercise CHO restriction following strenuous exercise combining continuous cycling exercise (CCE) and sprint interval exercise could affect the gene expression related to mitochondrial biogenesis and oxidative metabolism in human skeletal muscle.
Methods
In a randomized cross-over design, 8 recreationally active males performed two cycling exercise sessions separated by 4 weeks. Each session consisted of 60-min CCE and six 30-s all-out sprints, which was followed by ingestion of either a CHO or placebo beverage in the post-exercise recovery period. Muscle glycogen concentration and the mRNA levels of several genes related to mitochondrial biogenesis and oxidative metabolism were determined before, immediately after, and at 3 h after exercise.
Results
Compared to pre-exercise, strenuous cycling led to a severe muscle glycogen depletion (> 90%) and induced a large increase in PGC1A and PDK4 mRNA levels (~ 20-fold and ~ 10-fold, respectively) during the acute recovery period in both trials. The abundance of the other transcripts was not changed or was only moderately increased during this period. CHO restriction during the 3-h post-exercise period blunted muscle glycogen resynthesis but did not increase the mRNA levels of genes associated with muscle adaptation to endurance exercise, as compared with abundant post-exercise CHO consumption.
Conclusion
CHO restriction after a glycogen-depleting and metabolically-demanding cycling session is not effective for increasing the acute mRNA levels of genes involved in mitochondrial biogenesis and oxidative metabolism in human skeletal muscle.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Carbohydrate (CHO) is a crucial macronutrient used to fuel skeletal muscle during exercise and restore glycogen after exercise (Bergstrom et al. 1967). Muscle glycogen, which is the major source of endogenous CHO, is highly depleted following prolonged and exhausting exercise (Cheng et al. 2020; Greiwe et al. 1999), and enhancing its resynthesis with CHO supplementation can accelerate recovery (Bergstrom et al. 1967; Alghannam et al. 2016). In contrast, CHO restriction has been proposed to be a potential regulator of intracellular signaling pathways promoting endurance-training adaptations in skeletal muscle (e.g., mitochondrial biogenesis, substrate utilization, oxidative metabolism) (Hearris et al. 2018; Mata et al. 2019).
Over the last two decades, the “train-low” concept that consists of purposely reducing glycogen stores and/or restricting CHO intake to enhance training adaptations has been widely used by endurance athletes (Bartlett et al. 2015; Hearris et al. 2018). Some evidence indicates that a reduction of pre-exercise CHO availability (due to an initial glycogen-depleting exercise combined with restricted CHO intake) potentiates the exercise-induced activation of signaling pathways associated with endurance-training adaptations in skeletal muscle (Psilander et al. 2013; Yeo et al. 2010; Bartlett et al. 2013). One critical limitation of reducing pre-exercise CHO availability is the decline of physical capacity during a subsequent intense exercise session performed until exhaustion (Hearris et al. 2019). Thus, this strategy is not optimal during specific periodization phases when endurance athletes need to perform strenuous exercises at high intensities several times per week.
One option to avoid the reduction of physical capacity during intense training sessions while maximizing the acute molecular response to endurance exercise could be to subject the athletes to post-exercise CHO restriction for a few hours directly after a strenuous endurance exercise. With such an approach, muscle glycogen stores could be restored within 24 h if an appropriate CHO-enriched diet is ingested following the acute CHO-restricted period (Jensen et al. 2015), allowing the athletes to be physically recovered for the subsequent intense workout. Previous studies indicated that CHO restriction following prolonged continuous exercise that induces moderate muscle glycogen-depleting (~ 40–65%) does not affect the mRNA levels of genes involved in mitochondrial biogenesis and muscle metabolism during the acute 2–5 h post-exercise recovery period (Jensen et al. 2015; Pilegaard et al. 2005). It was recently proposed that the acute increased expression of exercise-responsive genes and the chronic training adaptations associated with “train-low” protocols could be optimal when muscle glycogen depletion is severe (Impey et al. 2018; Hammond et al. 2019). Despite the pronounced muscle glycogen depletion (~ 75%) directly observed after an exhaustive cycling session performed at moderate intensity, the expression of key regulators of muscle adaptation was not affected by low CHO intake during the recovery period (Mathai et al. 2008). Thus, acute CHO restriction after prolonged and moderate-intensity exercise that induced severe muscle glycogen depletion does not seem to increase the acute expression (2–5 h post exercise) of genes related to mitochondrial biogenesis and oxidative metabolism.
Peroxisome proliferator-activated γ-receptor coactivator 1α (PGC-1α) is a transcriptional coactivator that governs the expression of numerous genes involved in endurance exercise-induced muscle adaptation, thus orchestrating key processes, such as mitochondrial biogenesis and oxidative metabolism (Correia et al. 2015). Prolonged moderate-intensity continuous cycling exercises (CCE) induced an increase in PGC1A mRNA levels, a response that was not influenced by post-exercise CHO restriction during the early recovery phase (2–5 h) (Pilegaard et al. 2005; Jensen et al. 2015; Mathai et al. 2008). However, high mRNA levels of PGC1A were still observed when post-exercise CHO restriction was maintained for a longer period (i.e., 8 h) (Pilegaard et al. 2005). Furthermore, sprint interval exercise (SIE), which consists of brief supramaximal efforts interspaced with resting periods, is a highly metabolically-demanding exercise that also greatly increases PGC1A mRNA levels (Cochran et al. 2014). Therefore, it is conceivable that performing a strenuous exercise session consisting of CCE followed by SIE, and which results in a drastic muscle glycogen depletion (Cheng et al. 2020), could answer the question as to whether post-exercise CHO restriction enhances the acute expression of PGC1A and other mitochondrial biogenesis-related genes. In this context where both severe muscle glycogen depletion and high metabolic stress are induced, we hypothesized that post-exercise CHO restriction may be beneficial to potentiate the acute molecular response associated with stimulation of mitochondrial biogenesis and oxidative metabolism. To test this hypothesis, we determined the effect of post-exercise CHO restriction following an exercise session combining CCE and SIE on the acute gene expression related to exercise-induced muscle adaptations in humans.
Materials and methods
This work is a follow-up to a recently published study (Cheng et al. 2020). The information related to the experimental design, the exercise sessions, CHO intake and nutritional control has already been described in the previous article. In this latter study, we determined whether CHO ingestion could accelerate recovery of prolonged low-frequency force depression induced by a glycogen-depleting exercise session combining moderate-intensity and high-intensity sprint exercises.
Participants
As previously described (Cheng et al. 2020), a randomized crossover design was used, in which eleven healthy male participants initially volunteered to participate. The inclusion criteria used in the current study were: aged between 18 and 40 years, being healthy and physically active with normal blood pressure and without medication. The exclusion criteria were: suffering from any kind of disease, having an injury or any other conditions that would compromise the ability to perform the physical tests. Due to the difficulty of recruiting endurance athletes, we only included recreationally active males who regularly exercised up to 5 h/week. It is noteworthy that one participant withdrew during the study and two subjects needed to be excluded from the muscle biological analysis (due to low amount of biological material available or technical issues during RNA isolation). Therefore, eight participants [age: 32.4 ± 6.4 years; height: 185.8 ± 4.6 cm; body mass: 86.8 ± 9.9 kg; percentage body fat: 16.6 ± 5.0%; body mass index: 25.2 ± 3.2 kg/m2; VO2max: 49.9 ± 8.9 mL/kg/min] were finally included in this study. A similar sample size (i.e., N = 7–9) was used in previous studies that analyzed the effects on CHO restriction following exercise on the mRNA levels of genes involved in endurance-training adaptations in skeletal muscle (Pilegaard et al. 2005; Jensen et al. 2015; Mathai et al. 2008). The percentage body fat was measured using bio-electrical impedance analysis (Tanita TBF-300 UK Ltd, West Drayton, UK) and VO2max was determined as previously described (Cheng et al. 2020). The distribution of myosin heavy chain (MHC) isoforms (as described below) was: MHC1: 36.9 ± 13.6%; MHC2A: 54.2 ± 9.4%; MHC2X: 8.9 ± 8.6%. This study was approved by the Kaunas Regional Research Ethics Committee (no. BE-2-17) and was performed in accordance with the last revision of the Declaration of Helsinki.
Experimental design and exercise session
The experimental design was previously described (Cheng et al. 2020). Briefly, the experiments consisted of three visits to the laboratory. During the first visit, a maximal incremental cycling test was performed to determine VO2max and the intensity used during the subsequent prolonged moderate-intensity cycling exercise executed in visits 2–3. During this first visit, the participants were also familiarized with the equipment and protocols used during the subsequent two visits. The second visit was performed approximately a week after the first visit. The two experimental trials (visits 2–3) were completed in a random order and were separated by four weeks during which the participants maintained their regular physical activity. The participants were asked to refrain from any strenuous exercise during the last 3 days prior to each experimental session.
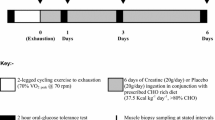
In visits 2–3, the participants arrived at the laboratory in the morning after a 10–12 h overnight fast and an initial biopsy was collected from the vastus lateralis muscle. An overview of the experimental protocol is presented in Fig. 1. It is noteworthy that neuromuscular testing of knee extensors (i.e. electrically evoked torque and maximal voluntary contraction) was executed before (1 test) and after exercise (7 tests). These tests were only performed on the dominant leg, and thus did not interfere with the muscle biological analyses since muscle biopsies were collected from the vastus lateralis muscle of the non-dominant leg (see below). These tests, performed to answer the aim of the initial study (Cheng et al. 2020), are not included in the current analysis. After muscle biopsy, a warm-up was performed, consisting of 8–10 min of cycling at 1 W/kg body mass, followed by light active stretching of the knee extensor muscles and by 2–3 short isometric knee extensions in the dynamometer chair. This latter exercise was used as a warm-up for the neuromuscular testing. After the completion of the baseline neuromuscular testing (~ 10 min), the participants performed a cycling session involving CCE followed by SIE. For a full description of the exercise session, refer to Cheng et al. (2020). In short, this session consisted of performing a 60-min continuous cycling exercise (i.e. CCE) at a power eliciting 60% VO2max followed by six 30-s all-out cycling sprints (SIE) interspaced with 4 min recovery. The 60-min CCE was performed on an electromechanically braked cycling ergometer (Ergoselect 200P, Ergoline, Medical Measurement Systems, Binz, Germany), and a short break (2–3 min) was allowed after 30 min of CCE during which the participants could drink water (up to 0.5 L). Heart rate (HR) and the rating of perceived exertion (RPE, 6–20 scale) were determined every 10 min during the CCE to evaluate the perceived effort level. The all-out cycling sprints were performed using a mechanically braked cycling ergometer (Monark 824E, Monark, Vansbro, Sweden) with a brake weight corresponding to 7.5% of the participant’s body mass. The peak HR of each cycling sprint and average power were obtained during the SIE. A second muscle biopsy was collected 2–3 min after the last cycling sprint. In a random order, the participants ingested a beverage containing either carbohydrate (CHO) or placebo (PLA) every 15 min during the 150-min period following the completion of the last sprint (post-exercise recovery period). A third biopsy was collected 180 min after the last sprint (i.e. 30 min after the last beverage ingestion). Capillary blood concentration of glucose and lactate was determined from the fingertip before exercise, immediately after CCE, immediately after SIE, and every 30 min during the 180-min recovery period, as previously described (Cheng et al. 2020). Additional neuromuscular testing was executed 15, 30, 60, 90, 120, 150 and 180 min after the completion of the last cycling sprint (not included in the analysis).

adapted from Cheng et al. (2020)
Overview of the experimental protocol performed during the two randomized trials. After a warm-up at low intensity (yellow bar), the participants performed a 60-min continuous cycling exercise (orange bar) followed by six 30-s all-out cycling sprints (light pink bar). From 15 to 150 min post-exercise, the subjects were given a carbohydrate (CHO) or a placebo (PLA) beverage. The neuromuscular testing performed and analyzed in the initial study (Cheng et al. 2020) is not included in this figure. This figure was
Carbohydrate intake during the recovery period and nutritional control
The CHO-enriched beverage contained glucose and fructose at a 2:1 ratio. CHO (final concentration: 100 g/L) was dissolved in a solution containing water and 1:20 lemon juice concentrate (22 g carbohydrate/L of concentrate; Solevita). The volume of CHO beverage was adjusted for each participant to provide 1.5 g/kg body mass/h CHO during the recovery period. An equal volume was ingested every 15 min following the last cycling sprint over a 150-min period to provide the adequate amount of CHO. The total amount of CHO ingested during the CHO trial (i.e. 150-min post-exercise period) was 328.9 ± 37.5 g. The rationale behind this nutritional approach was previously described (Cheng et al. 2020). The PLA beverage was prepared using a solution of water with lemon juice concentrate (1:20 dilution), in which 0.6 g/L of sodium saccharin (Hermesetas, Zurich, Switzerland) and 4 g/L of Suketter (containing sodium cyclamate and sodium saccharin; Cederroth, Upplands Väsby, Sweden) were dissolved. The total amount of CHO ingested during the PLA trial (i.e. 150-min post-exercise period) was negligible (3.6 ± 0.4 g). The volume of beverage consumed was identical for both CHO and PLA trials.
As described above, the participants performed the strenuous exercise sessions in the morning after a 10–12 h overnight fast. They were asked to take a similar meal on the evening before each exercise session and to refrain from any food and caffeine intakes on the morning before each exercise session. Overnight fasting was chosen for two main reasons: (1) to reduce the risks of gastrointestinal issues and vomiting commonly observed during or immediately after all-out cycling sprints; (2) to restrict CHO (that would have been available in the blood stream after breakfast) during exercise, thereby promoting large muscle glycogen depletion. A similar strategy (strenuous exercise after overnight fasting) was accompanied by very low muscle glycogen concentrations after exercise in untrained and trained participants (Hickner et al. 1997; Greiwe et al. 1999). It is also noteworthy that pre-exercise muscle glycogen concentration (see in the result section) was in the same range as that observed in the literature after overnight fast in a similar population (Blom et al. 1987; Maehlum et al. 1977).
Muscle biopsies
Needle biopsies from the non-dominant vastus lateralis muscle were collected by a trained physician, as previously described (Cheng et al. 2020). The non-dominant leg was not subjected to neuromuscular testing. Three biopsy samples (~ 10 mg each) were taken before exercise, 2–3 min after and 180 min after the last cycling sprint. The samples were immediately frozen in liquid nitrogen and stored at − 80 °C until further analysis.
Muscle glycogen measurements
Glycogen concentration was assessed using 5–10 mg of frozen muscle, as previously described (Cheng et al. 2020). Briefly, muscle samples were dissolved in 25 volumes of 2 M NaOH for 50 min at 95 °C, before being neutralized with an equal volume of 2 M HCl. The homogenates (1:50 dilution) were first diluted with distilled water (final dilution: 1:400), and then 5 µL of each diluted homogenate was loaded in duplicates into a 96-well plate. The samples were then analyzed using a fluorometric kit (ab65620, Abcam, Cambridge, UK) following the manufacturer’s instructions. Due to technical reasons, muscle glycogen concentrations could not be expressed in mmol glucosyl units/kg dry weight and were instead expressed as mmol glucosyl units/kg wet weight.
Distribution of myosin heavy chain (MHC) isoforms
The distribution of MHC isoforms was assessed using 5–10 mg of frozen muscle collected before exercise. Muscle samples were homogenized with a bead-homogenizer (TissueLyser LT, Qiagen, Sollentuna, Sweden) in ice-cold lysis buffer (20 µL/mg; pH 7.6) containing 20 mM Hepes, 150 mM NaCl, 5 mM EDTA, 25 mM KF, 1 mM Na3VO4, 5% glycerol, 0.5% Triton X-100, and EDTA-free protease inhibitor cocktail (1 tablet/10 mL buffer; # 04 693 159 001, Sigma-Aldrich, Stockholm, Sweden). Protein concentration of the homogenates was determined using BCA protein assay (#23227, Thermo Fisher Scientific, Stockholm, Sweden), and then adjusted to 2 µg/µL after dilution with the lysis buffer. Samples were then diluted to 10 ng/µL with loading buffer consisting of 125 mM Tris–HCl (pH 6.8), 1 mM EDTA, 20% glycerol, 5% SDS, 5% β-mercaptoethanol and bromophenol blue. Electrophoresis was performed using a Mini Protean III system (Bio-Rad Laboratories, Solna, Sweden). The separating gel consisted of 30% glycerol, 8% acrylamide, 0.16% bisacrylamide, 0.2 M Tris–HCl (pH 8.8), 0.1 M glycine, 0.4% SDS, 0.05% Temed and 1% ammonium persulfate. The stacking gel consisted of 30% glycerol, 4% acrylamide, 0.08% bisacrylamide, 70 mM Tris–HCl (pH 6.8), 4 mM EDTA, 0.4% SDS, 0.1% Temed and 1% ammonium persulfate. Protein lysates were heated at 95 °C for 10 min and 150 ng of protein was loaded into the gel. The gels were run at 4 °C during the whole migration period, including 40 min at 10 mA and 23 h 20 min at 140 V. The gels were silver-stained as described previously (Agbulut et al. 1996). The gels were scanned with an office scanner (CanoScan 9000F, Canon, The Netherlands, Amsterdam) and analysed with UN-SCAN-IT software (version 6.1, Silk Scientific Corporation, Orem, USA). The distribution of MHC isoforms was quantified using positive lane analysis and background correction (single region background value).
RNA isolation, cDNA synthesis and quantitative polymerase chain reaction (qPCR)
Muscle samples (5–10 mg) were disrupted with a bead-homogenizer (TissueLyser LT, Qiagen) in 75 µL RA1 lysis buffer (Macherey-Nage, Dueren, Germany) containing 1% β-mercaptoethanol, and 250 µL TRIzol reagent (Life Technologies, Stockholm, Sweden). Tissue homogenate was then mixed with 100 µL chloroform (Sigma-Aldrich) before centrifugation. The aqueous phase (200 µL) was collected and mixed with an equal volume of chloroform. After centrifugation, 170 µL of the aqueous phase was precipitated with an equal volume of isopropanol at − 20 °C for 10 min. After centrifugation and an extra precipitation step with isopropanol, RNA was washed twice with 70% ethanol to remove excess salt. The pellet was dried at room temperature for 10 min, and dissolved in 12 µL nuclease-free water. The RNA samples were incubated 10 min at 55 °C and then kept on ice. Total RNA concentration and purity were assessed by measuring the optical density (230, 260 and 280 nm) with a spectrophotometer (Nanodrop 2000, Thermo Fisher Scientific). RNA samples were treated with genomic DNA removal kit (Heat and Run DNase, ArcticZymes, Tromsø, Norway) according to the manufacturer’s instructions, and RNA concentration was assessed again.
Total RNA (1 µg) was reversed-transcribed using iScript Select cDNA synthesis kit (Bio-Rad Laboratories). Total RNA was converted into cDNA using a final reaction volume of 20 µL containing: 2 µL of oligodT, 2 µL of random hexamers, 4 µL of 5X-iscript select reaction mix, 1 µL of reverse transcriptase and 9 µL of nuclease-free water. The reaction mix was incubated at 25 °C for 5 min, at 42 °C for 60 min, and at 85 °C for 5 min. After this, the samples were cooled down on ice, aliquoted, and stored at − 80 °C until further analysis. qPCR (final volume: 20 µL) was performed with samples loaded in duplicate using 5 µL of diluted cDNA (1/20 dilution from stock cDNA mixture), 10 µL of Rotor-Gene SYBR Green RT-PCR Master Mix (Qiagen), and 1 µL (Biorad Laboratories) or 0.4 µL (reverse and forward primers at 20 µM) of primers (list of primers presented in Table 1). The sequences of the forward and reverse primers for total PGC1A, PGC1A transcripts from exon 1a (PGC1A-ex1a) and truncated PGC1A were kindly provided by Jorge Ruas’s lab (Karolinska Institutet, Sweden) and were previously used (Ruas et al. 2012). PGC1A-ex1a primers can detect PGC1A1 and NT-PGC1A-a, and truncated PGC1A primers can detect PGC1A4, NT-PGC1A-a and NT-PGC1A-c (not expressed in skeletal muscle) (Martinez-Redondo et al. 2015). PGC1A1 isoform and NT-PGC1A-a are important regulators of mitochondrial biogenesis whereas PGC1A4 isoform is induced after resistance exercise training and could promote muscle hypertrophy (Ruas et al. 2012; Martinez-Redondo et al. 2015). Several transcription factors associated with mitochondrial biogenesis and oxidative metabolism (Scarpulla et al. 2012), including nuclear respiratory factor 1 (NRF1), GA-binding protein transcription factor subunit alpha (GABPA, also called NRF2), mitochondrial transcription factor A (TFAM) and peroxisome proliferator-activated receptors alpha (PPARA) were also studied. The mRNA levels of three genes involved in redox homeostasis, a process closely connected to mitochondrial biogenesis (Ji et al. 2020), were assessed: Sirtuin 1 (SIRT1), nuclear factor erythroid 2-related factor 2 (NFE2L2) and superoxide dismutase 2 (SOD2). Finally, we determined the mRNA levels of two metabolic genes involved in substrate utilization (pyruvate dehydrogenase kinase 4, PDK4) and glucose transport (solute carrier family 2 member 4, SLC2A4, also called GLUT4).
qPCR was performed using a Rotor Gene Q thermocycler (Qiagen) for 40 cycles (95 °C for 5 s and 60 °C for 30 s) followed by melting curve analysis. qPCR efficiency was estimated for each primer pair by performing standard curves obtained from serial dilutions of a pooled sample. We initially tested two reference genes: RPLP0 (ribosomal protein lateral stalk subunit P0) and HPRT1 (hypoxanthine phosphoribosyltransferase 1). HPRT1 was selected and used for normalization because its expression was not affected by the experimental conditions (time and supplementation). The relative mRNA levels were calculated using the ΔΔCT method (Pfaffl 2001). The threshold cycle (CT) was calculated using the Rotor-Gene Q software (Qiagen), based on the qPCR conditions (auto-find threshold with slope correct adjustment) set up from the standard curves obtained for each gene. Since none of the target mRNAs were differently expressed between the two trials at baseline (pre-exercise), the mRNA levels were expressed as fold changes (FC) relative to pre-exercise values, which were set at 100%. This method is commonly used in studies with similar settings (i.e. cross-over design, post-exercise CHO restriction) (Pilegaard et al. 2005; Jensen et al. 2015; Mathai et al. 2008).
Statistical analysis
Data are presented as mean ± standard deviation (SD), and individual values are presented in Fig. 2a, b. All the statistical analyses were performed using GraphPad Prism (Graphpad Prism 8.0.2, San Diego, USA). As explained above, eight participants were included in the analysis of this study. Power output of the SIE was analyzed from only seven subjects because it was not recorded during the SIE of the PLA trial for one subject. In addition, one subject was excluded from the analysis of muscle glycogen (Fig. 2a, b) due to outlier values (i.e. > 2 SD) above the mean at post-exercise for the CHO trial. Similarly, one subject was excluded from the analysis of PDK4 mRNA (Fig. 6a) due to outlier values at the time points post-exercise and 3 h post-exercise for the CHO trial.

This figure was adapted from Cheng et al. (2020)
Muscle glycogen concentration (a), rate of muscle glycogen resynthesis (b), blood glucose concentration (c), and blood lactate concentration (d). Data are shown as means ± SD. Individual values are presented in a and b. ***P < 0.001: significant main effect of time. $P < 0.05, $$P < 0.01: significant differences between the CHO and PLA conditions. Pre-ex pre-exercise, Post-CE post-continuous cycling exercise, Post-SE, post-sprint cycling exercise.
Shapiro–Wilk tests were used to check normality before selecting the appropriate parametric or non-parametric statistical tests. Paired t tests were used to analyze the mean power, mean HR, peak HR, mean RPE and the rate of muscle glycogen resynthesis for both conditions. For the analysis of blood glucose concentration, and for the analysis of the mRNA levels of total PGC1A, TFAM, SIRT1 and GABPA, two-way repeated-measures analysis of variance (2-way RM ANOVA) tests were used to assess the effect of time, supplementation (CHO vs PLA), and time × supplementation interaction. When an interaction was observed, a Sidak multiple comparisons test was used to compare the PLA and CHO conditions. For the analysis of blood lactate and muscle glycogen concentration, as well as for the analysis of the mRNA levels of PGC1A-ex1a, truncated PGC1A, NFE2L2, NRF1, PDK4, SOD2, PPARA, SLC2A4, Friedman tests were used to assess the effect of time in both experimental trials. Then, Wilcoxon’s matched-pairs signed-rank tests with Bonferroni corrections were performed to compare the conditions (CHO vs. PLA) at each time point. The level of significance was set at P < 0.05.
Results
Strenuous glycogen-depleting cycling session
Participants performed the 60-min CCE at a power eliciting 60% VO2max (2.11 ± 0.40 W/kg body mass) in both the CHO and PLA experimental trials. During this exercise, mean RPE (15.3 ± 0.7 and 15.0 ± 1.1, respectively) and mean HR (150 ± 9 bpm and 147 ± 11 bpm, respectively) were similar in both the CHO and PLA conditions. During the SIE, the mean power output was similar in both the CHO and PLA trials (7.03 ± 0.68 W/kg body mass and 6.86 ± 0.55 W/kg body mass, respectively), while the average of the peak HR of each cycling sprint was slightly higher (2%) for the CHO trial compared to the PLA trial (168 ± 10 bpm and 164 ± 11 bpm, respectively; P < 0.05).
Before exercise, muscle glycogen concentration was not significantly different between the two randomized trials (CHO trial: 84.9 ± 32.3 mmol glucosyl units/kg wet weight; PLA trial: 99.4 ± 35.8 mmol glucosyl units/kg wet weight; P = 0.12) (Fig. 2a). After exercise, muscle glycogen concentration was severely decreased, reaching 2.7 ± 5.2 mmol glucosyl units/kg wet weight in the CHO condition and 9.6 ± 13.4 mmol glucosyl units/kg wet weight in the PLA condition. At 3 h post-exercise, muscle glycogen concentration was significantly higher in the CHO trial than in the PLA trial (42.1 ± 18.4 mmol glucosyl units/kg wet weight and 24.0 ± 15.4 mmol glucosyl units/kg wet weight, respectively; P < 0.05). The rate of muscle glycogen resynthesis during the 3-h recovery period was significantly higher in the CHO trial than the PLA trial (13.1 ± 5.6 and 4.8 ± 2.2 mmol glucosyl units/kg wet weight/h, respectively; Fig. 2b, P < 0.05), demonstrating that CHO ingestion promoted muscle glycogen resynthesis. In addition, blood glucose concentration, which was similar in both the CHO and PLA trials before exercise (4.76 ± 0.52 mM and 4.94 ± 0.47 mM, respectively), markedly increased in response to CHO ingestion only (time × supplementation interaction, P < 0.001), reaching the highest values at 60 min post-exercise (CHO trial: 9.91 ± 1.70 mM; PLA trial: 4.84 ± 0.66 mM; P < 0.01) (Fig. 2c). Blood glucose concentration was significantly higher in the CHO condition than in the PLA condition from 30 min post exercise (P < 0.01). Blood lactate concentration, which was similar in both the CHO and PLA experimental trials before exercise (1.16 ± 0.24 mM and 1.06 ± 0.27 mM, respectively), markedly increased after the SIE, reaching 16.35 ± 4.63 mM and 16.91 ± 5.66 mM in the CHO and PLA conditions, respectively (main effect of time, P < 0.001) (Fig. 2d). Blood lactate concentration progressively decreased to 2.15 ± 0.52 mM and 1.40 ± 0.43 mM 180 min post exercise in the CHO and PLA conditions, respectively. There were no significant differences in blood lactate concentration between the two trials at any time points.
mRNA levels of genes associated with mitochondrial biogenesis and oxidative metabolism
The mRNA levels of total PGC1A were highly increased after exercise (time effect: P < 0.001), resulting in a five to tenfold increase directly after exercise and a ~ 20-fold increase 3 h after exercise (Fig. 3a). PGC1A-ex1a mRNA increased 3.5- to 8-fold directly after exercise and 7- to 8.5-fold 3 h post exercise (time effect: P < 0.05) (Fig. 3b). A marked increase in truncated PGC1A mRNA levels (~ 20-fold) was observed 3 h post-exercise in both trials (time effect: P < 0.01) (Fig. 3c). CHO restriction in the PLA trial during the three hours following exercise did not affect the mRNA levels of total PGC1A, PGC1A-ex1a and truncated PGC1A.

mRNA levels of total PGC1A (a), PGC1A-ex1a (b), and truncated PGC1A (c). The results values are expressed as fold change (FC) relative to the pre-exercise values. Data are shown as means ± SD. *P < 0.05, **P < 0.01, ***P < 0.001: significant main effect of time
The mRNA levels of NRF1 (Fig. 4a), GABPA (Fig. 4b), and PPARA (Fig. 4c) were not significantly affected by time, while TFAM mRNA levels (Fig. 4d) slightly increased after exercise (~ 1.5- to 2-fold; time effect: P < 0.01). CHO restriction during the 3-h post-exercise period did not affect the abundance of NRF1, TFAM, and PPARA, while GABPA mRNA levels were slightly lower in the PLA than the CHO conditions at this time point (P < 0.05).

mRNA levels of transcription factors closely related to PGC1A. mRNA levels of NRF1 (a), GABPA (b), PPARA (c), and TFAM (d). The results values are expressed as fold change (FC) relative to the pre-exercise values. Data are shown as means ± SD. **P < 0.01: significant main effect of time. $P < 0.05, significant differences between the CHO and PLA conditions
The mRNA levels of SIRT1 (Fig. 5a) and NFE2L2 (Fig. 5b) were significantly increased during the recovery period (time effect: P < 0.01 and P < 0.05, respectively), while this result was only observed for SOD2 in the CHO trial (time effect: P < 0.05; Fig. 5c). CHO restriction during the three hours following exercise did not affect the mRNA abundance of SIRT1, NFE2L2 and SOD2.

mRNA levels of genes regulating redox homeostasis. mRNA levels of SIRT1 (a), NFE2L2 (b) and SOD2 (c). The results values are expressed as fold change (FC) relative to the pre-exercise values. Data are shown as means ± SD. *P < 0.05, **P < 0.01: significant main effect of time. #P < 0.05: significant main effect of time in the CHO condition only
PDK4 mRNA levels were increased ~ threefold directly after exercise and ~ tenfold 3 h post exercise (time effect: P < 0.001; Fig. 6a). SLC2A4 mRNA levels were not significantly affected by time (Fig. 6b). 3 h post exercise, the mRNA levels of these genes were not influenced by CHO restriction.

mRNA levels of genes associated with substrate utilization and glucose transport. mRNA levels of PDK4 (a) and SLC2A4 (b). The results values are expressed as fold change (FC) relative to the pre-exercise values. Data are shown as means ± SD. ***P < 0.001: significant main effect of time
Discussion
Reduced CHO availability has been proposed to be a potent metabolic regulator of intracellular signaling pathways promoting endurance-training adaptations in skeletal muscle (e.g., mitochondrial biogenesis, substrate utilization, oxidative metabolism) (Hearris et al. 2018; Mata et al. 2019). In the current study, we tested the hypothesis that CHO restriction following a severely glycogen-depleting and highly metabolically-demanding exercise would potentiate the acute molecular response associated with stimulation of mitochondrial biogenesis and oxidative metabolism adaptation in human skeletal muscle. Our results indicated that post-exercise CHO restriction following a strenuous cycling session that combined CCE and SIE did not amplify the mRNA levels of genes associated with muscle adaptation to endurance exercise.
In this study, we adopted an experimental exercise session that induced both severe muscle glycogen depletion and a high metabolic stress. Indeed, this exercise session led to more than 90% muscle glycogen depletion while blood lactate concentration was highly elevated after the last sprint. In addition, the mRNA levels of PGC1A, a central regulator of mitochondrial biogenesis and oxidative metabolism (Correia et al. 2015), were substantially increased 3 h post exercise (~ 20-fold). Intriguingly, the magnitude of the changes in PGC1A observed in the current study is much higher than that observed in previous studies which consisted of solely moderate-intensity exercise (Jensen et al. 2015; Pilegaard et al. 2005; Mathai et al. 2008), or high intensity interval exercise (HIIE) (Bartlett et al. 2013) (Perry et al. 2010), or SIE (Cochran et al. 2014). In our present study, these pronounced changes in mRNA levels are most likely the result of the combination of severe muscle glycogen depletion and metabolic stress caused by this strenuous exercise session (i.e. CCE following by SIE), although other factors could have also played a role (e.g. the time points of muscle sample collection, the training status of the participants, the form of activity, etc.). For instance, it has been reported that transient and large increases in PGC1A mRNA levels occur after HIIE, but the mRNA response to exercise was attenuated after several sessions as the muscle adapted to the exercise challenge (Perry et al. 2010).
Interestingly, the abundance of both PGC1A-ex1a and truncated PGC1A mRNA highly increased during the recovery period, but the changes observed 3 h after exercise for truncated PGC1A were of higher magnitude than those observed for PGC1A-ex1a. Since PGC1A-ex1a primers and truncated PGC1A primers can both detect NT-PGC1A-a (see in the method section), the large increased expression of truncated PGC1A observed at 3 h post exercise is most likely the result of PGC1A4 up-regulation. PGC1A1 isoform (and NT-PGC1A-a) specifically promotes mitochondrial biogenesis in response to endurance exercise, whereas overexpression of PGC1A4 isoform promotes muscle hypertrophy in mice and is induced in humans after resistance training (Ruas et al. 2012; Martinez-Redondo et al. 2015). However, some human studies have shown that the mRNA levels of truncated PGC1A isoforms, including PGC1A4, are highly increased in the early period (2–6 h) following endurance exercise, and are not preferentially increased in response to resistance exercise (Ydfors et al. 2013; Lundberg et al. 2014). Since a 6-week cycling SIE training intervention does not increase muscle fiber size (Joanisse et al. 2015), it seems that the role of PGC1A4 is not solely to induce muscle hypertrophy but could also be to promote endurance-training adaptations, such as angiogenesis (Ruas et al. 2012; Thom et al. 2014).
The main finding of this study is that CHO restriction following a severely glycogen-depleting and highly metabolically-demanding exercise does not increase the mRNA levels of numerous factors involved in mitochondrial biogenesis and oxidative metabolism in human skeletal muscle. From the different categories of transcripts studied (PGC1A, transcription factors closely related to PGC1A, regulators of redox homeostasis, and factors associated with substrate utilization and glucose transport), only GABPA mRNA levels were slightly lower in the PLA trial compared to the CHO trial 3 h post exercise. This result remains unclear, but the small differences observed may not be of biological relevance. In the current study, pre-exercise muscle glycogen stores were in the same range as those observed in a rested and fasted state in a similar population (i.e., physically active males) (Blom et al. 1987; Maehlum et al. 1977). The fasted state and training status (i.e., recreationally active) of the subjects, the absence of CHO supplementation during exercise and the exhausting characteristic of our exercise protocol most likely explain why muscle glycogen depletion was so substantial in our current study. In addition, PDK4, which is highly sensitive to low CHO availability, was shown to be upregulated in response to CHO restriction (Hammond et al. 2019; Cluberton et al. 2005). Here, PDK4 mRNA was substantially expressed after exercise, especially 3 h post exercise (~ tenfold), but no differences were found between the CHO and PLA trials. Our findings suggest that post-exercise CHO restriction did not further enhance mRNA levels of this metabolic gene because the stimuli associated with our exercise protocol (i.e. muscle severe glycogen depletion and high metabolic stress) were sufficient to maximize the expression of PDK4.
The muscle glycogen threshold hypothesis, whereby a low level of muscle glycogen during exercise (100–300 mmol glucosyl units/kg dry muscle weight) is especially potent in modulating the activation/expression of key molecular regulators of endurance-training adaptations has been recently proposed (Impey et al. 2018). In our study, muscle glycogen concentration was very low directly after exercise, and remained within the frame of 100–300 mmol glucosyl units/kg dry muscle weight after the short recovery period (3 h) in all participants of both the PLA and CHO trials [average of ~ 105 and 180 mmol glucosyl units/kg dry muscle weight, respectively; data converted from wet-weight values to dry-wet values using the conversion factor 4.325 (Murray and Rosenbloom 2018)]. These results may explain the absence of differential molecular response at 3-h post exercise between the two trials.
Reactive oxygen and nitrogen species (RONS) have been recently proposed to be a primary stimulus of mitochondrial biogenesis in skeletal muscle (Merry and Ristow 2016). RONS production highly increases in response to intense muscle contraction and intense exercise, at least in mice (Henríquez-Olguín et al. 2019; Place et al. 2015). Even though we did not directly measure RONS production after cycling sprints, our results indicate that the expression of the two redox sensitive genes SIRT1 and NFE2L2 was increased during the recovery period following exercise, but was not affected by post-exercise CHO restriction. Thus, it is likely that post-exercise CHO restriction does not enhance or suppress RONS-stimulated pathways involved in mitochondrial biogenesis.
Study limitations
Due to the difficulty of recruiting elite endurance athletes, this study was performed with recreationally active participants. Future experiments would be helpful to confirm our results in elite endurance athletes, since training status influences glycogen stores, as well as hormonal and metabolic changes in response to exercise (Hearris et al. 2018). In addition, collecting additional muscle samples at later time point during the recovery period (i.e. 5–8 h) would have been relevant to assess the expression of PGC1A when it potentially peaks (Pilegaard et al. 2005). This was not realized because our primary objective was to investigate the putative benefits of short post-exercise CHO restriction. Pre-exercise muscle glycogen concentration was slightly but not significantly lower in the CHO trial than the PLA trial. This slight difference might be explained by several parameters, including the absence of standardized meal on the evening before the exercise session, and the physical activity performed during the days preceding the experiments (although the participants were asked to refrain from any strenuous exercise during the last 3 days prior to each experimental session). Whether these limitations would have affected the results of our study is uncertain. Another limitation concerns the fact that changes in mRNA levels may not always reflect eventual changes at the translational level. Unfortunately, the assessment of protein levels was not possible due to the small size of the biopsy samples collected. Finally, a rather large variability was observed for the biological parameters analyzed in this study. Although including more participants may have increased the power of our analysis, when taken into context, our results are consistent with the majority of previous studies that showed no effect of CHO restriction following glycogen-depleting exercise on the acute mRNA levels of genes involved in mitochondrial biogenesis and muscle metabolism (Pilegaard et al. 2005; Jensen et al. 2015; Mathai et al. 2008).
Conclusion
We conclude that CHO restriction after a glycogen-depleting and metabolically-demanding cycling session does not increase the acute mRNA levels of genes involved in mitochondrial biogenesis and oxidative metabolism in human skeletal muscle. Our results indicate that the early molecular response to exercise was highly stimulated by the stimuli associated with our protocol (i.e. glycogen-depleting and metabolically-demanding exercise, fasted state, putative RONS production, plus the absence of CHO supplementation during exercise). Thus, the additional metabolic stress (i.e. post-exercise CHO restriction) had no additional benefit in amplifying the early gene response to exercise.
Abbreviations
- CCE:
-
Continuous cycling exercise
- CHO:
-
Carbohydrate
- GABPA:
-
GA-binding protein transcription factor, subunit alpha
- HPRT1:
-
Hypoxanthine phosphoribosyltransferase 1
- HIIE:
-
High-intensity interval exercise
- HR:
-
Heart rate
- MHC:
-
Myosin heavy chain
- NFE2L2:
-
Nuclear factor (erythroid-derived 2)-like 2
- NRF1:
-
Nuclear respiratory factor 1
- PDK4:
-
Pyruvate dehydrogenase kinase, isozyme 4
- PGC1α:
-
Peroxisome proliferator-activated γ-receptor coactivator 1alpha
- PLA:
-
Placebo
- PPARA:
-
Peroxisome proliferator-activated receptor alpha
- SIE:
-
Sprint interval exercise
- SIRT1:
-
Sirtuin 1
- SLC2A4:
-
Solute carrier family 2 member 4
- SOD2:
-
Superoxide dismutase 2, mitochondrial
- TFAM:
-
Transcription factor A, mitochondrial
References
Agbulut O, Li Z, Mouly V, Butler-Browne GS (1996) Analysis of skeletal and cardiac muscle from desmin knock-out and normal mice by high resolution separation of myosin heavy-chain isoforms. Biol Cell 88(3):131–135
Alghannam AF, Jedrzejewski D, Tweddle MG, Gribble H, Bilzon J, Thompson D, Tsintzas K, Betts JA (2016) Impact of muscle glycogen availability on the capacity for repeated exercise in man. Med Sci Sports Exerc 48(1):123–131. https://doi.org/10.1249/mss.0000000000000737
Bartlett JD, Louhelainen J, Iqbal Z, Cochran AJ, Gibala MJ, Gregson W, Close GL, Drust B, Morton JP (2013) Reduced carbohydrate availability enhances exercise-induced p53 signaling in human skeletal muscle: implications for mitochondrial biogenesis. Am J PhysiolRegulIntegr Comp Physiol 304(6):R450-458. https://doi.org/10.1152/ajpregu.00498.2012
Bartlett JD, Hawley JA, Morton JP (2015) Carbohydrate availability and exercise training adaptation: too much of a good thing? Eur J Sport Sci 15(1):3–12. https://doi.org/10.1080/17461391.2014.920926
Bergstrom J, Hermansen L, Hultman E, Saltin B (1967) Diet, muscle glycogen and physical performance. ActaPhysiolScand 71(2):140–150. https://doi.org/10.1111/j.1748-1716.1967.tb03720.x
Blom PC, Høstmark AT, Vaage O, Kardel KR, Maehlum S (1987) Effect of different post-exercise sugar diets on the rate of muscle glycogen synthesis. Med Sci Sports Exerc 19(5):491–496
Cheng AJ, Chaillou T, Kamandulis S, Subocius A, Westerblad H, Brazaitis M, Venckunas T (2020) Carbohydrates do not accelerate force recovery after glycogen-depleting followed by high-intensity exercise in humans. Scand J Med Sci Sports 30(6):998–1007. https://doi.org/10.1111/sms.13655
Cluberton LJ, McGee SL, Murphy RM, Hargreaves M (2005) Effect of carbohydrate ingestion on exercise-induced alterations in metabolic gene expression. J ApplPhysiol (1985) 99(4):1359–1363. https://doi.org/10.1152/japplphysiol.00197.2005
Cochran AJ, Percival ME, Tricarico S, Little JP, Cermak N, Gillen JB, Tarnopolsky MA, Gibala MJ (2014) Intermittent and continuous high-intensity exercise training induce similar acute but different chronic muscle adaptations. ExpPhysiol 99(5):782–791. https://doi.org/10.1113/expphysiol.2013.077453
Correia JC, Ferreira DM, Ruas JL (2015) Intercellular: local and systemic actions of skeletal muscle PGC-1s. Trends EndocrinolMetab 26(6):305–314. https://doi.org/10.1016/j.tem.2015.03.010
Greiwe JS, Hickner RC, Hansen PA, Racette SB, Chen MM, Holloszy JO (1999) Effects of endurance exercise training on muscle glycogen accumulation in humans. J ApplPhysiol (1985) 87(1):222–226. https://doi.org/10.1152/jappl.1999.87.1.222
Hammond KM, Sale C, Fraser W, Tang J, Shepherd SO, Strauss JA, Close GL, Cocks M, Louis J, Pugh J, Stewart C, Sharples AP, Morton JP (2019) Post-exercise carbohydrate and energy availability induce independent effects on skeletal muscle cell signalling and bone turnover: implications for training adaptation. J Physiol 597(18):4779–4796. https://doi.org/10.1113/jp278209
Hearris MA, Hammond KM, Fell JM, Morton JP (2018) Regulation of muscle glycogen metabolism during exercise: implications for endurance performance and training adaptations. Nutrients. https://doi.org/10.3390/nu10030298
Hearris MA, Hammond KM, Seaborne RA, Stocks B, Shepherd SO, Philp A, Sharples AP, Morton JP, Louis JB (2019) Graded reductions in preexercise muscle glycogen impair exercise capacity but do not augment skeletal muscle cell signaling: implications for CHO periodization. J ApplPhysiol (1985) 126(6):1587–1597. https://doi.org/10.1152/japplphysiol.00913.2018
Henríquez-Olguín C, Renani LB, Arab-Ceschia L, Raun SH, Bhatia A, Li Z, Knudsen JR, Holmdahl R, Jensen TE (2019) Adaptations to high-intensity interval training in skeletal muscle require NADPH oxidase 2. Redox Biol 24:101188. https://doi.org/10.1016/j.redox.2019.101188
Hickner RC, Fisher JS, Hansen PA, Racette SB, Mier CM, Turner MJ, Holloszy JO (1997) Muscle glycogen accumulation after endurance exercise in trained and untrained individuals. J ApplPhysiol (1985) 83(3):897–903. https://doi.org/10.1152/jappl.1997.83.3.897
Impey SG, Hearris MA, Hammond KM, Bartlett JD, Louis J, Close GL, Morton JP (2018) Fuel for the work required: a theoretical framework for carbohydrate periodization and the glycogen threshold hypothesis. Sports Med 48(5):1031–1048. https://doi.org/10.1007/s40279-018-0867-7
Jensen L, Gejl KD, Ortenblad N, Nielsen JL, Bech RD, Nygaard T, Sahlin K, Frandsen U (2015) Carbohydrate restricted recovery from long term endurance exercise does not affect gene responses involved in mitochondrial biogenesis in highly trained athletes. Physiol Rep. https://doi.org/10.14814/phy2.12184
Ji LL, Yeo D, Kang C, Zhang T (2020) The role of mitochondria in redox signaling of muscle homeostasis. J Sport Health Sci. https://doi.org/10.1016/j.jshs.2020.01.001
Joanisse S, McKay BR, Nederveen JP, Scribbans TD, Gurd BJ, Gillen JB, Gibala MJ, Tarnopolsky M, Parise G (2015) Satellite cell activity, without expansion, after nonhypertrophic stimuli. Am J PhysiolRegulIntegr Comp Physiol 309(9):R1101-1111. https://doi.org/10.1152/ajpregu.00249.2015
Lundberg TR, Fernandez-Gonzalo R, Norrbom J, Fischer H, Tesch PA, Gustafsson T (2014) Truncated splice variant PGC-1α4 is not associated with exercise-induced human muscle hypertrophy. ActaPhysiol (Oxf) 212(2):142–151. https://doi.org/10.1111/apha.12310
Maehlum S, Høstmark AT, Hermansen L (1977) Synthesis of muscle glycogen during recovery after prolonged severe exercise in diabetic and non-diabetic subjects. Scand J Clin Lab Investig 37(4):309–316. https://doi.org/10.3109/00365517709092634
Martinez-Redondo V, Pettersson AT, Ruas JL (2015) The hitchhiker’s guide to PGC-1alpha isoform structure and biological functions. Diabetologia 58(9):1969–1977. https://doi.org/10.1007/s00125-015-3671-z
Mata F, Valenzuela PL, Gimenez J, Tur C, Ferreria D, Domínguez R, Sanchez-Oliver AJ, MartínezSanz JM (2019) Carbohydrate availability and physical performance: physiological overview and practical recommendations. Nutrients. https://doi.org/10.3390/nu11051084
Mathai AS, Bonen A, Benton CR, Robinson DL, Graham TE (2008) Rapid exercise-induced changes in PGC-1alpha mRNA and protein in human skeletal muscle. J ApplPhysiol (1985) 105(4):1098–1105. https://doi.org/10.1152/japplphysiol.00847.2007
Merry TL, Ristow M (2016) Nuclear factor erythroid-derived 2-like 2 (NFE2L2, Nrf2) mediates exercise-induced mitochondrial biogenesis and the anti-oxidant response in mice. J Physiol 594(18):5195–5207. https://doi.org/10.1113/jp271957
Murray B, Rosenbloom C (2018) Fundamentals of glycogen metabolism for coaches and athletes. Nutr Rev 76(4):243–259. https://doi.org/10.1093/nutrit/nuy001
Perry CG, Lally J, Holloway GP, Heigenhauser GJ, Bonen A, Spriet LL (2010) Repeated transient mRNA bursts precede increases in transcriptional and mitochondrial proteins during training in human skeletal muscle. J Physiol 588(Pt 23):4795–4810. https://doi.org/10.1113/jphysiol.2010.199448
Pfaffl MW (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29(9):e45. https://doi.org/10.1093/nar/29.9.e45
Pilegaard H, Osada T, Andersen LT, Helge JW, Saltin B, Neufer PD (2005) Substrate availability and transcriptional regulation of metabolic genes in human skeletal muscle during recovery from exercise. Metabolism 54(8):1048–1055. https://doi.org/10.1016/j.metabol.2005.03.008
Place N, Ivarsson N, Venckunas T, Neyroud D, Brazaitis M, Cheng AJ, Ochala J, Kamandulis S, Girard S, Volungevicius G, Pauzas H, Mekideche A, Kayser B, Martinez-Redondo V, Ruas JL, Bruton J, Truffert A, Lanner JT, Skurvydas A, Westerblad H (2015) Ryanodine receptor fragmentation and sarcoplasmic reticulum Ca2+ leak after one session of high-intensity interval exercise. Proc Natl AcadSci USA 112(50):15492–15497. https://doi.org/10.1073/pnas.1507176112
Psilander N, Frank P, Flockhart M, Sahlin K (2013) Exercise with low glycogen increases PGC-1alpha gene expression in human skeletal muscle. Eur J ApplPhysiol 113(4):951–963. https://doi.org/10.1007/s00421-012-2504-8
Ruas JL, White JP, Rao RR, Kleiner S, Brannan KT, Harrison BC, Greene NP, Wu J, Estall JL, Irving BA, Lanza IR, Rasbach KA, Okutsu M, Nair KS, Yan Z, Leinwand LA, Spiegelman BM (2012) A PGC-1α isoform induced by resistance training regulates skeletal muscle hypertrophy. Cell 151(6):1319–1331. https://doi.org/10.1016/j.cell.2012.10.050
Scarpulla RC, Vega RB, Kelly DP (2012) Transcriptional integration of mitochondrial biogenesis. Trends EndocrinolMetab 23(9):459–466. https://doi.org/10.1016/j.tem.2012.06.006
Thom R, Rowe GC, Jang C, Safdar A, Arany Z (2014) Hypoxic induction of vascular endothelial growth factor (VEGF) and angiogenesis in muscle by truncated peroxisome proliferator-activated receptor γ coactivator (PGC)-1α. J BiolChem 289(13):8810–8817. https://doi.org/10.1074/jbc.M114.554394
Ydfors M, Fischer H, Mascher H, Blomstrand E, Norrbom J, Gustafsson T (2013) The truncated splice variants, NT-PGC-1α and PGC-1α4, increase with both endurance and resistance exercise in human skeletal muscle. Physiol Rep 1(6):e00140. https://doi.org/10.1002/phy2.140
Yeo WK, McGee SL, Carey AL, Paton CD, Garnham AP, Hargreaves M, Hawley JA (2010) Acute signalling responses to intense endurance training commenced with low or normal muscle glycogen. ExpPhysiol 95(2):351–358. https://doi.org/10.1113/expphysiol.2009.049353
Acknowledgements
This study was supported by grants from the Swedish Research Council for Sports Sciences for A.J.C. (P2017-0138, FO2017-0018, FO2018-0019). The authors thank Dr Paulo Jannig (Karolinska Institutet, Solna, Sweden) for useful discussions and comments related to the analysis of PGC1As.
Funding
Open Access funding provided by Örebro University.
Author information
Authors and Affiliations
Contributions
AJC, SK, MB, and TV were involved in the study design. SK, MB, AS, TC, CR and TV were involved in data collection, and data analysis was performed by AJC, TC, CR, SK, MB, and TV. CR and TC drafted the manuscript. AJC, TC, CR, SK, and TV edited and revised manuscript. SK, MB, AS, and TV assisted with recruitment of the subjects and data collection. All authors approved the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Communicated by Michael Lindinger.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ramos, C., Cheng, A.J., Kamandulis, S. et al. Carbohydrate restriction following strenuous glycogen-depleting exercise does not potentiate the acute molecular response associated with mitochondrial biogenesis in human skeletal muscle. Eur J Appl Physiol 121, 1219–1232 (2021). https://doi.org/10.1007/s00421-021-04594-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00421-021-04594-8