
Abstract
More than half a century of research on peroxisomes has revealed unique features of this ubiquitous subcellular organelle, which have often been in disagreement with existing dogmas in cell biology. About 50 peroxisomal enzymes have so far been identified, which contribute to several crucial metabolic processes such as β-oxidation of fatty acids, biosynthesis of ether phospholipids and metabolism of reactive oxygen species, and render peroxisomes indispensable for human health and development. It became obvious that peroxisomes are highly dynamic organelles that rapidly assemble, multiply and degrade in response to metabolic needs. However, many aspects of peroxisome biology are still mysterious. This review addresses recent exciting discoveries on the biogenesis, formation and degradation of peroxisomes, on peroxisomal dynamics and division, as well as on the interaction and cross talk of peroxisomes with other subcellular compartments. Furthermore, recent advances on the role of peroxisomes in medicine and in the identification of novel peroxisomal proteins are discussed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction: early milestones in peroxisome research
Peroxisomes (originally called microbodies) were discovered in 1954 using electron microscopy in mouse kidney (Rhodin 1954), 4 years before the first issue of this Journal (which in 1995 became Histochemistry and Cell Biology) was published. Since then the “Cinderella” among the subcellular organelles, which was once considered to be a “fossil organelle” and had been regarded as the cell’s “garbage pail”, has experienced a remarkable rise and turned into a dynamic and metabolically active cellular compartment essential for human health and development. De Duve and Baudhuin (1966) were the first who isolated peroxisomes from rat liver, and their biochemical studies led to the discovery of the colocalization of several H2O2-producing oxidases as well as catalase, an H2O2-degrading enzyme, in the matrix of peroxisomes (Fig. 1). On the basis of these findings, De Duve proposed the functional term “peroxisome”, which gradually replaced the former morphological designation, “microbody”, coined by Rhodin (Rhodin 1954). A specific cytochemical staining for peroxisomes in light and electron microscopy became available with the introduction of the alkaline 3, 3′-diaminobenzidine (DAB) reaction for catalase (Fahimi 1968, 1969; Novikoff and Goldfischer 1969) (Fig. 2). Subsequent morphological studies exploiting this cytochemical procedure revealed that peroxisomes, such as mitochondria, are ubiquitous eukaryotic organelles (Hruban et al. 1972). They disappear during the development of red blood cells and sperms (Luers et al. 2006), and appear to be absent in the Apicomplexa phylum and in amitochondriate parasites (Schluter et al. 2006) (see “Peroxisomes in silico ”). In contrast to mitochondria, peroxisomes (0.1–1 μm in diameter) are devoid of DNA, and have a single-limiting membrane surrounding a fine granular matrix, which may contain crystalline inclusions of matrix enzymes (Fig. 2). Pharmacological studies with hypolipidemic drugs and plasticizers (so-called peroxisome proliferators) led to the observation that peroxisomes can remarkably increase in number and size, especially in the livers of rodents (Hess et al. 1965; Svoboda and Azarnoff 1966; Fahimi et al. 1982; Reddy and Lalwani 1983). Such a “peroxisome proliferation” is often accompanied by an increase in the synthesis of peroxisomal enzymes, and can result in the formation of hepatic tumors, mainly in rodents (Reddy et al. 1980, 1982; Moody et al. 1991). This selective induction of peroxisomal genes by those compounds is mediated by peroxisome proliferator activated receptor-α (PPARα), which belongs to the family of nuclear transcription factors (Issemann and Green 1990) and acts as heterodimeric partner with retinoid X receptor by binding to the peroxisome proliferator response elements (PPREs). On the basis of similar pharmacological studies, a fatty acid β-oxidation system in peroxisomes was discovered (Fig. 1), which coexists and cooperates with the mitochondrial fatty acid β-oxidation system in animal cells (Lazarow and De Duve 1976; Wanders 2000). Interestingly, in plant cells (Cooper and Beevers 1969) and in eukaryotic microorganisms, peroxisomes are the only site of β-oxidation (Poirier et al. 2006) which renders them essential for the utilization of fat in these organisms. Their important role in lipid metabolism as well as in health and disease became obvious in the 1980s, when the major function of peroxisomes in the β-oxidation of very-long-chain fatty acids (VLCFA) and in the biosynthesis of ether glycerolipids (plasmalogens) was discovered (Hajra et al. 1979; Brown et al. 1982) (Fig. 1). Their absence in Zellweger syndrome, the first genetic neurodegenerative peroxisomal disorder, was reported by Goldfischer et al. (1973) and was later linked to abnormalities in lipid metabolism (Heymans et al. 1983). Since then, ongoing research on the biogenesis of peroxisomes and their metabolic functions has greatly improved our knowledge about their crucial role in several inherited disorders, which are often not compatible with life or normal development (Weller et al. 2003; Wanders and Waterham 2005; Steinberg et al. 2006; Wanders and Waterham 2006a) and in other pathophysiological conditions (Table 1 and “Mysterious protection: some recent observations on the relevance of peroxisomes in medicine”).

The major metabolic pathways in peroxisomes of the mammalian liver. The very-long-chain fatty acids (VLCFA) are transported by membrane proteins (e.g., the ABC transporter proteins PMP70 or ALDP) (see Table 1) into the matrix, where they are oxidized by the lipid β-oxidation enzymes. Multiple acyl-CoA oxidases and thiolases, as well as two distinct multifunctional (hydratase/3-hydroxyacyl-CoA dehydrogenase) enzymes coexist in peroxisomes (Poirier et al. 2006). The products of the β-oxidation can either serve as substrates for the biosynthesis of ether glycerolipids, cholesterol and bile acids or may exit the peroxisome for further oxidation in mitochondria (MITO). Peroxisomal β-oxidation and the activity of other peroxisomal oxidases result in the production of hydrogen peroxide, which is decomposed by catalase. Asterisks there are separate enzymes for bile acid intermediates, sER smooth endoplasmic reticulum, ALDP adrenoleukodystrophy protein

a Cytochemical localization of catalase in rat hepatic peroxisomes stained with the alkaline diamino-benzidine technique (Fahimi 1969). Note the uniform staining of the peroxisome matrix. Magnification, ×28,600. b Cytochemical localization of urate oxidase in rat liver using the cerium method (Angermuller and Fahimi 1986). Note the dark staining of the crystalline core (arrowheads). Magnification, ×50,400. c Immunocytochemical localization of catalase in rat liver using the protein A-gold technique (Fahimi et al. 1996). Note the diffuse labelling of the matrix with gold particles and the sparing of the core region. Magnification, ×72,000. d Immunocytochemical localization of urate oxidase in rat liver using the protein A-gold technique (Völkl et al. 1988). Note the exclusive labelling of the core with gold particles (arrowheads). Magnification, ×61,600. e An autophagic vacuole in rat liver from an animal treated with the peroxisome proliferator bezafibrate. Note the presence of a peroxisome, stained for catalase with the diamino-benzidine reaction, in the autophagic vacuole. Magnification, ×38,000. PO peroxisome, MITO mitochondrium
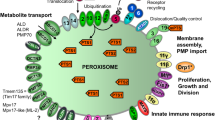
About 85 genes in Homo sapiens and 61 genes in Saccharomyces cerevisiae have been identified, which encode for peroxisomal proteins. Many of these proteins are metabolic enzymes (about 50 in mammalian peroxisomes), whereas some 32 proteins/genes, so-called peroxins (Pex), have been discovered, which are required for the biogenesis and maintenance of functional peroxisomes (32 in yeast, with approximately 20 mammalian and 23 plant homologs) (Kiel et al. 2006; Platta and Erdmann 2007a) (Fig. 3). Besides their essential catabolic (oxidation of pipecolic, phytanic and very-long-chain fatty acids) and anabolic (synthesis of plasmalogens, bile acids and cholesterol) functions in lipid metabolism (Fig. 1), peroxisomes play a key role in free radical detoxification, differentiation, development and morphogenesis from human to yeast. Although many peroxisomal enzymes and metabolic pathways have been well characterized (Table 2), research on peroxisomal metabolism is still continuing (van den Bosch et al. 1992; Wanders and Waterham 2006b). Noteworthy, peroxisomes in plants, yeasts and protozoa generally possess a far wider spectrum of activities than in vertebrates (e.g. penicillin biosynthesis in filamentous fungi, glyoxylate cycle, photorespiration, plant hormone biosynthesis/metabolism, and pathogen interaction in plants) (van der Klei et al. 2006; Kunze et al. 2006; Reumann and Weber 2006).

Schematic overview of peroxins and other poteins at the peroxisomal membrane. Cargo proteins containing the peroxisomal targeting signals PTS1 or PTS2 bind to the corresponding receptors Pex5p or Pex7p and form receptor-cargo complexes. The Pex7p–cargo complex requires accessory factors for import (Pex5pL, a long isoform of Pex5p, in mammals and plants, Pex18p and Pex21p in S. cerevisiae, Pex20p in Neurospora crassa, Yarrowia lipolytica, and Hansenula polymorpha). The receptor–cargo complex in yeast and mammals interacts with a docking site (Pex13p, Pex14p and Pex 17p). It is unclear whether the receptors translocate fully into the matrix (“extended shuttle hypothesis”) or remain associated with the translocation complex (“shuttle hypothesis”). The docking and RING complexes (Pex2p, Pex10p, Pex12p) involved in translocation in yeast and mammals are bridged by Pex8p in S. cerevisiae. Recycling of cargo-released Pex5p to the cytosol requires Pex4p, an ubiquitin-conjugating enzyme that is bound to Pex22p, and the AAA-ATPases Pex1p and Pex6p. Pex6p binds to Pex15p in S. cerevisiae or to Pex26p in humans. The DnaJ-like protein Djp1p assists in matrix protein import. Membrane assembly and insertion of peroxisomal membrane proteins (PMPs) (containing an mPTS) depends on Pex19p, Pex3p and Pex16p. Pex19p functions as a cycling receptor/chaperone, which binds the PMPs in the cytosol and interacts with Pex3p at the peroxisomal membrane. Pex11pα, Pex11pβ and Pex11pγ are the only peroxins known to be involved in the regulation of peroxisome size and number (proliferation) in mammals. In Y. lipolytica (Pex23p, and Pex24p) and S. cerevisiae (Pex25p, Pex27p-Pex32p) several other peroxins have been identified which influence the size and number or organization of peroxisomes. The division of peroxisomes is mediated by Fis1p and dynamin-like GTPases (DLP1/DRP1 in mammals, DRP3A in plants, Vps1p, Dnm1p in S. cerevisiae). Fis1p is the membrane receptor for DLP1 on peroxisomes (and mitochondria). Inp1p and Inp2p are involved in the inheritance and motility of peroxisomes in S. cerevisiae and Y. lipolytica. Inp2p is the membrane receptor for the type V myosin motor Myo2p on peroxisomes, which drives peroxisomes along actin filaments. The GTPase Rho1p binds to Pex25p and is involved in the recruitment of actin to peroxisomes in S. cerevisiae. Mammalian peroxisomes move along microtubules and bind dynein and kinesin motors by a yet unknown mechanism. Other membrane proteins are members of the PXMP34 and PXMP2/4 families, as well as the four ABC transporter proteins in mammals. Other PMPs are PMP22, PMP47, and the recently identified PMP52, as well as Mosc2 and ATAD1, an AAA-ATPase. Proteins with a dual localization to both peroxisomes and mitochondria are marked with asterisk. The ORF of YlPEX9 was misidentified; it corresponds to HsPEX26. ALD adrenoleukodystrophy protein, ALDR ALD-related protein
The biogenesis of peroxisomes
Protein import into peroxisomes: the mysterious membrane pore
Peroxisomes do not contain DNA or protein translation machinery, and all their proteins are encoded by nuclear genes. Majority of the peroxisomal proteins are synthesized on free polyribosomes in the cytoplasm and are imported post-translationally (Lazarow and Fujiki 1985) (Fig. 3). A major breakthrough in the elucidation of the mechanism of protein import into peroxisomes was the identification of the first peroxisomal targeting signal (PTS1) at the C-terminus of luciferase of the firefly Photinus pyralis (Keller et al. 1987; Gould et al. 1987). Luciferase actually localizes to peroxisomes in cells of the lantern organ of the firefly where it catalyzes the light-producing bioluminescent reaction. Today, we know that majority of the peroxisomal matrix proteins contain a C-terminal PTS1, and very few an N-terminal PTS2. It also became clear that the import into peroxisomes is a unique process, which differs substantially from the import mechanisms into the ER, mitochondria or chloroplasts. The peroxisomes import fully folded, co-factor bound and even oligomeric proteins by shuttling receptors (Leon et al. 2006). The PTS1- or PTS2-containing matrix proteins are recognized by soluble receptors (PTS1 by Pex5p, a tetra-tricopeptide repeat (TPR) domain protein, PTS2 by Pex7p, a WD40 domain protein, and its coreceptors) in the cytosol, which guide them to a docking site at the peroxisomal membrane (Fig. 3). After translocation of the receptor–cargo complex to the luminal side of the peroxisomal membrane, the cargo is released and the receptors shuttle back to the cytosol. Whereas the components of the import machinery (the importomer complex; composed of the docking and RING subcomplexes which in yeast are bridged by Pex8p or Pex3p) (Fig. 3) and their interactions have been quite well studied (Rayapuram and Subramani 2006; Platta and Erdmann 2007b), the mechanism of translocation of folded proteins across the membrane and the cargo release still remain mysterious (Gould and Collins 2002). A stable import channel or membrane pore, like in the nucleus, is missing in the peroxisomal membrane. As an alternative to a pore, a pinocytosis-like mechanism has also been proposed (McNew and Goodman 1996). Recently, the existence of a “transient pore” has been postulated, which might be dynamically formed by the import receptors themselves (Erdmann and Schliebs 2005). In this respect, it is also noteworthy that very little is known about the membrane channels and transporters of peroxisomes, which are involved in membrane permeability and solute transfer of metabolites (Antonenkov and Hiltunen 2006; Visser et al. 2007).
It has also been shown that Pex5p can be ubiquitinated (Erdmann and Schliebs 2005; Kragt et al. 2005b; Thoms and Erdmann 2006). Monoubiquitination of Pex5p appears to be a prerequisite for its dislocation from the peroxisomal membrane, and depends on the E2 enzyme Pex4p (Platta and Erdmann 2007a; Carvalho et al. 2007). Its function has been a mystery for over a decade; however, respective E3 enzymes are still elusive (the RING finger proteins Pex2p, Pex10p and Pex12p might be good candidates for the missing ubiquitin ligases). The membrane detachment/recycling of Pex5p is ATP dependent (but not the binding and translocation of Pex5p itself), and depends on AAA-ATPases Pex1p and Pex6p (Fig. 3). Polyubiquitination of Pex5p is believed to be part of a quality control process, which results in proteasomal degradation of dysfunctional receptor molecules (Platta and Erdmann 2007a). Noteworthy, these processes show a striking analogy to ERAD, the ER-associated degradation process (see also “Peroxisomes in silico”).
Interestingly, there is experimental evidence that manipulation of the peroxisomal import of catalase has an influence on aging. The alteration of the PTS1 of catalase to a more effective serine–lysine–leucine (SKL) sequence repolarized mitochondria and reduced cellular hydrogen peroxide levels and the number of senescent cells in a population (Terlecky et al. 2006; Koepke et al. 2007). Furthermore, targeting of catalase to mitochondria in transgenic mice has demonstrated to increase murine life span (Schriner et al. 2005).
The targeting and insertion of peroxisomal membrane proteins (PMPs) require other components than those involved in peroxisomal matrix import and is less well understood (Heiland and Erdmann 2005; Van Ael and Fransen 2006; Fujiki et al. 2006). Those PMPs, which are synthesized on free ribosomes in the cytosol, contain internal membrane targeting sequences (mPTS), which comprise a Pex19p-binding site and a membrane-anchoring sequence (either a transmembrane domain or a protein binding site) (Van Ael and Fransen 2006). Pex19p is suggested to function as a cycling receptor/chaperone, which binds the PMPs in the cytosol and is then recruited to the peroxisome by the membrane receptor Pex3p (Fujiki et al. 2006) (Fig. 3). In mammals, Pex16p is also required, which might function as a tethering factor for Pex3p, or as part of the putative membrane-insertion machinery. A loss of Pex3p, Pex16p, or Pex19p results in the absence of detectable peroxisomes/peroxisomal membranes, whereas reintroduction surprisingly leads to a de novo formation of peroxisomes from the ER (see below). Some PMPs (e.g., Pex3p and Pex16p) are supposed to be targeted indirectly to peroxisomes via the ER by an as yet unknown mechanism (see below).
Mysterious formation: growth and division, de novo formation, or both?
Since their discovery in 1954, there has been much speculation about the formation of peroxisomes. It was suggested that peroxisomes were precursors of mitochondria (Bernhard and Rouiller 1956), that they could arise from the Golgi complex (Rouiller and Jezequel 1963) or were specialized forms of lysosomes (Novikoff and Essner 1960). Whereas those concepts were abandoned with improvements in cytochemical methods, it was also proposed that peroxisomes formed from terminal cisternae of the ER (Novikoff and Shin 1964). However, the discovery of the synthesis of peroxisomal proteins on free polyribosomes in the cytoplasm (Goldman and Blobel 1978), their post-translational transport into peroxisomes, and the observations of interconnections between peroxisomes, led to the proposal of the “growth and division” model of peroxisome biogenesis with formation out of pre-existing organelles (Lazarow and Fujiki 1985). Recent discoveries have again challenged this classical view. The peroxins Pex3p, Pex19p and Pex16p are required to maintain the peroxisomal membrane (South and Gould 1999; Hettema et al. 2000; Heiland and Erdmann 2005), and their loss of function leads to the absence of peroxisomal membranes and thus, peroxisomes. Transexpression of Pex3, Pex19 or Pex16 and re-introduction of the missing genes has now been demonstrated to restore peroxisome formation in yeast and mammals, and lead to the postulation of a de novo synthesis of peroxisomes (Matsuzono et al. 1999; South and Gould 1999; Muntau et al. 2000; Titorenko and Rachubinski 2001; Faber et al. 2002; Geuze et al. 2003; Hoepfner et al. 2005; Kragt et al. 2005a; Haan et al. 2006; Kim et al. 2006). Pex3p and Pex19p have been observed to initially localize to the ER before maturing into import-competent peroxisomes (Hoepfner et al. 2005; Tam et al. 2005; Haan et al. 2006), indicating that the ER/nuclear envelope is the source of the newly synthesized membrane and organelle.
However, peroxisomes have also been reported to divide, and recently proteins have been identified in yeast, mammalian and plant cells, which play a role in peroxisomal fission. Among them are dynamin-like GTPases (DLPs) with mechanochemical properties (yeast Dnm1p and Vps1p, mammalian DLP1/Drp1, plant DRP3A), Fis1p, a membrane adaptor for DLPs, and Pex11 proteins (for recent reviews see Schrader and Fahimi 2006; Fagarasanu et al. 2007) (Fig. 3). Morphological observations show that growth and division of peroxisomes in mammalian cells is a multistep process including peroxisome elongation, constriction, and final fission (Koch et al. 2003, 2004, 2005) (Figs. 4, 5). Peroxisomes tend to have a characteristic, segmented, “beads on a string”-like appearance before they divide and distribute (Schrader et al. 1996), or when DLPs are non-functional (Hoepfner et al. 2001; Koch et al. 2003, 2004; Tanaka et al. 2006; Waterham et al. 2007) (Fig. 4). Pex11 proteins are implicated early on in the elongation step of peroxisomes, whereas Fis1p and DLPs are required for the final fission step (Thoms and Erdmann 2005; Schrader and Fahimi 2006; Fagarasanu et al. 2007; Kobayashi et al. 2007) (Fig. 5). Peroxisomal constriction can proceed independently of DLP1 (Koch et al. 2004), but the molecular mechanism is so far largely unknown. Interestingly, Fis1p and DLP1 (Dnm1p, DRP3A) are also involved in mitochondrial fission (Hoopins et al. 2007), and are shared by both organelles (Schrader 2006). These findings indicate that peroxisomes and mitochondria share some basic characteristics and are much closer than previously assumed, thus underlining the tight cooperation and cross talk between both organelles (Schrader and Yoon 2007) (Figs. 4, 5; and “Peroxisomes and mass spectrometry ”). Similar to mitochondria, peroxisomal morphology and dynamics might influence peroxisomal functions as well as developmental and physiological processes. Interestingly, a lethal defect in peroxisomal and mitochondrial fission, which appears to be based on a point mutation in the DLP1 gene, has recently been described (Waterham et al. 2007). These findings might point to a new class of diseases characterized by defects in peroxisomes and mitochondria (Table 1).

Fluorescence microscopy of peroxisomes (a, b) and mitochondria (a) in human hepatoma cells. Cells were stained with antibodies to the peroxisomal membrane protein PMP70 (a, green; b) and to mitochondrial manganese superoxide dismutase (MnSOD) (a, red). Peroxisomes exhibit spherical, rod-shaped and elongated, tubular morphologies, whereas mitochondria form an interconnected tubular network. During division peroxisomes appear like “beads on a string” (arrow) (Schrader et al. 1996). Boxed region shows higher magnification view. (b) Peroxisome morphology after silencing of DLP1 by siRNA. Note the highly elongated but constricted appearance of peroxisomes defective in final membrane fission (arrows). The formation of spherical peroxisomes by division is completely inhibited. N nucleus. Scale bars, 10 μm (a, b)

Model of peroxisome dynamics and interactions in mammalian cells. The majority of the peroxisomal matrix and membrane proteins (Class I PMPs) are synthesized on free polyribosomes in the cytosol and are post-translationally imported into pre-existing peroxisomes. Some membrane proteins (Class II PMPs, early peroxins, for example, Pex3p) are routed to peroxisomes via the ER or a pre-peroxisomal compartment, presumably by vesicular transport. A retrograde peroxisome-to-ER transport might exist. A novel vesicular mitochondria-to-peroxisome trafficking route has been described recently (Neuspiel et al. 2008), but its function is still mysterious. It is currently unknown, whether a retrograde peroxisome-to-mitochondrium transport exists, and whether peroxisome-derived vesicles contribute to the removal of damaged (e.g., by ROS production), misfolded or mistargeted proteins. Besides ER and mitochondria (Mito), peroxisomes are supposed to interact with the smooth ER (sER), with lipid droplets/bodies, the cytoskeleon, and other peroxisomes (Schrader et al. 2000). Peroxisomes can multiply by growth/elongation, constriction and final fission/division, forming spherical peroxisomes. Pex11pβ is involved in the elongation/tubulation of peroxisomes, whereas DLP1/Drp1 and Fis1p mediate peroxisomal fission (Schrader and Fahimi 2006). Fis1p is supposed to recruit cytosolic DLP1/Drp1 to the peroxisomal membrane. The involvement of other proteins (related to yeast Mdv1p/Caf4p) is likely. Components involved in the constriction of peroxisomes are presently unknown. Proper intracellular distribution of the peroxisomes formed by fission requires microtubules (MT) and motor proteins. In yeast and plants peroxisomes are distributed via the actin cytoskeleton. The consecutive steps are linked to each other, and may be triggered by the assembly of distinct machineries at the peroxisomal membrane
The physiological significance of the mechanism of de novo formation in comparison to the classical pathway of growth and division is still controversially discussed (Lazarow 2003; Kunau 2005; Haan et al. 2006; Mullen and Trelease 2006; Tabak et al. 2006). In order to investigate to what extent these pathways of peroxisome formation contribute to peroxisome numbers in yeast, Motley and Hettema (2007) have developed an elegant, fluorescence-based mating assay to follow the fate of existing and de novo-formed peroxisomes in S. cerevisiae. They provide evidence that in wild-type cells (grown on a nonfermentable carbon source) peroxisomes multiply by growth and division and do not form de novo. Only cells lacking peroxisomes as a result of a segregation defect were observed to form peroxisomes de novo out of the ER. In contrast to peroxisome fission, de novo formation was much slower and appeared to be independent of DLPs such as Dnm1p or Vps1p (Motley and Hettema 2007; Nagotu et al. 2007; Jourdain et al. 2008). In some yeasts, which possess only a few peroxisomes, de novo formation may therefore represent a rescue mechanism that becomes functional in case peroxisomes are lost (e.g., due to failure in inheritance). Several components involved in peroxisome inheritance in yeast have recently been discovered, for example the peroxisomal proteins Inp1p and Inp2p, which together with the type V myosin motor Myo2p and actin play a role in the retention and motility of peroxisomes (Fagarasanu et al. 2006, 2007) (Fig. 3).
However, there is now also firm evidence that some peroxisomal proteins are routed indirectly to peroxisomes via the ER (e.g. Pex3p) (Hoepfner et al. 2005; Tam et al. 2005; Kragt et al. 2005a; Haan et al. 2006; Kim et al. 2006; Mullen and Trelease 2006; Motley and Hettema 2007). This trafficking pathway is supposed to involve ER-derived vesicular or preperoxisomal structures, which do not require DLPs for exit from the ER (Motley and Hettema 2007; Nagotu et al. 2007), and are observed to fuse with pre-existing peroxisomes. Thus, a semi-autonomous model of peroxisome formation can be envisaged, whereby the ER supplies existing peroxisomes with essential membrane proteins (and lipids) to allow peroxisomal growth and division (Fig. 5). However, it is currently not understood how the peroxisomal proteins enter and leave the ER, how they are sorted and packaged, what the nature and composition of the ER-derived structures is, how fusion with peroxisomes is mediated, and if a retrograde, peroxisome-to-ER pathway exists (Fig. 5). Interestingly, such a retrograde transport pathway has recently been proposed in infected plant cells (McCartney et al. 2005; Mullen and Trelease 2006) and trypanosomes (Subramanya and Mensa-Wilmot 2006). Furthermore, a role of the Dsl1p secretory complex in regulating de novo synthesis of peroxisomes from the ER has been very recently proposed in S. cerevisiae (Perry and Rachubinski 2007).
Mysterious trafficking: a novel vesicular mitochondria-to-peroxisome pathway?
Besides vesicular ER-to peroxisome transport, even a transport route between mitochondria and peroxisomes might exist (Fig. 5). Neuspiel et al. (2008) have very recently identified a new outer membrane mitochondrial anchored protein ligase (MAPL) containing a RING finger domain. MAPL appeared to have a regulatory function in controlling mitochondrial morphology. Excitingly, MAPL was also incorporated within unique, vesicular structures, which emanated from the sides of mitochondria. These new mitochondrial-derived vesicles (MDVs) (70–100 nm in diameter) formed in a DLP1-independent manner, showed an increase in electron density around the surface, and often contained both outer and inner mitochondrial membranes, which appeared as two concentric circles. Remarkably, in live-cell experiments MAPL-YFP-positive vesicles were observed to fuse with a small subset of peroxisomes (Neuspiel et al. 2008). Furthermore, other populations of MDVs were discovered, which excluded MAPL, but contained TOM20 (which was absent form MAPL-positive MDVs). The TOM20-positive MDVs did not fuse with peroxisomes, and their fate is still under investigation. However, it might be possible that these vesicles contribute to the removal of damaged, misfolded, or mistargeted proteins from the mitochondria/mitochondrial membranes, and route them to the lysosomal/autophagic degradation pathway (Fig. 5). The physiological function of the new vesicular mitochondria-to-peroxisome transport pathway is also still mysterious. MAPL appears to be present at low levels in only a subpopulation of peroxisomes, and is not required for MDV formation at the mitochondria or targeting to peroxisomes. Silencing or overexpression of MAPL was not observed to change peroxisomal morphology, and thus, it might not play a role in peroxisomal growth and division (in contrast to its function on mitochondria). Peroxisomes and mitochondria are metabolically linked organelles, which cross talk and cooperate, and even share components of their division machinery (Schrader 2006; Schrader and Yoon 2007) (Figs. 1, 4). The MAPL-positive MDVs might therefore function in the transport of metabolites, lipids, or proteins to a peroxisome subpopulation. Interestingly, a dual localization to both peroxisomes and mitochondria has been observed for mitochondrial enzymes and some mitochondrial membrane proteins (e.g., Fis1p, Mosc2, and ATAD1, a member of the AAA-superfamily of ATPases) (Koch et al. 2005; Wiese et al. 2007) (see “Peroxisomes and mass spectrometry ”). However, Fis1p and the other proteins are found in the vast majority of peroxisomes (distinct from MAPL), and are presumably imported directly from the cytosol. An alternative function of the peroxisome-targeted MDVs might be the retrieval of peroxisomal (membrane) proteins, which have been mistargeted to mitochondria. Mistargeting of peroxisomal membrane proteins to mitochondria is often observed under different experimental conditions, for example when peroxisomal membrane insertion is affected (Sacksteder et al. 2000; Soukupova et al. 1999), and might be due to some overlap in the targeting information that is also recognized by the mitochondrial import machinery (Van Ael and Fransen 2006; Subramani 1998). Another interesting issue is the fusion event of a double-membrane-bound MDV with a single-membrane-bound peroxisome (Neuspiel et al. 2008). Such a fusion would likely result in the release of a single-membrane-bound vesicle (the former inner mitochondrial membrane) into the lumen of the peroxisome. Internal peroxisomal membrane structures have been described in yeast cells and in mammalian tissue (McNew and Goodman 1996), and very recently after deletion of the peroxisomal lipase Lpx1p in S. cerevisiae (Thoms et al. 2008). It will be very interesting to elucidate the fate of the putative vesicle remnant, and to identify the components of the putative MDV-peroxisome fusion machinery.
It is intriguing to speculate that peroxisomes themselves—like mitochondria—might be able to emenate vesicles from their membranes (which would then be PDVs, peroxisome-derived-vesicles) (Fig. 5). Budding events at the peroxisomal membrane have been occasionally observed (Jedd and Chua 2002). Furthermore, ADP-ribosylation factor (ARF) and coatomer have been localized to peroxisomes (Passreiter et al. 1998; Lay et al. 2005). Efforts to link ARF and coatomer recruitment to (ER-derived) peroxisome formation or growth and division have so far been inconclusive or are difficult to interpret (Lay et al. 2006). It might actually be possible that these components play only a minor, if any, role in peroxisome formation, but are instead involved in the generation of PDVs, which might either deliver metabolites, lipids, or (mistargeted) proteins to mitochondria (or to other subcellular compartments, e.g., to the ER; see “Mysterious formation: growth and division, de novo formation, or both?”), or might be involved in the removal of damaged (e.g., by peroxisomal ROS production), misfolded, or mistargeted (mitochondrial?) proteins from the peroxisomal membranes, and route them to the lysosomal/autophagic degradation pathway (Fig. 5). As the peroxisome appears to be connected to transport pathways coming from the ER and presumably also from mitochondria, there will certainly be some degree of mistargeting. Therefore, mechanisms must exist, which assist the peroxisome in getting rid of potentially mistargeted proteins, especially those in the peroxisomal membrane, which are not readily accessible to peroxisomal enzymes/proteases (e.g., membrane proteins mainly exposed to the cytoplasm). Besides vesicle formation, these mechanisms might also include polyubiquitination (Platta and Erdmann 2007a) or autophagy/pexophagy of the whole organelle (Sakai et al. 2006) (see below).
Mysterious removal: degradation and autophagy of peroxisomes
The number of peroxisomes in cells and their enzymatic equipment is modified depending on metabolic state and the environmental requirements. A well-known characteristic of peroxisomes is their inducibility by divergent chemicals and drugs which is mediated by the transcription factors referred to as “peroxisome proliferator activated receptors” (PPARs) (Issemann and Green 1990). Upon withdrawal of treatment, the peroxisome proliferation is reversed and the excess particles are removed by autophagy (Yokota 1993) (Fig. 2e), which in the case of yeast cells is also referred to as pexophagy (Sakai et al. 2006). Autophagy is a process regulated by ATG genes, which determine the sequestration and degradation of cell organelles and other cellular components within lysosomes or vacuoles. Two distinct mechanisms of sequestration by autophagy have been described: macroautophagy and microautophagy. Whereas in macroautophagy the cell components (e.g., peroxisomes) are first sequestered within autophagosomes, which in turn fuse with lysosomes/vacuoles, in microautophagy membrane events occur on the surface of the vacuole/lysosome which then engulf the cell components to be digested resulting in the formation of microautohagic bodies (Sakai et al. 1998a). From the numerous ATG genes (Klionsky et al. 2003) involved in general cellular autophagy, many have also been identified to participate in pexophagy (Schroder et al. 2007). On the other hand, much less is known about the peroxisomal components, which are involved in this process. In the yeast Hansenula polymorpha two peroxisomal membrane proteins, HpPex3p (Bellu et al. 2002) and HpPex14p (Bellu et al. 2001), appear to be important in the process of macropexophagy. In yeasts lacking HpPex14p, degradation of peroxisomal membranes is defective and it is suggested that the highly conserved N-terminal region of HpPex14p is necessary for macropexophagy. Since Pex14p has also been known to be a translocon and is involved in the formation of peroxisomes (Fig. 3), it is the only Pex protein that participates both in peroxisome formation and degradation (Zutphen et al. 2008). The yeast models Pichia pastoris and H. polymorpha have extensively been used for the analysis of molecular events and the morphological steps of pexophagy, and there are excellent recent reviews on this subject (e.g., Dunn et al. 2005; Sakai et al. 1998b, 2006; Platta and Erdmann 2007a).
In mammalian hepatocytes, the degradation of peroxisomes by autophagy was noted quite early by electron microscopic studies in rodents treated with hypolipidemic drugs (Leighton et al. 1975; Moody and Reddy 1976; Staubli et al. 1977) (Fig. 2e). Although the morphological aspects and the participation of lysosomes in this process were well characterized (Yokota 2003), its molecular aspects remained quite mysterious compared to yeast cells. Recently, Iwata et al. (2006) reported that in Atg7-deficient mice (lacking the essential gene for autophagy), the excess peroxisomes induced by phthalate ester could not be degraded within 1 week after discontinuation of the treatment. Moreover, the Atg7-deficient hepatocytes lacked the autophagosomes containing sequestered peroxisomes, thus confirming the role of autophagy in degradation of excess peroxisomes. Further studies are required to analyse the role of other genes and their products in autophagy in mammalian systems and the search has just started (Monastyrska and Klionsky 2006).
In addition to the autophagic degradation of excess peroxisomes discussed above, another unique aspect of turnover of this organelle deserves to be mentioned, namely, the possible role of 15-lipoxygenase in selective destruction of peroxisomal membranes in normal untreated animals. In reticulocytes, 15-lipoxygenase has been shown to bind selectively to the membranes of organelles and induce the diffusion of their contents (van Leyen et al. 1998). The importance of this process for the degradation of cell organelles in differentiating lens fibres (Bassnett and Mataic 1997) and in maturation of reticulocytes has been suggested (Schewe et al. 1975). This notion has been confirmed recently in Atg5−/− mice which show normal organelle degradation in lens fibres and erythroid cells inspite of inability to form autophagic vacuoles (Matsui et al. 2006). The presence of 15-lipoxygenase in the membranes of normal rat hepatocyte peroxisomes was demonstrated recently, and it was suggested that, similar to maturing reticulocytes and lens fibres, it could also be involved in the degradation of peroxisome membranes (Yokota et al. 2001). This notion is supported by the observation that 15-lipoxygenase was detected in only some but not all peroxisomes and that its presence correlated with the diffusion of catalase from some but not all peroxisomes in aldehyde fixed sections of rat liver. The latter finding was reported almost 30 years before and was contributed to the heterogeneous stability of peroxisome membranes (Fahimi 1974). Since the disruption of peroxisome membranes and the diffusion of catalase were both prevented by inhibitors of 15-lipoxygenase such as propyl gallate and esculetin, it was suggested that 15-lipoxygenase could participate in physiological degradation and turnover of peroxisomes in normal untreated animals, contrasting the role of autophagy in degradation of excess peroxisomes induced by treatment with chemicals and drugs (Yokota et al. 2001).
Mysterious protection: some recent observations on the relevance of peroxisomes in medicine
There are by now more than two dozens inborn metabolic disorders due to various dysfunctions or absence of peroxisomes and that number is growing (Schluter et al. 2007). Table 1 summarizes those disease conditions with the age of the onset, the basic metabolic defect and the genes involved. In addition to those genetic peroxisomal disorders, which have been addressed in recent excellent reviews (Weller et al. 2003; Wanders and Waterham 2005, 2006a; Faust et al. 2005; Steinberg et al. 2006), there is also increasing evidence that peroxisomes may be affected in some medical conditions associated with disorders of the lipid metabolism. Since peroxisomes and their proteins can be induced by a variety of drugs and chemicals, generally referred to as PPAR-agonists, there are emerging new possibilities for therapy of those conditions. Two such disorders are briefly discussed here: fatty liver disease and neuroinflammation.
Peroxisomes in fatty liver disease
The fatty liver disease (FLD) represents an excess accumulation of triglycerides in hepatic parenchymal cells. Whereas in the past excess ethanol consumption accounted for most cases, which were called alcoholic FLD (AFLD), in the last two decades, the non-alcoholic FLD (NAFLD), particularly in association with obesity, has emerged as the most common chronic liver condition in the western world (Adams et al. 2005). NAFLD is now present in 17–33% of the population in the United States, but has a world-wide distribution, and parallels the frequency of central adiposity, obesity, insulin resistance, metabolic syndrome and type 2 diabetes (Farrell and Larter 2006). In almost one-third of NAFLD cases, there is an associated steatohepatitis (NASH), which progresses to hepatic fibrosis and upon chronicity to liver cirrhosis. Many cases of the so-called cryptogenic liver cirrhosis are likely end-stage NASH (Neuschwander-Tetri and Caldwell 2003). Although insulin resistance is the underlying problem, in the pathogenesis of FLD both excess energy consumption as well as defective energy combustion contribute to a sequence of events which begins with hepatic steatosis and ends in liver cirrhosis and cancer (Reddy and Rao 2006). Whereas hepatic steatosis is considered to be innocuous and reversible, the progression to steatohepatitis is influenced by the severity and persistence of the causative agent and additional factors. According to the “two-hit” hypothesis, secondary insults that may severely damage the steatotic liver include reactive oxygen species, endotoxin, tumor necrosis factor-alpha (TNFα) and other cytokines (Day and James 1998). In this respect, it should be noted that inflammatory cytokines such as TNFα have been shown to suppress the hepatic peroxisomal catalase and the lipid β-oxidation enzymes, as well as the transcription factor PPARα, at both protein and mRNA levels (Beier et al. 1992, 1997). The reduction of the fatty acid β-oxidation enzymes in peroxisomes could contribute to the lipid accumulation in hepatocytes and to the severity of steatohepatitis. Indeed, mice lacking the peroxisomal acyl-CoA oxidase gene develop severe steatohepatitis, lipogranulomas and hepatocellular carcinomas (Fan et al. 1998). Some of the features of those AOX−/− mice resemble the spectrum of obesity associated human liver alterations justifying further detailed studies of peroxisomes in patients with morbid obesity and type 2 diabetes.
From the PPAR subfamily of nuclear transcription factors (Michalik et al. 2006), the PPARα functions mainly as a lipid sensor in the liver responding to the influx of fatty acids by up-regulation of the transcription of genes involved in β-oxidation of fatty acids in peroxisomes and mitochondria, as well as the microsomal ω-oxidation system. PPARα−/− mice cannot respond to increased fatty acid influx and develop severe hepatic steatosis (Hashimoto et al. 2000). Such PPARα–/− mice also develop severe steatohepatitis after feeding with a choline and methionine deficient diet (Kashireddy and Rao 2004). Interestingly, both agonists of PPARα and PPARδ have been found to improve the hepatic steatosis and prevent the inflammation associated with steatohepatitis induced by choline-methionine deficient diet in mice (Nagasawa et al. 2006). More importantly, in rats with severe steatohepatitis and liver fibrosis induced by a choline-methionine deficient diet, administration of PPARα agonists prevents not only the development of steatohepatitis but also reverses the fibrotic process in the liver (Rao et al. 2002). Thus, it seems that induction of fatty acid oxidation enzymes by activation of PPARα and PPARδ can prevent the development of hepatic steatosis. Indeed, in clinical studies, the PPARγ agonists rosiglitazone (Neuschwander-Tetri et al. 2003) and pioglitazone (Promrat et al. 2004) have been tried and found to improve significantly all histological and biochemical features of non-alcoholic steatohepatitis. This has also been proven in a placebo-controlled trial with pioglitazone justifying further long-term studies (Belfort et al. 2006). Thus, PPAR-agonists such as fibrates or thiazolidinediones offer the potential for treatment of not only the hepatic complications but also the underlying disease process of metabolic syndrome and type 2 diabetes with dyslipidaemia (Plutzky 2007; Staels 2007).
Peroxisomes and neuroinflammatory diseases
The severe structural and functional abnormalities of the central nervous system (CNS) seen in patients with peroxisome biogenesis disorders (PBDs) (Evrard et al. 1978; Powers et al. 1985) (Table 1) demonstrate the important role of peroxisomes in normal development and function of the brain. This is also confirmed in brain specific gene-knock out mouse models of PEX5 (Janssen et al. 2003) and PEX2 (Faust 2003), which exhibit neuronal migration defects and abnormalities in cerebellar histogenesis, respectively. Recently, in a mouse model of selective knock out of PEX5 in oligodendrocytes, the central role of peroxisomes in the myelination process was demonstrated (Kassmann et al. 2007). Although the mutant mice appeared normal at birth, within a few months they developed ataxia, tremor and died prematurely with widespread evidence of axonal degeneration and progressive subcortical demyelination. Moreover, severe inflammation accompanied the axonal degeneration with infiltration of B and CD8+ T cells in cerebral lesions resembling closely the neuroinflammatory lesions associated with human demyelinating disorders, particularly the multiple sclerosis.
Thus, it seems that peroxisomes in oligodendrocytes have a neuroprotective function preventing not only the axonal degeneration but also the associated process of neuroinflammation. The anti-inflammatory function of peroxisomes and the regulatory role of PPAR-activators in this process were discovered more than a decade ago (Devchand et al. 1996). Both, PPARα and PPARγ, are expressed on T cells, and their respective agonists inhibit the secretion of interleukin-2 and the proliferation of T cells (Cunard et al. 2002; Marx et al. 2002). Moreover, PPAR-agonists have been found to inhibit the progression of a variety of inflammatory diseases in experimental animals such as adjuvant arthritis in rats (Kawahito et al. 2000), inflammatory bowel disease (Su et al. 1999) and atherosclerosis in mice (Li et al. 2000). Experimental autoimmune encephalomyelitis (EAE) is an autoimmune disorder in mice characterized by inflammation of the CNS and severe demyelination which are the typical features of multiple sclerosis in humans and both PPARγ and PPARα agonists have been shown to exert therapeutic effects in the mouse model (Niino et al. 2001; Diab et al. 2002; Xu et al. 2007). Moreover, in a report of a single patient with severe multiple sclerosis, pioglitazone, a PPARγ activator, was found to substantially improve the patients condition (Pershadsingh et al. 2004). Those observations have now stimulated the call for large-scale clinical trials of PPAR-agonists for therapy of multiple sclerosis (Racke et al. 2006; Niino 2007). Since an inflammatory process in the brain is also observed in other severe conditions affecting the CNS such as Alzheimer’s and Parkinson’s disease, PPAR-agonists have also been tried (Landreth 2007; Dehmer et al. 2004) with promising results, thus justifying future trials of those drugs in various neurological disorders associated with neuroinflammation. It is of interest that not all effects of those receptor-agonists are mediated via the activation of PPAR, since some therapeutic effects are also observed with gemfibrozil, a PPARα agonist, in PPARα−/− mice with autoimmune encephalomyelitis (Dasgupta et al. 2007) suggesting receptor independent mechanisms of action. This is, however, mostly new territory where therapeutic benefits of those drugs remain to be discovered. The exact knowledge of the structure and function of peroxisomes and their heterogeneity in the CNS, as reported recently (Ahlemeyer et al. 2007) is a prerequisite for such future studies.
Mysterious players: identifying novel peroxisomal proteins
Peroxisomes in silico
The rapid advances in genome sequencing allow computational approaches (comparative genomics) to predict peroxisomal proteins. These in silico analyses take advantage of the conserved targeting sequences in peroxisomal proteins (i.e. PTS1, PTS2, see “Introduction ” and Fig. 3). PTS domain-based comparisons across genomes from yeast to humans and plants have been performed, predicting large numbers of putative peroxisomal proteins (Emanuelsson et al. 2003; Kurochkin et al. 2005; Kamada et al. 2003; Reumann et al. 2004, database AraPerox: http://www.araperox.uni-goettingen.de/; Hawkins et al. 2007). Some of the predictions have already been confirmed experimentally, others still await validation. There are, however, limitations, because many proteins of peroxisomes cannot currently be predicted from genome sequences. These include peripheral and integral membrane proteins, proteins that are imported in a piggy-back fashion, or matrix proteins with internal PTS1-like peptides. On the other hand, predicted PTSs can be masked by protein conformation, multimer formation, or posttranslational regulatory mechanisms.
Mysterious rise
Recently, computational approaches on the basis of proteomes and sequenced genomes of different organisms have been used to investigate the evolutionary origin of peroxisomes (Schluter et al. 2006; Gabaldon et al. 2006; Schluter et al. 2007, peroxisome database: http://www.peroxisomedb.org/). The peroxisomal proteins Pex3p, Pex19p, Pex10p, and Pex12p have been identified as markers for unequivocal in silico peroxisome detection (Schluter et al. 2006). The Apicomplexa phylum (containing for example, Plasmodium falciparum, Cryptosporidium parvu) has been detected as the first group of organisms devoid of peroxisomes in the presence of mitochondria. The absence of peroxisomes had so far only been documented in amitochondriate parasites, such as Encephalitozoon cuniculi, Girardia lamblia, or Entamoeba histolytica. Interestingly, a high degree of similarity among membrane proteins and between components of the matrix import machinery and the endoplasmic reticulum/proteasome degradation system has been observed (see “Mysterious formation: growth and division, de novo formation, or both? ”) (Schluter et al. 2006; Gabaldon et al. 2006). Most of the conserved peroxisome biogenesis and maintenance proteins are apparently eukaryotic innovations with no prokaryotic counterpart. This is consistent with findings that report an ER-dependent formation of peroxisomes (see “Mysterious formation: growth and division, de novo formation, or both? ”) (Tabak et al. 2006), and indicates that peroxisomes do not have an endosymbiotic origin. In contrast, many peroxisomal matrix enzymes were found to have prokaryotic homologues. These might have been recruited individually from pools existing within the primitive eukaryote (e.g., from mitochondria), but an en bloc recruitment of this coherent collection of enzymes can also be envisaged (de Duve 2007). It is still an open (but interesting, and hotly debated) question, when the peroxisomes first appeared in the evolution of the eukaryotic cell, and if they might have been present before the adoption of mitochondria (Cavalier-Smith 1997).
Peroxisomes and mass spectrometry
The recent technical advances in two-dimensional gel electrophoresis (2D-GEP), liquid chromatography (LC), as well as mass spectrometrical (MS) peptide identification and quantification laid the foundation for progress in organellar proteome studies, and have also been exploited to unravel the peroxisomal proteome. These studies depend not only on the availability of complete genome information or large EST collections of the species of interest for protein identification but also on the purity of the isolated organelle fraction. Due to the fragile nature of peroxisomes, the isolation of intact peroxisomes in high purity and sufficient quantity is difficult. However, sophisticated protocols have been developed for the isolation of mammalian peroxisomes, for fungi and yeast, as well as for plant peroxisomes (e.g., Völkl et al. 1996, 1999; Luers et al. 1998; Weber et al. 2004; Distel and Kragt 2006; Reumann et al. 2007).
One-dimensional (1D) and 2D-GEP of peroxisomes from rat liver followed by LC/tandem MS or MS resulted in the identification of more than 50 known constituents as well as novel peroxisomal proteins, among them a new isoform of Lon protease (one of the first peroxisomal proteases discovered) (Kikuchi et al. 2004), microsomal gluthatione-S-transferase (Islinger et al. 2006), as well as the known microsomal proteins aldehyde dehydrogense, cytochrome b5 and its corresponding reductase (Kikuchi et al. 2004; Islinger et al. 2006; Wiese et al. 2007). An early high throughput characterization of the yeast peroxisomal membrane proteome was performed by Schafer et al. (2001) using oleate-induced cultures. In order to differentiate between proteins that are contaminants of low abundance and those that have specifically enriched in the purified peroxisome fractions, efforts have been made to study relative protein quantities. Aitchinson and coworkers introduced a relative quantitative MS-based proteomics approach (ICAT, isotope-coded affinity tagging) to determine the enrichment or depletion of proteins detected in two peroxisomal membrane fractions from S. cerevisiae that differed in their degree of purity, and revealed a role for the GTPase Rho1p in actin organization on the peroxisomal membrane (Marelli et al. 2004; Saleem et al. 2006). In another quantitative MS-based proteomics approach (iTRAQ, isobaric Tag for Relative and Absolute Quantitation) with peroxisome matrix and membrane subfractions from bezafibrate treated rats, Islinger et al. (2007) identified 134 individual proteins, including 15 new candidates, among them the new peroxisomal membrane proteins PMP52 (unknown function, related to PMP24), the iron–sulfur protein Mosc2 (MOCO sulphurase C-terminal domain containing 2 protein), and splice variants of the acyl-CoA binding protein ACBD5, as well as a trypsin-like serine protease (Tysnd1) of the peroxisomal matrix (Fig. 3). The mouse variant of this protease has been identified and characterized in detail (Kurochkin et al. 2007). Interestingly, Tysnd1 removes the leader peptide from PTS2 proteins and specifically processes PTS1 proteins involved in peroxisomal β-oxidation. Proteomic characterization of mouse kidney peroxisomes by tandem MS and protein correlation profiling has also led to the identification of 15 new peroxisomal candidates including Zadh2 (Zinc-binding alcohol dehydrogensase domain containing protein 2), Acad11 (acyl-CoA dehydrogenase family member 11), the aforementioned ACBD5, PMP52, Mosc2 (Wiese et al. 2007), as well as nudix hydrolase 19 (RP2) (Ofman et al. 2006). In addition, a quantitative proteomic comparison of mouse peroxisomes from liver and kidney has been performed (Mi et al. 2007b), as well as an age-related subproteomic analysis (Mi et al. 2007a). With the development of a purification method for leaf peroxisomes, Reumann et al. (2007) were able to perform a comprehensive proteome analysis of Arabidopsis leave peroxisomes. They identified 42 novel proteins that had previously not been assigned to plant peroxisomes revealing novel targeting peptides, metabolic pathways, and defense mechanisms (Reumann et al. 2007). All these studies have contributed to a more complete picture of the peroxisome, and clearly demonstrate that we are not looking at an isolated structure, but rather on a dynamic compartment that is linked to (and might interact with) other subcellular structures (e.g. the ER, mitochondria, chloroplasts, the cytoskeleton, lipid droplets/bodies and other peroxisomes), and even shares proteins with them (in particular with the ER and mitochondria).
Although mass spectrometry can generally be more accurate than antibody-based detection methods, a validation of the proteins predicted to be peroxisomal is often required. The peroxisomal localization of some of the identified novel proteins has been experimentally confirmed by expression studies with tagged proteins, and by antibody-based approaches such as immunofluorescence microscopy and immunoblotting (e.g. Marelli et al. 2004; Islinger et al. 2006, 2007; Wiese et al. 2007).
In addition, large-scale localization studies have been performed in which the whole genome or a subset of the organisms’ proteome was systematically tagged with GFP. The subcellular localizations of the GFP fusion proteins and their dynamics can then be addressed under live cell conditions, and several fusion proteins with peroxisomal localization have been identified by this approach (Cutler et al. 2000; Kumar et al. 2002; Huh et al. 2003; Natter et al. 2005).
Nevertheless, a lot of large-scale data are awaiting validation and contextualization. The great challenge for the future will be to provide a comprehensive and more complete picture of the relevant peroxisomal proteins, their abundance, functions, interactions and dynamic changes (Saleem et al. 2006). This can only be achieved by incorporating data from multiple sources, such as localization (see above) and interaction studies (e.g., two hybrid studies, analyses of immunoisolated protein complexes), metabolite interactions, and microarray expression analyses.
Concluding remarks
This review has highlighted recent novel discoveries in the peroxisome field. Ongoing studies on peroxisome biogenesis, formation and protein import have so far revealed unique features of peroxisomes, which have often been in disagreement with existing dogmas in cell biology. Contrary to mitochondria and ER, peroxisomal proteins can be imported in a completely folded or even oligomeric state, presumably via the formation of a transient membrane pore (Erdmann and Schliebs 2005). Recent findings also demonstrate that peroxisomes can be formed de novo from the ER or a subdomain of the ER (Hoepfner et al. 2005) in addition to growth and division of pre-existing peroxisomes (Yan et al. 2005; Schrader 2006; Fagarasanu et al. 2007). Furthermore, they appear to have an ER origin in evolutionary history (Gabaldon et al. 2006; Schluter et al. 2006). The physiological significance of the mechanism of de novo formation in comparison to the classical pathway of growth and division has again been challenged (Motley and Hettema 2007) and awaits further approval. In contrast to peroxisome division, de novo formation appears to be independent of dynamin-like proteins (Motley and Hettema 2007; Nagotu et al. 2007) and thus, the molecular machinery for de novo formation remains to be identified. Many new proteins affecting peroxisome growth, number, division, inheritance and turnover have been characterized and much has been learned about their putative function, localization, and possible interaction with other proteins. The discovery that dynamin-related proteins like Vps1, DLP1/Drp1 and DLP/Drp-receptor proteins like Fis1p are required for peroxisomal fission has opened the field for the molecular characterization of the peroxisomal division machinery (Schrader and Fahimi 2006). Interestingly, peroxisomes and mitochondria share components of their fission machinery, underlining the tight cooperation and cross talk between both organelles (Schrader 2006; Schrader and Yoon 2007). It is still an open question, how the dual targeting of these components is mediated, and if organelle-specific components exist which distinctly regulate the assembly of the division machineries on both organelles. Furthermore, the mechanism of peroxisome constriction and of membrane phospholipid transfer during growth or de novo synthesis remains to be discovered. Another important question is, if and how peroxisomal morphology and dynamics influence peroxisomal functions as well as developmental and physiological processes. An improved understanding of the signalling pathways and the PPAR-mediated mechanism of peroxisome proliferation will further stimulate the investigation of the risks and benefits of peroxisome proliferators as therapeutical agents, for example in fatty liver disease or neuroinflammatory diseases.
There is firm evidence now for a semi-autonomous model of peroxisome formation, whereby the ER supplies existing peroxisomes with essential membrane proteins to allow growth and division. It will be a great challenge for the future to elucidate this trafficking pathway, which is supposed to involve ER-derived vesicular or pre-peroxisomal structures. Surprisingly, besides vesicular ER-to peroxisome transport, even a vesicular transport route between mitochondria and peroxisomes might exist (Neuspiel et al. 2008). It will therefore be of significant interest to unravel to what extend, why and how peroxisomes interact and exchange with other subcellular compartments, and whether they are themselves involved in the formation of vesicles. It is clear now that we are not looking at an isolated structure, but rather on a dynamic compartment that is linked to other subcellular structures, interacts with them and even shares proteins. There is no doubt that peroxisomes, which are still among the more mysterious subcellular compartments in eukaryotic cells, will reveal further surprises in the near future.
Abbreviations
- ALDP:
-
Adrenoleukodystrophy protein
- CNS:
-
Central nervous system
- ER:
-
Endoplasmic reticulum
- FLD:
-
Fatty liver disease
- PBD:
-
Peroxisome biogenesis disorder
- PEX:
-
Peroxin
- PMP:
-
Peroxisomal membrane protein
- PPAR:
-
Peroxisome proliferator activated receptor
- PTS:
-
Peroxisomal targeting signal
- ROS:
-
Reactive oxygen species
- VLCFA:
-
Very-long-chain fatty acids
References
Adams LA, Angulo P, Lindor KD (2005) Nonalcoholic fatty liver disease. CMAJ 172:899–905
Ahlemeyer B, Neubert I, Kovacs WJ, Baumgart-Vogt E (2007) Differential expression of peroxisomal matrix and membrane proteins during postnatal development of mouse brain. J Comp Neurol 505:1–17
Angermuller S, Fahimi HD (1986) Ultrastructural cytochemical localization of uricase in peroxisomes of rat liver. J Histochem Cytochem 34:159–165
Antonenkov VD, Hiltunen JK (2006) Peroxisomal membrane permeability and solute transfer. Biochim Biophys Acta 1763:1697–1706
Bassnett S, Mataic D (1997) Chromatin degradation in differentiating fiber cells of the eye lens. J Cell Biol 137:37–49
Beier K, Volkl A, Fahimi HD (1992) Suppression of peroxisomal lipid beta-oxidation enzymes of TNF-alpha. FEBS Lett 310:273–276
Beier K, Volkl A, Fahimi HD (1997) TNF-alpha downregulates the peroxisome proliferator activated receptor-alpha and the mRNAs encoding peroxisomal proteins in rat liver. FEBS Lett 412:385–387
Belfort R, Harrison SA, Brown K, Darland C, Finch J, Hardies J, Balas B, Gastaldelli A, Tio F, Pulcini J, Berria R, Ma JZ, Dwivedi S, Havranek R, Fincke C, DeFronzo R, Bannayan GA, Schenker S, Cusi K (2006) A placebo-controlled trial of pioglitazone in subjects with nonalcoholic steatohepatitis. N Engl J Med 355:2297–2307
Bellu AR, Komori M, van der Klei IJ, Kiel JA, Veenhuis M (2001) Peroxisome biogenesis and selective degradation converge at Pex14p. J Biol Chem 276:44570–44574
Bellu AR, Salomons FA, Kiel JA, Veenhuis M, Van Der Klei IJ (2002) Removal of Pex3p is an important initial stage in selective peroxisome degradation in Hansenula polymorpha. J Biol Chem 277:42875–42880
Bernhard W, Rouiller C (1956) Microbodies and the problem of mitochondrial regeneration in liver cells. J Biophys Biochem Cytol 2:355–360
Brown FR III, McAdams AJ, Cummins JW, Konkol R, Singh I, Moser AB, Moser HW (1982) Cerebro-hepato-renal (Zellweger) syndrome and neonatal adrenoleukodystrophy: similarities in phenotype and accumulation of very long chain fatty acids. Johns Hopkins Med J 151:344–351
Carvalho AF, Pinto MP, Grou CP, Alencastre IS, Fransen M, Sá-Miranda C, Azevedo JE (2007) Ubiquitination of mammalian Pex5p, the peroxisomal import receptor. J Biol Chem 282:31267–31272
Cavalier-Smith T (1997) Cell and genome coevolution: facultative anaerobiosis, glycosomes and kinetoplastan RNA editing. Trends Genet 13:6–9
Cooper TG, Beevers H (1969) Beta oxidation in glyoxysomes from castor bean endosperm. J Biol Chem 244:3514–3520
Cunard R, Ricote M, DiCampli D, Archer DC, Kahn DA, Glass CK, Kelly CJ (2002) Regulation of cytokine expression by ligands of peroxisome proliferator activated receptors. J Immunol 168:2795–2802
Cutler SR, Ehrhardt DW, Griffitts JS, Somerville CR (2000) Random GFP: cDNA fusions enable visualization of subcellular structures in cells of Arabidopsis at a high frequency. Proc Natl Acad Sci USA 97:3718–3723
Dasgupta S, Roy A, Jana M, Hartley DM, Pahan K (2007) Gemfibrozil ameliorates relapsing-remitting experimental autoimmune encephalomyelitis independent of peroxisome proliferator-activated receptor-alpha. Mol Pharmacol 72:934–946
Day CP, James OF (1998) Steatohepatitis: a tale of two “hits”? Gastroenterology 114:842–845
De Duve C (2007) The origin of eukaryotes: a reappraisal. Nat Rev Genet 8:395–403
De Duve C, Baudhuin P (1966) Peroxisomes (microbodies and related particles). Physiol Rev 46:323–357
Dehmer T, Heneka MT, Sastre M, Dichgans J, Schulz JB (2004) Protection by pioglitazone in the MPTP model of Parkinson’s disease correlates with I kappa B alpha induction and block of NF kappa B and iNOS activation. J Neurochem 88:494–501
Devchand PR, Keller H, Peters JM, Vazquez M, Gonzalez FJ, Wahli W (1996) The PPARalpha-leukotriene B4 pathway to inflammation control. Nature 384:39–43
Diab A, Deng C, Smith JD, Hussain RZ, Phanavanh B, Lovett-Racke AE, Drew PD, Racke MK (2002) Peroxisome proliferator-activated receptor-gamma agonist 15-deoxy-Delta(12,14)-prostaglandin J(2) ameliorates experimental autoimmune encephalomyelitis. J Immunol 168:2508–2515
Distel B, Kragt A (2006) Purification of yeast peroxisomes. Methods Mol Biol 313:21–26
Dunn WA Jr, Cregg JM, Kiel JA, van der Klei IJ, Oku M, Sakai Y, Sibirny AA, Stasyk OV, Veenhuis M (2005) Pexophagy: the selective autophagy of peroxisomes. Autophagy 1:75–83
Emanuelsson O, Elofsson A, von Heijne G, Cristobal S (2003) In silico prediction of the peroxisomal proteome in fungi, plants and animals. J Mol Biol 330:443–456
Erdmann R, Schliebs W (2005) Opinion: peroxisomal matrix protein import—the transient pore model. Nat Rev Mol Cell Biol 6:738–742
Evrard P, Caviness VS Jr, Prats-Vinas J, Lyon G (1978) The mechanism of arrest of neuronal migration in the Zellweger malformation: an hypothesis bases upon cytoarchitectonic analysis. Acta Neuropathol 41:109–117
Faber KN, Haan GJ, Baerends RJ, Kram AM, Veenhuis M (2002) Normal peroxisome development from vesicles induced by truncated Hansenula polymorpha Pex3p. J Biol Chem 277:11026–11033
Fagarasanu M, Fagarasanu A, Rachubinski RA (2006) Sharing the wealth: peroxisome inheritance in budding yeast. Biochim Biophys Acta 1763:1669–1677
Fagarasanu A, Fagarasanu M, Rachubinski RA (2007) Maintaining peroxisome populations: a story of division and inheritance. Annu Rev Cell Dev Biol 23:321–344
Fahimi HD (1968) Cytochemical localization of peroxidase activity in rat hepatic microbodies (peroxisomes). J Histochem Cytochem 16:547–550
Fahimi HD (1969) Cytochemical localization of peroxidatic activity of catalase in rat hepatic microbodies (peroxisomes). J Cell Biol 43:275–288
Fahimi HD (1974) Effect of buffer storage on fine structure and catalase cytochemistry of peroxisomes. J Cell Biol 63:675–683
Fahimi HD, Reinicke A, Sujatta M, Yokota S, Ozel M, Hartig F, Stegmeier K (1982) The short- and long-term effects of bezafibrate in the rat. Ann N Y Acad Sci 386:111–135
Fahimi HD, Reich D, Volkl A, Baumgart E (1996) Contributions of the immunogold technique to investigation of the biology of peroxisomes. Histochem Cell Biol 106:105–114
Fan CY, Pan J, Usuda N, Yeldandi AV, Rao MS, Reddy JK (1998) Steatohepatitis, spontaneous peroxisome proliferation and liver tumors in mice lacking peroxisomal fatty acyl-CoA oxidase. Implications for peroxisome proliferator-activated receptor alpha natural ligand metabolism. J Biol Chem 273:15639–15645
Farrell GC, Larter CZ (2006) Nonalcoholic fatty liver disease: from steatosis to cirrhosis. Hepatology 43:S99–S112
Faust PL (2003) Abnormal cerebellar histogenesis in PEX2 Zellweger mice reflects multiple neuronal defects induced by peroxisome deficiency. J Comp Neurol 461:394–413
Faust PL, Banka D, Siriratsivawong R, Ng VG, Wikander TM (2005) Peroxisome biogenesis disorders: the role of peroxisomes and metabolic dysfunction in developing brain. J Inherit Metab Dis 28:369–383
Fujiki Y, Matsuzono Y, Matsuzaki T, Fransen M (2006) Import of peroxisomal membrane proteins: the interplay of Pex3p- and Pex19p-mediated interactions. Biochim Biophys Acta 1763:1639–1646
Gabaldon T, Snel B, van Zimmeren F, Hemrika W, Tabak H, Huynen MA (2006) Origin and evolution of the peroxisomal proteome. Biol Direct 1:8
Geuze HJ, Murk JL, Stroobants AK, Griffith JM, Kleijmeer MJ, Koster AJ, Verkleij AJ, Distel B, Tabak HF (2003) Involvement of the endoplasmic reticulum in peroxisome formation. Mol Biol Cell 14:2900–2907
Goldfischer S, Moore CL, Johnson AB, Spiro AJ, Valsamis MP, Wisniewski HK, Ritch RH, Norton WT, Rapin I, Gartner LM (1973) Peroxisomal and mitochondrial defects in the cerebro-hepato-renal syndrome. Science 182:62–64
Goldman BM, Blobel G (1978) Biogenesis of peroxisomes: intracellular site of synthesis of catalase and uricase. Proc Natl Acad Sci USA 75:5066–5070
Gould SJ, Collins CS (2002) Opinion: peroxisomal-protein import—is it really that complex? Nat Rev Mol Cell Biol 3:382–389
Gould SG, Keller GA, Subramani S (1987) Identification of a peroxisomal targeting signal at the carboxy terminus of firefly luciferase. J Cell Biol 105:2923–2931
Haan GJ, Baerends RJ, Krikken AM, Otzen M, Veenhuis M, Klei IJ (2006) Reassembly of peroxisomes in Hansenula polymorpha pex3 cells on reintroduction of Pex3p involves the nuclear envelope. FEMS Yeast Res 6:186–194
Hajra AK, Burke CL, Jones CL (1979) Subcellular localization of acyl coenzyme A: dihydroxyacetone phosphate acyltransferase in rat liver peroxisomes (microbodies). J Biol Chem 254:10896–10900
Hashimoto T, Cook WS, Qi C, Yeldandi AV, Reddy JK, Rao MS (2000) Defect in peroxisome proliferator-activated receptor alpha-inducible fatty acid oxidation determines the severity of hepatic steatosis in response to fasting. J Biol Chem 275:28918–28928
Hawkins J, Mahony D, Maetschke S, Wakabayashi M, Teasdale RD, Boden M (2007) Identifying novel peroxisomal proteins. Proteins 69:606–616
Heiland I, Erdmann R (2005) Biogenesis of peroxisomes: topogenesis of the peroxisomal membrane and matrix proteins. Febs J 272:2362–2372
Hess R, Staubli W, Riess W (1965) Nature of the hepatomegalic effect produced by ethyl-chlorophenoxy-isobutyrate in the rat. Nature 208:856–858
Hettema EH, Girzalsky W, van Den Berg M, Erdmann R, Distel B (2000) Saccharomyces cerevisiae pex3p and pex19p are required for proper localization and stability of peroxisomal membrane proteins. Embo J 19:223–233
Heymans HS, Schutgens RB, Tan R, van den Bosch H, Borst P (1983) Severe plasmalogen deficiency in tissues of infants without peroxisomes (Zellweger syndrome). Nature 306:69–70
Hoepfner D, van den Berg M, Philippsen P, Tabak HF, Hettema EH (2001) A role for Vps1p, actin, and the Myo2p motor in peroxisome abundance and inheritance in Saccharomyces cerevisiae. J Cell Biol 155:979–990
Hoepfner D, Schildknegt D, Braakman I, Philippsen P, Tabak HF (2005) Contribution of the endoplasmic reticulum to peroxisome formation. Cell 122:85–95
Hogenboom S, Tuyp JJ, Espeel M, Koster J, Wanders RJ, Waterham HR (2004) Mevalonate kinase is a cytosolic enzyme in humans. J Cell Sci 117:631–639
Hoopins S, Lackner L, Nunnari J (2007) The machines that divide and fuse mitochondria. Annu Rev Biochem 76:751–780
Hruban Z, Vigil EL, Slesers A, Hopkins E (1972) Microbodies: constituent organelles of animal cells. Lab Invest 27:184–191
Huh WK, Falvo JV, Gerke LC, Carroll AS, Howson RW, Weissman JS, O’Shea EK (2003) Global analysis of protein localization in budding yeast. Nature 425:686–691
Islinger M, Luers GH, Zischka H, Ueffing M, Volkl A (2006) Insights into the membrane proteome of rat liver peroxisomes: microsomal glutathione-S-transferase is shared by both subcellular compartments. Proteomics 6:804–816
Islinger M, Luers GH, Li KW, Loos M, Volkl A (2007) Rat liver peroxisomes after fibrate treatment: a survey using quantitative mass spectrometry. J Biol Chem 282:23055–23069
Issemann I, Green S (1990) Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature 347:645–650
Iwata J, Ezaki J, Komatsu M, Yokota S, Ueno T, Tanida I, Chiba T, Tanaka K, Kominami E (2006) Excess peroxisomes are degraded by autophagic machinery in mammals. J Biol Chem 281:4035–4041
Janssen A, Gressens P, Grabenbauer M, Baumgart E, Schad A, Vanhorebeek I, Brouwers A, Declercq PE, Fahimi D, Evrard P, Schoonjans L, Collen D, Carmeliet P, Mannaerts G, Van Veldhoven P, Baes M (2003) Neuronal migration depends on intact peroxisomal function in brain and in extraneuronal tissues. J Neurosci 23:9732–9741
Jedd G, Chua NH (2002) Visualization of peroxisomes in living plant cells reveals acto-myosin-dependent cytoplasmic streaming and peroxisome budding. Plant Cell Physiol 43:384–392
Jourdain I, Sontam D, Johnson C, Dillies C, Hyams JS (2008) Dynamin-dependent biogenesis, cell cycle regulation and mitochondrial association of peroxisomes in fission yeast. Traffic 9:353–365
Kamada T, Nito K, Hayashi H, Mano S, Hayashi M, Nishimura M (2003) Functional differentiation of peroxisomes revealed by expression profiles of peroxisomal genes in Arabidopsis thaliana. Plant Cell Physiol 44:1275–1289
Kashireddy PV, Rao MS (2004) Lack of peroxisome proliferator-activated receptor alpha in mice enhances methionine and choline deficient diet-induced steatohepatitis. Hepatol Res 30:104–110
Kassmann CM, Lappe-Siefke C, Baes M, Brugger B, Mildner A, Werner HB, Natt O, Michaelis T, Prinz M, Frahm J, Nave KA (2007) Axonal loss and neuroinflammation caused by peroxisome-deficient oligodendrocytes. Nat Genet 39:969–976
Kawahito Y, Kondo M, Tsubouchi Y, Hashiramoto A, Bishop-Bailey D, Inoue K, Kohno M, Yamada R, Hla T, Sano H (2000) 15-deoxy-delta(12,14)-PGJ(2) induces synoviocyte apoptosis and suppresses adjuvant-induced arthritis in rats. J Clin Invest 106:189–197
Keller GA, Gould S, Deluca M, Subramani S (1987) Firefly luciferase is targeted to peroxisomes in mammalian cells. Proc Natl Acad Sci USA 84:3264–3268
Kiel JA, Veenhuis M, van der Klei IJ (2006) PEX genes in fungal genomes: common, rare or redundant. Traffic 7:1291–1303
Kikuchi M, Hatano N, Yokota S, Shimozawa N, Imanaka T, Taniguchi H (2004) Proteomic analysis of rat liver peroxisome: presence of peroxisome-specific isozyme of Lon protease. J Biol Chem 279:421–428
Kim PK, Mullen RT, Schumann U, Lippincott-Schwartz J (2006) The origin and maintenance of mammalian peroxisomes involves a de novo PEX16-dependent pathway from the ER. J Cell Biol 173:521–532
Klionsky DJ, Cregg JM, Dunn WA Jr, Emr SD, Sakai Y, Sandoval IV, Sibirny A, Subramani S, Thumm M, Veenhuis M, Ohsumi Y (2003) A unified nomenclature for yeast autophagy-related genes. Dev Cell 5:539–545
Kobayashi S, Tanaka A, Fujiki Y (2007) Fis1, DLP1, and Pex11p coordinately regulate peroxisome morphogenesis. Exp Cell Res 313:1675–1686
Koch A, Thiemann M, Grabenbauer M, Yoon Y, McNiven MA, Schrader M (2003) Dynamin-like protein 1 is involved in peroxisomal fission. J Biol Chem 278:8597–8605
Koch A, Schneider G, Luers GH, Schrader M (2004) Peroxisome elongation and constriction but not fission can occur independently of dynamin-like protein 1. J Cell Sci 117:3995–4006
Koch A, Yoon Y, Bonekamp NA, McNiven MA, Schrader M (2005) A role for fis1 in both mitochondrial and peroxisomal fission in Mammalian cells. Mol Biol Cell 16:5077–5086
Koepke JI, Nakrieko KA, Wood CS, Boucher KK, Terlecky LJ, Walton PA, Terlecky SR (2007) Restoration of peroxisomal catalase import in a model of human cellular aging. Traffic 8:1590–1600
Kragt A, Voorn-Brouwer T, van den Berg M, Distel B (2005a) Endoplasmic reticulum-directed Pex3p routes to peroxisomes and restores peroxisome formation in a Saccharomyces cerevisiae pex3Delta strain. J Biol Chem 280:34350–34357
Kragt A, Voorn-Brouwer T, van den Berg M, Distel B (2005b) The Saccharomyces cerevisiae peroxisomal import receptor Pex5p is monoubiquitinated in wild type cells. J Biol Chem 280:7867–7874
Kumar A, Agarwal S, Heyman JA, Matson S, Heidtman M, Piccirillo S, Umansky L, Drawid A, Jansen R, Liu Y, Cheung KH, Miller P, Gerstein M, Roeder GS, Snyder M (2002) Subcellular localization of the yeast proteome. Genes Dev 16:707–719
Kunau WH (2005) Peroxisome biogenesis: end of the debate. Curr Biol 15:R774–R776
Kunze M, Pracharoenwattana I, Smith SM, Hartig A (2006) A central role for the peroxisomal membrane in glyoxylate cycle function. Biochim Biophys Acta 1763:1441–1452
Kurochkin IV, Nagashima T, Konagaya A, Schonbach C (2005) Sequence-based discovery of the human and rodent peroxisomal proteome. Appl Bioinformatics 4:93–104
Kurochkin IV, Mizuno Y, Konagaya A, Sakaki Y, Schonbach C, Okazaki Y (2007) Novel peroxisomal protease Tysnd1 processes PTS1- and PTS2-containing enzymes involved in beta-oxidation of fatty acids. Embo J 26:835–845
Landreth G (2007) Therapeutic use of agonists of the nuclear receptor PPARgamma in Alzheimer’s disease. Curr Alzheimer Res 4:159–164
Lay D, B LG, Heid H, Gorgas K, Just WW (2005) Binding and functions of ADP-ribosylation factor on mammalian and yeast peroxisomes. J Biol Chem 280:34489–34499
Lay D, Gorgas K, Just WW (2006) Peroxisome biogenesis: where Arf and coatomer might be involved. Biochim Biophys Acta 1763:1678–1687
Lazarow PB (2003) Peroxisome biogenesis: advances and conundrums. Curr Opin Cell Biol 15:489–497
Lazarow PB, De Duve C (1976) A fatty acyl-CoA oxidizing system in rat liver peroxisomes; enhancement by clofibrate, a hypolipidemic drug. Proc Natl Acad Sci USA 73:2043–2046
Lazarow PB, Fujiki Y (1985) Biogenesis of peroxisomes. Annu Rev Cell Biol 1:489–530
Leighton F, Coloma L, Koenig C (1975) Structure, composition, physical properties, and turnover of proliferated peroxisomes: a study of the trophic effects of Su-13437 on rat liver. J Cell Biol 67:281–309
Leon S, Goodman JM, Subramani S (2006) Uniqueness of the mechanism of protein import into the peroxisome matrix: transport of folded, co-factor-bound and oligomeric proteins by shuttling receptors. Biochim Biophys Acta 1763:1552–1564
Li AC, Brown KK, Silvestre MJ, Willson TM, Palinski W, Glass CK (2000) Peroxisome proliferator-activated receptor gamma ligands inhibit development of atherosclerosis in LDL receptor-deficient mice. J Clin Invest 106:523–531
Luers GH, Hartig R, Mohr H, Hausmann M, Fahimi HD, Cremer C, Völkl A (1998) Immuno-isolation of highly purified peroxisomes using magnetic beads and continuous immunomagnetic sorting. Electrophoresis 19:1205–1210
Luers GH, Thiele S, Schad A, Volkl A, Yokota S, Seitz J (2006) Peroxisomes are present in murine spermatogonia and disappear during the course of spermatogenesis. Histochem Cell Biol 125:693–703
Marelli M, Smith JJ, Jung S, Yi E, Nesvizhskii AI, Christmas RH, Saleem RA, Tam YY, Fagarasanu A, Goodlett DR, Aebersold R, Rachubinski RA, Aitchison JD (2004) Quantitative mass spectrometry reveals a role for the GTPase Rho1p in actin organization on the peroxisome membrane. J Cell Biol 167:1099–1112
Marx N, Kehrle B, Kohlhammer K, Grub M, Koenig W, Hombach V, Libby P, Plutzky J (2002) PPAR activators as antiinflammatory mediators in human T lymphocytes: implications for atherosclerosis and transplantation-associated arteriosclerosis. Circ Res 90:703–710
Matsui M, Yamamoto A, Kuma A, Ohsumi Y, Mizushima N (2006) Organelle degradation during the lens and erythroid differentiation is independent of autophagy. Biochem Biophys Res Commun 339:485–489
Matsuzono Y, Kinoshita N, Tamura S, Shimozawa N, Hamasaki M, Ghaedi K, Wanders RJ, Suzuki Y, Kondo N, Fujiki Y (1999) Human PEX19: cDNA cloning by functional complementation, mutation analysis in a patient with Zellweger syndrome, and potential role in peroxisomal membrane assembly. Proc Natl Acad Sci USA 96:2116–2121
McCartney AW, Greenwood JS, Fabian MR, White KA, Mullen RT (2005) Localization of the tomato bushy stunt virus replication protein p33 reveals a peroxisome-to-endoplasmic reticulum sorting pathway. Plant Cell 17:3513–3531
McNew JA, Goodman JM (1996) The targeting and assembly of peroxisomal proteins: some old rules do not apply. Trends Biochem Sci 21:54–58
Mi J, Garcia-Arcos I, Alvarez R, Cristobal S (2007a) Age-related subproteomic analysis of mouse liver and kidney peroxisomes. Proteome Sci 5:19
Mi J, Kirchner E, Cristobal S (2007b) Quantitative proteomic comparison of mouse peroxisomes from liver and kidney. Proteomics 7:1916–1928
Michalik L, Auwerx J, Berger JP, Chatterjee VK, Glass CK, Gonzalez FJ, Grimaldi PA, Kadowaki T, Lazar MA, O’Rahilly S, Palmer CN, Plutzky J, Reddy JK, Spiegelman BM, Staels B, Wahli W (2006) International Union of Pharmacology. LXI. Peroxisome proliferator-activated receptors. Pharmacol Rev 58:726–741
Monastyrska I, Klionsky DJ (2006) Autophagy in organelle homeostasis: peroxisome turnover. Mol Aspects Med 27:483–494
Moody DE, Reddy JK (1976) Morphometric analysis of the ultrastructural changes in rat liver induced by the peroxisome proliferator SaH 42–348. J Cell Biol 71:768–780
Moody DE, Reddy JK, Lake BG, Popp JA, Reese DH (1991) Peroxisome proliferation and nongenotoxic carcinogenesis: commentary on a symposium. Fundam Appl Toxicol 16:233–248
Motley AM, Hettema EH (2007) Yeast peroxisomes multiply by growth and division. J Cell Biol 178:399–410
Mullen RT, Trelease RN (2006) The ER-peroxisome connection in plants: development of the “ER semi-autonomous peroxisome maturation and replication” model for plant peroxisome biogenesis. Biochim Biophys Acta 1763:1655–1668
Muntau AC, Mayerhofer PU, Paton BC, Kammerer S, Roscher AA (2000) Defective peroxisome membrane synthesis due to mutations in human PEX3 causes Zellweger syndrome, complementation group G. Am J Hum Genet 67:967–975
Nagasawa T, Inada Y, Nakano S, Tamura T, Takahashi T, Maruyama K, Yamazaki Y, Kuroda J, Shibata N (2006) Effects of bezafibrate, PPAR pan-agonist, and GW501516, PPARdelta agonist, on development of steatohepatitis in mice fed a methionine- and choline-deficient diet. Eur J Pharmacol 536:182–191
Nagotu S, Saraya R, Otzen M, Veenhuis M, van der Klei IJ (2007) Peroxisome proliferation in Hansenula polymorpha requires Dnm1p which mediates fission but not de novo formation. Biochim Biophys Acta. doi:10.1016/j.bbamcr.2007.10.018
Natter K, Leitner P, Faschinger A, Wolinski H, McCraith S, Fields S, Kohlwein SD (2005) The spatial organization of lipid synthesis in the yeast Saccharomyces cerevisiae derived from large scale green fluorescent protein tagging and high resolution microscopy. Mol Cell Proteomics 4:662–672
Neuschwander-Tetri BA, Caldwell SH (2003) Nonalcoholic steatohepatitis: summary of an AASLD single topic conference. Hepatology 37:1202–1219
Neuschwander-Tetri BA, Brunt EM, Wehmeier KR, Oliver D, Bacon BR (2003) Improved nonalcoholic steatohepatitis after 48 weeks of treatment with the PPAR-gamma ligand rosiglitazone. Hepatology 38:1008–1017
Neuspiel M, Schauss AC, Braschi E, Zunino R, Rippstein P, Rachubinski RA, Andrade-Navarro MA, McBride HM (2008) Cargo-selected transport from the mitochondria to peroxisomes is mediated by vesicular carriers. Curr Biol 18:102–108. doi:10.1016/j.cub.2007.12.038
Niino M (2007) Peroxisome proliferator-activated receptor agonists as potential therapeutic agents in multiple sclerosis. Mini Rev Med Chem 7:1129–1135
Niino M, Iwabuchi K, Kikuchi S, Ato M, Morohashi T, Ogata A, Tashiro K, Onoe K (2001) Amelioration of experimental autoimmune encephalomyelitis in C57BL/6 mice by an agonist of peroxisome proliferator-activated receptor-gamma. J Neuroimmunol 116:40–48
Novikoff AB, Essner E (1960) The liver cell: some new approaches to its study. Am J Med 29:102–131
Novikoff AB, Goldfischer S (1969) Visualization of peroxisomes (microbodies) and mitochondria with diaminobenzidine. J Histochem Cytochem 17:675–680
Novikoff AB, Shin WY (1964) The endoplasmic reticulum in the Golgi zone and its relations to microbodies, Golgi apparatus, and autophagic vacuoles in rat liver cells. J Microsc (Paris) 3:187–206
Ofman R, Speijer D, Leen R, Wanders RJ (2006) Proteomic analysis of mouse kidney peroxisomes: identification of RP2p as a peroxisomal nudix hydrolase with acyl-CoA diphosphatase activity. Biochem J 393:537–543
Passreiter M, Anton M, Lay D, Frank R, Harter C, Wieland FT, Gorgas K, Just WW (1998) Peroxisome biogenesis: involvement of ARF and coatomer. J Cell Biol 141:373–383
Perry RJ, Rachubinski RA (2007) The yeast Dsl1p secretory complex is involved in peroxisome biogenesis. In: 47th ASCB annual meeting, Washington 183 (abstract)
Pershadsingh HA, Heneka MT, Saini R, Amin NM, Broeske DJ, Feinstein DL (2004) Effect of pioglitazone treatment in a patient with secondary multiple sclerosis. J Neuroinflammation 1:3
Platta HW, Erdmann R (2007a) Peroxisomal dynamics. Trends Cell Biol 17:474–484
Platta HW, Erdmann R (2007b) The peroxisomal protein import machinery. FEBS Lett 581:2811–2819
Plutzky J (2007) Preventing type 2 diabetes and cardiovascular disease in metabolic syndrome: the role of PPARalpha. Diab Vasc Dis Res 4(Suppl 3):S12–S14
Poirier Y, Antonenkov VD, Glumoff T, Hiltunen JK (2006) Peroxisomal beta-oxidation–a metabolic pathway with multiple functions. Biochim Biophys Acta 1763:1413–1426
Powers JM, Moser HW, Moser AB, Upshur JK, Bradford BF, Pai SG, Kohn PH, Frias J, Tiffany C (1985) Fetal cerebrohepatorenal (Zellweger) syndrome: dysmorphic, radiologic, biochemical, and pathologic findings in four affected fetuses. Hum Pathol 16:610–620
Promrat K, Lutchman G, Uwaifo GI, Freedman RJ, Soza A, Heller T, Doo E, Ghany M, Premkumar A, Park Y, Liang TJ, Yanovski JA, Kleiner DE, Hoofnagle JH (2004) A pilot study of pioglitazone treatment for nonalcoholic steatohepatitis. Hepatology 39:188–196
Racke MK, Gocke AR, Muir M, Diab A, Drew PD, Lovett-Racke AE (2006) Nuclear receptors and autoimmune disease: the potential of PPAR agonists to treat multiple sclerosis. J Nutr 136:700–703
Rao MS, Papreddy K, Musunuri S, Okonkwo A (2002) Prevention/reversal of choline deficiency-induced steatohepatitis by a peroxisome proliferator-activated receptor alpha ligand in rats. In Vivo 16:145–152
Rayapuram N, Subramani S (2006) The importomer: a peroxisomal membrane complex involved in protein translocation into the peroxisome matrix. Biochim Biophys Acta 1763:1613–1619
Reddy JK, Lalwani ND (1983) Carcinogenesis by hepatic peroxisome proliferators: evaluation of the risk of hypolipidemic drugs and industrial plasticizers to humans. CRC Critical Rev Tox 12:1–58
Reddy JK, Rao MS (2006) Lipid metabolism and liver inflammation: II. Fatty liver disease and fatty acid oxidation. Am J Physiol Gastrointest Liver Physiol 290:G852–G858
Reddy JK, Azarnoff DL, Hignite CE (1980) Hypolipidaemic hepatic peroxisome proliferators form a novel class of chemical carcinogens. Nature 283:397–398
Reddy JK, Warren JR, Reddy MK, Lalwani ND (1982) Hepatic and renal effects of peroxisome proliferators: biological implications. Ann N Y Acad Sci 386:81–110
Reumann S, Weber AP (2006) Plant peroxisomes respire in the light: some gaps of the photorespiratory C(2) cycle have become filled-Others remain. Biochim Biophys Acta 1763:1496–1510
Reumann S, Ma C, Lemke S, Babujee L (2004) AraPerox: a database of putative Arabidopsis proteins from plant peroxisomes. Plant Physiol 136:2587–2608
Reumann S, Babujee L, Ma C, Wienkoop S, Siemsen T, Antonicelli GE, Rasche N, Luder F, Weckwerth W, Jahn O (2007) Proteome analysis of arabidopsis leaf peroxisomes reveals novel targeting peptides, metabolic pathways, and defense mechanisms. Plant Cell 19(10):3170–3193
Rhodin J (1954) Correlation of ultrastructural organization and function in normal experimentally changed convoluted tubule cells of the mouse kidney. Ph.D. thesis. Stockholm, Aktiebolaget Godvil
Rouiller C, Jezequel AM (1963) Electron microscopy of the liver. In: Rouiller C, Jezequel AM (eds) The liver: morphology, biochemistry, physiology. Academic Press, New York, pp 195–264
Sacksteder KA, Jones JM, South ST, Li X, Liu Y, Gould SJ (2000) PEX19 binds multiple peroxisomal membrane proteins, is predominantly cytoplasmic, and is required for peroxisome membrane synthesis. J Cell Biol 148:931–944
Sakai Y, Koller A, Rangell LK, Keller GA, Subramani S (1998a) Peroxisome degradation by microautophagy in Pichia pastoris: identification of specific steps and morphological intermediates. J Cell Biol 141:625–636
Sakai Y, Yurimoto H, Matsuo H, Kato N (1998b) Regulation of peroxisomal proteins and organelle proliferation by multiple carbon sources in the methylotrophic yeast, Candida boidinii. Yeast 14:1175–1187
Sakai Y, Oku M, van der Klei IJ, Kiel JA (2006) Pexophagy: autophagic degradation of peroxisomes. Biochim Biophys Acta 1763:1767–1775
Saleem RA, Smith JJ, Aitchison JD (2006) Proteomics of the peroxisome. Biochim Biophys Acta 1763:1541–1551
Schafer H, Nau K, Sickmann A, Erdmann R, Meyer HE (2001) Identification of peroxisomal membrane proteins of Saccharomyces cerevisiae by mass spectrometry. Electrophoresis 22:2955–2968
Schewe T, Halangk W, Hiebsch C, Rapoport SM (1975) A lipoxygenase in rabbit reticulocytes which attacks phospholipids and intact mitochondria. FEBS Lett 60:149–152
Schluter A, Fourcade S, Ripp R, Mandel JL, Poch O, Pujol A (2006) The evolutionary origin of peroxisomes: an ER-peroxisome connection. Mol Biol Evol 23:838–845
Schluter A, Fourcade S, Domenech-Estevez E, Gabaldon T, Huerta-Cepas J, Berthommier G, Ripp R, Wanders RJ, Poch O, Pujol A (2007) PeroxisomeDB: a database for the peroxisomal proteome, functional genomics and disease. Nucleic Acids Res 35:D815–D822
Schrader M (2006) Shared components of mitochondrial and peroxisomal division. Biochim Biophys Acta 1763:531–541
Schrader M, Fahimi HD (2006) Growth and division of peroxisomes. Int Rev Cytol 255:237–290
Schrader M, Yoon Y (2007) Mitochondria and peroxisomes: are the ‘Big Brother’ and the ‘Little Sister’ closer than assumed? Bioessays 29:1105–1114
Schrader M, Burkhardt JK, Baumgart E, Luers G, Spring H, Volkl A, Fahimi HD (1996) Interaction of microtubules with peroxisomes: tubular and spherical peroxisomes in HepG2 cells and their alterations induced by microtubule-active drugs. Eur J Cell Biol 69:24–35
Schrader M, King SJ, Stroh TA, Schroer TA (2000) Real time imaging reveals a peroxisomal reticulum in living cells. J Cell Sci 113:3663–3671
Schriner SE, Linford NJ, Martin GM, Treuting P, Ogburn CE, Emond M, Coskun PE, Ladiges W, Wolf N, Van Remmen H, Wallace DC, Rabinovitch PS (2005) Extension of murine life span by overexpression of catalase targeted to mitochondria. Science 308:1909–1911
Schroder LA, Glick BS, Dunn WA (2007) Identification of pexophagy genes by restriction enzyme-mediated integration. Methods Mol Biol 389:203–218
Soukupova M, Sprenger C, Gorgas K, Kunau WH, Dodt G (1999) Identification and characterization of the human peroxin PEX3. Eur J Cell Biol 78:357–374
South ST, Gould SJ (1999) Peroxisome synthesis in the absence of preexisting peroxisomes. J Cell Biol 144:255–266
Spinazzola A, Viscomi C, Fernandez-Vizarra E, Carrara F, D’Adamo P, Calvo S, Marsano RM, Donnini C, Weiher H, Strisciuglio P, Parini R, Sarzi E, Chan A, Dimauro S, Rotig A, Gasparini P, Ferrero I, Mootha VK, Tiranti V, Zeviani M (2006) MPV17 encodes an inner mitochondrial membrane protein and is mutated in infantile hepatic mitochondrial DNA depletion. Nat Genet 38:570–575
Staels B (2007) PPAR agonists and the metabolic syndrome. Therapie 62:319–326
Staubli W, Schweizer W, Suter J, Weibel ER (1977) The proliferative response of hepatic peroxidomes of neonatal rats to treatment with SU-13 437 (nafenopin). J Cell Biol 74:665–689
Steinberg SJ, Dodt G, Raymond GV, Braverman NE, Moser AB, Moser HW (2006) Peroxisome biogenesis disorders. Biochim Biophys Acta 1763:1733–1748
Su CG, Wen X, Bailey ST, Jiang W, Rangwala SM, Keilbaugh SA, Flanigan A, Murthy S, Lazar MA, Wu GD (1999) A novel therapy for colitis utilizing PPAR-gamma ligands to inhibit the epithelial inflammatory response. J Clin Invest 104:383–389
Subramani S (1998) Components involved in peroxisome import, biogenesis, proliferation, turnover, and movement. Physiol Rev 78:171–188
Subramanya S, Mensa-Wilmot K (2006) Regulated cleavage of intracellular glycosylphosphatidylinositol in a trypanosome. Peroxisome-to-endoplasmic reticulum translocation of a phospholipase C. Febs J 273:2110–2126
Svoboda DJ, Azarnoff DL (1966) Response of hepatic microbodies to a hypolipidemic agent, ethyl chlorophenoxyisobutyrate (CPIB). J Cell Biol 30:442–450
Tabak HF, Hoepfner D, Zand A, Geuze HJ, Braakman I, Huynen MA (2006) Formation of peroxisomes: present and past. Biochim Biophys Acta 1763:1647–1654
Tam YY, Fagarasanu A, Fagarasanu M, Rachubinski RA (2005) Pex3p initiates the formation of a preperoxisomal compartment from a subdomain of the endoplasmic reticulum in Saccharomyces cerevisiae. J Biol Chem 280:34933–34939
Tanaka A, Kobayashi S, Fujiki Y (2006) Peroxisome division is impaired in a CHO cell mutant with an inactivating point-mutation in dynamin-like protein 1 gene. Exp Cell Res 312:1671–1684
Terlecky SR, Koepke JI, Walton PA (2006) Peroxisomes and aging. Biochim Biophys Acta 1763:1749–1754
Thoms S, Erdmann R (2005) Dynamin-related proteins and Pex11 proteins in peroxisome division and proliferation. Febs J 272:5169–5181
Thoms S, Erdmann R (2006) Peroxisomal matrix protein receptor ubiquitination and recycling. Biochim Biophys Acta 1763:1620–1628
Thoms S, Debelyy MO, Nau K, Meyer HE, Erdmann R (2008) Lpx1p is a peroxisomal lipase required for normal peroxisome morphology. Febs J 275:504–514
Titorenko VI, Rachubinski RA (2001) The life cycle of the peroxisome. Nat Rev Mol Cell Biol 2:357–368
Van Ael E, Fransen M (2006) Targeting signals in peroxisomal membrane proteins. Biochim Biophys Acta 1763:1629–1638
van den Bosch H, Schutgens RB, Wanders RJ, Tager JM (1992) Biochemistry of peroxisomes. Annu Rev Biochem 61:157–197
van der Klei IJ, Yurimoto H, Sakai Y, Veenhuis M (2006) The significance of peroxisomes in methanol metabolism in methylotrophic yeast. Biochim Biophys Acta 1763:1453–1462
van Leyen K, Duvoisin RM, Engelhardt H, Wiedmann M (1998) A function for lipoxygenase in programmed organelle degradation. Nature 395:392–395
Visser WF, van Roermund CW, Ijlst L, Waterham HR, Wanders RJ (2007) Metabolite transport across the peroxisomal membrane. Biochem J 401:365–375
Völkl A, Baumgart E, Fahimi HD (1988) Localization of urate oxidase in the crystalline cores of rat liver peroxisomes by immunocytochemistry and immunoblotting. J Histochem Cytochem 36:329–336
Völkl A, Baumgart E, Fahimi HD (1996) Isolation and characterization of peroxisomes. In: Graham J, Rickwood D (eds) Subcellular fractionation: a practical approach. Oxford University Press, Oxford, pp 143–167
Völkl A, Mohr H, Fahimi HD (1999) Peroxisome subpopulations of the rat liver: isolation by immune free flow electrophoresis. J Histochem Cytochem 47:1111–1118
Wanders RJ (2000) Peroxisomes, lipid metabolism, and human disease. Cell Biochem Biophys 32:89–106
Wanders RJ, Waterham HR (2005) Peroxisomal disorders: I. Biochemistry and genetics of peroxisome biogenesis disorders. Clin Genet 67:107–133
Wanders RJ, Waterham HR (2006a) Peroxisomal disorders: the single peroxisomal enzyme deficiencies. Biochim Biophys Acta 1763:1707–1720
Wanders RJ, Waterham HR (2006b) Biochemistry of mammalian peroxisomes revisited. Annu Rev Biochem 75:295–332
Waterham HR, Koster J, van Roermund CW, Mooyer PA, Wanders RJ, Leonard JV (2007) A lethal defect of mitochondrial and peroxisomal fission. N Engl J Med 356:1736–1741
Weber G, Islinger M, Weber P, Eckerskorn C, Volkl A (2004) Efficient separation and analysis of peroxisomal membrane proteins using free-flow isoelectric focusing. Electrophoresis 25:1735–1747
Weller S, Gould SJ, Valle D (2003) Peroxisome biogenesis disorders. Annu Rev Genomics Hum Genet 4:165–211
Wiese S, Gronemeyer T, Ofman R, Kunze M, Grou CP, Almeida JA, Eisenacher M, Stephan C, Hayen H, Schollenberger L, Korosec T, Waterham HR, Schliebs W, Erdmann R, Berger J, Meyer HE, Just W, Azevedo JE, Wanders RJ, Warscheid B (2007) Proteomic characterization of mouse kidney peroxisomes by tandem mass spectrometry and protein correlation profiling. Mol Cell Proteomics 6(12):2045–2057
Xu J, Racke MK, Drew PD (2007) Peroxisome proliferator-activated receptor-alpha agonist fenofibrate regulates IL-12 family cytokine expression in the CNS: relevance to multiple sclerosis. J Neurochem 103:1801–1810
Yan M, Rayapuram N, Subramani S (2005) The control of peroxisome number and size during division and proliferation. Curr Opin Cell Biol 17:376–383
Yokota S (1993) Formation of autophagosomes during degradation of excess peroxisomes induced by administration of dioctyl phthalate. Eur J Cell Biol 61:67–80
Yokota S (2003) Degradation of normal and proliferated peroxisomes in rat hepatocytes: regulation of peroxisomes quantity in cells. Microsc Res Tech 61:151–160
Yokota S, Oda T, Fahimi HD (2001) The role of 15-lipoxygenase in disruption of the peroxisomal membrane and in programmed degradation of peroxisomes in normal rat liver. J Histochem Cytochem 49:613–622
Zutphen T, Veenhuis M, van der Klei IJ (2008) Pex14 is the sole component of the peroxisomal translocon that is required for pexophagy. Autophagy 4:63–66
Acknowledgments
We thank Ronald Wanders, Alfred Völkl, Markus Islinger, and Georg Luers for helpful suggestions, and members of the laboratories for stimulating discussions and comments on the manuscript. We apologize to those whose work has not been cited due to space limitations. Supported by the German Research Foundation (DFG) (SCHR 518/6-1) and the Portuguese Foundation for Science and Technology (FCT) (PTDC/BIA-BCM/71932/2006).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Schrader, M., Fahimi, H.D. The peroxisome: still a mysterious organelle. Histochem Cell Biol 129, 421–440 (2008). https://doi.org/10.1007/s00418-008-0396-9
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00418-008-0396-9