
Abstract
Purpose
Hydroxychloroquine (HCQ) is used in the treatment of several diseases, such as malaria, Sjögren's disease, Covid-19, and rheumatoid arthritis. However, HCQ induces retinal pigment epithelium death via the excessive increase of cytosolic (cROS) and mitochondrial (mROS) free oxygen radical production. The transient receptor potential melastatin 2 (TRPM2) cation channel is stimulated by ADP-ribose (ADPR), cROS, and mROS, although it is inhibited by curcumin (CRC). We aimed to investigate the modulating action of CRC on HCQ-induced TRPM2 stimulation, cROS, mROS, apoptosis, and death in an adult retinal pigment epithelial 19 (ARPE19) cell line model.
Material and methods
ARPE19 cells were divided into four groups: control (CNT), CRC (5 µM for 24 h), HCQ (60 µM for 48 h), and CRC + HCQ groups.
Results
The levels of cell death (propidium iodide positive cell numbers), apoptosis markers (caspases -3, -8, and -9), oxidative stress (cROS and mROS), mitochondria membrane depolarization, TRPM2 current density, and intracellular free Ca2+ and Zn2+ fluorescence intensity were upregulated in the HCQ group after stimulation with hydrogen peroxide and ADPR, but their levels were downregulated by treatments with CRC and TRPM2 blockers (ACA and carvacrol). The HCQ-induced decrease in retinal live cell count and cell viability was counteracted by treatment with CRC.
Conclusion
HCQ-mediated overload Ca2+ influx and retinal oxidative toxicity were induced in an ARPE19 cell line through the stimulation of TRPM2, although they were attenuated by treatment with CRC. Hence, CRC may be a potential therapeutic antioxidant for TRPM2 activation and HCQ treatment-induced retinal oxidative injury and apoptosis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.

Introduction
Hydroxychloroquine sulphate (HCQ) has been used as an antimalarial drug for over 50 years. HCQ has also been used in the treatment of inflammatory diseases, including rheumatoid arthritis, systemic lupus erythematosus, Covid-19, and Sjögren’s disease [1]. Recently, this drug has also been indicated to be a potential treatment for various tumours when administered in a high dose as an adjuvant chemotherapeutic agent [2]. HCQ-mediated retinal toxicity is a well-documented phenomenon [3]. To date, there has been limited information on the exact mechanism of HCQ-induced eye toxicity in the literature. Since eyes have a high melanin concentration, HCQ is known to deposit specifically in tissues with high melanin content, such as the retinal pigment epithelium (RPE) [4, 5]. The RPE is a single layer of eye cells that is essential for clearing debris from the photoreceptor outer segment of the retina. In in vitro experiments, treatment with HCQ has led to the degradation of the retina–blood barrier by changing the lysosome pH and monolayer permeability of the RPE [4, 5]. These changes cause significant vision loss via secondary changes in the overlying photoreceptors and neuroretinal cells [6]. The mechanisms of HCQ-mediated RPE death and toxicity have not been definitively elucidated. There is evidence that treatment with HCQ induces oxidative stress and cell death in human retinal pigment epithelial 19 (ARPE19) cells [7], although such treatment also plays an antioxidant and antiapoptotic role via transient receptor potential (TRP) vanilloid 1 (TRPV1) channel inhibition in human monocytes [8]. In ARPE19 cells, moreover, treatment with chloroquine increased cell death and reactive oxygen species (ROS) production [9]. In contrast, caspase-3 (CAS/3) activity, ROS production, and apoptosis levels remained unchanged, although the ARPE19 cell death rate was reduced through treatment with antioxidants (trolox and N-acetyl cysteine) [10]. The use of the antioxidant curcumin (CRC) may have similar protective effects in ARPE19 cells.
Long-term retinal damage can be caused by the increased generation of cytoplasmic (cROS) and mitochondrial (mROS) free reactive oxygen species [8, 11]. Apoptosis and cell death are stimulated by the overproduction of cROS and mROS [11,12,13]. This makes antioxidants unable to combat excessive cROS and mROS formation. Excessive cytosolic Ca2+ (cCa2+)-mediated increases in mitochondrial membrane potential (mMP) damage retinal cells [14]. Chloroquine-mediated cROS and mROS generation also cause apoptosis in retina cells, including ARPE19 cells [9], although a conflicting finding was reported [8]. Stimulation of the transient receptor potential melastatin 2 (TRPM2) channels in ARPE19 cells by a number of drugs, including bevacizumab [11] and cisplatin [15], has been reported to cause adverse oxidative and apoptotic effects. However, these negative effects have been counteracted by the use of TRPM2 channel antagonists, including carvacrol (CRV) and N-(p-amylcinnamoyl)anthranilic acid (ACA) [12, 13, 16], as well as antioxidants, such as CRC and glutathione [11, 15]. The involvement of TRPM2 has not been reported in HCQ-mediated overload cCa2+ influx and mROS generation in ARPE19 cells.
Mammalian cells contain 28 members of the TRP superfamily, including TRPM2 [17]. ADP-ribose (ADPR), NAD+, and hydrogen peroxide (H2O2) are the major activators of the Ca2+-permeable TRPM2 channel [18,19,20]. ACA and CRV are nonspecific antagonists of TRPM2 [21, 22]. The influx of Ca2+ modulates the physiological functions of retinal cells [17]. However, increased mROS production and apoptosis are induced by elevated Ca2+ accumulation within the mitochondria via the activation of TRPM2 in ARPE19 cells [12, 13, 15, 16]. CRC is a bright-yellow material produced by plants of the Curcuma longa genus. Recently, it was discovered that CRC regulates apoptosis, cROS, and mROS in rat hepatocytes [23], SH-SY5Y neuroblastoma cells [24, 25], renal collecting duct cells [26], and mouse optic nerves [15] by inhibiting the TRPM2 channel. However, it is unclear how CRC influences harmful HCQ-induced oxidative and apoptotic activity via the activation of TRPM2 in ARPE19 cells.
In this study, we investigated the potential role of TRPM2 in detrimental HCQ-mediated oxidative and apoptotic activity in ARPE19 cells. Treatment with CRC may help counteract these detrimental effects by inhibiting TRPM2 in these cells.
Material and methods
Cell culture
ARPE-19 cells were derived from the retina of a 19-year-old male. Although they lack some physiological characteristics of the RPE, ARPE-19 cells are regarded as a reliable model for investigating RPE cell metabolism [9, 11, 12]. The detrimental oxidative and apoptotic effects of medicines such as HCQ and chloroquine have been experimentally studied using ARPE19 cells as a model of the human RPE [9, 11, 12]. Recently, it was also reported that a TRPM2 channel naturally exists in ARPE19 cells [16, 27]. These factors led us to choose the cells used in the current investigation, which were a gift from Professor Suat Erdoğan (Trakya University, Edirne, Turkey). The cells were incubated at 37 °C in a cell culture incubator (NB-203QS, South Korea) with a low-glucose (1 g/l) medium mixture (DMEM/HAM's F12 mixture, Capricorn, Germany) supplemented with a combination of foetal bovine serum (10%) and antibiotics (penicillin/streptomycin) (1%). The cells were incubated at a concentration of 1 × 106 cells/flask in T25 flasks with filter caps (n = 6).
Determination of a nontoxic HCQ concentration in an ARPE19 cell line
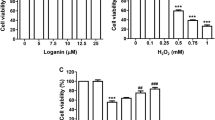
Chloroquine and HCQ have been shown to be nontoxic at concentrations between 5 and 80 μM for 48 h in a variety of cell lines [8, 28, 29]. In the initial phase of the present work, we investigated the nontoxic level of HCQ in ARPE19 cells. For 48 h, the cells were exposed to doses of HCQ of 10, 20, 30, 40, 50, 60, 70, and 80 μM (Fig. 1A). Compared to the control (CNT) group, the levels of cell viability in the group administered 60 μM of HCQ were not lower (p ≥ 0.05). However, these levels were considerably reduced in the presence of 70 or 80 μM of HCQ compared to the CNT group (p ≤ 0.05). The cells were therefore exposed to dosages of 60 μM of HCQ for 48 h.

The determination of nontoxic HCQ concentration and the effects of CRC treatment (5 µM for 24 h) on the HCQ (60 µM for 48 h)-induced decreases of the live ARPE19 count and viability. (Mean ± SD and n = 3–6). A. The nontoxic HCQ concentration was determined in the plate reader by using the MTT analysis. In the four groups (CNT, CRC, HCQ, and HCQ + CRC), the live cell count (B), debris amount (C), and cell viability (D) were determined in the electronic cell counter (CASY) by using the CASY tone solution. (*p ≤ 0.05 vs CNT and CRC. ×p ≤ 0.05 vs HCQ)
Study groups
The CNT cells were kept in cell culture conditions without treatment for 72 h. The cells in the CRC groups were incubated with 5 μM of CRC (Cat no: C1386, Sigma-Aldrich Chemical Co., MO, USA) for 24 h [30, 31]. The cells in the HCQ (Cat no: H0915, HCQ, Sigma-Aldrich) groups were exposed to 60 µM of HCQ for 48 h. In the HCQ + CRC groups, the cells were kept in a 48-h HCQ incubation period, followed by a 24-h CRC incubation period. In addition to the T25 flasks, 35-mm Mattek dishes with bottom glasses (Istanbul, Turkey) were used to incubate the cells (1 × 103/ml) in an incubator (NB-203QS) to be analysed with a laser confocal microscope (LSM-800, Carl-Zeiss Foundation, Oberkochen, Germany) and for patch-clamp electrophysiological investigation. The attached cells were used for the analyses. For analyses using a CASY Cell Counter and automatic plate reader (Infinite 200 PRO, Tecan Life Sci, Austria GmbH, Groedig, Austria), the cells in the 25 T flasks were used.
Determination of live cell count, debris amount, and cell viability
Using a CASY Cell Counter Electronic System (Model TT, Roche Innovatis AG, Routlingen, Germany), we analyzed the live cell count, debris (death cell) amount, and cell viability in the groups of ARPE19 cells after inducing four groups (CNT, CRC, HCQ, and HCQ + CRC. In the analyses, viable cells are detected with their entire cell volume. In CASY live cell count analyses, the conductivity between two electrodes is measured. An electrical pulse that corresponds to the volume of the cell is produced as the cell passes via the pore. In CASYtone buffer solution, the cells are suspended before the analyses. The unit of 106/per ml was used for expressing the debris amount and live cell count, while the unit of percentage (%) was used to express the viability of the cells.
The assays of cell viability and apoptosis
In addition to the CASY cell viability analyses, the cell viability analyses were repeated in the cells by using the MTT test [11, 12]. For the analyses, the ARPE19 cells were seeded in ninety-six-well plates at an initial density of 1 × 106 cells/well and were treated with 100 µl of 5 mg/ml of MTT in PBS was added to each well, and incubated for 4 h at 37 °C. 0.5 ml of dimethyl sulfoxide (DMSO; Sigma-Aldrich) was added to dissolve the formazan crystals. A microplate reader (Infinite PRO 200) was used to measure the absorbance at 492 nm after shaking the 96-well plate for 1 min.
An APOPercentage stain (Biocolor Ltd. Northern Ireland) was used to measure the amount of apoptosis triggered by HCQ in the ARPE19 cells at 550 nm in a spectrophotometer (UV-1800, Shimadzu Corporation, Kyoto, Japan). The dye of APOPercentage that is specifically internalized by cells going through apoptosis is used in the experiment. In the analyses, necrotic cells are not stained because they are unable to hold onto the dye.
In the MTT and apoptosis analyses, the 100% value, as the standard to assess cytotoxicity, was obtained from the optical density (OD) values assayed in untreated cells.
The assays of caspase -3 (CAS/3), caspase -8 (CAS/8), caspase -9 (CAS/9), cROS, and mMP
Following the appropriate incubation periods of six groups, the culture medium was removed and cells were washed twice with PBS. The three main fluorogenic substrates of passive caspases are Ac-DEVD-AMC (for CAS/3), Ac-IETD-AFC (for CAS/8), and Ac-LEHD-AFC (for CAS/9), and they were purchased from Bachem AG. (Bubendorf, Switzerland). The active CAS/3, CAS/8, and CAS/9 may specifically cleave AMC and AFCs when they are exposed to apoptotic cell lysates. At excitation wavelengths of 360–400 nm and emission wavelengths of 460–505 nm, the free AMC and AFCs were examined in the Infinite 200 PRO [32].
In the cytoplasm of living cells, oxidants convert the non-fluorescent form of cROS, 2',7' -Dichlorofluorescin diacetate (DCFH-DA) (Abcam, Istanbul, Turkey), into the fluorescent form (DCF) [33]. Using the excitation (at 495 nm)/emission (at 527 nm) rate, its fluorescent form (DCF) was detected in the black plates and dark after the cells were treated with 1 µM DCFH-DA for 30–45 min. Fluorescent signals were measured using the Infinite 200 PRO microplate reader.
In the caspase and DCFH-DA, the 100% value, as the standard to assess the fluorescence levels, was obtained from the OD values assayed in untreated cells.
The ARPE19 cells were stained with 5 µM 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolo-carbocyanine iodide (JC1) staining solution for 30–45 min at 37 °C [34]. Following incubation, the JC1 solution and medium were removed and 500 µl extracellular buffer with Ca2+ was added to suspend of the cells. The samples were then analyzed by the Infinite 200 PRO. Although JC1 monomers were measured in the cells at 525 nm, the fluorescence intensity of JC1 aggregates was found at 590 nm after stimulating (488 nm) the cells. The JC1 dye statistics that were derived from the emission ratios (590/ 525) were displayed as a percentage of the control.
Cell death rate analyses
Hoechst (33342) dye can easily enter the nucleus of living cells, and it exhibits blue color following the laser activation (405 nm). Propidium iodide (PI), a dye that can accumulate in damaged nuclei, causes cells to stain red when stimulated with a laser (561 nm). The cell death rate was calculated using the Hoechst 33342 (Cell Signaling Technology, Ankara, Turkey) and PI (Cell Signaling Technology). The cells were incubated at dark and 37 ℃ with Hoechst 33342 (8.1 µM) and PI (1.5 µM) for 15–20 min. After washing the dyes by using 1xPBS, The 2.5D, blue (Hoechst), red (PI), and merge images were captured using the LSM-800. The total number of ARPE19 cell death was manually counted, and the percentage change of the PI-positive cells was calculated. Black/white bright field (BF) images of the cells were recorded in an Axio Observer 7 microscope by using a CDD camera with high resolution (Zeiss).
The analyses of cROS, mROS, and mMP by using the confocal laser scanning microscope (LSM-800)
The production of mitochondrial ROS (mROS) was evaluated in the LSM-800 by using the Mito-SOX Red Mitochondrial Superoxide Indicator (ThermoFisher Sci., GmbH, Dreieich, Germany). In addition to the plate reader analyses, we repeated DCFH-DA and JC1 analyses in the LSM-800 by recording the ARPE19 green and orange images. The ARPE19 cells were incubated in the medium of dishes with Mito-SOX Red (1 µM), DCFH-DA (1 µM), and JC1 (5 µM) for 30–45 min. Following the incubations, the dyes were removed, and 500 µl extracellular buffer with Ca2+ was added to the cells in the dishes with bottom glasses. The cells in the dishes were analyzed in the LSM-800 by using the appropriate laser stimulations [11, 12]. The fluorescence intensity changes of Mito-SOX Red, DCFH-DA, JC1, merge, and 2.5D were recorded in the area (12 µm2) of each cell for the calibration of intensity, and their changes were calculated in the ZEN program. Data of MitoSOX, JC1, and DCFH-DA were presented as the changes of arbitrary unit (a.u).
The analyses of cytosolic free Zn2+ (cZn2+) concentration
FluoZin-3 is a selective indicator of cZn2+ concentration, although N,N,N'N'-tetrakis(-)(2-pyridylmethyl)-ethylenediamine (TPEN) is a cZn2+ chelator. The ARPE19 cells in the dishes were stained with 1 µM FluoZin-3 (Catalog no: # F24194, ThermoFisher Sci.) and (Catalog no: # 4309, Tocris, Istanbul, Turkey) for 30–45 min [35]. After washing the dyes by using the extracellular solution, the green and 2.5 images of cells were recorded in the LSM-800 after the laser excitation (488 nm) (Objective: 20x). The laser wavelengths of FluoZin-3 fluorescence were kept at 493 nm (excitation) and 513 nm (emission) in the cells. The changes of fluorescence intensity were recorded in the LSM-800 and Axio Observer 7 microscope by using the ZEN program, and the mean data of cZn2+ were shown a.u.
Electrophysiology (patch-clamp) analyses
Cationic currents were recorded in the whole-cell patch-clamp mode [26]. The patch pipettes were produced using borosilicate capillary tubes and a PC-10 puller (Narishige Group, Tokyo, Japan). Patch pipettes were filled with a solution containing potassium gluconate (140 mM), glucose (10 mM), EGTA (5 mM), HEPES (10 mM), NaCl (10 mM), and CaCl2 (1) (310 mOsm/l, pH 7.3 with KOH). The ADPR (1 mM) in the cytosolic solution (via path pipette) was administrated into the cells. The bath solution contained (in mM/l) CaCl2 (2.5), HEPES (10), NaCl (130), KH2PO4 (2), KCl (5), MgCl2 (1), and glucose (10) and adjusted to 300 mOsm/l (with NaOH) and pH 7.26. In addition to the extracellular ACA (25 µM), the currents of TRPM2 were inhibited by using N-methyl-D-glucamine (NMDG+ as a Na+ free patch chamber solution). Because TRPM2 is activated in the presence of high cCa2+ concentration, a high cCa2+ concentration (886 nM) was employed in the records rather than the standard cCa2+ concentration (50–100 nM) [36]. The ARPE19 cells were held at -60 mV and ramps from − 200 mV to + 200 mV (400 ms) were applied at a frequency (0.2 Hz) using the HEKA USB 10 amplifier and Patch-master software (Lamprecht, Germany). The experiments were performed at room temperature. The results for the acquired current density were expressed as current (pA)/ cell capacitance (pF).
The measurement of cytosolic free Ca2+ (cCa2+) concentration
In the images of ARPE19 that were stained with 1 µM Fluo 3/AM (at dark for 60 min) (ThermoFisher Sci.), we measured the cCa2+ concertation [16, 25]. Axio Observer 7 inverted microscope with 20 × objective (Carl-Zeiss) was attached to the LSM-800 setup for the laser stimulation of the Fluo 3/AM in the labeled cells. The extracellular solution was applied for removing the Fluo 3/AM dye from the cells prior to adding the TRPM2 stimulator (1 mM and H2O2) to the glass bottom dishes. For antagonizing the TRPM2 channel, 200 µM CRV was used in the LSM-800 experiments of Fluo 3/AM [22]. A specific computer with high resolution was used to measure the changes in the fluorescence intensity in the images taken by ARPE19 using a specific ZEN application (version ZEN 3.2 blue edition and Carl-Zeiss). The fluorescence intensity result was expressed using the a.u.
Statistical analysis
All results were expressed as the means ± standard deviation (SD) of the indicated number of experiments by using the one-way ANOVA of the SPSS program (Version 24.0). The presence of statistical significance (p ≤ 0.05) level was calculated by using the Student’s t test, and it was indicated by using the asterisks (*, x, a, b, and c).
Results
Cell viability and live cell count
We investigated the effects of HCQ and CRC on live ARPE19 count and viability. Although the amount of debris (Fig. 1C) was higher in the HCQ group than in the CNT and CRC groups, the live cell count (Fig. 1B) and cell viability (Fig. 1D) were lower. However, the live cell count and viability were increased in the HCQ + CRC group compared to the HCQ group (p ≤ 0.05), whereas the amount of debris was decreased in the HCQ + CRC group through treatment with CRC (p ≤ 0.05).
CRC counteracted the HCQ-induced increase of ARPE19 death and apoptosis via decreases in caspase and TRPM2 activation
We suspected that the antioxidant CRC would play a protective role against cell death (PI-positive cell) in ARPE19 cells following CRC treatment after detecting a decline in cell viability and live cell count in the HCQ group. The bright field (BF) (Fig. 2A), death (PI), live (Hoechst), merge (Fig. 2B), 2.5D (visual perception) (Fig. 2C), and PI-positive cell numbers (Fig. 2D) were analysed using an LSM-800.

The HCQ (60 µM for 48 h)-induced ARPE19 death was downregulated by the treatments of CRC (5 µM for 24 h) and TRPM2 antagonist (CRV and 200 µM for 24 h). (N = 13–15 and mean ± SD). The bright field (BF) (A) images were recorded in the Axio Observer 7 inverted microscope with 20 × objective by using the CDD camera. The red (PI), Hoechst (blue), and merge (C) images were recorded in the LSM-800 setup attached with 20 × objective. D. The percentage changes of PI positive cells. *p ≤ 0.05 vs CNT and CRC. ×p ≤ 0.05 vs HCQ)
In the HCQ group, there was an increase in the proportion of PI-positive cells compared to the CNT and CRC groups (p ≤ 0.05). However, the percentages of PI-positive cells were lower in the HCQ + CRC and HCQ + CRV groups than in the HCQ group (p ≤ 0.05).
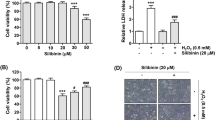
The accumulated data indicate that ARPE19 cell death is caused by the upregulation of apoptosis via increases in CAS/3, CAS/8, and CAS/9 activation [11, 12, 16]. After observing the protective roles of CRC and TRPM2 antagonist (CRV) on retina cell death in the ARPE19 cells, we suspected an increase in apoptosis and caspases in these cells. The MTT level (Fig. 3A) was decreased in the HCQ group, although this level was increased through treatment with CRC and CRV (p ≤ 0.05). The levels of apoptosis (Fig. 3B), CAS/3 (Fig. 3C), CAS/8 (Fig. 3D), and CAS/9 (Fig. 3E) were increased in the HCQ groups (p ≤ 0.05), although they were decreased in the HCQ + CRC and HCQ + CRV groups through treatments with CRC and CRV (p ≤ 0.05).

The HCQ (60 µM for 48 h)-induced changes of MTT, apoptosis, CAS/3, CAS/8, and CAS/9 were modulated in the ARPE19 cells by the treatments of CRC (5 µM for 24 h) and CRV (200 µM for 24 h). (N = 6 and mean ± SD). A. The level of call viability was determined by using the MTT assay. B. The level of apoptosis was manually determined in the plate reader (Infinite PRO 200). The activities of CAS/3 (C), CAS/8 (D), and CAS/9 (E) were assayed in the plate reader (Infinite PRO 200) by using the caspase substrates. The cells were stimulated by the TRPM2 agonist (H2O2 and 1 mM). (*p ≤ 0.05 vs CNT and CRC. ×p ≤ 0.05 vs HCQ)
Treatment with CRC and TRPM2 anatagonist diminished HCQ-induced increases in mROS, mMP, and cROS
Until now, no research has been done on how HCQ causes changes in mROS, mMP, and cROS in ARPE19 cells by activating TRPM2. We wondered whether increased TRPM2 activation in the ARPE19 cells was responsible for the upregulation of mROS, mMP, and cROS.
Normal (Fig. 4A) and 2.5D (Fig. 4B) images of mROS, mMP, and cROS were recorded with a confocal microscope (LSM-800) using MitoSOX Red, JC1, and DCFH-DA, respectively. In addition to the LSM-800 analysis, JC1 (Fig. 4F), and DCFH-DA (Fig. 4G) analyses were also performed in the microplate reader after H2O2 stimulation. The mean fluorescence intensities of MitoSOX Red (Fig. 4C), JC1 (Fig. 4D and 4F), and DCFH-DA (Fig. 4E and 4G) were markedly upregulated in the HCQ group compared to the CNT and CRC groups (p ≤ 0.05). However, treatment with CRC and TRPM2 antagonist (CRV) diminished the action of HCQ via decreases in MitoSOX Red (Fig. 4C), JC1 (Fig. 4D and 4F), and DCFH-DA (Fig. 4E and 4G) in the cells (p ≤ 0.05). Although their increases were downregulated in the HCQ + CRC and HCQ + CRV groups by the CRC and CRV treatments, TRPM2 stimulation after HCQ upregulation was correlated with increased mROS, mMP, and cROS.

The HCQ (60 µM for 48 h)-induced increases of mROS, mMP, and cROS concentrations were downregulated in the ARPE19 by the treatments of CRC (5 µM for 24 h) and TRPM2 antagonist (CRV, 200 µM for 24 h). (Mean ± SD). After staining the cells with MitoSOX Red (1 µM), JC1 (5 µM), and DCFH-DA (1 µM) dyes, the normal (A) and 2.5D (B) images of MitoSOX Red, JC1 and DCFH-DA were captured in the LSM-800 with the × 20 objective. The mean fluorescence intensities of MitoTracker (C), JC1 (D), and DCFH-DA (E) were measured in the images by using the ZEN program. Arbitrary unit: a.u. The JC1 (F) and DCFH-DA (G) analyses were also repeated in the microplate reader. (*p ≤ 0.05 vs CNT and CRC. ×p ≤ 0.05 vs HCQ)
The HCQ-induced increase in cZn2+ was attenuated in the ARPE19 cells by treatment with CRC, CRV, and TPEN
In several cells, HCQ and cZn2+ have a direct correlation [37, 38]. Bonded and free Zn2+ have opposite effects on cells. Bonded Zn2+ works as an antioxidant in many cells as a co-factor of antioxidant enzymes [39]. However, the mMP and mROS levels of ARPE19 are increased by the excessive influx of cZn2+ into the mitochondria [30, 40]. Referring to the ARPE19 cells treated with CRC, TRPM2 antagonist (CRV), and cZn2+ chelator (TPEN), we aimed to understand the mechanism underlying the HCQ-induced increase in cZn2+ concentration.
Using the ZEN programme, variations in the fluorescence intensity of cZn2+ (FluoZin-3) in the recorded cells as observed with the LSM-800 were quantified (Fig. 5A). Treatment with HCQ resulted in an increase in cZn2+ content in the cells (p ≤ 0.05). However, the use of CRC, CRV, and TPEN therapy reduced the effect of HCQ by preventing the upregulation of cZn2+ in the cells (p ≤ 0.05) (Fig. 5B).

The treatment of CRC (5 µM for 24 h), CRV (200 µM for 24 h), and TPEN (1 µM for 30–45 min) diminished HCQ-induced cZn2+ increase in the ARPE19. (N = 20–30 and mean ± SD). The FluoZin-3 green and 2.5D images (A) were recorded in the LSM-800 setup attached with the inverted microscope (Axio Observer 7) and 20 × objective. B. The fluorescence intensity changes of FluoZin-3 as arbitrary unit (a.u.). (*p ≤ 0.05 vs CNT and CRC. ×p ≤ 0.05 vs HCQ)
Incubation with CRC attenuated ADPR-induced TRPM2 current density in ARPE19 cells
TRPM2 activation can be investigated using the patch-clamp technique, which is highly useful. We investigated the HCQ-mediated activation of TRPM2 in the cells using cytosolic (in patch pipettes) 1-mM ADPR stimulation. In the analyses, we used the whole cell configurations of the cells to record the currents (Fig. 6A and 6B). The patch pipettes were produced in a puller using borosilicate capillary tubes (Fig. 6J). There were limited currents (0.05 nA) in the cells of the CNT group without ADPR stimulation (Fig. 6C). In the CNT groups, TRPM2 was activated up to 0.9 nA (Fig. 6D) by ADPR, although it was gated up to 2.0 nA in the HCQ group (Fig. 6E). Hence, the current density was higher in the HCQ group (255 pA/pF) than in the CNT group (100 pA/pF) (p ≤ 0.05) (Fig. 6I). The TRPM2 currents caused by the ADPR in the CNT and HCQ groups were downregulated to CNT levels through treatment with NMDG+ and ACA (Fig. 6D and 6E). Treatment with HCQ accelerated TRPM2 activation in the ARPE19 cells, and their activation times were longer in the CNT group (1.15 min) than in the HCQ group (0.82 min) (p ≤ 0.05) (Fig. 6H). In the CRC (0.05 nA) (Fig. 6F) and HCQ + CRC (0.05 nA) (Fig. 6G) groups, there were limited currents (as in the control group) after ADPR stimulation. The TRPM2 currents were lower in the CRC + ADPR (6.76 pA/pF) and HCQ + CRC + ADPR (9.96 pA/pF) groups than in the CNT + ADPR (100 pA/pF) and HCQ + ADPR (255 pA/pF) groups (p ≤ 0.05) (Fig. 6I).

Treatment with HCQ (60 µM for 48 h) increased the ADPR-induced activation of TRPM2 in the ARPE19, but treatment with CRC (5 µM for 24 h) decreased the activation. (Mean ± SD and n = 4–6). A. The image of whole cell (W.C.) configuration in the cells of patch-chamber and voltage-clamp (at -60 mV). B. The image of W.C. configuration between cell and path pipette. C. The current records of control cell without ADPR stimulation. The cytosolic (via patch pipette) ADPR (1 mM)-induced TRPM2 current and current (I)/ voltage (V) rate changes in the CNT (D) and HCQ (E) groups were inhibited by ACA (25 µM) and NMDG+. F and G. CRC and HCQ + CRC, respectively. H. The TRPM2 activation times in the CNT and HCQ after ADPR. I. The mean current density changes in the four groups (CNT, CRC, HCQ, and HCQ + CRC) with/without ADPR and ACA. J. Patch-clamp puller (pipette maker). (Δp ≤ 0.05 vs. CNT (without ADPR). *p ≤ 0.05 vs. CNT + ADPR. ×p ≤ 0.05 vs. HCQ)
Treatment with CRC downregulated the HCQ-mediated upregulation of cCa2+ concentration via the modulation of TRPM2 in ARPE19 cells
In addition to the patch-clamp investigations, we also investigated the potential protective effects of CRC on the elevation of HCQ-induced TRPM2 stimulation and cCa2+ levels in the cells.
Figure 7A presents images of Fluo-3/AM (cCa2+) in the four groups (CNT, CRC, HCQ, and HCQ + CRC). Following the stimulation of H2O2 (1 mM) in the four groups (CNT, CRC, HCQ, and HCQ + CRC), the concentration of cCa2+ was significantly (p ≤ 0.05) upregulated (Fig. 7B), while the administration of CRV in the H2O2 + CRV groups significantly (p ≤ 0.05) downregulated the concentration of cCa2+ (Fig. 7C).

The HCQ-induced increase of cCa2+ was downregulated via the inhibition of TRPM2 in the ARPE19 by the CRC treatment. (n = 12–15). After staining the ARPE19 cells with the cCa2+ indicator dye (1 µM Fluo 3/AM for 60 min), the TRPM2 stimulators (H2O2 and 1 mM) and blockers (CRV and 200 µM) were applied to the captured ARPE19 cells in four groups (CNT, CRC, HCQ, and HCQ + CRC) in the LSM-800 attached with Axio Observer 7 microscope and objective (20x). In the images of captured cells, the fluorescence intensity changes of Fluo 3/AM as arbitrary unit (a.u.) were determined by using the ZEN program. A. The images of the Fluo 3/AM in the four groups. B and C. The representation of mean fluorescence intensities in the four group after the treatment of H2O2 or CRV. D. The images of HCQ. E and F. The mean fluorescence intensity changes of Fluo 3/AM in the groups during the 396 s. G. The images of HCQ + CRV. H and I. The mean fluorescence intensity of Fluo 3/AM in the group of HCQ + CRV. (*p ≤ 0.05 vs. (-H2O2) group. *p ≤ 0.05 vs. CNT without H2O2. xp ≤ 0.05 vs. CNT with H2O2. ◊p ≤ 0.05 vs. + H2O2 group. xp ≤ 0.05 vs. HCQ. ap ≤ 0.05 vs. HCQ + H2O2)
Images of cCa2+ from the HCQ, HCQ + H2O2, and HCQ + H2O2 + CRV (Fig. 7D) groups were continually recorded for 396 s, and their concentrations are shown in the Fig. 7E and 7F. The Fluo-3/AM fluorescence intensity was higher in the HCQ + H2O2 group (Fig. 7E and 7F) than in the HCQ + CRV + H2O2 group. In addition, the images of cCa2+ from the HCQ + CRC, HCQ + CRC + H2O2, and HCQ + CRC + H2O2 + CRV groups (Fig. 7G) were continually recorded for 396 s, and their concentrations are shown in Fig. 7H and 7I. There was no stimulator action of H2O2 in the HCQ + CRC group (p ≥ 0.05) (Fig. 7G).

Possible defence mechanisms of curcumin (CRC) against hydroxychloroquine (HCQ)-initiated apoptosis, oxidative stress, and TRPM2 activation in ARPE19 cells. DNA damage (via PARP-1 activation)-induced ADP-ribose (ADPR) and oxidative stress stimulate the NUDT9 region of TRPM2 in the C domain, although treatment with ACA and carvacrol inhibits TRPM2 channel activity in ARPE19 cells. The accumulation of free Ca2+ and Zn2+ in mitochondria caused by HCQ raises the mitochondrial membrane potential (mMP). Increased oxidative stress (cytosolic and mitochondrial ROS) and apoptotic factor (caspase (CAS)/3, CAS/8, and CAS/9) activity are induced by increased TRPM2-mediated Ca2+ influx. This stimulates apoptotic signalling pathways, which ultimately leads to cell death. Through the downregulation of TRPM2 activity, treatment with CRC decreases HCQ-induced increases in apoptosis, death, and ROS generation in ARPE19 cells. (↑) Increase. (↓) Decrease
Discussion
Excessive Ca2+ influx-mediated oxidative stress is a key factor in the initiation of RPE oxidative injury and apoptosis [27, 28, 31, 41, 42]. RPE cells normally perform processes that result in high levels of oxidative stress, such as phagocytosis and the deterioration of photoreceptor outer segments. These cells also tolerate continued exposure to light [43]. Chloroquine and HCQ therapies have a low risk of causing significant retinopathy [44, 45]. Although most retinal injuries are repairable, irreversible damage, such as vision loss, can develop in some circumstances. Oxidative stress further increases during treatment with HCQ [7]. ARPE19 pathology is associated with dysfunctional cCa2+ signalling pathways, which result in increased numbers of ROS-producing mitochondria and the accumulation of cCa2+ through TRPM2 stimulation in the RPE cells [15, 16, 25, 27]. All these factors aggravate oxidative stress, induce apoptosis, and activate TRPM2, which contribute to the induction of RPE oxidative injury.
We aimed to investigate the protective effect of CRC treatment on HCQ-induced oxidative stress and apoptosis in ARPE19 cells by blocking TRPM2. The stimulation of TRPM2 with ADPR and H2O2 increased the levels of debris, apoptosis, apoptotic markers (CAS/3, CAS/8, and CAS/9), cell death (PI positive cell number), ROS (cROS and mROS), TRPM2 currents, and cCa2+ concentration in the HCQ group, although these levels were decreased by incubation with CRC and TRPM2 antagonists (ACA and CRV). The HCQ-induced ARPE19 viability and live count were also increased by these treatments (Fig. 8). The data clearly indicated that HCQ-mediated TRPM2 stimulation caused ARPE19 cell death and oxidative toxicity, although they were decreased via the TRPM2 inhibition caused by CRC treatment.
By means of DNA repair processes (via the activity of poly [ADP-ribose] polymerase -1 (PARP-1) and mitochondrial oxidative stress generation), ADPR is produced in the nuclei of cells [17, 18]. The enzyme ADPR hydrolase, which is present in the C-terminal NUDT9 domain of TRPM2, is necessary for activating TRPM2 [17, 18]. Thus, ADPR participates in the regulation of the TRPM2 channel in a variety of cells, including ARPE19 [15, 16, 25, 27], although treatment with CRC alters the action of ADPR in several cell lines [15, 23,24,25]. The current patch-clamp results show that cytosolic ADPR stimulation activated TRPM2 in the HCQ group. DNA damage and an increase in mitochondrial ROS (mROS) are two effects of HCQ treatment [46, 47]. After stimulation with H2O2 and ADPR in the patch-clamp and Fluo 3/AM analyses, the TRPM2 was further gated in the HCQ group compared to the CNT and CRC groups. Analyses of several cells, notably ARPE19, have shown that CRC and CRV have antioxidant and TRPM2 blocker activities [15, 16, 25, 27]. In the current investigation, the antioxidants CRV and CRC had similar effects on TRPM2 modulation in ARPE19 cells, and the HCQ-induced rise in cCa2+ was completely counterbalanced by the treatment with CRC and CRV via the reduction of TRPM2 activation in the HCQ + CRV and HCQ + CRV groups.
The activation of caspases such as CAS/3, CAS/8, and CAS/9 is closely related to the induction of apoptosis and cell death. Under typical cellular metabolic and stress conditions, caspase activation and apoptosis both play crucial roles in ARPE19 death [15, 16, 25, 27]. Several recent studies have examined this issue, but the exact mechanism of action underlying the combined effects of HCQ and apoptosis is still not entirely understood [9, 10]. We found that HCQ treatment reduced the viability of ARPE19 cells. However, this reduction in ARPE19 viability was modified by pretreatment with CRC. There is evidence that the activation of apoptotic pathways causes HCQ-induced retinal oxidative damage and neurotoxicity in ARPE19 cells [7]. It has been hypothesised that increases in excessive Ca2+ influx (via the stimulation of TRPM2)-mediated mROS and cROS formation kill ARPE19 cells via the accumulation of CAS/3, CAS/8, and CAS/9 [15, 16, 25, 27]. The subsequent activation of TRPM2 through the stimulation of apoptosis results in the death of ARPE19 cells [11, 12]. Inhibiting CAS/3, CAS/8, and CAS/9 activity by inhibiting TRPM2 may, therefore, help protect the retina against HCQ toxicity. In line with these findings, the administration of HCQ in the present investigation led to elevations in CAS/3, CAS/8, and CAS/9 activity in ARPE19 cells, as well as an increase in cell death (PI positive cell number). However, using CRC and TRPM2 antagonists (ACA and CRV) attenuated this impact, indicating that CRC protects ARPE19 cells against HCQ-induced retinal toxicity and death by blocking TRPM2 channel signalling pathways. In accordance with the present results, the chloroquine-mediated cell death rate was decreased in ARPE19 cells through treatment with antioxidants (trolox and N-acetyl cysteine) [9]. It has also been reported that apoptosis and mROS levels were increased in ARPE19 cells through treatment with chloroquine [10].
Due to its high rate of oxygen consumption and exposure to light, the retina is a crucial target for cROS and mROS [48]. Furthermore, HCQ-induced RPE toxicity appears to be linked to oxidative stress. Administering HCQ into lysosomes leads to a rise in pH, which simultaneously causes Fe2+ release and a decrease in lysosomal hydrolase activity [9, 46, 49]. Fenton’s reaction and oxidative stress cause excessive ROS production [17]. By expanding the mitochondrial permeability transition pore and harming mMP, increased Ca2+ influx caused by the activation of TRPM2 enhances the production of cROS and mROS [17]. In several cells, TRPM2 suppression-mediated reductions of cROS and mROS by treatment with antioxidants such as glutathione and N-acetyl cysteine controlled Ca2+ influx by regulating the permeability transition pores in the mitochondria [15, 23,24,25]. Although CRC suppressed the production of cROS (DCFH-DA) and mROS (MitoSOX), we investigated whether HCQ-induced apoptosis would occur after an increase in ROS. Additionally, treatment with CRC via the suppression of TRPM2 inhibits the control of apoptosis and mMP under oxidative stress [15, 23,24,25]. Based on the finding that exposure to HCQ increased cROS and mROS through the activation of TRPM2, while treatment for CRC eliminated these effects through the suppression of TRPM2, our study supports this connection. Numerous studies have shown that apoptotic signalling inhibition adversely affects cell fate [15, 23,24,25], which is consistent with our findings using both inhibitors.
In conclusion, our data indicate that exposure to HCQ increases TRPM2 activity in ARPE19 cells. This indicates that it might help to initiate and spread apoptosis and mitochondrial oxidative stress. Although several issues remain unclarified in this study, our research demonstrates the key role of CRC and TRPM2 inhibition in controlling oxidative stress and apoptosis via mMP, cROS, MROS, and caspase signalling. There are hundreds of papers in medicine across the globe on the benefits of CRC for several disorders such as cisplatin-induced optic nerve injury [15] and albumin-induced kidney cell injury [26], and interestingly we have not reached a point where it can be specifically be used to target without being vague. Further studies of HCQ-induced oxidative retinopathy in in vivo experimental animal models are required to elucidate the protective effects of CRC.
Data availability
The data analyses in the current study were performed in the BSN Health, Analyses, Innov., Consul., Org., Agricul. and Industry Ltd (Isparta, Turkey). https://www.bsnsaglik.com.tr/ The details of current analyses are available from the Professor M. Nazıroğlu on reasonable request.
Abbreviations
- ACA:
-
N-(p-amylcinnamoyl)anthranilic acid
- ADPR:
-
ADP-ribose
- ARPE19:
-
Adult retinal pigment epithelial 19
- BF:
-
Bright field
- Ca2+ :
-
Calcium ion
- CNT:
-
Control
- CAS/3:
-
Caspase -3
- CAS/8:
-
Caspase -8
- CAS/9:
-
Caspase -9
- LSM-800:
-
Confocal laser scanning microscope
- CRV:
-
Carvacrol
- cCa2+ :
-
Cytosolic free calcium ion
- cROS:
-
Cytosolic free reactive oxygen radicals
- cZn2+ :
-
Cytosolic free Zn2+
- HCQ:
-
Hydroxychloroquine
- ROS :
-
Free reactive oxygen radicals
- mROS:
-
Mitochondrial free reactive oxygen radicals
- mMP:
-
Mitochondrial membrane potential
- PARP-1:
-
Poly [ADP-ribose] polymerase -1
- RPE:
-
Retinal pigment epithelium
- TRP :
-
Transient receptor potential
- TRPM2 :
-
Transient receptor potential melastatin 2
References
Dima A, Jurcut C, Arnaud L (2021) Hydroxychloroquine in systemic and autoimmune diseases: where are we now? Joint Bone Spine 88(3):105143. https://doi.org/10.1016/j.jbspin.2021.105143
Ferreira PMP, Sousa RWR, Ferreira JRO, Militão GCG, Bezerra DP (2021) Chloroquine and hydroxychloroquine in antitumor therapies based on autophagy-related mechanisms. Pharmacol Res 168:105582. https://doi.org/10.1016/j.phrs.2021.105582
Hsu ST, Ponugoti A, Deaner JD, Vajzovic L (2021) Update on retinal drug toxicities. Curr Ophthalmol Rep 9(4):168–177. https://doi.org/10.1007/s40135-021-00277-x
Doyno C, Sobieraj DM, Baker WL (2021) Toxicity of chloroquine and hydroxychloroquine following therapeutic use or overdose. Clin Toxicol (Phila) 59(1):12–23. https://doi.org/10.1080/15563650.2020.1817479
Ding HJ, Denniston AK, Rao VK, Gordon C (2016) Hydroxychloroquine-related retinal toxicity. Rheumatology (Oxford) 55(6):957–967. https://doi.org/10.1093/rheumatology/kev357
Korthagen NM, Bastiaans J, van Meurs JC, van Bilsen K, van Hagen PM, Dik WA (2015) Chloroquine and hydroxychloroquine increase retinal pigment epithelial layer permeability. J Biochem Mol Toxicol 29(7):299–304. https://doi.org/10.1002/jbt.21696
Dhillon B, Singh S, Keifer J, Kumar U, Shaikh S, Ho S, Seal S (2022) Ameliorating hydroxychloroquine induced retinal toxicity through cerium oxide nanoparticle treatments. J Biomater Appl 36(6):1033–1041. https://doi.org/10.1177/08853282211030150
Güzel M, Akpınar O (2021) Hydroxychloroquine attenuates acute inflammation (LPS)-induced apoptosis via inhibiting TRPV1 channel/ROS signaling pathways in human monocytes. Biology (Basel) 10(10):967. https://doi.org/10.3390/biology10100967
Nguyen Hoang AT, Lee H, Lee SJ (2022) Casein kinase I inhibitor D4476 influences autophagy and apoptosis in chloroquine-induced adult retinal pigment epithelial-19 cells. Exp Eye Res 218:109004. https://doi.org/10.1016/j.exer.2022.109004
Yoon YH, Cho KS, Hwang JJ, Lee SJ, Choi JA, Koh JY (2010) Induction of lysosomal dilatation, arrested autophagy, and cell death by chloroquine in cultured ARPE-19 cells. Invest Ophthalmol Vis Sci 51(11):6030–6037. https://doi.org/10.1167/iovs.10-5278
Özkaya D, Nazıroğlu M (2021) Bevacizumab induces oxidative cytotoxicity and apoptosis via TRPM2 channel activation in retinal pigment epithelial cells: Protective role of glutathione. Graefes Arch Clin Exp Ophthalmol 259(6):1539–1554. https://doi.org/10.1007/s00417-021-05074-7
Özkaya D, Shu X, Nazıroğlu M (2021) Deletion of mitochondrial translocator protein (TSPO) gene decreases oxidative retinal pigment epithelial cell death via modulation of TRPM2 channel. Biology (Basel) 10(5):382. https://doi.org/10.3390/biology10050382
Özkaya D, Nazıroğlu M, Vanyorek L, Muhamad S (2021) Involvement of TRPM2 channel on hypoxia-induced oxidative injury, inflammation, and cell death in retinal pigment epithelial cells: modulator action of selenium nanoparticles. Biol Trace Elem Res 199(4):1356–1369. https://doi.org/10.1007/s12011-020-02556-3
Bhattacharyya S, Sturgis J, Maminishkis A, Miller SS, Bonilha VL (2022) Oxidation of DJ-1 cysteines in retinal pigment epithelium function. Int J Mol Sci 23(17):9938. https://doi.org/10.3390/ijms23179938
Özkaya D, Nazıroğlu M (2020) Curcumin diminishes cisplatin-induced apoptosis and mitochondrial oxidative stress through inhibition of TRPM2 channel signaling pathway in mouse optic nerve. J Recept Signal Transduct Res 40(2):97–108. https://doi.org/10.1080/10799893.2020.1720240
Daldal H, Nazıroğlu M (2022) Carvacrol protects the ARPE19 retinal pigment epithelial cells against high glucose-induced oxidative stress, apoptosis, and inflammation by suppressing the TRPM2 channel signaling pathways. Graefes Arch Clin Exp Ophthalmol 260(8):2567–2583. https://doi.org/10.1007/s00417-022-05731-5
Nazıroğlu M (2007) New molecular mechanisms on the activation of TRPM2 channels by oxidative stress and ADP-ribose. Neurochem Res 32(11):1990–2001
Perraud AL, Fleig A, Dunn CA, Bagley LA, Launay P, Schmitz C et al (2001) ADP-ribose gating of the calcium-permeable LTRPC2 channel revealed by Nudix motif homology. Nature 411(6837):595–599. https://doi.org/10.1038/35079100
Hara Y, Wakamori M, Ishii M, Maeno E, Nishida M, Yoshida T et al (2002) LTRPC2 Ca2+-permeable channel activated by changes in redox status confers susceptibility to cell death. Mol Cell 9(1):163–173. https://doi.org/10.1016/s1097-2765(01)00438-5
Nazıroğlu M, Lückhoff A (2008) A calcium influx pathway regulated separately by oxidative stress and ADP-Ribose in TRPM2 channels: single channel events. Neurochem Res 33(7):1256–1262. https://doi.org/10.1007/s11064-007-9577-5
Kraft R, Grimm C, Frenzel H, Harteneck C (2006) Inhibition of TRPM2 cation channels by N-(p-amylcinnamoyl)anthranilic acid. Br J Pharmacol 148(3):264–273. https://doi.org/10.1038/sj.bjp.0706739
Nazıroğlu M (2022) A novel antagonist of TRPM2 and TRPV4 channels: carvacrol. Metab Brain Dis 37(3):711–728. https://doi.org/10.1007/s11011-021-00887-1
Kheradpezhouh E, Barritt GJ, Rychkov GY (2016) Curcumin inhibits activation of TRPM2 channels in rat hepatocytes. Redox Biol 7:1–7. https://doi.org/10.1016/j.redox.2015.11.001
Öz A, Çelik Ö (2016) Curcumin inhibits oxidative stress-induced TRPM2 channel activation, calcium ion entry and apoptosis values in SH-SY5Y neuroblastoma cells: Involvement of transfection procedure. Mol Membr Biol 33(3–5):76–88. https://doi.org/10.1080/09687688.2017.1318224
Güzel M, Nazıroğlu M, Akpınar O, Çınar R (2021) Interferon gamma-mediated oxidative stress induces apoptosis, neuroinflammation, zinc ion influx, and TRPM2 channel activation in neuronal cell line: modulator role of curcumin. Inflammation 44(5):1878–1894. https://doi.org/10.1007/s10753-021-01465-4
Nazıroğlu M, Çiğ B, Yazğan Y, Schwaerzer GK, Theilig F, Pecze L (2019) Albumin evokes Ca2+-induced cell oxidative stress and apoptosis through TRPM2 channel in renal collecting duct cells reduced by curcumin. Sci Rep 9(1):12403. https://doi.org/10.1038/s41598-019-48716-x
MeléndezGarcía R, Arredondo Zamarripa D, Arnold E, Ruiz-Herrera X, NoguezImm R, Baeza Cruz G et al (2016) Prolactin protects retinal pigment epithelium by inhibiting sirtuin 2-dependent cell death. EBioMedicine 7:35–49. https://doi.org/10.1016/j.ebiom.2016.03.048
Bhattarai N, Korhonen E, Toppila M, Koskela A, Kaarniranta K, Mysore Y, Kauppinen A (2020) Resvega alleviates hydroquinone-induced oxidative stress in ARPE-19 cells. Int J Mol Sci 21(6):2066. https://doi.org/10.3390/ijms21062066
Chen D, Xie J, Fiskesund R, Dong W, Liang X, Lv J, Jin X et al (2018) Chloroquine modulates antitumor immune response by resetting tumor-associated macrophages toward M1 phenotype. Nat Commun 9(1):873. https://doi.org/10.1038/s41467-018-03225-9
Shanmuganathan S, Sumantran VN, Angayarkanni N (2017) Epigallocatechin gallate & curcumin prevent transforming growth factor beta 1-induced epithelial to mesenchymal transition in ARPE-19 cells. Indian J Med Res 146(Suppl):S85–S96. https://doi.org/10.4103/ijmr.IJMR_1583_15
Bardak H, Uğuz AC, Bardak Y (2017) Curcumin regulates intracellular calcium release and inhibits oxidative stress parameters, VEGF, and caspase-3/-9 levels in human retinal pigment epithelium cells. Physiol Int 104(4):301–315. https://doi.org/10.1556/2060.104.2017.4.3
Bejarano I, Espino J, Marchena AM, Barriga C, Paredes SD, Rodríguez AB, Pariente JA (2011) Melatonin enhances hydrogen peroxide-induced apoptosis in human promyelocytic leukaemia HL-60 cells. Mol Cell Biochem 353(1–2):167–176. https://doi.org/10.1007/s11010-011-0783-8
Kalyanaraman B, Darley-Usmar V, Davies KJ, Dennery PA, Forman HJ, Grisham MB et al (2012) Measuring reactive oxygen and nitrogen species with fluorescent probes: challenges and limitations. Free Radic Biol Med 52(1):1–6. https://doi.org/10.1016/j.freeradbiomed.2011.09.030
Sivandzade F, Bhalerao A, Cucullo L (2019) Analysis of the mitochondrial membrane potential using the cationic JC-1 dye as a sensitive fluorescent probe. Bio Protoc 9(1): e3128. https://doi.org/10.21769/BioProtoc
Cho KS, Yoon YH, Choi JA, Lee SJ, Koh JY (2012) Induction of autophagy and cell death by tamoxifen in cultured retinal pigment epithelial and photoreceptor cells. Invest Ophthalmol Vis Sci 53(9):5344–5353. https://doi.org/10.1167/iovs.12-9827
McHugh D, Flemming R, Xu SZ, Perraud AL, Beech DJ (2003) Critical intracellular Ca2+ dependence of transient receptor potential melastatin 2 (TRPM2) cation channel activation. J Biol Chem 278(13):11002–11006. https://doi.org/10.1074/jbc.M210810200
Campos-Blázquez JP, Schuth N, Garay E, Clark AH, Vogelsang U, Nachtegaal M, Contreras RG, Quintanar L, Missirlis F (2022) Chloroquine disrupts zinc storage granules in primary Malpighian tubule cells of Drosophila melanogaster. Metallomics. 14(10):mfac075. https://doi.org/10.1093/mtomcs/mfac075
Hoang BX, Han BA (2020) possible application of hinokitiol as a natural zinc ionophore and anti-infective agent for the prevention and treatment of COVID-19 and viral infections. Med Hypotheses 145:110333. https://doi.org/10.1016/j.mehy.2020.110333
Olechnowicz J, Tinkov A, Skalny A, Suliburska J (2018) Zinc status is associated with inflammation, oxidative stress, lipid, and glucose metabolism. J Physiol Sci 68(1):19–31. https://doi.org/10.1007/s12576-017-0571-7
Daldal H, Nazıroğlu M (2022) Rituximab attenuated lipopolysaccharide-induced oxidative cytotoxicity, apoptosis, and inflammation in the human retina cells via modulating the TRPM2 signaling pathways. Ocul Immunol Inflamm 30(6):1315–1328. https://doi.org/10.1080/09273948.2022.2075400
Zhu X, Wang K, Zhou F, Zhu L (2018) Paeoniflorin attenuates atRAL-induced oxidative stress, mitochondrial dysfunction and endoplasmic reticulum stress in retinal pigment epithelial cells via triggering Ca2+/CaMKII-dependent activation of AMPK. Arch Pharm Res 41(10):1009–1018. https://doi.org/10.1007/s12272-018-1059-6
Li X, Zhao H, Wang Q, Liang H, Jiang X (2015) Fucoidan protects ARPE-19 cells from oxidative stress via normalization of reactive oxygen species generation through the Ca2+-dependent ERK signaling pathway. Mol Med Rep 11(5):3746–3752. https://doi.org/10.3892/mmr.2015.3224
Qin S, De Vries GW (2008) alpha2 But not alpha1 AMP-activated protein kinase mediates oxidative stress-induced inhibition of retinal pigment epithelium cell phagocytosis of photoreceptor outer segments. J Biol Chem 283(11):6744–6751. https://doi.org/10.1074/jbc.M708848200
Gaynes BI, Torczynski E, Varro Z, Grostern R, Perlman J (2008) Retinal toxicity of chloroquine hydrochloride administered by intraperitoneal injection. J Appl Toxicol 28(7):895–900. https://doi.org/10.1002/jat.1353
Kellner U, Kellner S, Weinitz S (2008) Chloroquine retinopathy: lipofuscin- and melanin-related fundus autofluorescence, optical coherence tomography and multifocal electroretinography. Doc Ophthalmol 116(2):119–127. https://doi.org/10.1007/s10633-007-9105-6
Qu X, Sheng J, Shen L, Su J, Xu Y, Xie Q, Wu Y, Zhang X, Sun L (2017) Autophagy inhibitor chloroquine increases sensitivity to cisplatin in QBC939 cholangiocarcinoma cells by mitochondrial ROS. PLoS One 12(3):e0173712. https://doi.org/10.1371/journal.pone.0173712
Ovejero-Sánchez M, Rubio-Heras J, Vicente de la Peña MDC, San-Segundo L, Pérez-Losada J, González-Sarmiento R, Herrero AB (2022) Chloroquine-induced DNA damage synergizes with nonhomologous end joining inhibition to cause ovarian cancer cell cytotoxicity. Int J Mol Sci 23(14):7518. https://doi.org/10.3390/ijms23147518
Eshaq RS, Wright WS, Harris NR (2014) Oxygen delivery, consumption, and conversion to reactive oxygen species in experimental models of diabetic retinopathy. Redox Biol 2:661–666. https://doi.org/10.1016/j.redox.2014.04.006
Ganguli A, Choudhury D, Datta S, Bhattacharya S, Chakrabarti G (2014) Inhibition of autophagy by chloroquine potentiates synergistically anti-cancer property of artemisinin by promoting ROS dependent apoptosis. Biochimie 107 Pt B:338–349. https://doi.org/10.1016/j.biochi.2014.10.001
Acknowledgments
The study was summarized form the M.D. thesis of Dr. A. Ertuğrul.
Funding
The study was performed with a financial support of Scientific Research Unit (BAP) of Suleyman Demirel University. (Project No: TTU-2022–8532. Project owner: Dr. Dilek Özkaya).
Author information
Authors and Affiliations
Contributions
Dr. A. Ertuğrul: The design, data analyses, and critical revision of present study. Dr. D. Özkaya: The design, manuscript preparation, conception, project application, data analyses, and critical revision of present study. Dr. M. Nazıroğlu: Data analyses and preparation of figures in the manuscript. The manuscript submission was approved by the authors.
Corresponding author
Ethics declarations
Ethical approval
None of the authors used samples from experimenters who were humans or from animals in the current investigation.
Conflicts of interest
None declared.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article received support from the mentorship team.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Ertuğrul, A., Özkaya, D. & Nazıroğlu, M. Curcumin attenuates hydroxychloroquine-mediated apoptosis and oxidative stress via the inhibition of TRPM2 channel signalling pathways in a retinal pigment epithelium cell line. Graefes Arch Clin Exp Ophthalmol 261, 2829–2844 (2023). https://doi.org/10.1007/s00417-023-06082-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00417-023-06082-5