
Abstract
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease characterized by the progressive degeneration of both upper and lower motor neurons. A defining histopathological feature in approximately 97% of all ALS cases is the accumulation of phosphorylated trans-activation response (TAR) DNA-binding protein 43 protein (pTDP-43) aggregates in the cytoplasm of neurons and glial cells within the central nervous system. Traditionally, it was believed that the accumulation of TDP-43 aggregates and subsequent neurodegeneration primarily occurs in motor neurons. However, contemporary evidence suggests that as the disease progresses, other systems and brain regions are also affected. Despite this, there has been a limited number of clinical studies assessing the non-motor symptoms in ALS patients. These studies often employ various outcome measures, resulting in a wide range of reported frequencies of non-motor symptoms in ALS patients. The importance of assessing the non-motor symptoms reflects in a fact that they have a significant impact on patients’ quality of life, yet they frequently go underdiagnosed and unreported during clinical evaluations. This review aims to provide an up-to-date overview of the current knowledge concerning non-motor symptoms in ALS. Furthermore, we address their diagnosis and treatment in everyday clinical practice.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease characterized by progressive degeneration of both upper and lower motor neurons. Common clinical signs of ALS include progressive muscle wasting, weakness, dysarthria, dysphagia, and ultimately, respiratory failure. The initial presentation of ALS varies depending on the site of onset. Limb (spinal) onset is the most common (approximately 65% of cases), followed by bulbar onset (about 30%), and, less commonly, respiratory onset (about 5%) [1]. The worldwide incidence rates of ALS vary depending on the region, with estimates ranging from 0.5 to 3.6 cases per 100,000 individuals [2].
ALS is a complex disorder believed to result from a combination of genetic and environmental factors. While the majority of cases are sporadic (sALS), approximately 5–10% are familial (fALS), with a Mendelian inheritance pattern [3]. Over 30 genes have been implicated in ALS pathogenesis. The most common genetic mutations associated with fALS are found in the Superoxide dismutase 1 (SOD1) gene, Chromosome 9 open reading frame 72 (C9orf72) gene, Trans-activation response (TAR) DNA-binding protein 43 (TARDBP) gene, and Fused in sarcoma (FUS) gene, collectively accounting for about 70% of fALS cases [4].
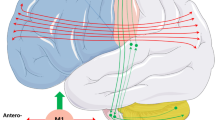
The accumulation of phosphorylated 43-kDa TDP protein (pTDP-43) aggregates in the cytoplasm of neurons and glial cells within the central nervous system (CNS) is a defining histopathological feature observed in approximately 97% of all cases of ALS [5]. Exceptions do exist, such as in cases of ALS with SOD1 [6] or FUS [7] mutations, where other types of protein aggregates are observed. First, it was assumed that accumulation of TDP-43 aggregates and subsequent neurodegeneration occurs primarily in motor neurons. However, more recent evidence suggests that other systems and brain regions are also affected as the disease progresses. The degeneration starts in a focal manner (typically aligning with the region of symptom onset) and subsequently spreads throughout the CNS, affecting not only the motor regions but also non-motor regions of CNS [8,9,10,11].
Parkinson’s disease, another neurodegenerative disorder characterized by the accumulation of toxic protein aggregates (in this case, α-synuclein), has undergone extensive evaluation of its non-motor symptoms. This evaluation has not only led to improvements in the quality of life (QoL) of patients but has also enhanced our understanding of the underlying disease mechanisms. Regarding ALS, only 1% of publications have focused on non-motor symptoms [12]. The frequency of non-motor symptoms in ALS varies widely between studies, ranging from 5 to 80%. They significantly affect patients' QoL, often going underdiagnosed and unreported during clinical evaluations [13]. The gross classification of non-motor symptoms in ALS encompasses four main categories: neuropsychiatric symptoms, autonomic symptoms, vascular symptoms, and gastrointestinal symptoms [13].
The term “non-motor symptoms” in ALS often conceals a certain level of misunderstanding. What exactly falls under this category? Should we consider dysphagia, sialorrhea, or alterations to the sense of taste due to riluzole therapy as non-motor symptoms? For instance, Shojaie et al., in their recent paper on non-motor symptoms in ALS, discussed how these symptoms can stem directly from neuromuscular weakness (such as sialorrhea), indirectly from weakness (such as pain due to immobility), as side effects of therapy (like alterations to taste from riluzole), or from neurodegeneration occurring outside the corticobulbar and corticospinal motor system [14]. It is clear that exact distinction between what falls under this term can be challenging.
This review aims to provide an up-to-date overview of the current knowledge regarding non-motor symptoms in ALS, as well as their diagnosis and treatment in everyday clinical practice. We focus on pain, fatigue, sleep disorders and restless legs syndrome, autonomic dysfunction, and, finally, cognitive and neuropsychiatric symptoms, metabolic abnormalities and weight loss.
Pain
With the exception of some earlier reports [15], pain has been utterly neglected for a long time since ALS was considered purely a disease of motor neurons. However, awareness of the presence of pain in ALS patients emerged in the past decades and years, given its significant negative influence on the QoL of ALS patients and their caregivers [16,17,18,19,20]. The pathogenesis and characteristics of pain in ALS are still not entirely understood.
Frequency of pain in ALS
Reported pain frequency among ALS patients varies widely, with rates ranging from 15 to 85% [18, 19, 21,22,23,24,25,26,27]. These variations can be attributed to differences in study designs and the use of various pain assessment instruments [28]. The number of ALS patients included in these studies also varies significantly, ranging from seven to 2092 patients. Furthermore, some individuals may not report pain because they perceive it as a minor symptom compared to other aspects of ALS [20]. In the two most recent studies, both employing self-constructed questionnaires, pain was one of the most common non-motor symptoms in patients with ALS [14, 29].
The latest meta-analysis focused on pain in ALS, conducted in 2021 [30], included 21 articles, all of which were observational studies, comprising 14 cross-sectional studies, six cohort studies, and one case–control study. The findings revealed that between half and two-thirds of ALS patients experience pain, with a pooled prevalence of 60% (95% confidence interval [CI] = 50–69%). However, it is important to note that there was a substantial heterogeneity in the results (I2 = 94%, p < 0.001). The lowest heterogeneity was observed for studies using validated measures (I2 = 82%, p < 0.002), which was still quite high.
Characteristics of pain in ALS patients
Primary versus secondary pain
Primary pain originates from damage to the nervous system and can be categorized into neuropathic pain and pain due to cramps and spasticity. Secondary pain arises due to non-neuronal damage and is nociceptive in nature [31]. Chio et al. discussed in their review that most of the pain experienced by ALS patients seems to result from the motor impairment itself but that not all types of pain can be explained this way [28].
Neuropathic pain is caused by a lesion or disease of the somatosensory nervous system [32]. Some evidence of the involvement of the sensory cortex as the part of the neurodegeneration was found in post-mortem studies of ALS patients [33, 34], as well as, in numerous neuroimaging [35,36,37,38,39] and electrophysiological studies [40,41,42,43,44,45,46,47,48]. Neuroimaging and neurophysiological studies have also identified alterations in the spinal sensory ascending pathways in as many as 85% of ALS patients [43, 44, 49,50,51]. These findings have been further supported by pathological studies in both humans and mouse models, which have reported that up to 50% of ALS patients exhibit degeneration of the dorsal columns [51,52,53,54,55]. Furthermore, dorsal roots and peripheral nerves, as well as small sensory nerve fibers can be affected in ALS [22, 51, 56,57,58,59,60,61,62,63,64,65]. Yet, neuropathic pain appears to be relatively uncommon in ALS. Depending on the pain assessment tool used, studies have shown a prevalence of neuropathic pain in ALS ranging from 0 to 9% [19, 66,67,68]. The global prevalence of neuropathic pain is believed to range from 6.9 to 10% [69], which does not considerably differ from the prevalence of pain in ALS.
The occurrence of muscle cramps in ALS patients is attributed to the instability of the affected motor units and is commonly linked to muscle denervation [70]. These cramps, marked by sudden and involuntary muscle contractions originating from peripheral nerves [70], tend to be more frequent in patients with limb-onset ALS and in older individuals with the disease [71]. Spasticity, on the other hand, is a velocity-dependent increase in muscle tone due to the loss of inhibitory control of upper motor neurons [72]. Clinically, it leads to exaggerated tendon tap reflexes and an increased resistance of a muscle to stretching, stiffness, fine motor control difficulties and gait problems [72, 73]. In the research conducted by Verschueren et al., spasticity was observed in 36% of a sample of 150 ALS patients. Among those with spasticity, 42.5% reported spasticity-related pain, with the majority of these patients describing their pain as mild [74].
Secondary pain in ALS arises from alterations in non-neuronal tissues, such as connective tissue, bones, and joints. These changes result from muscle atrophy, weakness, and prolonged immobility and lead to musculoskeletal pain [28]. Joint pain is a common manifestation in ALS patients, and it typically occurs when weakened and wasted muscles can no longer provide adequate support to the joints. The shoulders and hips are the most frequently affected joints [18, 28, 75]. Furthermore, immobility in ALS can cause skin pressure and decubitus ulcers, occasionally leading to perceived pain [28]. Patients on mechanical ventilation, especially those using non-invasive ventilation, may also experience skin issues, often around the nasal bridge due to mask interfaces [31].
Pain severity
Studies show substantial variability in reporting pain severity. For instance, Hanisch et al. [25] found that the majority of participants reported mild pain (58.0%), whereas Pizzimenti et al. [76] reported a high prevalence of very severe pain (65.4%) in ALS patients. However, a recent meta-analysis revealed that slightly over three-quarters of ALS patients reported experiencing moderate pain, with 17.5% experiencing severe pain. Mild and very severe pain were less common, reported in fewer than 2.0% of cases [30].
Progression of pain throughout the disease
There is a substantial lack of longitudinal studies examining pain in ALS. Wigand et al. conducted a longitudinal study in which they examined pain with the Brief Pain Inventory (BPI) at three different time points in 151 ALS patients from three German centers [77]. They found that approximately half of the ALS patients had pain at the baseline assessment. Furthermore, 70% of 40 patients reported pain at the third survey. Adelman et al. investigated the agreement between 69 end-stage ALS patients and their family caregivers concerning various indicators of physical and psychological well-being at the end of life, including the assessment of pain [78]. Patients were asked to rate their current pain using a Visual Analogue Scale (VAS) by answering the question, 'How much pain is the patient feeling?'. The authors discovered a significant increase in pain levels on the VAS, with scores rising by 1 point (from 2.3 to 3.3; p < 0.003) during the last assessment. Fifty-four of the patients had undergone at least two assessments, with a median number of study assessments being 3. Caress et al. conducted a 21-month follow-up study involving 41 ALS patients, revealing that cramps were experienced by 95% of patients during the course of the disease [71]. Cramps typically emerged early in the disease, with a decreasing trend observed in subsequent years (mean number of cramps in the first year was 46.3 ± 95.7, in the second year was 37.6 ± 62.5, and in the third year was 24.1 ± 31.7). However, it is worth noting that this trend did not reach statistical significance.
Other available studies are predominantly cross-sectional in nature, which may not be ideal for assessing the natural history of pain in ALS. Moreover, these studies have yielded conflicting results. Some have indicated a correlation between pain and the progression of functional impairment, suggesting that pain becomes more frequent in the later stages of the disease [18, 79, 80]. Conversely, other studies have reported no significant differences in pain frequency between early and late-stage disease, and have found no clear correlations between pain and the course or severity of the disease [19, 81].
Treatment
To treat pain effectively, it is crucial to comprehend its characteristics and its nature (primary or secondary pain) [82]. This highlights the significance of accurate pain assessment because administering inappropriate pain medications, for example using antiepileptic drugs in patients with nociceptive pain, can rather potentially exacerbate discomfort due to side effects. This means that pain treatment should be individualized. Pharmacological and non-pharmacological treatment options stand in disposal as a treatment of pain in ALS patients. Pharmacological treatments are primarily employed for neuropathic and primary pain, often in conjunction with non-pharmacological treatment, whereas non-pharmacological strategies are typically more effective for addressing secondary pain [28].
Pharmacological treatment options
Nociceptive (secondary) pain
A recent Cochrane review concluded that there is a lack of evidence from randomized controlled trials when it comes to managing pain in ALS [83]. According to this Cochrane review from 2017, treatment of nociceptive (secondary) pain in ALS should follow the 1990 World Health Organization Analgesic Ladder [83]. This approach entails the recommendation of nonsteroidal anti-inflammatory drugs (NSAIDs) for managing mild pain, and for moderate to severe pain, a combination of NSAIDs and either weak or potent opioids [84, 85]. The concern regarding respiratory depression in ALS patients receiving opioid treatment seems to be exaggerated, and a low-dose opioid therapy should typically be safe when used in conjunction with noninvasive ventilation, according to Dorst et al. [86].
Neuropathic pain
Neuropathic pain should be treated according to the guidelines. In two surveys conducted across 18 European ALS centers, gabapentin, pregabalin, and tricyclic antidepressants were the most frequently employed medications for managing neuropathic pain in ALS patients [87, 88]. There have been just two surveys conducted on the utilization of cannabinoids for neuropathic and nociceptive pain in ALS [89, 90]. While these surveys suggest that cannabis may have potential effectiveness in pain reduction and could potentially complement the action of opioids, it's important to note that they primarily serve an epidemiological perspective and do not offer substantial clinical guidance [28]. Urbi et al. published the study protocol in 2019 for the ongoing EMERALD trial which assesses the effects of cannabis-based medicine extract on spasticity, pain, weight loss and QoL, as secondary outcome measures [91].
Muscle cramps
Quinine sulfate is a commonly prescribed drug in European countries for managing cramps but has not been approved by the U.S. Food and Drug Administration (FDA) [28, 31]. Also, quinine sulfate is effective for idiopathic muscle cramps, but there is still no randomized trials conducted in ALS [92]. Of note is also that quinine sulfate carries the potential for severe side effects, including thrombocytopenia, cinchonism, myocardial toxicity and interactions with other drugs [31, 88, 93]. On the other hand, the effectiveness of sodium channel blocker, mexiletine, for muscle cramps in ALS, was demonstrated in two randomized clinical studies at a dosage of 2 × 150 mg [94, 95]. Alternatively, another sodium channel blocker, ranolazine, can be used [84], but it has only been studied in myotonia so far [96]. In cases where treatment is not effective, cannabinoids may be considered [84]. Several other substances, such as baclofen, memantine, vitamin E, and L-threonine, have not demonstrated notable effects in alleviating muscle cramps among ALS patients [97]. Shakuyakukanzoto (TJ-68), a traditional Japanese medicine used to treat muscle cramps, is undergoing evaluation in a two-site, double-blind, randomized clinical trial for its effectiveness in alleviating muscle cramps in 22 ALS patients [98].
Spasticity
Baclofen, tizanidine, benzodiazepines, dantrolene, and carbamazepine are among the options for managing spasticity [88]. However, it is important to note that there have been no controlled clinical trials specifically demonstrating the efficacy of these medications in addressing spasticity in ALS patients. Given that these medications can lead to significant side effects, such as weakness, daytime drowsiness, or excessive fatigue, a cautious approach to their initiation, titration, and discontinuation is essential [31]. In a placebo-controlled randomized phase 2 trial involving 60 ALS patients, the use of a delta-9-tetrahydrocannabinol and cannabidiol (THC/CBD) spray showed promise in reducing spasticity symptoms in patients with motor neuron disease and had an acceptable safety and tolerability profile [99]. Additionally, in cases of focal spasticity that is resistant to standard treatments, botulinum toxin A may be a viable therapeutic option [100].
Non-pharmacological treatment options
Physical and occupational therapy should be prescribed to ALS patients to prevent secondary complications such as pain and contractures [84]. A growing body of evidence suggests that the inclusion of flexibility exercises in the management of individuals with neuromuscular conditions is a valuable strategy for preventing the development of painful contractures that could disrupt their daily lives [101]. Less frequently employed approaches for managing secondary forms of pain encompass warm and cold compress therapy, transcutaneous electrical nerve stimulation and acupuncture [28]. Assistive devices, like wheelchairs, special bedding and mattresses, splints, canes and walkers can reduce pain from limited mobility, prevent joint contractures and falling accidents [28].
Future directions
We still have limited knowledge about the natural history of pain in ALS, primarily due to the absence of longitudinal studies. Furthermore, ALS is a substantially clinically and genetically heterogeneous disease [102], which underscores the need for extensive cohort studies to draw meaningful conclusions about pain in ALS. Understanding how pain varies in different ALS subtypes or genetic backgrounds is crucial. To address this gap, future research should adopt a multicenter and longitudinal approach, involving a substantial number of ALS patients, and utilize validated measures. Moreover, it is evident that certain ALS patients may concurrently experience various pain mechanisms. This underscores the significance of prioritizing the development of new standardized measures that can comprehensively encompass and address these various pain mechanisms in ALS. There is a complete lack of randomized clinical trials assessing specific pharmacological treatment options in treatment of pain in ALS. General practitioners and neurologists should be more educated about the presence and mechanisms of pain in ALS. Future clinical trials should also utilize validated screening methods to assess the pain development under investigated medication.
Fatigue
Fatigue is defined as an overwhelming sense of tiredness, lack of energy, and a feeling of exhaustion [103], and it has been reported in a wide range of both neurological [104,105,106,107,108] and non-neurological diseases [109,110,111,112,113]. It can be classified into peripheral and central fatigue. Regarding neuromuscular disorders, peripheral fatigue emerges as a direct consequence of diminished muscle endurance attributable to nerve, muscle, or neuromuscular junction dysfunctions. In contrast, central fatigue presents as an all-encompassing feeling of lethargy and reduced vitality, regardless of muscle weakness or pain, frequently hindering both mental and physical activities [114].
Fatigue is a largely overlooked clinical concern in ALS, often evading the notice of healthcare professionals who provide care for individuals with ALS. Nonetheless, it is very important to assess this non-motor symptom in ALS patients, since it can seriously lower the QoL of the patients [115,116,117]. In ALS, both central and peripheral fatigue play a significant role. Dysfunction of lower motor neurons results in motor units' inability to sustain a given level of activity, leading to peripheral fatigue. In contrast, impairments at the spinal and/or cortical level reduce voluntary drive, causing central fatigue [118]. Beyond the physical factors contributing to fatigue, ALS patients also contend with associated factors that can exacerbate fatigue symptoms. These may encompass depression, sleep disturbances, respiratory issues, and weight loss [118]. Additionally, the medications often prescribed to ALS patients, such as those used to manage spasticity (e.g., baclofen), antidepressants, and anticholinergic drugs have the potential to further intensify fatigue symptoms [118].
Prevalence of fatigue in ALS
Hamad et al. recently conducted a meta-analysis, which included 11 studies (eight cross-sectional and three longitudinal) that analyzed fatigue using a validated tool with a specific cutoff value [119]. This meta-analysis encompassed a total of 1072 ALS patients. The pooled prevalence of fatigue was 48% (95% CI 40–57%). The included studies exhibited significant heterogeneity (I2 = 85%, p < 0.01). The study by Vogt et al. reported the highest prevalence of fatigue at 76.7%, based on an assessment of 121 patients with a mean ALSFRS-R score of 27.8 ± 9.5, using the Fatigue Severity Scale (FSS) [117]. In contrast, An et al. found the lowest reported prevalence of fatigue at 32.6% in 175 included ALS patients, with a mean ALSFRS-R score of 39.5 ± 1.5, also using the FSS [120]. Hamad et al. further reported that the prevalence of fatigue was higher in studies with lower ALSFRS-R scores (< 30) compared to studies with higher ALSFRS-R scores (≥ 30). The pooled prevalence of fatigue was 62% (95% CI 43–79%) and 43% (95% CI 37–49%), respectively [119].
Factors associated with fatigue in ALS patients
Fatigue exhibits a negative association with the ALSFRS-R score [115, 120,121,122,123]. Hamad et al. proposed that this correlation may be attributed to reduced functionality, diminished QoL, higher pain levels, advanced disease progression, and muscle weakness [119]. Moreover, various studies have revealed a negative correlation between fatigue and QoL [115,116,117], forced vital capacity [121] and sleep quality [121]. On the other side, a positive association has been observed between fatigue and pain [16, 115], sleepiness [120, 121] and depression [121, 122]. Hamad et al. found no significant correlations between fatigue and sample size, gender distribution (number of males), duration of disease, or publication year [119].
Treatment of fatigue in ALS
Pharmacological treatment of fatigue in ALS
Currently, there is no established evidence-based treatment for fatigue in ALS due to the limited and low-quality evidence available from randomized controlled trials [124]. However, the 2012 European Federation of the Neurological Societies (EFNS) guidelines suggest that modafinil may be a consideration for treating debilitating fatigue in ALS [125]. Modafinil is an FDA-approved treatment for fatigue and excessive daytime sleepiness in narcolepsy. The precise mechanism by which modafinil reduces fatigue in neurological conditions remains uncertain. Some observations suggest that modafinil may promote wakefulness through the activation of the histaminergic system [126] or by increasing glutamate levels and decreasing gamma-aminobutyric acid (GABA) levels in CNS, particularly in regions that control the sleep-wakefulness cycle [127]. In ALS, an open-label study showed a decrease in fatigue severity by 17%, and sleepiness by 45% following a 2-week course of modafinil [128]. Furthermore, in a small placebo-controlled trial in 32 ALS patients, modafinil showed a response rate of 86% (compared to 14% in placebo group), and the number needed to treat was 1.6 [129]. However, according to the latest Cochrane review from 2018, it is still uncertain whether modafinil could be of benefit [124]. Furthermore, Rosenfeld et al. measured fatigue during isometric contraction in their multicenter, double-blinded study and found no significant improvement after 9 months of treatment with creatine monohydrate [130]. In a small randomized double-blind, crossover trial conducted by Bertorini et al., involving 13 patients diagnosed with motor neuron disease, the drug amifampridine (voltage-gated potassium channel blocker) exhibited a modest improvement in subjective fatigue scores following 4 weeks of treatment compared to a placebo [131]. Finally, it is crucial to consider discontinuing medications if fatigue is recognized as a potential side effect of drug therapy, as recommended by the latest guidelines from the European Academy of Neurology (EAN) [85].
Non-pharmacological treatment of fatigue in ALS
In healthy individuals, physical activity enhances the effectiveness of the neuromuscular system and reduces fatigue [132]. In the context of ALS, several interventions, such as treadmill ambulation [133], muscular exercise [134], and repetitive transcranial magnetic stimulation [135], have been examined as potential strategies to alleviate fatigue. Sanjak et al. investigated repetitive rhythmic exercise through supported treadmill ambulation training, performed three times a week for 8 weeks by nine ALS patients [133]. The study found no significant changes in the FSS score. Drory et al. conducted a study involving 25 ALS patients, where some were randomly assigned to a daily exercise regimen, while the rest adhered to their typical activity levels [134]. The exercise group showed minimal changes in the FSS score, while the control group experienced an increase. However, the difference between the two groups did not reach statistical significance. Zanette et al. assessed the effects of repetitive transcranial magnetic stimulation in 10 ALS patients and reported no significant alterations in FSS scores after a 2-week period. A Cochrane review has also concluded that there is insufficient evidence that breathing exercises, resistance exercise, or repetitive transcranial magnetic stimulation are of benefit in ALS [124].
Future directions
Given that behavioral symptoms like apathy are commonly observed in ALS [136], it is crucial for future research to explore how these symptoms intersect with fatigue and contribute to its severity. While earlier studies used a cross-sectional approach to examine fatigue in ALS, there is a clear need for longitudinal studies to uncover its natural history in ALS. Moreover, employing comprehensive assessment tools, such as the Multidimensional Fatigue Inventory (MFI), which examine both the physical and mental aspects of fatigue, would be more beneficial than using unidimensional ones like the FSS.
Autonomic dysfunction in ALS
As previously mentioned, ALS is now acknowledged as a multisystem disorder, involving impairment of the autonomic nervous system as well [137,138,139]. Dysautonomia in ALS is often overlooked in routine clinical practice. However, studies have yielded conflicting results, with some reporting a relatively high frequency of autonomic dysfunction [139], while others report a much lower frequency [140]. The study conducted by Piccione et al. on 132 ALS patients revealed that one-third of patients experienced autonomic symptoms. Among these symptoms, urinary and gastrointestinal issues were the most prevalent. However, the degree of autonomic impairment was generally mild in the majority of cases (85%), moderate in 15%, while none of the patients demonstrated severe generalized autonomic failure. Notably, patients with predominantly upper motor neuron affection had more severe autonomic impairment [139].
Affection of the urogenital system
Symptoms of lower urinary tract involvement have been reported to occur in ALS patients with a prevalence ranging from 4 to almost 45% [141,142,143,144]. While medication usage and decreased mobility may play a role in exacerbating urinary symptoms in ALS [145], it has been suggested that neurogenic bladder is the primary cause in the majority of cases [144]. Among these symptoms, urgency urinary incontinence emerges as the most prevalent presentation, leading to an increased burden of disease [141, 143, 144]. A post void residual (PVR) of > 50 ml was found in 24–35% of patients [144, 146], and this was found to correlate with an increased ALSFRS-R and lower limb affection. In a study by Arlandis et al., urodynamic studies on 10 ALS patients revealed that detrusor overactivity with obstruction, primarily due to non-relaxing external sphincter (five patients) or bladder neck (two patients), was the most common cause of increased PVR [144]. Vázquez-Costa et al. observed that patients reporting early (< 2 years after disease onset) lower urinary symptoms, especially neurogenic bladder disorders, have a worse survival rate than patients with later onset [145]. Interestingly, clinically significant lower urinary tract symptoms appear to be independent of age, phenotype, disability, cognitive or behavioral impairment, or disease progression, while female sex appeared to be a protective factor [145].
The underlying mechanisms of urinary symptoms in ALS are not well understood. Various central and peripheral nervous system structures are involved in the two phases of micturition: storage and voiding phase. Generally, suprapontine (predominantly storage symptoms), spinal (infrapontine–suprasacral) (both storage and voiding symptoms) or sacral/infrasacral lesions (predominantly voiding symptoms) can cause the neurogenic bladder. Vázquez-Costa et al. conducted a study which revealed that the majority of ALS patients experiencing lower urinary symptoms reported both storage and voiding symptoms, suggesting that an infrapontine–suprasacral lesion may be the underlying cause of these symptoms in motor neuron disease [145]. Moreover, earlier studies have identified the involvement of both the sacral intermediolateral nucleus and Onuf's nucleus (distinct group of neurons located in the ventral part of the anterior horn of the sacral region of the spinal cord), which play critical roles in autonomic bladder function [147, 148].
Various treatment options are available for managing lower urinary symptoms, ranging from conservative approaches (behavioral therapy, antimuscarinic agents, desmopressin, onabotulinumtoxin A injections into the detrusor, β3-adrenoceptor agonists, and tibial neuromodulation) to surgical interventions (sacral neuromodulation, bladder augmentation, sacral deafferentation/anterior root stimulation, and continent/incontinent urinary diversion) [149]. According to a study by Samara et al., catheterization, oxybutynin, and doxazosin were identified as the most effective interventions and medications for treating urinary symptoms in ALS, according to the patients [150].
Gastrointestinal symptoms
Symptoms of bowel movement dysfunction are common in ALS patients [141, 150]. Constipation is the most commonly reported issue by patients, and its frequency tends to increase with disease progression. In a study performed by Samara et al., approximately 30% of the patients had obstipation, increasing to 60% on the second follow-up appointment 6–12 months after diagnosis [150]. Bowel incontinence was uncommon, reported by only 9% of patients, and this prevalence did not change as the disease advanced [141, 150]. In patients on ventilators who are in total locked-in states and live beyond respiratory failure, loss of anal sphincter function has been described [151]. This is further confirmed by de Carvalho et al. who showed affection of the external anal sphincter by single fiber electromyography, despite loss of muscle fiber density in semimembranosus-semitendinosus muscles [146]. Additionally, it has been shown that ALS patients have a delayed colonic transport time. Toepfer et al. have shown by multiple-ingestion single-radiograph technique that patients exhibit a significantly decreased right and left colonic transit [152].
ALS can cause gastrointestinal symptoms through various pathophysiological pathways. Neurons in the Onuf’s nucleus have been found to atrophy in ALS patients [147,148,149]. This finding could potentially contribute to the loss of control of the external anal sphincter. Furthermore, there is evidence suggesting that the enteric nervous system may be affected by ALS. In the TDP-43 A315T mouse model, a decrease in nitric oxide synthase (NOS) neurons in the myenteric plexus has been observed, contributing to intestinal dysmotility [153].
Treatment of bowel movement disorder primarily revolves around managing constipation. The most effective medications were found to be docusate sodium salts and polyethylene glycol. Non-medication-based treatments included an increase in dietary fiber and fluid intake [150].
Cardiovascular symptoms
Already in the early stages of disease, ALS patients exhibit reduced heart rate variability (HRV) and an increased resting heart rate [154,155,156]. A decreased HRV coefficient of variation has been shown to precede unexpected death in ALS [157]. Especially in ventilated patients, circulatory collapse following an autonomic storm has been reported as a common cause of death. These patients exhibited nightly hypotension without a corresponding increase in tachycardia, ultimately leading to circulatory collapse and death [158]. Cardiac magnetic resonance tomography revealed reduced ejection volumes in the left and right heart in ALS patients compared to healthy controls. Rosenbohm et al. showed that ALS patients had an increased T1 enhancement in cardiac magnetic resonance tomography in 77% of the patients compared to 27% of controls [159].
Cardiovascular symptoms in ALS could be attributed to an imbalance in the sympathetic and parasympathetic nervous system. A study conducted by Tanaka et al., utilizing [123I] MIBG scintigraphy to indicate cardiac sympathetic activity, revealed that some ALS patients exhibit sympathetic hyperactivity at the time of diagnosis [160]. Sympathetic affection was linked to disease progression and worse outcomes. Additionally, norepinephrine serum and cerebrospinal fluid levels are elevated in ALS patients [161, 162]. The elevation of norepinephrine levels in ALS patients is a subject of dispute, with some suggesting it may be secondary to factors like respiratory distress [163], while others argue it may be primary and linked to the pathophysiology of ALS [162]. In addition to a hyperactive sympathetic state, there appears to be a parasympathetic dysfunction. ALS patients had a significant decreased cross-sectional area (CSA) of the vagal nerve compared to controls [138]. This combination of parasympathetic hypofunction and sympathetic hyperfunction could explain the increase in resting heart rate loss of HRV and thereby sudden circulation failure in late-stage ALS patients.
Treatment options of cardiovascular symptoms in ALS are very limited. A clinical trial investigated the effects of intrathecally administered brain-derived neurotrophic factor (BDNF) on autonomic functions in 10 patients with ALS. The trial did not show success, and the authors concluded that autonomic nervous system function deteriorates alongside poorer motor performance, independently from treatment with BDNF [164]. In another study, tamsulosin hydrochloride was evaluated for its effect on decreasing serum norepinephrine levels in ALS patients. Forty-one ALS patients received an oral dose of 0.2 mg/day tamsulosin for 4 weeks, resulting in a significant reduction of serum norepinephrine levels. However, no significant differences were observed in HRV or blood pressure [162].
Cardiovascular comorbidities in ALS
Retrospective cohort studies have revealed substantial disparities in the occurrence of specific concurrent comorbidities among individuals affected by ALS when compared to the general population [165]. For example, German cohort studies analyzing comorbidities prior to ALS diagnosis discovered that although cardiovascular risk factors were the most prevalent among ALS patients, their occurrence remained notably higher in the general population compared to the ALS-affected cohort [166, 167]. Many studies proposed the existence of a potential shared mechanism connecting a favorable cardiovascular fitness profile and susceptibility to ALS [168, 169]. Xu et al. conducted a recent systemic review encompassing 17 studies to explore the prevalence and impacts of cardiovascular diseases on ALS. They found substantial regional variations in the prevalence of cardiovascular conditions [170]. Hypertension was highest in France (57%) [171], Portugal (48%) [172, 173], while the Netherlands [174] and Poland [175] reported lower rates at approximately 26% and 23% respectively. Chinese ALS patients had even lower prevalence of hypertension at 15% [176]. In Germany, around 10% and 5% of ALS patients experienced cardiac arrhythmia and heart failure respectively [166, 177]. Conversely, in the United States, coronary heart disease afflicted 24% of ALS patients [178], whereas Denmark and the Netherlands exhibited notably lower rates ranging from 4 to 5% [174, 179]. Remarkably, Danish ALS patients showed the highest prevalence of heart diseases, reaching almost 24% [179]. Xu et al. concluded that hypertension could notably decrease the survival of ALS patients, while coronary heart disease could significantly elevate the risk of developing ALS and therefore suggested to pay special attention to this subset of ALS patients in routine clinical practice [170]. Kim et al. have released a preprint (pending peer-review) proposing that treatment with riluzole is linked to a reduced incidence of heart failure. This might suggest a potential preventive strategy for early management [180].
Future directions
Limited understanding exists regarding whether autonomic symptoms in ALS are secondary manifestations due to muscle weakness, dysphagia, or respiratory issues, or if they arise from primary neurodegeneration affecting non-motor brain regions. Often, these symptoms are overlooked in routine clinical practice. Conducting thorough examinations of these symptoms can offer valuable insights into disease mechanisms and contribute to enhancing QoL for patients. Do compensatory mechanisms effectively obscure the clinical presentation of autonomic symptoms? Certainly, there is a significant need for multicenter studies with a longitudinal design, involving a large ALS patient cohort, for comprehensive assessment of autonomic nervous system involvement in ALS. The development of a self-reported, ALS-specific questionnaire assessing the autonomic nervous system should be considered for the future, both for everyday practice as well as clinical trials. Additionally, there is a necessity for further research into treatment options targeting the sympathetic and parasympathetic imbalance to prevent sudden death in patients with advanced ALS.
Sleep disorders
Sleep disorders can be generally categorized into six groups: insomnia, sleep-related breathing disorders, central disorders of hypersomnia, circadian rhythm sleep–wake disorders, parasomnias, and sleep-related movement disorders [181]. Sleep disturbances are common in neurodegenerative disorders such as Alzheimer’s, Parkinson’s, and Huntington’s diseases, and this trend likely extends to ALS [182]. Sleep disorders are frequently observed and can emerge as an early sign of ALS, yet they might remain undetected until the disease progresses to its later stages [183]. Overall, 50–63% of patients with ALS have poor sleep quality [184]. In ALS, research has primarily focused on sleep-related breathing disorders, followed by insomnia, parasomnias, and sleep-related movement disorders. Notably, hypersomnia or circadian rhythm sleep–wake disorders have not been assessed in ALS so far. Sleep disorders significantly impact the health-related QoL, psychological well-being, and day-to-day functioning of affected individuals and therefore require more thorough investigation [121, 183, 185,186,187].
Hypothalamus and ALS
The hypothalamus, known for regulating sleep cycles and the endocrine system, has shown evidence of involvement in ALS, with observed hypothalamic atrophy and pTDP-43 aggregation in morphological and pathological studies [188, 189]. Disruptions in hormone systems, including growth hormone/insulin-like growth hormone-1, melanocortin and the hypothalamic–pituitary–adrenal axis, are also noted in ALS [190,191,192,193,194,195]. While Gnoni et al. hypothesized that hypothalamic dysfunction may play a key role in the sleep disturbances exhibited by ALS patients, the limited number of studies prevents definitive conclusions [196]. It appears that not all sleep disturbances in ALS can be solely linked to hypothalamic pathology, as some might be a result of disease-related factors.
Sleep-related breathing disorders
In ALS patients, sleep-related breathing disorders primarily involve nocturnal hypoventilation and obstructive sleep apnea, with central sleep apnea being a rare occurrence [197]. The majority of ALS patients experience respiratory muscle weakness due to bilateral degeneration of phrenic nerve motor neurons, leading to progressive diaphragm weakness. This heightens the risk of developing the most common sleep-related breathing disorder, nocturnal hypoventilation, particularly during rapid eye movement (REM) sleep, where muscle atonia occurs [197]. Obstructive sleep apnea is less common in ALS and less prevalent in patients with severe bulbar dysfunction, probably due to tongue atrophy [198, 199]. Also, sleep apnea often accompanies nocturnal hypoventilation [198]. In Boentert et al.'s study, encompassing 250 non-ventilated ALS patients, out of 3309 recorded apnoeic events, 71.3% were classified as obstructive, 23.3% as central, and 5.4% as mixed type [198]. Symptoms of sleep-related breathing disorder comprise sleep fragmentation, nonrestorative sleep, morning headache, daytime fatigue, and excessive sleepiness [199]. Regular evaluation of these symptoms during patient visits helps assess the need for polysomnographic testing. Apneas, hypopneas, and compromised gas exchange correlate with decreased sleep efficiency, frequent shifts in sleep stages, arousals from sleep, and a decrease in N3 (deep) or REM sleep [197]. The recognition of obstructive sleep apnea during diagnostic sleep assessments has been associated with reduced survival rates in ALS. This underscores the significance of early detection of obstructive sleep apnea in individuals with ALS [200].
Parasomnias and insomnias
The parasomnias can be divided into non-rapid eye movement (NREM) related (confusional arousal, sleepwalking, and sleep terrors), rapid eye movement (REM) related, and other [181]. There isn't any compelling evidence suggesting that NREM parasomnias occur frequently among patients with ALS. REM parasomnias are usually linked to α-synucleinopathies (such as Parkinson’s disease, dementia with Lewy bodies, and multisystem atrophy) [201]. However, REM parasomnias can be a crucial feature in some tauopathies (such as anti-IgLON5 disease) as well [202]. Some pathological studies found Bunina bodies and inclusions of pTDP-43 protein in potentially REM controlling brainstem areas, e.g. in locus coeruleus [203] and in the reticular formation of severely affected ALS patients [204]. There's a general lack of understanding regarding REM behavioral disorders in ALS, with the prevailing belief that it might manifest in only a limited subset of patients [199]. Two studies observed a lower REM atonia index and a higher frequency of chin/leg movements per hour of sleep among ALS patients compared to a healthy control group [205, 206]. Notably, both of these measures exhibited a significant correlation with the ALSFRS-R score, indicating a link between these sleep-related parameters and the severity of ALS symptoms.
Insomnia disorders can be generally divided into chronic insomnia disorders, short-term insomnia disorders and others [181]. The clinical criteria defining chronic insomnia involve reported difficulties in falling or staying asleep, adequate opportunities for sleep, and resulting daytime consequences [181]. The prevalence of insomnia in ALS patients is estimated to range between 48 and 69% [185, 187, 207]. It is worth noting that these studies relied on self-reported measures rather than employing the aforementioned diagnostic criteria for defining an insomnia disorder. Insomnia in ALS patients can also arise from factors such as immobility, muscle cramps and nocturnal pain, and difficulty in adjusting positions in bed, emotional distress associated with the disease, and the impacts of medications used in treatment [199].
Restless legs syndrome
Restless legs syndrome (RLS) is a common neurological sensorimotor sleep disorder characterized by an irresistible urge to move the legs, often accompanied by uncomfortable or unpleasant sensations [208]. These symptoms typically occur during periods of rest or inactivity, particularly in the evening or at night, and can be partially or completely relieved by movement [208]. The discomfort and restlessness often lead to sleep disturbance, which can significantly impact an individual's QoL and daily functioning [208]. The exact cause of RLS is not entirely understood, but it is believed to involve both genetic and environmental factors. Certain conditions, such as iron deficiency, anemia, pregnancy, end-stage renal disease, diabetes mellitus, and peripheral neuropathy, can exacerbate or contribute to RLS. Many neurodegenerative diseases, including Parkinson's disease, spinocerebellar ataxias, Huntington's disease, and hereditary spastic paraparesis, have been associated with a higher occurrence of RLS [209,210,211,212]. So far, only four studies assessed this non-motor symptom in ALS patients [213,214,215,216].
Epidemiology and characteristics of RLS in ALS
Among the general adult population, reported prevalence rates for RLS typically range between 5 and 15%, primarily observed in studies within Caucasian populations. Conversely, in regions like Asia and South America, a lower prevalence is noted, with estimates ranging from 1.6 to 2.0% [217,218,219]. RLS prevalence in ALS cohorts worldwide varies from 14.6 to 25% [199]. These studies consistently demonstrate a higher prevalence of RLS in ALS patients compared to the general population or control groups. However, there are exceptions; for instance, one study in a small ALS-cohort found a prevalence of only 5% in an Indian ALS cohort [185]. Sun et al. observed no significant differences between genetic and non- genetic ALS patients (32 ALS genes were screened by whole exome sequencing) [216].
Previous research indicates varying degrees of increased risk for RLS in ALS patients compared to control subjects: 4.1-fold [214], 12.7-fold [216], and 19-fold [213]. Additionally, Lo Coco et al. noted that ALS patients reported a notably shorter history of RLS symptoms and a higher frequency of symptom occurrence compared to the control group [214]. Limousin et al. discovered that RLS disturbances preceded ALS onset in 26% of their patients [215], a contrast to Liu et al.'s findings where nearly all patients reported RLS symptoms following ALS onset [213]. Concerning RLS intensity, Limousin et al. observed that RLS severity was rated as moderate or severe in almost all their patients (92%) [215].
Factors associated with RLS in ALS
Previous studies have linked the presence of RLS in ALS patients with several factors: higher ALSFRS-R scores indicating increased functional disability [214], insomnia [214], older age [215], lower limb function scores on the ALSFRS-R scale [213], excessive daytime sleepiness (EDS) and anxiety [213]. Liu et al. proposed a plausible explanation for the association between RLS and more severe leg dysfunction, suggesting progressive spinal cord dysfunction as a potential cause. They theorized that since dopamine acts within the spinal cord to modulate sensory and motor functions, its involvement might intersect with the RLS pathway [213].
Boentert suggested that ALS patients might have a higher likelihood of experiencing RLS due to factors like immobilization, mild sensory neuropathy, or even small fiber neuropathy [197]. This suggestion finds support in Limousin et al.'s study, where they observed an association between the “turning in bed and adjusting the bedclothes” subscale of the ALSFRS-R score and RLS symptoms, suggesting that immobility in bed may worsen leg discomfort in RLS patients [215]. Additionally, Boentert proposed actively inquiring about RLS symptoms in ALS patients.
Treatment of sleep disorders in ALS
Treatment of RLS in ALS
Customizing treatment approaches is crucial, taking into account the patient’s overall health, disease stage, and presence of other non-motor symptoms. Before initiating any treatment, confirming the presence of RLS is vital, given that symptoms such as pain, cramps, or edema often overlap with RLS [199]. Additionally, it is essential to check for and address iron deficiency following standard recommendations [220]. Stating non-pharmacological methods as the initial approach for those with occasional or mild RLS symptoms can offer significant benefits and, at times, might serve as the sole treatment required [208]. Techniques like massage, stretching, walking, engaging in cognitive distractions such as games or puzzles, and even taking moderate (cold or warm) temperature baths can effectively alleviate RLS symptoms [208]. Suggesting these non-pharmacological approaches as supplementary therapies can be beneficial, potentially reducing the need for higher doses of pharmacological treatments [208].
Approved medications for RLS treatment include, pramipexole (0.375–0.5 mg), ropinirole (3.0–4.0 mg), rotigotine (2.0–3.0 mg), and gabapentin (600–1200 mg). However, none of these treatment options were examined in ALS patients. In the authors’ opinion, it seems crucial to consider the presence of other non-motor symptoms, such as chronic nociceptive or neuropathic pain, when assessing RLS treatment in ALS patients. For instance, gabapentin may be preferred for treating RLS in ALS patients experiencing chronic pain. Additionally, several off-label treatment options, such as pregabalin (150–450 mg), tramadol (200–300 mg), oxycodone (10–40 mg), and methadone (10–30 mg), exist for RLS [208]. These off-label treatments not only address RLS but can also aid in managing chronic pain. Their potential augmentation in treating chronic pain should be considered for selected ALS patients [199].
Future directions
Similar to other non-motor symptoms in ALS, no longitudinal study assessing sleep disorders in ALS patients has been conducted to date. Consequently, our understanding of the natural history of sleep disorders in ALS remains sparse. The efficacy of available treatments for treating sleep disorders in ALS patients has not been explored. Future studies should adopt a multicentric and longitudinal approach to investigate this. Both use of self-reported measures (such as PSQI and ESS) and video polysomnography are needed to fully understand this non-motor symptom in ALS.
Regarding RLS, it might be both misdiagnosed and underdiagnosed in ALS, as limb pain and cramps could be mistaken for RLS. A distinguishing feature could be its rhythmic occurrence, unlike spasms or pain, which do not follow a circadian rhythm. Healthcare providers should be aware about this symptom in ALS as it can be treated. Left unaddressed, RLS can disrupt nighttime sleep, exacerbate depression, and diminish overall QoL [221].
Cognitive and neuropsychiatric symptoms
Cognitive and behavioural dysfunction in ALS
Cognitive and behavioural abnormalities have long been known to be concomitant with ALS [222,223,224], especially relating to behavioural and verbal variants of the frontotemporal dementia (FTD) spectrum [223]. However, in ALS patients, cognitive dysfunction is not confined solely to the FTD phenotype. One meta-analysis revealed that every cognitive domain, except visuoperception, is affected in ALS [224]. The cognitive spectrum in ALS exhibits significant heterogeneity, encompassing deficits in executive function, attention, verbal fluency, naming, language, social cognition, visuospatial abilities, verbal memory, and other cognitive domains [225]. However, the primary cognitive domains affected in ALS are executive, language and slightly less observed—memory impairment [226]. The prevalence of cognitive impairment (CI) varies between 30 and 75%, and it correlates with later disease stages, patients' genotype/phenotype, and has an adverse impact on patient survival. Similar to that, behavioural dysfunction is prevalent in as many as half of ALS patients [225]. Apathy is the predominant behavioural change, present in as many as 60% of patients. Features typical of FTD, like disinhibition, decreased empathy, stereotyped behaviours, and dietary changes, are also commonly found in ALS and are often associated with declines in social cognition [227]. Interestingly, one study showed that ALS patients with behavioural impairment (ALSbi) experienced more pronounced deterioration in motor function compared to ALS patients with cognitive impairment (ALSci) [228]. The indication that ALS with a bulbar site of onset is more frequently associated with cognitive decline [223] is a subject of dispute. More to that, when the speed of diagnostic neuropsychological tests was adjusted for dysarthria, no significant difference between spinal and bulbar onset was found [224]. Detecting and monitoring CI in ALS early on is essential not just for prognostic reasons, but also because it could have significant implications for future clinical trials [229].
Diagnosis of cognitive impairment in ALS
Diagnosing CI in ALS patients involves using specific neuropsychological screening batteries, such as the Edinburgh Cognitive and Behavioral ALS Screen (ECAS) and the ALS Cognitive Behavioral Screen (ALS-CBS). These tests are thoroughly standardized, available in several languages, and can be performed in short time [230,231,232,233,234,235,236]. Additionally, screening tests such as the Mini-Mental State Examination (MMSE) and the Montreal Cognitive Assessment (MOCA), which are more applicable for primary caregivers, have been shown to effectively screen for CI in ALS patients [224]. Some other neuropsychological assessment tools, such as Arrows and Colors Cognitive Test [237], Sydney Language Battery [238], Test for Reception of Grammar [239], Frontal Assessment Battery [240] and Dimensional Apathy Scale [241] can also be useful [226].
Neuroimaging techniques serve as an essential adjunct to diagnostics. While ALS patients without CI commonly display cerebral atrophy primarily in the primary motor cortex, ALSci exhibit more widespread grey matter atrophy. This involves not only bilateral involvement of the primary motor cortex but also extends to frontotemporoparietal regions, the somatosensory area, limbic cortex, thalamus, striatum, pallidum, hippocampus, entorhinal cortex, cingulate cortex, amygdala, bilateral cerebellum, basal ganglia, and various other brain regions [225, 242]. In magnetic resonance imaging (MRI) studies of ALSci patients carrying the C9orf72 mutations, an increased pattern of cortical and subcortical damage has been observed. Additionally, there is evidence of more extensive damage to the mediodorsal and pulvinar nuclei in ALSci, rather than an evenly distributed involvement of the thalamus [243]. This thalamo-cortico-striatal atrophy pattern is specific for the C9orf72 genotype. In addition to the current standard of care utilizing MRI scans, novel FDG-PET imaging has demonstrated the capability to differentiate between ALS-FTD, ALSci, and ALS without CI [244, 245]. In ALS-FTD there is a significant FDG-hypometabolism in the frontal lobe compared to ALS without CI. Patients with ALSci show an intermediate metabolism compared to ALS without CI and ALS-FTD. Interestingly, ALS-FTD displays a distinctive pattern with a relative hypermetabolism in the cerebellum [245].
While the level of the neurodegeneration marker neurofilament light chain (NfL) in serum correlates with ALS phenotype and disease progression, studies have not found a significant correlation with CI in ALS [246]. Serum NfL levels in patients with ALSci do not show a significant difference compared to ALS patients without CI. However, pTau levels are elevated in ALSci compared to ALS without CI in cerebrospinal fluid [247].
Moreover, genetic testing serves as a crucial tool for predicting the development of FTD symptoms in ALS patients, given that the genotype in familial ALS determines the disease phenotype [248,249,250]. Table 1 provides an overview of some ALS gene mutations, their frequency, and their association with the FTD spectrum.
Treatment of cognitive impairment in ALS
Currently there are only limited treatment possibilities for CI in ALS. Treatments generally follow the regime applied for other forms of CI, with a focus on occupational therapy and speech therapy. There is evidence suggesting that patients with bulbar symptoms may benefit from early access to communication devices, enhancing patient acceptance and proficiency with the device as cognitive and motor skills decline, thereby facilitating better interpersonal participation [251]. A clinical trial showed efficacy of dextromethorphan/quinidine for treatment of pathological crying and laughing, a common feature in ALS-FTD and ALS as signs of pseudobulbar affection [252]. In a small case series, pathological crying also responded well to the treatment with selective serotonin reuptake inhibitors (SSRI) [253].
Novel gene therapies using antisense oligonucleotides (ASOs) may show potential in the treatment not only of motor symptoms but also of CI in ALS patients [254,255,256]. Of the currently established ASOs (anti-SOD1 and anti-FUS) only the FUS gene is currently a viable target linked to the ALS-FTD spectrum. There has been promising first data of ASOs targeting C9orf72 and with it, the most frequent cause for familiar ALS-FTD. In a first-in-human trial conducted in 2022, a patient underwent intrathecal treatment with anti-C9orf72 ASOs, resulting in target engagement and a substantial reduction of PolyGP. However, levels of NfL and phosphorylated Neurofilament Heavy Chain (pNFH) in serum and cerebrospinal fluid did not exhibit a significant decrease [257]. Additionally, two clinical trials, one initiated by Biogen (https://investors.biogen.com/newsreleases/news-release-details/biogen-and-ionis-announce-topline-phase-1-study-results) and the other by Wave Pharmaceuticals (https://www.thepharmaletter.com/article/wave-life-sciences-endswve-004-program), were terminated due to neurotoxicity and/or insufficient clinical efficacy.
CI in ALS is a common cause for increase of morbidity in ALS patients, for which primary care givers should regularly screen to quickly adapt the necessary treatment options. While there is currently no direct treatment for CI in ALS, the emergence of novel gene therapeutics offers hope for the future, at least for familial cases. Considering the predictive value of the genotype in developing ALS-FTD, we suppose that genetic testing should be offered to every ALS patient. This approach aims to enhance disease management and, if applicable, facilitate appropriate treatment strategies. Further research is essential, as novel therapeutics may only be applicable to a fraction of ALS patients with the right genotype and as the primary readout of studies was the ALSFRS-R, which does not reflect CI in patients.
Depression in ALS
Depression is highly prevalent among ALS patients, with a meta-analysis indicating that approximately 34% (27–41%) of individuals diagnosed with ALS experience depressive symptoms [258]. This increased frequency of depressive symptoms is also shared with the caregivers of patients. In a prospective cohort study involving 33 ALS patients and their families, it was observed that 13% of the patients and 29% of the relatives experienced symptoms of depression after the diagnosis of ALS. Interestingly, there seems to be no correlation between physical disability and the frequency of depression and mental well-being in ALS patients [259, 260]. However, this correlation is observed in caregivers of the patients. In this context, there is an increase in caregiver burden corresponding to the escalating physical disabilities associated with disease progression [261, 262]. This observation could not be confirmed in a study by Chen et al., where no correlations were found between the decrease in ALSFRS-R and the severity of depression in either patients or caregivers.
Depression not only negatively impacts QoL but is also associated with a shorter survival time [263]. This can be partly explained by an increase in loss of appetite and weight loss, which, in itself, is a negative prognostic factor for ALS [264]. Furthermore, ALS patients have a higher risk of suicide than the general population, with a particularly pronounced risk in the early days after diagnosis [265].
Pathophysiology of depression in ALS
The pathophysiology underlying depression in ALS patients is unfortunately poorly understood. Depression solely attributed to the diagnosis can only partially account for the prevalence, especially considering that ALS is a multi-system disorder affecting various parts of CNS. Currently, there are no available studies providing a conclusive explanation for depression in ALS. A study by Benbrika et al. indicated that patients with elevated cortical thinning at the time of diagnosis more frequently exhibited depressive or anxious symptoms. However, over the course of the study, there was no exacerbation of psychological symptoms despite an increase in cortical thinning [266].
Treatment of depression in ALS
Due to the heavy reduction in QoL treatment of depression in ALS is of high importance. Depression in ALS is usually treated with a combination of psychotherapy and pharmacological intervention. Although there is no clinical evidence supporting the pharmacological treatment of depression in ALS, there is a general consensus on the use of SSRIs or tricyclic antidepressants. These medications have shown efficacy in managing depressive symptoms in patients with other major and potentially life-threatening comorbidities, such as cancer [267]. There is limited evidence for a benefit of cognitive behavioral therapy in ALS patients suffering from depression [268]. Gould et al. showed the feasibility of engaging people living with motor neuron disease in Acceptance and Commitment Therapy, an acceptance-based behaviour therapy, which was positively received by this particular population. Moreover, despite the expected deterioration in disease-related functioning and health status, anxiety levels and psychological QoL exhibited improvements over the 6-month period. A fully powered randomized controlled trial is underway to evaluate the clinical and cost-effectiveness of Acceptance and Commitment Therapy for people living with motor neuron disease [269].
Despite experiencing similar rates of depressive symptoms as ALS patients, their caregivers receive, on average, less frequent treatment for depression, whether psychotherapeutically or pharmacologically. This discrepancy should be considered by clinicians treating ALS patients, as caregiver burden is correlated with their depression and anxiety. Providing sufficient support to caregivers is crucial for enhancing patient well-being [270].
Future directions
CI continues to be a source for increased morbidity in ALS patients especially for patients suffering from ALS-FTD. However, more widespread awareness and more routinely performed genetic testing will allow faster diagnosis, thereby leading to faster access to the necessary support in the primary care setting. It remains to be seen how novel treatment possibilities affect the cognitive decline. With the current advent of gene therapies for several types of familiar ALS having the potential to significantly slow the progress of neurodegeneration there needs to be future research focused on the CI in the addition to the motor symptoms. Routinely testing, also in everyday practice, using the available bedside test or if possible self-reported questionnaires will allow for easy assessing of the ALS cohort and thereby facilitate the future clinical studies needed to slow CI.
Samra et al. discovered in a substantial cohort of patients with FTD that incorporating a Global Motor Score into the CDR® plus NACC FTLD scale (one of the main rating scales currently used for FTD [271]) resulted in the development of a new CDR® plus NACC FTLD-M scale. This adaptation led to a more precise assessment of disease severity compared to the original scale [272]. Inspired by their approach, one could consider employing principal component analysis to incorporate cognitive features into an expanded version of the ALS-FRS-R scale. This holistic approach would maybe enable a more comprehensive evaluation of disease severity in ALS patients, encompassing both motor and cognitive dimensions.
Psychiatric symptoms in fatal diseases are common but often underdiagnosed and thereby naturally undertreated. Regularly testing and assessing the need for psychological support of ALS patients and their caregivers is crucial for early recognition of the need for both medical and therapy-based psychiatric treatment. However, with very limited evidence for the effectiveness of using antidepressants in ALS, there needs to be future, if possible multicentric, clinical trials in this area of research. Improving the treatment of the psychological impact of an ALS diagnosis, for both patients and caregivers, can lead to an increase in QoL and a decrease in the burden of the disease.
Metabolic abnormalities and weight loss
Weight loss is a major challenge in handling ALS patients. More than half of patients report significant weight loss at the time of diagnosis [273, 274], with those presenting with a bulbar onset reporting a higher percentage loss of body weight [275]. Interestingly, weight loss has also been observed in presymptomatic gene carriers of ALS [276], with evidence suggesting that it can precede the onset of weakness by more than a decade [177, 277,278,279]. Weight stability is a prognostic factor for overall survival, and a decrease in body weight is strongly correlated with the risk of death in a dose–response relationship. This correlation holds true for weight loss before diagnosis, but neither body mass index (BMI) before nor at the point of diagnosis shows the same correlation [280]. For every 10% weight loss, there is an increase in mortality of 16.5–23%, with a median survival of 14.7–30.5 months for patients with weight loss and 22.5–48.8 months for those without [275, 281]. This effect is particularly pronounced in female patients, who experience increased weight loss after diagnosis [282].
Physiology of weight loss in ALS
Weight loss in ALS results from various factors, broadly categorized into reduced caloric intake and increased energy expenditure. Dysphagia primarily contributes to weight loss in ALS, but other factors such as anorexia, depression, cognitive impairment, polypharmacy, and difficulties in meal preparation due to weakness also can lead to reduced caloric intake in ALS patients [283]. Between 25 and 66% of ALS patients report a loss of appetite, resulting in weight loss independent of other factors like dysphagia [264, 284,285,286]. This loss of appetite correlates with disease progression, especially with the bulbar subscore of the ALSFRS-R, but is independent of anthropometric measures such as weight, BMI, fat mass, or fat-free mass [284].
The reduction in caloric intake is exacerbated by hypermetabolism observed in 50–80% of ALS patients [287, 288]. Hypermetabolic ALS patients have an increased resting energy expenditure (REE) of approximately 1500 kcal/24 h compared to 1230 kcal/24 h in normal subjects [289, 290]. Additionally, ALS patients also experience increased energy expenditure due to weakened skeletal muscles, nonfunctional muscular activity, and pseudobulbar motor activities [291]. Metabolic alterations in ALS may be partially attributed to mitochondrial dysfunction observed in SOD1 and C9orf72 mouse models [292, 293]. This dysfunction results in reduced ATP production and increased oxidative stress, contributing to the metabolic changes seen in the disease. Furthermore, there appears to be an impairment in glucose metabolism in ALS mouse models. Borges et al. showed that in SOD1 G93A mice the pentose phosphate pathway is impaired. The intermediate ribose 5-phosphate has been shown to be decreased [294].
In addition, a loss of metabolic flexibility was found in astrocytes of both C9orf72 and sporadic ALS patients [295]. This is attributed to a decrease in glycogen phosphorylase and phosphoglucomutase, resulting in a reduction of glycogen metabolism during times of high energy demand. Besides glycogen metabolism, it has been demonstrated that glycolysis is impaired in ALS patients.
Borges et al. demonstrated reduced total and 1−13C labeled lactate and alanine levels in cortex and spinal cord of SOD1 G93A mouse model using nuclear magnetic resonance spectroscopy. In their study, no reduction in 1−13C-labeled glucose was found, suggesting a decrease in glucose metabolism [296]. In humans, autopsy analysis of 33 ALS patients showed TDP-43 pathology in the hypothalamus, with those having notably reduced BMI exhibiting TDP-43 pathology in the lateral hypothalamic area [297], suggesting that pathology in this region may contribute to metabolic disturbances and weight loss in ALS.
Dupuis et al. conducted a combined mouse and human study and found that ALS is associated with alterations in the melanocortin system [195]. Despite the administration of pioglitazone, ALS patients in the study did not demonstrate weight gain, implying potential disruptions in the hypothalamic melanocortin pathway. However, patients did show reduced glycaemia and liver enzyme levels, along with increased adiponectin levels, which are efficacy markers in the periphery. This suggests that ALS patients did not merely fail to respond to the drug. Pioglitazone typically suppresses hypothalamic neurons that produce proopiomelanocortin (POMC), thereby increasing food intake [298]. The authors suggested that the already diminished melanocortin tone in ALS may hinder the effectiveness of pioglitazone in silencing POMC neurons. In presymptomatic SOD1 G86R mice, Dupuis et al. found a decrease of POMC and an increase of endogenous melanocortin inhibitor agouti-related peptide, fitting to the loss of pioglitazone effect. Furthermore, the same group of authors found that melanin concentrating hormone (MCH)-positive neurons are affected in lateral hypothalamic area in both ALS patients and three ALS mouse models (SOD1G86R, SOD1G93A and B6-FusΔNLS1Ldup/Crl). Continuous intracerebroventricular delivery of MCH (1.2 µg/d) induced weight gain in male Sod1G86R mice, increasing food intake, restoring expression of the appetite-related neuropeptide AgRP (agouti-related protein), and altering the respiratory exchange ratio. The authors also observed pTDP-43 pathology and neurodegeneration in the lateral hypothalamic area in autopsy studies of 17 sporadic ALS cases. These findings were suggestive that the loss of hypothalamic MCH neurons contributes to metabolic changes, including weight loss and decreased appetite.
Treatment of non-dysphagia-related weight loss
Stabilizing the weight of ALS patients has been the main goal in treatment of metabolic changes in ALS. Non-invasive ventilation (NIV) has been shown to significantly reduce the REE in ALS patients, even in non-hypermetabolic patients exhibiting respiratory insufficiency due to ventilatory dysfunction [299]. This reduction in energy expenditure is postulated to be due to the elimination of the energy burden on inspiratory neck muscles. Loss of appetite has been investigated as a secondary outcome in a randomized controlled trial using THC compared to placebo. In this study, 27 ALS patients received 5 mg THC twice per day; however, there was no improvement in loss of appetite in the study group [300]. Weight loss and the consequent disease progression continue to pose significant challenges in the treatment of ALS patients, with currently available therapeutic options proving inadequate in addressing these issues. As of now, only the early implementation of NIV has been shown to counteract the increase in energy expenditure and should be evaluated as soon as patients show ventilatory affection.
Weight loss plays a crucial role in the progression of ALS and should be continuously monitored. Metabolic changes, such as an increased REE, aggravates the risk of malnutrition, which is already present due to dysphagia and reduced caloric intake. Strict weight monitoring should be performed to adjust necessary treatments, such as an early use of NIV. Further research in the future is needed to better understand and be able to influence the metabolic part of the weight loss equation in ALS.
In summary, it is vital to assess whether energy intake is insufficient relative to energy expenditure in ALS patients. If this discrepancy exists, efforts should be made to augment energy intake, either through oral route or by considering placement of a percutaneous endoscopic gastrostomy (PEG) tube.
Future directions
Hypermetabolism undoubtedly plays a crucial role in the pathogenesis of ALS. While significant strides have been made in understanding this issue over the past two decades, further research is needed to fully comprehend its impact on disease progression and whether it serves as a pivotal trigger for neurodegeneration. Translational studies are essential for the development of effective treatments targeting metabolism in ALS.
Conclusion
ALS is a multisystem disorder characterized by neurodegeneration affecting both motor and non-motor regions of the brain. While motor symptoms are relatively well-known, non-motor symptoms remain enigmatic, often overlooked, and consequently undertreated. Despite their association with lower QoL, the understanding, diagnosis, and treatment of these symptoms lag behind. Comprehensive awareness and recognition of these non-motor symptoms in ALS are crucial for accurate diagnosis and effective intervention. This review underscores the importance of shedding light on these “less-explored” aspects of ALS, emphasizing their impact on QoL and the necessity for improved diagnostic tools. Addressing the existing knowledge gaps in non-motor symptoms of ALS, the review underscores the necessity for multicentric, prospective, and longitudinal studies to unravel their natural history. Additionally, the urgency for developing efficient, self-reported measures for fast diagnosis and monitoring of non-motor symptoms in clinical practice is highlighted, aiming to guide timely and tailored interventions. The overarching goal is to enhance our understanding of non-motor symptoms in ALS and pave the way for more effective management strategies.
Data Availability
Not applicable.
Abbreviations
- ALS:
-
Amyotrophic lateral sclerosis
- ALSci:
-
ALS patients with cognitive impairment
- ALSbi:
-
ALS patients with behavioural impairment
- ALSFRS-R:
-
ALS Functional Rating Scale Score-Revised
- ASOs:
-
Antisense oligonucleotides
- BDNF:
-
Brain-derived neurotrophic factor
- BMI:
-
Body mass index
- C9orf72 :
-
Chromosome 9 open reading frame 72 Gene
- CI:
-
Cognitive impairment
- CNS:
-
Central nervous system
- CSA:
-
Cross-sectional area
- EFNS:
-
European Federation of the Neurological Societies
- fALS:
-
Familial amyotrophic lateral sclerosis
- FDA:
-
Food and Drug Administration
- FSS:
-
Fatigue Severity Scale
- FTD:
-
Frontotemporal dementia
- FUS :
-
Fused in sarcoma Gene
- GABA:
-
Gamma-aminobutyric acid
- HRV:
-
Heart rate variability
- MRI:
-
Magnetic resonance imaging
- NIV:
-
Non-invasive ventilation
- NSAIDs:
-
Nonsteroidal anti-inflammatory drugs
- PEG:
-
Percutaneous endoscopic gastrostomy
- pTAU:
-
Hyperphosphorylated Tau protein
- pTDP-43:
-
Phosphorylated 43-kDa TDP protein
- PVR:
-
Post void residual
- QoL:
-
Quality of life
- REE:
-
Resting energy expenditure
- REM:
-
Rapid eye movement
- RLS:
-
Restless legs syndrome
- sALS:
-
Sporadic amyotrophic lateral sclerosis
- SOD1 :
-
Superoxide dismutase 1 Gene
- SSRI:
-
Selective serotonin reuptake inhibitors
- TARDBP :
-
Trans-activation response (TAR) DNA-binding protein 43 Gene
- TDP-43:
-
Transactive response DNA binding protein 43
- THC/CBD:
-
Delta-9-tetrahydrocannabinol and cannabidiol
- VAS:
-
Visual Analogue Scale
References
Feldman EL, Goutman SA, Petri S, Mazzini L, Savelieff MG, Shaw PJ, Sobue G (2022) Amyotrophic lateral sclerosis. Lancet 400:1363–1380
Chio A, Logroscino G, Traynor BJ, Collins J, Simeone JC, Goldstein LA, White LA (2013) Global epidemiology of amyotrophic lateral sclerosis: a systematic review of the published literature. Neuroepidemiology 41:118–130
Hardiman O, Al-Chalabi A, Chio A, Corr EM, Logroscino G, Robberecht W, Shaw PJ, Simmons Z, van den Berg LH (2017) Amyotrophic lateral sclerosis. Nat Rev Dis Primers 3:17071
Chio A, Battistini S, Calvo A, Caponnetto C, Conforti FL, Corbo M, Giannini F, Mandrioli J, Mora G, Sabatelli M, Consortium I, Ajmone C, Mastro E, Pain D, Mandich P, Penco S, Restagno G, Zollino M, Surbone A (2014) Genetic counselling in ALS: facts, uncertainties and clinical suggestions. J Neurol Neurosurg Psychiatry 85:478–485
Kabashi E, Valdmanis PN, Dion P, Spiegelman D, McConkey BJ, Vande Velde C, Bouchard JP, Lacomblez L, Pochigaeva K, Salachas F, Pradat PF, Camu W, Meininger V, Dupre N, Rouleau GA (2008) TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet 40:572–574
Mackenzie IR, Bigio EH, Ince PG, Geser F, Neumann M, Cairns NJ, Kwong LK, Forman MS, Ravits J, Stewart H, Eisen A, McClusky L, Kretzschmar HA, Monoranu CM, Highley JR, Kirby J, Siddique T, Shaw PJ, Lee VM, Trojanowski JQ (2007) Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann Neurol 61:427–434
Verbeeck C, Deng Q, Dejesus-Hernandez M, Taylor G, Ceballos-Diaz C, Kocerha J, Golde T, Das P, Rademakers R, Dickson DW, Kukar T (2012) Expression of Fused in sarcoma mutations in mice recapitulates the neuropathology of FUS proteinopathies and provides insight into disease pathogenesis. Mol Neurodegener 7:53
Ravits JM, La Spada AR (2009) ALS motor phenotype heterogeneity, focality, and spread: deconstructing motor neuron degeneration. Neurology 73:805–811
Braak H, Brettschneider J, Ludolph AC, Lee VM, Trojanowski JQ, Del Tredici K (2013) Amyotrophic lateral sclerosis–a model of corticofugal axonal spread. Nat Rev Neurol 9:708–714
Ludolph AC, Brettschneider J (2015) TDP-43 in amyotrophic lateral sclerosis - is it a prion disease? Eur J Neurol 22:753–761
Del Tredici K, Braak H (2022) Neuropathology and neuroanatomy of TDP-43 amyotrophic lateral sclerosis. Curr Opin Neurol 35:660–671
Shojaie A, Rota S, Al Khleifat A, Ray Chaudhuri K, Al-Chalabi A (2023) Non-motor symptoms in amyotrophic lateral sclerosis: lessons from Parkinson’s disease. Amyotroph Lateral Scler Frontotemporal Degener 2023:1–10
Fang T, Jozsa F, Al-Chalabi A (2017) Nonmotor symptoms in amyotrophic lateral sclerosis: a systematic review. Int Rev Neurobiol 134:1409–1441
Shojaie A, Al Khleifat A, Opie-Martin S, Sarraf P, Al-Chalabi A (2024) Non-motor symptoms in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener 25:61–66
Drake ME Jr (1983) Chronic pain syndrome in amyotrophic lateral sclerosis. Arch Neurol 40:453–454
An R, Wu Y, Li Y, Li X, Ai S, Xu Y, He C (2022) Pain-related factors and their impact on quality of life in Chinese patients with amyotrophic lateral sclerosis. Front Neurosci 16:897598
Kong Z, Chen P, Jiang J, Wang X, Wang Y, Shi Y, Zhao B, Zhu J (2021) Pain characteristics in amyotrophic lateral sclerosis patients and its impact on quality of life: a prospective observational study in a northern city of China. Ann Palliat Med 10:1668–1674
Chio A, Canosa A, Gallo S, Moglia C, Ilardi A, Cammarosano S, Papurello D, Calvo A (2012) Pain in amyotrophic lateral sclerosis: a population-based controlled study. Eur J Neurol 19:551–555
Wallace VC, Ellis CM, Burman R, Knights C, Shaw CE, Al-Chalabi A (2014) The evaluation of pain in amyotrophic lateral sclerosis: a case controlled observational study. Amyotroph Lateral Scler Frontotemporal Degener 15:520–527
Akerblom Y, Jakobsson Larsson B, Zetterberg L, Asenlof P (2020) The multiple faces of pain in motor neuron disease: a qualitative study to inform pain assessment and pain management. Disabil Rehabil 42:2123–2132
de Castro-Costa CM, Oria RB, Machado-Filho JA, Franco MT, Diniz DL, Giffoni SD, Santos TJ, da Cunha FM, de Bruin VS, Teixeira CA (1999) Amyotrophic lateral sclerosis. Clinical analysis of 78 cases from Fortaleza (northeastern Brazil). Arq Neuropsiquiatr 57:761–774
Truini A, Biasiotta A, Onesti E, Di Stefano G, Ceccanti M, La Cesa S, Pepe A, Giordano C, Cruccu G, Inghilleri M (2015) Small-fibre neuropathy related to bulbar and spinal-onset in patients with ALS. J Neurol 262:1014–1018
Dalla Bella E, Lombardi R, Porretta-Serapiglia C, Ciano C, Gellera C, Pensato V, Cazzato D, Lauria G (2016) Amyotrophic lateral sclerosis causes small fiber pathology. Eur J Neurol 23:416–420
Stephens HE, Lehman E, Raheja D, Yang C, Walsh S, McArthur DB, Simmons Z (2015) Pain in amyotrophic lateral sclerosis: Patient and physician perspectives and practices. Amyotroph Lateral Scler Frontotemporal Degener 17:21–29
Hanisch F, Skudlarek A, Berndt J, Kornhuber ME (2015) Characteristics of pain in amyotrophic lateral sclerosis. Brain Behav 5:e00296
Hoffman AJ, Jensen MP, Abresch RT, Carter GT (2005) Chronic pain in persons with neuromuscular disease. Phys Med Rehabil Clin N Am 16:1099–1112, xii
Abe Y, Miyashita M, Ito N, Shirai Y, Momose Y, Ichikawa Y, Tsuji S, Kazuma K (2008) Attitude of outpatients with neuromuscular diseases in Japan to pain and use of analgesics. J Neurol Sci 267:22–27
Chio A, Mora G, Lauria G (2017) Pain in amyotrophic lateral sclerosis. Lancet Neurol 16:144–157
Beswick E, Forbes D, Johnson M, Newton J, Dakin R, Glasmcher S, Abrahams S, Carson A, Chandran S, Pal S (2024) Non-motor symptoms in motor neuron disease: prevalence, assessment and impact. Brain Commun 6:fcad336
Hurwitz N, Radakovic R, Boyce E, Peryer G (2021) Prevalence of pain in amyotrophic lateral sclerosis: a systematic review and meta-analysis. Amyotroph Lateral Scler Frontotemporal Degener 22:449–458
Kwak S (2022) Pain in amyotrophic lateral sclerosis: a narrative review. J Yeungnam Med Sci 39:181–189
Haanpaa ML, Backonja MM, Bennett MI, Bouhassira D, Cruccu G, Hansson PT, Jensen TS, Kauppila T, Rice AS, Smith BH, Treede RD, Baron R (2009) Assessment of neuropathic pain in primary care. Am J Med 122:S13-21
Mochizuki Y, Mizutani T, Shimizu T, Kawata A (2011) Proportional neuronal loss between the primary motor and sensory cortex in amyotrophic lateral sclerosis. Neurosci Lett 503:73–75
Oyanagi K, Mochizuki Y, Nakayama Y, Hayashi K, Shimizu T, Nagao M, Hashimoto T, Yamazaki M, Matsubara S, Komori T (2015) Marked preservation of the visual and olfactory pathways in ALS patients in a totally locked-in state. Clin Neuropathol 34:267–274
Han J, Ma L (2010) Study of the features of proton MR spectroscopy ((1)H-MRS) on amyotrophic lateral sclerosis. J Magn Reson Imaging 31:305–308
Rajagopalan V, Pioro EP (2021) Corticospinal tract and related grey matter morphometric shape analysis in ALS phenotypes: a fractal dimension study. Brain Sci 2021:11
Agosta F, Valsasina P, Absinta M, Riva N, Sala S, Prelle A, Copetti M, Comola M, Comi G, Filippi M (2011) Sensorimotor functional connectivity changes in amyotrophic lateral sclerosis. Cereb Cortex 21:2291–2298
Agosta F, Canu E, Inuggi A, Chio A, Riva N, Silani V, Calvo A, Messina S, Falini A, Comi G, Filippi M (2014) Resting state functional connectivity alterations in primary lateral sclerosis. Neurobiol Aging 35:916–925
Sala A, Iaccarino L, Fania P, Vanoli EG, Fallanca F, Pagnini C, Cerami C, Calvo A, Canosa A, Pagani M, Chio A, Cistaro A, Perani D (2019) Testing the diagnostic accuracy of [18F]FDG-PET in discriminating spinal- and bulbar-onset amyotrophic lateral sclerosis. Eur J Nucl Med Mol Imaging 46:1117–1131
Matheson JK, Harrington HJ, Hallett M (1986) Abnormalities of multimodality evoked potentials in amyotrophic lateral sclerosis. Arch Neurol 43:338–340
Dasheiff RM, Drake ME, Brendle A, Erwin CW (1985) Abnormal somatosensory evoked potentials in amyotrophic lateral sclerosis. Electroencephalogr Clin Neurophysiol 60:306–311
Bosch EP, Yamada T, Kimura J (1985) Somatosensory evoked potentials in motor neuron disease. Muscle Nerve 8:556–562
Constantinovici A (1993) Abnormal somatosensory evoked potentials in amyotrophic lateral sclerosis. Rom J Neurol Psychiatry 31:273–278
Radtke RA, Erwin A, Erwin CW (1986) Abnormal sensory evoked potentials in amyotrophic lateral sclerosis. Neurology 36:796–801
Ogata K, Tobimatsu S, Furuya H, Kira J (2001) Sporadic amyotrophic lateral sclerosis showing abnormal somatosensory evoked potentials: a report of three cases. Fukuoka Igaku Zasshi 92:242–250
Shimizu T, Bokuda K, Kimura H, Kamiyama T, Nakayama Y, Kawata A, Isozaki E, Ugawa Y (2018) Sensory cortex hyperexcitability predicts short survival in amyotrophic lateral sclerosis. Neurology 90:e1578–e1587
Shimizu T, Nakayama Y, Funai A, Morishima R, Hayashi K, Bokuda K, Nakata Y, Isozaki E (2020) Progressive deterioration of sensory cortex excitability in advanced amyotrophic lateral sclerosis with invasive ventilation. Amyotroph Lateral Scler Frontotemporal Degener 21:147–149
Khalili-Ardali M, Wu S, Tonin A, Birbaumer N, Chaudhary U (2021) Neurophysiological aspects of the completely locked-in syndrome in patients with advanced amyotrophic lateral sclerosis. Clin Neurophysiol 132:1064–1076
Iglesias C, Sangari S, El Mendili MM, Benali H, Marchand-Pauvert V, Pradat PF (2015) Electrophysiological and spinal imaging evidences for sensory dysfunction in amyotrophic lateral sclerosis. BMJ Open 5:e007659
Cohen-Adad J, El Mendili MM, Morizot-Koutlidis R, Lehericy S, Meininger V, Blancho S, Rossignol S, Benali H, Pradat PF (2013) Involvement of spinal sensory pathway in ALS and specificity of cord atrophy to lower motor neuron degeneration. Amyotroph Lateral Scler Frontotemporal Degener 14:30–38
Hammad M, Silva A, Glass J, Sladky JT, Benatar M (2007) Clinical, electrophysiologic, and pathologic evidence for sensory abnormalities in ALS. Neurology 69:2236–2242
Schoeberl F, Abicht A, Kuepper C, Voelk S, Sonnenfeld S, Tonon M, Schaub A, Scholz V, Kleinle S, Erdmann H, Wolf DA, Reilich P (2022) Sensory neuropathy due to RFC1 in a patient with ALS: More than a coincidence? J Neurol 269:2774–2777
Guo YS, Wu DX, Wu HR, Wu SY, Yang C, Li B, Bu H, Zhang YS, Li CY (2009) Sensory involvement in the SOD1-G93A mouse model of amyotrophic lateral sclerosis. Exp Mol Med 41:140–150
Sabado J, Casanovas A, Tarabal O, Hereu M, Piedrafita L, Caldero J, Esquerda JE (2014) Accumulation of misfolded SOD1 in dorsal root ganglion degenerating proprioceptive sensory neurons of transgenic mice with amyotrophic lateral sclerosis. Biomed Res Int 2014:852163
Rubio MA, Herrando-Grabulosa M, Gaja-Capdevila N, Vilches JJ, Navarro X (2022) Characterization of somatosensory neuron involvement in the SOD1(G93A) mouse model. Sci Rep 12:7600
Pugdahl K, Fuglsang-Frederiksen A, de Carvalho M, Johnsen B, Fawcett PR, Labarre-Vila A, Liguori R, Nix WA, Schofield IS (2007) Generalised sensory system abnormalities in amyotrophic lateral sclerosis: a European multicentre study. J Neurol Neurosurg Psychiatry 78:746–749
Theys PA, Peeters E, Robberecht W (1999) Evolution of motor and sensory deficits in amyotrophic lateral sclerosis estimated by neurophysiological techniques. J Neurol 246:438–442
Gregory R, Mills K, Donaghy M (1993) Progressive sensory nerve dysfunction in amyotrophic lateral sclerosis: a prospective clinical and neurophysiological study. J Neurol 240:309–314
Pegat A, Bouhour F, Mouzat K, Vial C, Pegat B, Leblanc P, Broussolle E, Millecamps S, Lumbroso S, Bernard E (2019) Electrophysiological characterization of C9ORF72-associated amyotrophic lateral sclerosis: a retrospective study. Eur Neurol 82:106–112
Pugdahl K, Fuglsang-Frederiksen A, Johnsen B, de Carvalho M, Fawcett PR, Labarre-Vila A, Liguori R, Nix WA, Schofield IS (2008) A prospective multicentre study on sural nerve action potentials in ALS. Clin Neurophysiol 119:1106–1110
Isak B, Tankisi H, Johnsen B, Pugdahl K, Torvin MA, Finnerup NB, Christensen PB, Fuglsang-Frederiksen A (2016) Involvement of distal sensory nerves in amyotrophic lateral sclerosis. Muscle Nerve 54:1086–1092
Luigetti M, Conte A, Del Grande A, Bisogni G, Romano A, Sabatelli M (2012) Sural nerve pathology in ALS patients: a single-centre experience. Neurol Sci 33:1095–1099
Weis J, Katona I, Muller-Newen G, Sommer C, Necula G, Hendrich C, Ludolph AC, Sperfeld AD (2011) Small-fiber neuropathy in patients with ALS. Neurology 76:2024–2029
Isak B, Pugdahl K, Karlsson P, Tankisi H, Finnerup NB, Furtula J, Johnsen B, Sunde N, Jakobsen J, Fuglsang-Frederiksen A (2017) Quantitative sensory testing and structural assessment of sensory nerve fibres in amyotrophic lateral sclerosis. J Neurol Sci 373:329–334
Bombaci A, Lupica A, Pozzi FE, Remoli G, Manera U, Di Stefano V (2023) Sensory neuropathy in amyotrophic lateral sclerosis: a systematic review. J Neurol 2023:1
Moisset X, Cornut-Chauvinc C, Clavelou P, Pereira B, Dallel R, Guy N (2016) Is there pain with neuropathic characteristics in patients with amyotrophic lateral sclerosis? A cross-sectional study. Palliat Med 30:486–494
An R, Li Y, He X, Li C, Li X, Xu Y, He C (2021) The evaluation of pain with nociceptive and neuropathic characteristics from three different perspectives in amyotrophic lateral sclerosis patients: a case controlled observational study in southwestern China. Neural Plast 2021:5537892
Lopes LCG, Galhardoni R, Silva V, Jorge FMH, Yeng LT, Callegaro D, Chadi G, Teixeira MJ, Ciampi de Andrade D (2018) Beyond weakness: Characterization of pain, sensory profile and conditioned pain modulation in patients with motor neuron disease: A controlled study. Eur J Pain 22:72–83
van Hecke O, Austin SK, Khan RA, Smith BH, Torrance N (2014) Neuropathic pain in the general population: a systematic review of epidemiological studies. Pain 155:654–662
Miller TM, Layzer RB (2005) Muscle cramps. Muscle Nerve 32:431–442
Caress JB, Ciarlone SL, Sullivan EA, Griffin LP, Cartwright MS (2016) Natural history of muscle cramps in amyotrophic lateral sclerosis. Muscle Nerve 53:513–517
Dietz V, Sinkjaer T (2012) Spasticity. Handb Clin Neurol 109:197–211
Mayer NH (1997) Clinicophysiologic concepts of spasticity and motor dysfunction in adults with an upper motoneuron lesion. Muscle Nerve Suppl 6:S1-13
Verschueren A, Grapperon AM, Delmont E, Attarian S (2021) Prevalence of spasticity and spasticity-related pain among patients with Amyotrophic Lateral Sclerosis. Rev Neurol (Paris) 177:694–698
Ho DT, Ruthazer R, Russell JA (2011) Shoulder pain in amyotrophic lateral sclerosis. J Clin Neuromuscul Dis 13:53–55
Pizzimenti A, Aragona M, Onesti E, Inghilleri M (2013) Depression, pain and quality of life in patients with amyotrophic lateral sclerosis: a cross-sectional study. Funct Neurol 28:115–119
Wigand B, Schlichte I, Schreiber S, Heitmann J, Meyer T, Dengler R, Petri S, Haghikia A, Vielhaber S, Vogt S (2022) Characteristics of pain and the burden it causes in patients with amyotrophic lateral sclerosis - a longitudinal study. Amyotroph Lateral Scler Frontotemporal Degener 23:284–291
Adelman EE, Albert SM, Rabkin JG, Del Bene ML, Tider T, O’Sullivan I (2004) Disparities in perceptions of distress and burden in ALS patients and family caregivers. Neurology 62:1766–1770
Raheja D, Stephens HE, Lehman E, Walsh S, Yang C, Simmons Z (2016) Patient-reported problematic symptoms in an ALS treatment trial. Amyotroph Lateral Scler Frontotemporal Degener 17:198–205
Ganzini L, Johnston WS, Hoffman WF (1999) Correlates of suffering in amyotrophic lateral sclerosis. Neurology 52:1434–1440
Rivera I, Ajroud-Driss S, Casey P, Heller S, Allen J, Siddique T, Sufit R (2013) Prevalence and characteristics of pain in early and late stages of ALS. Amyotroph Lateral Scler Frontotemporal Degener 14:369–372
Nalamachu S (2013) An overview of pain management: the clinical efficacy and value of treatment. Am J Manag Care 19:s261-266
Ng L, Khan F, Young CA, Galea M (2017) Symptomatic treatments for amyotrophic lateral sclerosis/motor neuron disease. Cochrane Database Syst Rev 1:CD011776
Petri S, Grehl T, Grosskreutz J, Hecht M, Hermann A, Jesse S, Lingor P, Loscher W, Maier A, Schoser B, Weber M, Ludolph AC (2023) Guideline “Motor neuron diseases” of the German Society of Neurology (Deutsche Gesellschaft fur Neurologie). Neurol Res Pract 5:25
Van Damme P, Al-Chalabi A, Andersen PM, Chio A, Couratier P, De Carvalho M, Hardiman O, Kuzma-Kozakiewicz M, Ludolph A, McDermott CJ, Mora JS, Petri S, Probyn K, Reviers E, Salachas F, Silani V, Tysnes OB, van den Berg LH, Villanueva G, Weber M (2024) European Academy of Neurology (EAN) guideline on the management of amyotrophic lateral sclerosis in collaboration with European Reference Network for Neuromuscular Diseases (ERN EURO-NMD). Eur J Neurol:e16264
Dorst J, Ludolph AC, Huebers A (2018) Disease-modifying and symptomatic treatment of amyotrophic lateral sclerosis. Ther Adv Neurol Disord 11:1756285617734734
Chio A, Silani V, Italian ALSSG (2001) Amyotrophic lateral sclerosis care in Italy: a nationwide study in neurological centers. J Neurol Sci 191:145–150
Borasio GD, Shaw PJ, Hardiman O, Ludolph AC, Sales Luis ML, Silani V, European ALSSG (2001) Standards of palliative care for patients with amyotrophic lateral sclerosis: results of a European survey. Amyotroph Lateral Scler Other Motor Neuron Disord 2:159–164
Amtmann D, Weydt P, Johnson KL, Jensen MP, Carter GT (2004) Survey of cannabis use in patients with amyotrophic lateral sclerosis. Am J Hosp Palliat Care 21:95–104
Carter GT, Abood ME, Aggarwal SK, Weiss MD (2010) Cannabis and amyotrophic lateral sclerosis: hypothetical and practical applications, and a call for clinical trials. Am J Hosp Palliat Care 27:347–356
Urbi B, Broadley S, Bedlack R, Russo E, Sabet A (2019) Study protocol for a randomised, double-blind, placebo-controlled study evaluating the Efficacy of cannabis-based Medicine Extract in slowing the disease pRogression of Amyotrophic Lateral sclerosis or motor neurone Disease: the EMERALD trial. BMJ Open 9:e029449
El-Tawil S, Al Musa T, Valli H, Lunn MP, El-Tawil T, Weber M (2010) Quinine for muscle cramps. Cochrane Database Syst Rev:CD005044
Hogan DB (2015) Quinine: not a safe drug for treating nocturnal leg cramps. CMAJ 187:237–238
Oskarsson B, Moore D, Mozaffar T, Ravits J, Wiedau-Pazos M, Parziale N, Joyce NC, Mandeville R, Goyal N, Cudkowicz ME, Weiss M, Miller RG, McDonald CM (2018) Mexiletine for muscle cramps in amyotrophic lateral sclerosis: a randomized, double-blind crossover trial. Muscle Nerve 2018:1
Weiss MD, Macklin EA, McIlduff CE, Vucic S, Wainger BJ, Kiernan MC, Goutman SA, Goyal NA, Rutkove SB, Ladha SS, Chen IA, Harms MB, Brannagan TH, Lacomis D, Zivkovic S, Ma M, Wang LH, Simmons Z, Rivner MH, Shefner JM, Cudkowicz ME, Atassi N, Mexiletine ALSSG (2021) Effects of mexiletine on hyperexcitability in sporadic amyotrophic lateral sclerosis: preliminary findings from a small phase II randomized controlled trial. Muscle Nerve 63:371–383
Lorusso S, Kline D, Bartlett A, Freimer M, Agriesti J, Hawash AA, Rich MM, Kissel JT, David Arnold W (2019) Open-label trial of ranolazine for the treatment of paramyotonia congenita. Muscle Nerve 59:240–243
Baldinger R, Katzberg HD, Weber M (2012) Treatment for cramps in amyotrophic lateral sclerosis/motor neuron disease. Cochrane Datab Syst Rev 2012:CD004157
Mitsumoto H, Cheung K, Oskarsson B, Andrews HF, Jang GE, Andrews JA, Shah JS, Fernandes JA, McElhiney M, Santella RM (2023) Randomized double-blind personalized N-of-1 clinical trial to test the safety and potential efficacy of TJ-68 for treating muscle cramps in amyotrophic lateral sclerosis (ALS): study protocol for a TJ-68 trial. Trials 24:449
Riva N, Mora G, Soraru G, Lunetta C, Ferraro OE, Falzone Y, Leocani L, Fazio R, Comola M, Comi G, Group CS (2019) Safety and efficacy of nabiximols on spasticity symptoms in patients with motor neuron disease (CANALS): a multicentre, double-blind, randomised, placebo-controlled, phase 2 trial. Lancet Neurol 18:155–164
Marvulli R, Megna M, Citraro A, Vacca E, Napolitano M, Gallo G, Fiore P, Ianieri G (2019) Botulinum toxin type a and physiotherapy in spasticity of the lower limbs due to amyotrophic lateral sclerosis. Toxins (Basel) 2019:11
Krivickas LS (2003) Exercise in neuromuscular disease. J Clin Neuromuscul Dis 5:29–39
Swinnen B, Robberecht W (2014) The phenotypic variability of amyotrophic lateral sclerosis. Nat Rev Neurol 10:661–670
Krupp LB, Pollina DA (1996) Mechanisms and management of fatigue in progressive neurological disorders. Curr Opin Neurol 9:456–460
Merkies IS, Kieseier BC (2016) Fatigue, pain, anxiety and depression in guillain-barre syndrome and chronic inflammatory demyelinating polyradiculoneuropathy. Eur Neurol 75:199–206
de Vries JM, Hagemans ML, Bussmann JB, van der Ploeg AT, van Doorn PA (2010) Fatigue in neuromuscular disorders: focus on Guillain–Barre syndrome and Pompe disease. Cell Mol Life Sci 67:701–713
Manjaly ZM, Harrison NA, Critchley HD, Do CT, Stefanics G, Wenderoth N, Lutterotti A, Muller A, Stephan KE (2019) Pathophysiological and cognitive mechanisms of fatigue in multiple sclerosis. J Neurol Neurosurg Psychiatry 90:642–651
Acciarresi M, Bogousslavsky J, Paciaroni M (2014) Post-stroke fatigue: epidemiology, clinical characteristics and treatment. Eur Neurol 72:255–261
Siciliano M, Trojano L, Santangelo G, De Micco R, Tedeschi G, Tessitore A (2018) Fatigue in Parkinson’s disease: a systematic review and meta-analysis. Mov Disord 33:1712–1723
Seifert O, Baerwald C (2019) Impact of fatigue on rheumatic diseases. Best Pract Res Clin Rheumatol 33:101435
Jiang M, Ma Y, Yun B, Wang Q, Huang C, Han L (2020) Exercise for fatigue in breast cancer patients: an umbrella review of systematic reviews. Int J Nurs Sci 7:248–254
Vis R, van de Garde EMW, Grutters JC, Korenromp IHE (2020) The effects of pharmacological interventions on quality of life and fatigue in sarcoidosis: a systematic review. Eur Respir Rev 2020:29
Skoie IM, Dalen I, Ternowitz T, Jonsson G, Kvivik I, Norheim K, Omdal R (2017) Fatigue in psoriasis: a controlled study. Br J Dermatol 177:505–512
Riis PT, Sigsgaard V, Boer J, Jemec GBE (2018) A pilot study of fatigue in patients with hidradenitis suppurativa. Br J Dermatol 178:e42–e43
Peric SZ, Cornblath DR (2020) Fatigue in chronic inflammatory demyelinating polyradiculoneuropathy. Muscle Nerve 62:649–651
Alencar MA, Soares BL, Rangel MFA, Abdo JS, Almeida RAP, Araujo CM, Souza LC, Gomes GC (2022) Fatigue in amyotrophic lateral sclerosis and correlated factors. Arq Neuropsiquiatr 80:1045–1051
Sandstedt P, Johansson S, Ytterberg C, Ingre C, Holmqvist LW, Kierkegaard M (2016) Predictors of health-related quality of life in people with amyotrophic lateral sclerosis. J Neurol Sci 370:269–273
Vogt S, Schreiber S, Pfau G, Kollewe K, Heinze HJ, Dengler R, Petri S, Vielhaber S, Brinkers M (2020) Dyspnea as a fatigue-promoting factor in als and the role of objective indicators of respiratory impairment. J Pain Symptom Manag 60:430-438 e431
Abraham A, Drory VE (2012) Fatigue in motor neuron diseases. Neuromuscul Disord 22(Suppl 3):S198-202
Hamad AA, Amer BE, Abbas NB, Alnajjar AZ, Meshref M (2023) Prevalence and correlates of fatigue in amyotrophic lateral sclerosis: a systematic review and meta-analysis. Neurol Sci 2023:1
An R, Li C, Li X, Wu Y, He X, Ai S, Xu Y, He C (2022) Fatigue in Chinese patients with amyotrophic lateral sclerosis: associated factors and impact on quality of life. Front Neurol 13:806577
Lo Coco D, La Bella V (2012) Fatigue, sleep, and nocturnal complaints in patients with amyotrophic lateral sclerosis. Eur J Neurol 19:760–763
McElhiney MC, Rabkin JG, Gordon PH, Goetz R, Mitsumoto H (2009) Prevalence of fatigue and depression in ALS patients and change over time. J Neurol Neurosurg Psychiatry 80:1146–1149
Panitz S, Kornhuber M, Hanisch F (2015) The checklist individual strength (CIS20-R) in patients with amyotrophic lateral sclerosis—a longitudinal study. Acta Neurol Scand 131:372–380
Gibbons C, Pagnini F, Friede T, Young CA (2018) Treatment of fatigue in amyotrophic lateral sclerosis/motor neuron disease. Cochrane Database Syst Rev 1:CD011005
Diagnosis ETFo, Management of Amyotrophic Lateral S, Andersen PM, Abrahams S, Borasio GD, de Carvalho M, Chio A, Van Damme P, Hardiman O, Kollewe K, Morrison KE, Petri S, Pradat PF, Silani V, Tomik B, Wasner M, Weber M (2012) EFNS guidelines on the clinical management of amyotrophic lateral sclerosis (MALS)—revised report of an EFNS task force. Eur J Neurol 19:360–375
Ferraro L, Antonelli T, O’Connor WT, Tanganelli S, Rambert F, Fuxe K (1997) The antinarcoleptic drug modafinil increases glutamate release in thalamic areas and hippocampus. NeuroReport 8:2883–2887
Ferraro L, Antonelli T, Tanganelli S, O’Connor WT, Perez de la Mora M, Mendez-Franco J, Rambert FA, Fuxe K (1999) The vigilance promoting drug modafinil increases extracellular glutamate levels in the medial preoptic area and the posterior hypothalamus of the conscious rat: prevention by local GABAA receptor blockade. Neuropsychopharmacology 20:346–356
Carter GT, Weiss MD, Lou JS, Jensen MP, Abresch RT, Martin TK, Hecht TW, Han JJ, Weydt P, Kraft GH (2005) Modafinil to treat fatigue in amyotrophic lateral sclerosis: an open label pilot study. Am J Hosp Palliat Care 22:55–59
Rabkin JG, Gordon PH, McElhiney M, Rabkin R, Chew S, Mitsumoto H (2009) Modafinil treatment of fatigue in patients with ALS: a placebo-controlled study. Muscle Nerve 39:297–303
Rosenfeld J, King RM, Jackson CE, Bedlack RS, Barohn RJ, Dick A, Phillips LH, Chapin J, Gelinas DF, Lou JS (2008) Creatine monohydrate in ALS: effects on strength, fatigue, respiratory status and ALSFRS. Amyotroph Lateral Scler 9:266–272
Bertorini TE, Rashed H, Zeno M, Tolley EA, Igarashi M, Li YD (2011) Effects of 3–4 diaminopyridine (DAP) in motor neuron diseases. J Clin Neuromuscul Dis 12:129–137
Behm DG, St-Pierre DM (1998) The effects of strength training and disuse on the mechanisms of fatigue. Sports Med 25:173–189
Sanjak M, Bravver E, Bockenek WL, Norton HJ, Brooks BR (2010) Supported treadmill ambulation for amyotrophic lateral sclerosis: a pilot study. Arch Phys Med Rehabil 91:1920–1929
Drory VE, Goltsman E, Reznik JG, Mosek A, Korczyn AD (2001) The value of muscle exercise in patients with amyotrophic lateral sclerosis. J Neurol Sci 191:133–137
Zanette G, Forgione A, Manganotti P, Fiaschi A, Tamburin S (2008) The effect of repetitive transcranial magnetic stimulation on motor performance, fatigue and quality of life in amyotrophic lateral sclerosis. J Neurol Sci 270:18–22
Kutlubaev MA, Caga J, Xu Y, Areprintseva DK, Pervushina EV, Kiernan MC (2023) Apathy in amyotrophic lateral sclerosis: systematic review and meta-analysis of frequency, correlates, and outcomes. Amyotroph Lateral Scler Frontotemporal Degener 24:14–23
Weise D, Menze I, Metelmann MCF, Woost TB, Classen J, Otto Pelz J (2022) Multimodal assessment of autonomic dysfunction in amyotrophic lateral sclerosis. Eur J Neurol 29:715–723
Papadopoulou M, Bakola E, Papapostolou A, Stefanou MI, Moschovos C, Salakou S, Zis P, Zouvelou V, Kimiskidis VK, Chroni E, Tsivgoulis G (2022) Autonomic dysfunction in amyotrophic lateral sclerosis: A neurophysiological and neurosonology study. J Neuroimaging 32:710–719
Piccione EA, Sletten DM, Staff NP, Low PA (2015) Autonomic system and amyotrophic lateral sclerosis. Muscle Nerve 51:676–679
McCluskey L, Vandriel S, Elman L, Van Deerlin VM, Powers J, Boller A, Wood EM, Woo J, McMillan CT, Rascovsky K, Grossman M (2014) ALS-Plus syndrome: non-pyramidal features in a large ALS cohort. J Neurol Sci 345:118–124
Nubling GS, Mie E, Bauer RM, Hensler M, Lorenzl S, Hapfelmeier A, Irwin DE, Borasio GD, Winkler AS (2014) Increased prevalence of bladder and intestinal dysfunction in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener 15:174–179
Baltadzhieva R, Gurevich T, Korczyn AD (2005) Autonomic impairment in amyotrophic lateral sclerosis. Curr Opin Neurol 18:487–493
Lopes de Carvalho ML, Motta R, Battaglia MA, Brichetto G (2011) Urinary disorders in amyotrophic lateral sclerosis subjects. Amyotroph Lateral Scler 12:352–355
Arlandis S, Vazquez-Costa JF, Martinez-Cuenca E, Sevilla T, Boronat F, Broseta E (2017) Urodynamic findings in amyotrophic lateral sclerosis patients with lower urinary tract symptoms: Results from a pilot study. Neurourol Urodyn 36:626–631
Vazquez-Costa JF, Arlandis S, Hervas D, Martinez-Cuenca E, Cardona F, Perez-Tur J, Broseta E, Sevilla T (2017) Clinical profile of motor neuron disease patients with lower urinary tract symptoms and neurogenic bladder. J Neurol Sci 378:130–136
Carvalho M, Schwartz MS, Swash M (1995) Involvement of the external anal sphincter in amyotrophic lateral sclerosis. Muscle Nerve 18:848–853
Konno H, Yamamoto T, Iwasaki Y, Iizuka H (1986) Shy-Drager syndrome and amyotrophic lateral sclerosis. Cytoarchitectonic and morphometric studies of sacral autonomic neurons. J Neurol Sci 73:193–204
Kihira T, Yoshida S, Yoshimasu F, Wakayama I, Yase Y (1997) Involvement of Onuf’s nucleus in amyotrophic lateral sclerosis. J Neurol Sci 147:81–88
Panicker JN, Fowler CJ, Kessler TM (2015) Lower urinary tract dysfunction in the neurological patient: clinical assessment and management. Lancet Neurol 14:720–732
Samara VC, Jerant P, Gibson S, Bromberg M (2021) Bowel, bladder, and sudomotor symptoms in ALS patients. J Neurol Sci 427:117543
Hayashi H, Kato S (1989) Total manifestations of amyotrophic lateral sclerosis. ALS in the totally locked-in state. J Neurol Sci 93:19–35
Toepfer M, Schroeder M, Klauser A, Lochmuller H, Hirschmann M, Riepl RL, Pongratz D, Muller-Felber W (1997) Delayed colonic transit times in amyotrophic lateral sclerosis assessed with radio-opaque markers. Eur J Med Res 2:473–476
Herdewyn S, Cirillo C, Van Den Bosch L, Robberecht W, Vanden Berghe P, Van Damme P (2014) Prevention of intestinal obstruction reveals progressive neurodegeneration in mutant TDP-43 (A315T) mice. Mol Neurodegener 9:24
Pisano F, Miscio G, Mazzuero G, Lanfranchi P, Colombo R, Pinelli P (1995) Decreased heart rate variability in amyotrophic lateral sclerosis. Muscle Nerve 18:1225–1231
Iscan D, Karaaslan MB, Deveci OS, Akilli Eker R, Koc F (2021) The importance of heart rate variability in predicting cardiac autonomic dysfunction in patients with amyotrophic lateral sclerosis. Int J Clin Pract 75:e14536
Pavlovic S, Stevic Z, Milovanovic B, Milicic B, Rakocevic-Stojanovic V, Lavrnic D, Apostolski S (2010) Impairment of cardiac autonomic control in patients with amyotrophic lateral sclerosis. Amyotroph Lateral Scler 11:272–276
Pinto S, Pinto A, De Carvalho M (2012) Decreased heart rate variability predicts death in amyotrophic lateral sclerosis. Muscle Nerve 46:341–345
Shimizu T, Hayashi H, Kato S, Hayashi M, Tanabe H, Oda M (1994) Circulatory collapse and sudden death in respirator-dependent amyotrophic lateral sclerosis. J Neurol Sci 124:45–55
Rosenbohm A, Schmid B, Buckert D, Rottbauer W, Kassubek J, Ludolph AC, Bernhardt P (2017) Cardiac findings in amyotrophic lateral sclerosis: a magnetic resonance imaging study. Front Neurol 8:479
Tanaka Y, Yamada M, Koumura A, Sakurai T, Hayashi Y, Kimura A, Hozumi I, Inuzuka T (2013) Cardiac sympathetic function in the patients with amyotrophic lateral sclerosis: analysis using cardiac [123I] MIBG scintigraphy. J Neurol 260:2380–2386
Bertel O, Malessa S, Sluga E, Hornykiewicz O (1991) Amyotrophic lateral sclerosis: changes of noradrenergic and serotonergic transmitter systems in the spinal cord. Brain Res 566:54–60
Ohno T, Shimizu T, Kato S, Hayashi H, Hirai S (2001) Effect of tamsulosin hydrochloride on sympathetic hyperactivity in amyotrophic lateral sclerosis. Auton Neurosci 88:94–98
Yamashita A, Koike Y, Takahashi A, Hirayama M, Murakami N, Sobue G (1997) Relationship between respiratory failure and plasma noradrenaline levels in amyotrophic lateral sclerosis. Clin Auton Res 7:173–177
Beck M, Flachenecker P, Magnus T, Giess R, Reiners K, Toyka KV, Naumann M (2005) Autonomic dysfunction in ALS: a preliminary study on the effects of intrathecal BDNF. Amyotroph Lateral Scler Other Motor Neuron Disord 6:100–103
Zarei S, Carr K, Reiley L, Diaz K, Guerra O, Altamirano PF, Pagani W, Lodin D, Orozco G, Chinea A (2015) A comprehensive review of amyotrophic lateral sclerosis. Surg Neurol Int 6:171
Korner S, Kollewe K, Ilsemann J, Muller-Heine A, Dengler R, Krampfl K, Petri S (2013) Prevalence and prognostic impact of comorbidities in amyotrophic lateral sclerosis. Eur J Neurol 20:647–654
Gosswald A, Schienkiewitz A, Nowossadeck E, Busch MA (2013) Prevalence of myocardial infarction and coronary heart disease in adults aged 40–79 years in Germany: results of the German Health Interview and Examination Survey for Adults (DEGS1). Bundesgesundheitsblatt Gesundheitsforschung Gesundheitsschutz 56:650–655
Visser AE, Seelen M, Hulsbergen A, de Graaf J, van der Kooi AJ, Raaphorst J, Veldink JH, van den Berg LH (2017) Exploring the fitness hypothesis in ALS: a population-based case-control study of parental cause of death and lifespan. J Neurol Neurosurg Psychiatry 88:550–556
Turner MR, Wotton C, Talbot K, Goldacre MJ (2012) Cardiovascular fitness as a risk factor for amyotrophic lateral sclerosis: indirect evidence from record linkage study. J Neurol Neurosurg Psychiatry 83:395–398
Xu K, Ji H, Hu N (2022) Cardiovascular comorbidities in amyotrophic lateral sclerosis: a systematic review. J Clin Neurosci 96:43–49
Moreau C, Brunaud-Danel V, Dallongeville J, Duhamel A, Laurier-Grymonprez L, de Reuck J, Wiart AC, Perez T, Richard F, Amouyel P, Bordet R, Defebvre L, Destee A, Devos D (2012) Modifying effect of arterial hypertension on amyotrophic lateral sclerosis. Amyotroph Lateral Scler 13:194–201
Pereira M, Gromicho M, Henriques A, Pronto-Laborinho AC, Grosskreutz J, Kuzma-Kozakiewicz M, Petri S, Uysal H, Swash M, de Carvalho M (2021) Cardiovascular comorbidities in amyotrophic lateral sclerosis. J Neurol Sci 421:117292
Mandrioli J, Ferri L, Fasano A, Zucchi E, Fini N, Moglia C, Lunetta C, Marinou K, Ticozzi N, Drago Ferrante G, Scialo C, Soraru G, Trojsi F, Conte A, Falzone YM, Tortelli R, Russo M, Sansone VA, Mora G, Silani V, Volanti P, Caponnetto C, Querin G, Monsurro MR, Sabatelli M, Chio A, Riva N, Logroscino G, Messina S, Calvo A (2018) Cardiovascular diseases may play a negative role in the prognosis of amyotrophic lateral sclerosis. Eur J Neurol 25:861–868
Sutedja NA, van der Schouw YT, Fischer K, Sizoo EM, Huisman MH, Veldink JH, Van den Berg LH (2011) Beneficial vascular risk profile is associated with amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry 82:638–642
Pawlukowska W, Baumert B, Golab-Janowska M, Meller A, Machowska-Sempruch K, Welnicka A, Paczkowska E, Rotter I, Machalinski B, Nowacki P (2020) The relationship between selected demographic factors and speech organ dysfunction in sporadic ALS patients. Medicina (Kaunas) 2020:56
Lu J, Lu Y, Wang X, Li X, Linderman GC, Wu C, Cheng X, Mu L, Zhang H, Liu J, Su M, Zhao H, Spatz ES, Spertus JA, Masoudi FA, Krumholz HM, Jiang L (2017) Prevalence, awareness, treatment, and control of hypertension in China: data from 1.7 million adults in a population-based screening study (China PEACE Million Persons Project). Lancet 390:2549–2558
Diekmann K, Kuzma-Kozakiewicz M, Piotrkiewicz M, Gromicho M, Grosskreutz J, Andersen PM, de Carvalho M, Uysal H, Osmanovic A, Schreiber-Katz O, Petri S, Korner S (2020) Impact of comorbidities and co-medication on disease onset and progression in a large German ALS patient group. J Neurol 267:2130–2141
Armon C, Kurland LT, O’Brien PC, Mulder DW (1991) Antecedent medical diseases in patients with amyotrophic lateral sclerosis. A population-based case-controlled study in Rochester, Minn, 1925 through 1987. Arch Neurol 48:283–286
Trabjerg BB, Garton FC, van Rheenen W, Fang F, Henderson RD, Mortensen PB, Agerbo E, Wray NR (2020) ALS in Danish Registries: heritability and links to psychiatric and cardiovascular disorders. Neurol Genet 6:e398
KIM K, Kim S, Katana M, Terentyev D, Radwański PB, Munger M (2023) Riluzole is Associated With Reduced Risk of Heart Failure. medRxiv:2023.2012.2028.23300552
Sateia MJ (2014) International classification of sleep disorders-third edition: highlights and modifications. Chest 146:1387–1394
Hood S, Amir S (2017) Neurodegeneration and the circadian clock. Front Aging Neurosci 9:170
Ahmed RM, Newcombe RE, Piper AJ, Lewis SJ, Yee BJ, Kiernan MC, Grunstein RR (2016) Sleep disorders and respiratory function in amyotrophic lateral sclerosis. Sleep Med Rev 26:33–42
Lucia D, McCombe PA, Henderson RD, Ngo ST (2021) Disorders of sleep and wakefulness in amyotrophic lateral sclerosis (ALS): a systematic review. Amyotroph Lateral Scler Frontotemporal Degener 22:161–169
Panda S, Gourie-Devi M, Sharma A (2018) Sleep disorders in amyotrophic lateral sclerosis: a questionnaire-based study from India. Neurol India 66:700–708
Diaz-Abad M, Buczyner JR, Venza BR, Scharf SM, Kwan JY, Lubinski B, Russell JW (2018) Poor sleep quality in patients with amyotrophic lateral sclerosis at the time of diagnosis. J Clin Neuromuscul Dis 20:60–68
Lo Coco D, Mattaliano P, Spataro R, Mattaliano A, La Bella V (2011) Sleep-wake disturbances in patients with amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry 82:839–842
Liu S, Ren Q, Gong G, Sun Y, Zhao B, Ma X, Zhang N, Zhong S, Lin Y, Wang W, Zheng R, Yu X, Yun Y, Zhang D, Shao K, Lin P, Yuan Y, Dai T, Zhang Y, Li L, Li W, Zhao Y, Shan P, Meng X, Yan C (2022) Hypothalamic subregion abnormalities are related to body mass index in patients with sporadic amyotrophic lateral sclerosis. J Neurol 269:2980–2988
Gorges M, Vercruysse P, Muller HP, Huppertz HJ, Rosenbohm A, Nagel G, Weydt P, Petersen A, Ludolph AC, Kassubek J, Dupuis L (2017) Hypothalamic atrophy is related to body mass index and age at onset in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry 88:1033–1041
Ozdinler PH, Macklis JD (2006) IGF-I specifically enhances axon outgrowth of corticospinal motor neurons. Nat Neurosci 9:1371–1381
Chung JY, Kim HJ, Kim M (2015) The protective effect of growth hormone on Cu/Zn superoxide dismutase-mutant motor neurons. BMC Neurosci 16:1
Gasperi M, Castellano AE (2010) Growth hormone/insulin-like growth factor I axis in neurodegenerative diseases. J Endocrinol Invest 33:587–591
Spataro R, Volanti P, Vitale F, Meli F, Colletti T, Di Natale A, La Bella V (2015) Plasma cortisol level in amyotrophic lateral sclerosis. J Neurol Sci 358:282–286
Monachelli GG, Meyer M, Rodriguez G, Garay L, Sica RE, De Nicola AF, Gonzalez Deniselle MC (2011) Progesterone and cortisol levels in sporadic amyotrophic lateral sclerosis (sALS): correlation with prognostic factors. Horm Mol Biol Clin Investig 6:167–173
Vercruysse P, Sinniger J, El Oussini H, Scekic-Zahirovic J, Dieterle S, Dengler R, Meyer T, Zierz S, Kassubek J, Fischer W, Dreyhaupt J, Grehl T, Hermann A, Grosskreutz J, Witting A, Van Den Bosch L, Spreux-Varoquaux O, Group GAS, Ludolph AC, Dupuis L (2016) Alterations in the hypothalamic melanocortin pathway in amyotrophic lateral sclerosis. Brain 139:1106–1122
Gnoni V, Zoccolella S, Giugno A, Urso D, Tamburrino L, Filardi M, Logroscino G (2023) Hypothalamus and amyotrophic lateral sclerosis: potential implications in sleep disorders. Front Aging Neurosci 15:1193483
Boentert M (2020) Sleep and sleep disruption in amyotrophic lateral sclerosis. Curr Neurol Neurosci Rep 20:25
Boentert M, Glatz C, Helmle C, Okegwo A, Young P (2018) Prevalence of sleep apnoea and capnographic detection of nocturnal hypoventilation in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry 89:418–424
Boentert M (2019) Sleep disturbances in patients with amyotrophic lateral sclerosis: current perspectives. Nat Sci Sleep 11:97–111
Quaranta VN, Carratu P, Damiani MF, Dragonieri S, Capozzolo A, Cassano A, Resta O (2017) The prognostic role of obstructive sleep apnea at the onset of amyotrophic lateral sclerosis. Neurodegener Dis 17:14–21
Iranzo A, Fernandez-Arcos A, Tolosa E, Serradell M, Molinuevo JL, Valldeoriola F, Gelpi E, Vilaseca I, Sanchez-Valle R, Llado A, Gaig C, Santamaria J (2014) Neurodegenerative disorder risk in idiopathic REM sleep behavior disorder: study in 174 patients. PLoS ONE 9:e89741
Sabater L, Gaig C, Gelpi E, Bataller L, Lewerenz J, Torres-Vega E, Contreras A, Giometto B, Compta Y, Embid C, Vilaseca I, Iranzo A, Santamaria J, Dalmau J, Graus F (2014) A novel non-rapid-eye movement and rapid-eye-movement parasomnia with sleep breathing disorder associated with antibodies to IgLON5: a case series, characterisation of the antigen, and post-mortem study. Lancet Neurol 13:575–586
Iwanaga K, Wakabayashi K, Honma Y, Takahashi H (1997) Amyotrophic lateral sclerosis: occurrence of Bunina bodies in the locus ceruleus pigmented neurons. Clin Neuropathol 16:23–26
Hayashi K, Mochizuki Y, Takeuchi R, Shimizu T, Nagao M, Watabe K, Arai N, Oyanagi K, Onodera O, Hayashi M, Takahashi H, Kakita A, Isozaki E (2016) Clinicopathological characteristics of patients with amyotrophic lateral sclerosis resulting in a totally locked-in state (communication Stage V). Acta Neuropathol Commun 4:107
Puligheddu M, Congiu P, Arico D, Rundo F, Borghero G, Marrosu F, Fantini ML, Ferri R (2016) Isolated rapid eye movement sleep without atonia in amyotrophic lateral sclerosis. Sleep Med 26:16–22
Lo Coco D, Puligheddu M, Mattaliano P, Congiu P, Borghero G, Fantini ML, La Bella V, Ferri R (2017) REM sleep behavior disorder and periodic leg movements during sleep in ALS. Acta Neurol Scand 135:219–224
Choquer M, Blasco H, Plantier L, Beltran S, Bakkouche SE, Corcia P, Limousin N (2021) Insomnia is frequent in amyotrophic lateral sclerosis at the time of diagnosis. Sleep Biol Rhythms 19:121–126
Gossard TR, Trotti LM, Videnovic A, St Louis EK (2021) Restless legs syndrome: contemporary diagnosis and treatment. Neurotherapeutics 18:140–155
Diaconu S, Irincu L, Ungureanu L, Ciopleias B, Tint D, Falup-Pecurariu C (2023) Restless Legs Syndrome in Parkinson's Disease. J Pers Med 2023:13
Alonso-Navarro H, Garcia-Martin E, Agundez JAG, Jimenez-Jimenez FJ (2019) Association between restless legs syndrome and other movement disorders. Neurology 92:948–964
Hogl B, Stefani A (2017) Restless legs syndrome and periodic leg movements in patients with movement disorders: Specific considerations. Mov Disord 32:669–681
Sperfeld AD, Unrath A, Kassubek J (2007) Restless legs syndrome in hereditary spastic paraparesis. Eur Neurol 57:31–35
Liu S, Shen D, Tai H, Su N, Ding Q, Fu H, Zhang K, Wang Z, Liu M, Huang Y, Cui L (2018) Restless legs syndrome in chinese patients with sporadic amyotrophic lateral sclerosis. Front Neurol 9:735
Lo Coco D, Piccoli F, La Bella V (2010) Restless legs syndrome in patients with amyotrophic lateral sclerosis. Mov Disord 25:2658–2661
Limousin N, Blasco H, Corcia P, Arnulf I, Praline J (2011) The high frequency of restless legs syndrome in patients with amyotrophic lateral sclerosis. Amyotroph Lateral Scler 12:303–306
Sun X, Zhao X, Liu Q, Liu S, Zhang K, Wang ZL, Yang X, Shang L, Huang Y, Cui L, Zhang X (2020) Study on sleep-wake disorders in patients with genetic and non-genetic amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry 2020:1
Castillo PR, Kaplan J, Lin SC, Fredrickson PA, Mahowald MW (2006) Prevalence of restless legs syndrome among native South Americans residing in coastal and mountainous areas. Mayo Clin Proc 81:1345–1347
Nomura T, Inoue Y, Kusumi M, Uemura Y, Nakashima K (2008) Prevalence of restless legs syndrome in a rural community in Japan. Mov Disord 23:2363–2369
Cho SJ, Hong JP, Hahm BJ, Jeon HJ, Chang SM, Cho MJ, Lee HB (2009) Restless legs syndrome in a community sample of Korean adults: prevalence, impact on quality of life, and association with DSM-IV psychiatric disorders. Sleep 32:1069–1076
Allen RP, Picchietti DL, Auerbach M, Cho YW, Connor JR, Earley CJ, Garcia-Borreguero D, Kotagal S, Manconi M, Ondo W, Ulfberg J, Winkelman JW, International Restless Legs Syndrome Study G (2018) Evidence-based and consensus clinical practice guidelines for the iron treatment of restless legs syndrome/Willis–Ekbom disease in adults and children: an IRLSSG task force report. Sleep Med 41:27–44
Garcia-Borreguero D, Williams AM (2014) An update on restless legs syndrome (Willis–Ekbom disease): clinical features, pathogenesis and treatment. Curr Opin Neurol 27:493–501
Zago S, Lorusso L, Aiello EN, Ugolini M, Poletti B, Ticozzi N, Silani V (2022) Cognitive and behavioral involvement in ALS has been known for more than a century. Neurol Sci 43:6741–6760
Chio A, Moglia C, Canosa A, Manera U, Vasta R, Brunetti M, Barberis M, Corrado L, D’Alfonso S, Bersano E, Sarnelli MF, Solara V, Zucchetti JP, Peotta L, Iazzolino B, Mazzini L, Mora G, Calvo A (2019) Cognitive impairment across ALS clinical stages in a population-based cohort. Neurology 93:e984–e994
Beeldman E, Raaphorst J, Klein Twennaar M, de Visser M, Schmand BA, de Haan RJ (2016) The cognitive profile of ALS: a systematic review and meta-analysis update. J Neurol Neurosurg Psychiatry 87:611–619
Jellinger KA (2023) The Spectrum of Cognitive Dysfunction in Amyotrophic Lateral Sclerosis: An Update. Int J Mol Sci 2023:24
De Marchi F, Carrarini C, De Martino A, Diamanti L, Fasano A, Lupica A, Russo M, Salemme S, Spinelli EG, Bombaci A, Sig N (2021) Cognitive dysfunction in amyotrophic lateral sclerosis: Can we predict it? Neurol Sci 42:2211–2222
Pender N, Pinto-Grau M, Hardiman O (2020) Cognitive and behavioural impairment in amyotrophic lateral sclerosis. Curr Opin Neurol 33:649–654
Woolley S, Goetz R, Factor-Litvak P, Murphy J, Hupf J, Garofalo DC, Lomen-Hoerth C, Andrews H, Heitzman D, Bedlack R, Katz J, Barohn R, Sorenson E, Oskarsson B, Filho AF, Kasarskis E, Mozaffar T, Nations S, Swenson A, Koczon-Jaremko A, Christodoulou G, Mitsumoto H (2018) Longitudinal screening detects cognitive stability and behavioral deterioration in ALS patients. Behav Neurol 2018:5969137
Benatar M, Turner MR, Wuu J (2023) Presymptomatic amyotrophic lateral sclerosis: from characterization to prevention. Curr Opin Neurol 36:360–364
Saxon JA, Thompson JC, Harris JM, Ealing J, Hamdalla H, Chaouch A, Young C, Blackburn D, Majeed T, Gall C, Richardson AMT, Langheinrich T, Jones M, Snowden JS (2020) The Edinburgh Cognitive and Behavioral ALS Screen (ECAS) in frontotemporal dementia. Amyotroph Lateral Scler Frontotemporal Degener 21:606–613
Watanabe Y, Ogino M, Ichikawa H, Hanajima R, Nakashima K (2021) The Edinburgh cognitive and behavioural ALS screen (ECAS) for Japanese ALS and FTD patients. Amyotroph Lateral Scler Frontotemporal Degener 22:66–72
Woolley SC, York MK, Moore DH, Strutt AM, Murphy J, Schulz PE, Katz JS (2010) Detecting frontotemporal dysfunction in ALS: utility of the ALS cognitive behavioral screen (ALS-CBS). Amyotroph Lateral Scler 11:303–311
Turon-Sans J, Gascon-Bayarri J, Rene R, Rico I, Gamez C, Paipa A, Povedano M (2016) Cognitive impairment in ALS patients and validation of the Spanish version of the ALS-CBS test. Amyotroph Lateral Scler Frontotemporal Degener 17:221–227
Niven E, Newton J, Foley J, Colville S, Swingler R, Chandran S, Bak TH, Abrahams S (2015) Validation of the Edinburgh Cognitive and Behavioural Amyotrophic Lateral Sclerosis Screen (ECAS): a cognitive tool for motor disorders. Amyotroph Lateral Scler Frontotemporal Degener 16:172–179
Aiello EN, Solca F, Greco LC, La Tona A, Torre S, Carelli L, Morelli C, Doretti A, Colombo E, Messina S, Pain D, Radici A, Lizio A, Casiraghi J, Cerri F, Brugnera A, Compare A, Woolley S, Murphy J, Tremolizzo L, Appollonio I, Verde F, Sansone VA, Lunetta C, Silani V, Ticozzi N, Poletti B (2022) Standardization of the Italian ALS-CBS caregiver behavioral questionnaire. Front Psychol 13:1107001
Abrahams S, Newton J, Niven E, Foley J, Bak TH (2014) Screening for cognition and behaviour changes in ALS. Amyotroph Lateral Scler Frontotemporal Degener 15:9–14
Poletti B, Carelli L, Faini A, Solca F, Meriggi P, Lafronza A, Ciringione L, Pedroli E, Ticozzi N, Ciammola A, Cipresso P, Riva G, Silani V (2018) The Arrows and Colors Cognitive Test (ACCT): a new verbal-motor free cognitive measure for executive functions in ALS. PLoS ONE 13:e0200953
Savage S, Hsieh S, Leslie F, Foxe D, Piguet O, Hodges JR (2013) Distinguishing subtypes in primary progressive aphasia: application of the Sydney language battery. Dement Geriatr Cogn Disord 35:208–218
Bishop DV (1982) Comprehension of spoken, written and signed sentences in childhood language disorders. J Child Psychol Psychiatry 23:1–20
Dubois B, Slachevsky A, Litvan I, Pillon B (2000) The FAB: a frontal assessment battery at bedside. Neurology 55:1621–1626
Radakovic R, Stephenson L, Newton J, Crockford C, Swingler R, Chandran S, Abrahams S (2017) Multidimensional apathy and executive dysfunction in amyotrophic lateral sclerosis. Cortex 94:142–151
Castelnovo V, Canu E, De Mattei F, Filippi M, Agosta F (2023) Basal ganglia alterations in amyotrophic lateral sclerosis. Front Neurosci 17:1133758
Nigri A, Umberto M, Stanziano M, Ferraro S, Fedeli D, Medina Carrion JP, Palermo S, Lequio L, Denegri F, Agosta F, Filippi M, Valentini MC, Canosa A, Calvo A, Chio A, Bruzzone MG, Moglia C (2023) C9orf72 ALS mutation carriers show extensive cortical and subcortical damage compared to matched wild-type ALS patients. Neuroimage Clin 38:103400
Renard D, Collombier L, Castelnovo G, Fourcade G, Kotzki PO, LaBauge P (2011) Brain FDG-PET changes in ALS and ALS-FTD. Acta Neurol Belg 111:306–309
Canosa A, Pagani M, Cistaro A, Montuschi A, Iazzolino B, Fania P, Cammarosano S, Ilardi A, Moglia C, Calvo A, Chio A (2016) 18F-FDG-PET correlates of cognitive impairment in ALS. Neurology 86:44–49
Verde F, Milone I, Colombo E, Maranzano A, Solca F, Torre S, Doretti A, Gentile F, Manini A, Bonetti R, Peverelli S, Messina S, Maderna L, Morelli C, Poletti B, Ratti A, Silani V, Ticozzi N (2023) Phenotypic correlates of serum neurofilament light chain levels in amyotrophic lateral sclerosis. Front Aging Neurosci 15:1132808
Gong Z, Gao L, Lu Y, Wang Z (2022) CSF p-tau as a potential cognition impairment biomarker in ALS. Front Neurol 13:991143
Brenner D, Yilmaz R, Muller K, Grehl T, Petri S, Meyer T, Grosskreutz J, Weydt P, Ruf W, Neuwirth C, Weber M, Pinto S, Claeys KG, Schrank B, Jordan B, Knehr A, Gunther K, Hubers A, Zeller D, Kubisch C, Jablonka S, Sendtner M, Klopstock T, de Carvalho M, Sperfeld A, Borck G, Volk AE, Dorst J, Weis J, Otto M, Schuster J, Del Tredici K, Braak H, Danzer KM, Freischmidt A, Meitinger T, Strom TM, Ludolph AC, Andersen PM, Weishaupt JH, German ALSnMNDNET (2018) Hot-spot KIF5A mutations cause familial ALS. Brain 141:688–697
Ghasemi M, Brown RH Jr (2018) Genetics of amyotrophic lateral sclerosis. Cold Spring Harb Perspect Med 2018:8
Nguyen HP, Van Broeckhoven C, van der Zee J (2018) ALS genes in the genomic era and their implications for FTD. Trends Genet 34:404–423
Geronimo A, Stephens HE, Schiff SJ, Simmons Z (2015) Acceptance of brain-computer interfaces in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener 16:258–264
Patatanian E, Casselman J (2014) Dextromethorphan/quinidine for the treatment of pseudobulbar affect. Consult Pharm 29:264–269
McCullagh S, Feinstein A (2000) Treatment of pathological affect: variability of response for laughter and crying. J Neuropsychiatry Clin Neurosci 12:100–102
Boros BD, Schoch KM, Kreple CJ, Miller TM (2022) Antisense oligonucleotides for the study and treatment of ALS. Neurotherapeutics 19:1145–1158
Korobeynikov VA, Lyashchenko AK, Blanco-Redondo B, Jafar-Nejad P, Shneider NA (2022) Antisense oligonucleotide silencing of FUS expression as a therapeutic approach in amyotrophic lateral sclerosis. Nat Med 28:104–116
Miller TM, Cudkowicz ME, Genge A, Shaw PJ, Sobue G, Bucelli RC, Chio A, Van Damme P, Ludolph AC, Glass JD, Andrews JA, Babu S, Benatar M, McDermott CJ, Cochrane T, Chary S, Chew S, Zhu H, Wu F, Nestorov I, Graham D, Sun P, McNeill M, Fanning L, Ferguson TA, Fradette S, Valor, Group OLEW (2022) Trial of antisense oligonucleotide tofersen for SOD1 ALS. N Engl J Med 387:1099–1110
Tran H, Moazami MP, Yang H, McKenna-Yasek D, Douthwright CL, Pinto C, Metterville J, Shin M, Sanil N, Dooley C, Puri A, Weiss A, Wightman N, Gray-Edwards H, Marosfoi M, King RM, Kenderdine T, Fabris D, Bowser R, Watts JK, Brown RH Jr (2022) Suppression of mutant C9orf72 expression by a potent mixed backbone antisense oligonucleotide. Nat Med 28:117–124
Heidari ME, Nadali J, Parouhan A, Azarafraz M, Tabatabai SM, Irvani SSN, Eskandari F, Gharebaghi A (2021) Prevalence of depression among amyotrophic lateral sclerosis (ALS) patients: a systematic review and meta-analysis. J Affect Disord 287:182–190
Larsson BJ, Nordin K, Nygren I (2023) Symptoms of anxiety and depression in patients with amyotrophic lateral sclerosis and their relatives during the disease trajectory. J Neurol Sci 455:122780
Thakore NJ, Pioro EP (2016) Depression in ALS in a large self-reporting cohort. Neurology 86:1031–1038
Longinetti E, Mariosa D, Larsson H, Ye W, Ingre C, Almqvist C, Lichtenstein P, Piehl F, Fang F (2017) Neurodegenerative and psychiatric diseases among families with amyotrophic lateral sclerosis. Neurology 89:578–585
Hillemacher T, Grassel E, Tigges S, Bleich S, Neundorfer B, Kornhuber J, Hecht MJ (2004) Depression and bulbar involvement in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord 5:245–249
De Marchi F, Sarnelli MF, Solara V, Bersano E, Cantello R, Mazzini L (2019) Depression and risk of cognitive dysfunctions in amyotrophic lateral sclerosis. Acta Neurol Scand 139:438–445
Wang Y, Ye S, Chen L, Tang L, Fan D (2021) Loss of appetite in patients with amyotrophic lateral sclerosis is associated with weight loss and anxiety/depression. Sci Rep 11:9119
Silva-Moraes MH, Bispo-Torres AC, Barouh JL, Lucena PH, Armani-Franceschi G, Dorea-Bandeira I, Vieira F, Miranda-Scippa A, Quarantini LC, Lucena R, Bandeira ID (2020) Suicidal behavior in individuals with amyotrophic lateral sclerosis: a systematic review. J Affect Disord 277:688–696
Benbrika S, Doidy F, Carluer L, Mondou A, Pelerin A, Eustache F, Viader F, Desgranges B (2021) Longitudinal study of cognitive and emotional alterations in amyotrophic lateral sclerosis: clinical and imaging data. Front Neurol 12:620198
Ostuzzi G, Matcham F, Dauchy S, Barbui C, Hotopf M (2018) Antidepressants for the treatment of depression in people with cancer. Cochrane Database Syst Rev 4:CD011006
Gould RL, Coulson MC, Brown RG, Goldstein LH, Al-Chalabi A, Howard RJ (2015) Psychotherapy and pharmacotherapy interventions to reduce distress or improve well-being in people with amyotrophic lateral sclerosis: a systematic review. Amyotroph Lateral Scler Frontotemporal Degener 16:293–302
Gould RL, Thompson BJ, Rawlinson C, Kumar P, White D, Serfaty MA, Graham CD, McCracken LM, Bursnall M, Bradburn M, Young T, Howard RJ, Al-Chalabi A, Goldstein LH, Lawrence V, Cooper C, Shaw PJ, McDermott CJ (2022) A randomised controlled trial of acceptance and commitment therapy plus usual care compared to usual care alone for improving psychological health in people with motor neuron disease (COMMEND): study protocol. BMC Neurol 22:431
Grabler MR, Weyen U, Juckel G, Tegenthoff M, Mavrogiorgou-Juckel P (2018) Death anxiety and depression in amyotrophic lateral sclerosis patients and their primary caregivers. Front Neurol 9:1035
Miyagawa T, Brushaber D, Syrjanen J, Kremers W, Fields J, Forsberg LK, Heuer HW, Knopman D, Kornak J, Boxer A, Rosen H, Boeve B, Consortium AL (2020) Use of the CDR(R) plus NACC FTLD in mild FTLD: Data from the ARTFL/LEFFTDS consortium. Alzheimers Dement 16:79–90
Samra K, MacDougall AM, Peakman G, Bouzigues A, Bocchetta M, Cash DM, Greaves CV, Convery RS, van Swieten JC, Jiskoot L, Seelaar H, Moreno F, Sanchez-Valle R, Laforce R, Graff C, Masellis M, Tartaglia C, Rowe JB, Borroni B, Finger E, Synofzik M, Galimberti D, Vandenberghe R, de Mendonca A, Butler CR, Gerhard A, Ducharme S, Le Ber I, Tiraboschi P, Santana I, Pasquier F, Levin J, Otto M, Sorbi S, Rohrer JD, Russell LL, Genetic FTDI (2023) Motor symptoms in genetic frontotemporal dementia: developing a new module for clinical rating scales. J Neurol 270:1466–1477
Goutman SA, Boss J, Iyer G, Habra H, Savelieff MG, Karnovsky A, Mukherjee B, Feldman EL (2023) Body mass index associates with amyotrophic lateral sclerosis survival and metabolomic profiles. Muscle Nerve 67:208–216
Marin B, Desport JC, Kajeu P, Jesus P, Nicolaud B, Nicol M, Preux PM, Couratier P (2011) Alteration of nutritional status at diagnosis is a prognostic factor for survival of amyotrophic lateral sclerosis patients. J Neurol Neurosurg Psychiatry 82:628–634
Janse van Mantgem MR, van Eijk RPA, van der Burgh HK, Tan HHG, Westeneng HJ, van Es MA, Veldink JH, van den Berg LH (2020) Prognostic value of weight loss in patients with amyotrophic lateral sclerosis: a population-based study. J Neurol Neurosurg Psychiatry 91:867–875
Xia K, Witzel S, Witzel C, Klose V, Fan D, Ludolph AC, Dorst J (2023) Mutation-specific metabolic profiles in presymptomatic amyotrophic lateral sclerosis. Eur J Neurol 30:87–95
Peter RS, Rosenbohm A, Dupuis L, Brehme T, Kassubek J, Rothenbacher D, Nagel G, Ludolph AC (2017) Life course body mass index and risk and prognosis of amyotrophic lateral sclerosis: results from the ALS registry Swabia. Eur J Epidemiol 32:901–908
Mariosa D, Beard JD, Umbach DM, Bellocco R, Keller J, Peters TL, Allen KD, Ye W, Sandler DP, Schmidt S, Fang F, Kamel F (2017) Body mass index and amyotrophic lateral sclerosis: a study of US military veterans. Am J Epidemiol 185:362–371
Kandler K, Witzel S, Eder K, Rothenbacher D, Nagel G, Peter RS, Schuster J, Dorst J, Rosenbohm A, Ludolph AC, Group ALSRS (2022) Phenotyping of the thoracic-onset variant of amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry 93:563–565
Moglia C, Calvo A, Grassano M, Canosa A, Manera U, D’Ovidio F, Bombaci A, Bersano E, Mazzini L, Mora G, Chio A, Piemonte, Valle d’Aosta Register for ALS (2019) Early weight loss in amyotrophic lateral sclerosis: outcome relevance and clinical correlates in a population-based cohort. J Neurol Neurosurg Psychiatry 90:666–673
Wang MD, Little J, Gomes J, Cashman NR, Krewski D (2017) Identification of risk factors associated with onset and progression of amyotrophic lateral sclerosis using systematic review and meta-analysis. Neurotoxicology 61:101–130
Shimizu T, Nakayama Y, Matsuda C, Haraguchi M, Bokuda K, Ishikawa-Takata K, Kawata A, Isozaki E (2019) Prognostic significance of body weight variation after diagnosis in ALS: a single-centre prospective cohort study. J Neurol 266:1412–1420
Ludolph A, Dupuis L, Kasarskis E, Steyn F, Ngo S, McDermott C (2023) Nutritional and metabolic factors in amyotrophic lateral sclerosis. Nat Rev Neurol 19:511–524
Mezoian T, Belt E, Garry J, Hubbard J, Breen CT, Miller L, Levine-Weinberg M, Nalipinski P, Sullivan S, Chan J, Wills AM (2020) Loss of appetite in amyotrophic lateral sclerosis is associated with weight loss and decreased calorie consumption independent of dysphagia. Muscle Nerve 61:230–234
Sarmet M, Kabani A, Maragakis NJ, Mehta AK (2022) Appetite and quality of life in amyotrophic lateral sclerosis: a scoping review. Muscle Nerve 66:653–660
Ngo ST, van Eijk RPA, Chachay V, van den Berg LH, McCombe PA, Henderson RD, Steyn FJ (2019) Loss of appetite is associated with a loss of weight and fat mass in patients with amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener 20:497–505
Bouteloup C, Desport JC, Clavelou P, Guy N, Derumeaux-Burel H, Ferrier A, Couratier P (2009) Hypermetabolism in ALS patients: an early and persistent phenomenon. J Neurol 256:1236–1242
Cattaneo M, Jesus P, Lizio A, Fayemendy P, Guanziroli N, Corradi E, Sansone V, Leocani L, Filippi M, Riva N, Corcia P, Couratier P, Lunetta C (2022) The hypometabolic state: a good predictor of a better prognosis in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry 93:41–47
Fayemendy P, Marin B, Labrunie A, Boirie Y, Walrand S, Achamrah N, Coeffier M, Preux PM, Lautrette G, Desport JC, Couratier P, Jesus P (2021) Hypermetabolism is a reality in amyotrophic lateral sclerosis compared to healthy subjects. J Neurol Sci 420:117257
Jesus P, Fayemendy P, Marin B, Nicol M, Sourisseau H, Boirie Y, Walrand S, Achamrah N, Coeffier M, Preux PM, Lautrette G, Couratier P, Desport JC (2020) Increased resting energy expenditure compared with predictive theoretical equations in amyotrophic lateral sclerosis. Nutrition 77:110805
Kasarskis EJ, Mendiondo MS, Matthews DE, Mitsumoto H, Tandan R, Simmons Z, Bromberg MB, Kryscio RJ, Group ALSNNS (2014) Estimating daily energy expenditure in individuals with amyotrophic lateral sclerosis. Am J Clin Nutr 99:792–803
Manfredi G, Xu Z (2005) Mitochondrial dysfunction and its role in motor neuron degeneration in ALS. Mitochondrion 5:77–87
Gao J, Wang L, Yan T, Perry G, Wang X (2019) TDP-43 proteinopathy and mitochondrial abnormalities in neurodegeneration. Mol Cell Neurosci 100:103396
Tefera TW, Bartlett K, Tran SS, Hodson MP, Borges K (2019) Impaired pentose phosphate pathway in the spinal cord of the hSOD1(G93A) mouse model of amyotrophic lateral sclerosis. Mol Neurobiol 56:5844–5855
Allen SP, Hall B, Woof R, Francis L, Gatto N, Shaw AC, Myszczynska M, Hemingway J, Coldicott I, Willcock A, Job L, Hughes RM, Boschian C, Bayatti N, Heath PR, Bandmann O, Mortiboys H, Ferraiuolo L, Shaw PJ (2019) C9orf72 expansion within astrocytes reduces metabolic flexibility in amyotrophic lateral sclerosis. Brain 142:3771–3790
Tefera TW, Borges K (2019) Neuronal glucose metabolism is impaired while astrocytic TCA cycling is unaffected at symptomatic stages in the hSOD1(G93A) mouse model of amyotrophic lateral sclerosis. J Cereb Blood Flow Metab 39:1710–1724
Cykowski MD, Takei H, Schulz PE, Appel SH, Powell SZ (2014) TDP-43 pathology in the basal forebrain and hypothalamus of patients with amyotrophic lateral sclerosis. Acta Neuropathol Commun 2:171
Diano S, Liu ZW, Jeong JK, Dietrich MO, Ruan HB, Kim E, Suyama S, Kelly K, Gyengesi E, Arbiser JL, Belsham DD, Sarruf DA, Schwartz MW, Bennett AM, Shanabrough M, Mobbs CV, Yang X, Gao XB, Horvath TL (2011) Peroxisome proliferation-associated control of reactive oxygen species sets melanocortin tone and feeding in diet-induced obesity. Nat Med 17:1121–1127
Georges M, Morelot-Panzini C, Similowski T, Gonzalez-Bermejo J (2014) Noninvasive ventilation reduces energy expenditure in amyotrophic lateral sclerosis. BMC Pulm Med 14:17
Weber M, Goldman B, Truniger S (2010) Tetrahydrocannabinol (THC) for cramps in amyotrophic lateral sclerosis: a randomised, double-blind crossover trial. J Neurol Neurosurg Psychiatry 81:1135–1140
Acknowledgements
This article originated from the Third ALS Young Investigators Academy, hosted at the Klinikum rechts der Isar of the Technical University in Munich on 08–09 September 2023, with generous support of ITF Pharma.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
BB, M-B.B., J.H.-B. and SP were all involved in the conceptualization, writing the original draft of the manuscript, review and editing. All authors have read and agreed to the last version of the manuscript.
Corresponding author
Ethics declarations
Conflicts of interest
BB, M-B.B. and J.H.-B received compensation for travel expenses from ITF Pharma GmbH outside of the submitted work. SP has received speaker fees, non-financial support and research support from Biogen, Roche, AL-S Pharma, Amylyx, Cytokinetics, Ferrer, ITF-Pharma, and Sanofi and served on advisory boards of Amylyx, Biogen, Roche, Zambon and ITF Pharma outside of the submitted work.
Ethical standards
The manuscript does not contain clinical studies or patient data.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bjelica, B., Bartels, MB., Hesebeck-Brinckmann, J. et al. Non-motor symptoms in patients with amyotrophic lateral sclerosis: current state and future directions. J Neurol 271, 3953–3977 (2024). https://doi.org/10.1007/s00415-024-12455-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-024-12455-5