
Abstract
The term ‘neuromyelitis optica spectrum disorders’ (NMOSD) is used as an umbrella term that refers to aquaporin-4 immunoglobulin G (AQP4-IgG)-positive neuromyelitis optica (NMO) and its formes frustes and to a number of closely related clinical syndromes without AQP4-IgG. NMOSD were originally considered subvariants of multiple sclerosis (MS) but are now widely recognized as disorders in their own right that are distinct from MS with regard to immunopathogenesis, clinical presentation, optimum treatment, and prognosis. In part 1 of this two-part article series, which ties in with our 2014 recommendations, the neuromyelitis optica study group (NEMOS) gives updated recommendations on the diagnosis and differential diagnosis of NMOSD. A key focus is on differentiating NMOSD from MS and from myelin oligodendrocyte glycoprotein antibody-associated encephalomyelitis (MOG-EM; also termed MOG antibody-associated disease, MOGAD), which shares significant similarity with NMOSD with regard to clinical and, partly, radiological presentation, but is a pathogenetically distinct disease. In part 2, we provide updated recommendations on the treatment of NMOSD, covering all newly approved drugs as well as established treatment options.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The term ‘neuromyelitis optica spectrum disorders’ (NMOSD) is used to refer to inflammatory disorders of the central nervous system (CNS) that predominantly affect the optic nerves, the spinal cord, and, less frequently, the brainstem, causing acute attacks of optic neuritis, myelitis, and brainstem encephalitis [92, 93, 228]. In the majority of cases, NMOSD is associated with pathogenic immunoglobulin G (IgG) antibodies to aquaporin-4 (AQP4), the most common water channel in the CNS [89, 124, 125]. The discovery of AQP4-IgG in 2004/2005 had significant implications for the nosology, diagnosis, and treatment of NMOSD [228, 230]. The neuromyelitis optica study group (NEMOS; see www.nemos-net.de), founded in 2008 as a nation-wide network of primary, secondary, and tertiary care centers, seeks to broaden the understanding of NMOSD and clinically related (but pathogenetically distinct) disorders, such as myelin oligodendrocyte glycoprotein antibody-associated encephalomyelitis (MOG-EM; also termed MOG antibody-associated disease, MOGAD) [77, 134]. The group has published several national and international multicenter investigations and practical recommendations on the diagnosis and treatment of NMOSD and MOG-EM/MOGAD [6, 9, 10, 16, 59, 75, 76, 83, 84, 86, 87, 112,113,114,115, 154, 177, 178, 180, 200, 212, 213]. Tying in with the recommendations published by the group in 2014 [213], this two-part article series gives updated recommendations on the diagnosis (part 1) and therapy (part 2) of NMOSD.
Methods
A core working group consisting of representatives of 12 German neurological university centers (Charité—Universitätsmedizin Berlin, Ruhr University Bochum, Heinrich-Heine University Düsseldorf, University of Hamburg, Hannover Medical School, University of Heidelberg, University of Leipzig, Ludwig Maximilian University Munich, Technical University of Munich, University of Münster, University of Tübingen, University of Ulm), all of which are members of NEMOS, wrote a first draft, based on expert discussion during NEMOS meetings, with the Heidelberg and Hannover centers jointly responsible. The draft was then revised in several rounds based on expert comments and circulated to 121 study group members, representing 32 university and 30 non-university neurological institutions, for further comments and final approval (see Acknowledgements for a list of all study group members). Further revisions were made by the core working group following peer review, and the final manuscript was again sent to all study group members for comments and final approval.
When to consider NMOSD?—Hints from epidemiology
NMOSD must be considered as a potential differential diagnosis in all patients presenting with CNS inflammation of putative autoimmune etiology, especially if they have optic neuritis (ON), myelitis, or brainstem encephalitis, irrespective of sex, age, and ethnicity.
Significant heterogeneity exists among epidemiological studies with regard to inclusion criteria and methodology; in particular, not all studies have differentiated between AQP4-IgG-positive and AQP4-IgG-negative patients. However, some risk factors have been identified. NMOSD has been shown to predominantly affect females, to be more common in some non-Caucasian populations, and to start in adulthood in the majority of cases.
The incidence showed a peak in middle-aged adults in most studies (e.g., ~ 40 years among AQP4-IgG-positive and 38.5 years among AQP4-IgG-negative patients in a large European cohort [87]). However, a diagnosis of NMOSD must be given serious consideration also in the elderly and in children. Late-onset (LO) NMOSD (> 60 years) accounted for 20–28% of all incident NMOSD cases in some (mixed AQP4-IgG-positive and AQP4-IgG-negative) cohorts [158], and the disease may even start only at very old age (> 75 years). LO-NMOSD has been associated with a less favorable prognosis [28, 119, 189, 191]. This compares to a median age at clinical disease onset of around ~ 30 years in multiple sclerosis (MS) and adult MOG-EM/MOGAD.
While NMOSD must be considered in both sexes, there is a strong female preponderance irrespective of ethnicity, with a female:male ratio of up to 10:1 among AQP4-IgG-seropositive patients, who account for the vast majority of NMOSD, and up to 3:1 in seronegative patients [16, 84, 87]. Among AQP4-IgG-positive patients of fertile age, the sex ratio may even exceed 20:1 [16]. Accordingly, female patients with NMOSD are significantly more likely to be AQP4-IgG-positive than male patients [16]. In contrast, MOG-EM/MOGAD affects women and men in almost equal measure. MS shows a female preponderance similar to that reported for seronegative NMOSD.
Although a diagnosis of NMOSD must be taken into account irrespective of ethnic origin, the overall incidence and prevalence of NMOSD is thought to be higher among non-whites than among whites [19, 39], with the highest rates found for patients of African descent in some studies [39, 158]. This is in contrast to MS, which is most common in Caucasians of northern European ancestry. Reported incidence and prevalence estimates for NMOSD vary substantially among studies from different regions of the world (0.28–0.73/100,000 person-years and 0.52–10/100,000 persons, respectively) [56, 82, 143, 158], suggesting differences in genetic background and environmental factors across the populations analyzed [39, 220]. However, differences in study methodology and inclusion criteria may play a role as well. The few studies that reported antibody-specific incidences all found a much higher incidence of AQP4-IgG-positive than AQP4-IgG-negative NMOSD [156, 157, 190]; in line with this, AQP4-IgG-positive patients accounted for most cases of NMO or NMOSD in major cross-sectional studies.
The proportion of patients with NMOSD among patients with inflammatory demyelinating disorders (IDD) varies among populations. While NMOSD accounts only for a small proportion of white adult patients with IDD, most of whom have classic MS, rates are much higher in some (but not all [188]) Asian populations [19, 142, 208, 210, 220], at least in part reflecting the well-known lower prevalence of MS in Asia [147, 210]. NMOSD was initially thought to be more common than MOG-EM/MOGAD among adult patients with IDD [55, 106] and less frequent than MOG-EM/MOGAD among children with IDD [15, 34, 188]; however, recent data suggest that cases of MOG-EM/MOGAD may outnumber those of both AQP4-IgG-positive and AQP4-IgG-negative NMOSD also in adults, in both Asian and non-Asian populations [121, 188]. This change may possibly result from the recent rise in awareness regarding MOG-EM/MOGAD and the increasing availability of serological tests for MOG-IgG.
Certain human leukocyte antigen (HLA) types are associated with an increased risk of developing AQP4-IgG-positive NMOSD (e.g., HLA-DRB1*03 in Europe, Brazil, and India [odds ratio 1.79–9.23] [2]; HLA-DPB1*0501 in southern Han Chinese; HLA-DPB1*0501 and HLA-DRB1*1602 in Japanese; and HLA- DRB1*04:04 and HLA- DRB1*10:01 in Muslim Arab Israelis) [82]. However, HLA typing is not required to make a diagnosis of NMOSD and is currently not recommended outside the context of scientific studies. Other risk factors are less well established (e.g., vitamin D deficiency, smoking, low number of infections in early life) [82]. In contrast to MS, no convincing evidence for an increase in incidence with increasing latitude has been observed in NMOSD [19, 208, 210, 232].
When to consider NMOSD?—Clinical characteristics
NMOSD must be considered as a differential diagnosis in all patients presenting with acute attacks of transverse myelitis and/or (unilateral or, less frequently, bilateral) acute ON, the two clinical hallmarks of NMOSD, but also in patients presenting with suspected encephalitis or brainstem encephalitis. Myelitis and ON may occur either simultaneously or, far more often, successively. The combination of myelitis and ON gave the disease its name [82, 87].
Patients with suspected myelitis must be examined for sensory, motor (including respiratory), bladder/bowel, and sexual dysfunction. Moreover, pain should be assessed, ideally using structured questionnaires in addition to clinical examination. Pain, as an immediate or long-term sequela of myelitis, is highly prevalent in both AQP4-IgG-positive and AQP4-IgG-negative NMOSD [8, 9, 17, 104, 162] and includes neuropathic pain [9], painful tonic spasms [22, 104, 129], neuropathic pruritus [35, 54], and spasticity-associated pain [9]; dysesthesia (often girdle-like in the case of thoracic myelitis) and hyperalgesia/allodynia are frequent [162].
ON mostly leads to blurred vision, decline in visual acuity, headache, retrobulbar pain/pain upon eye movement, phosphenes, a relative afferent pupillary defect (if unilateral or asymmetrical), i.e., a positive Marcus-Gunn sign/swinging flashlight test, sometimes optic disk swelling/papilledema visible upon fundoscopy (a finding much more frequent in MOG-EM/MOGAD), visual field deficiency, and/or color desaturation [165]. Both eyes must be examined, since both optic nerves may be affected simultaneously with only minor or even subclinical damage possible in one eye. However, bilateral ON, which is rare in MS, is possibly more common in MOG-EM/MOGAD than in NMOSD, at least at onset [84, 87, 170].
Extra-opticospinal symptoms must not lead to premature exclusion of NMOSD. Following the discovery of AQP4-IgG, the clinical spectrum was found to be much broader than initially thought and to include, although more rarely, supratentorial and brainstem encephalitis. The latter often affects the dorsal medulla oblongata, causing intractable acute or subacute nausea, vomiting and/or hiccups (either in combination or isolated; constant or episodic), the so-called area postrema syndrome (APS) [197]. APS is much less common in AQP4-IgG-negative patients with NMOSD [87] and in MOG-EM/MOGAD [62, 84]. The diagnosis should be made according to the criteria proposed by Shosha et al. [197]. These require persistence of symptoms for at least 48 h if magnetic resonance imaging (MRI) shows no evidence of acute area postrema involvement (considering both the rarity of APS as a cause of vomiting and hiccups in the general population and the broad spectrum of differential diagnoses), whereas shorter duration is deemed sufficient if MRI shows such involvement. Moreover, demonstration of a lack of complete resolution after symptomatic therapy (antiemetics, IV fluid, hiccup treatments) and exclusion of other etiologies (gastrointestinal/mediastinal, metabolic [e.g., liver or kidney dysfunction, hyponatremia], CNS tumor, stroke, migraine, psychiatric eating disorders, etc.) is required. Symptom severity may be quantified using the recently proposed APS severity scale [197]. However, a broad spectrum of brainstem symptoms other than APS (oculomotor disturbances, facial palsy/numbness, ataxia, etc.) has also been described in patients with NMOSD, and all patients should be thoroughly examined for possible brainstem involvement [87, 117]. It should also be taken into account that NMOSD rarely causes diencephalic lesions that may cause hypersomnolence/narcolepsy or a syndrome of inadequate antidiuretic hormone secretion (SIADH). Patients should also be screened for signs of depression, fatigue, and cognitive impairment, and, if these are found, neuropsychological/neuropsychiatric consultation should be considered [9, 23, 60, 149]. Cerebral manifestations may also include headache, and, very rarely, epileptic seizures (with the latter being more frequent in MOG-EM/MOGAD than in AQP4-IgG-positive NMOSD) [8, 52, 152]. Extra-opticospinal symptoms are relatively rare at onset both in AQP4-IgG-positive and in AQP4-IgG-negative patients (e.g., 4% in a German cohort [87] and 9% in a Chinese study [127]), but many patients develop one of them at least once over the course of disease.
NMOSD takes a relapsing course in virtually all AQP4-IgG-positive patients but may be monophasic in AQP4-IgG-negative patients [87]. A diagnosis of monophasic NMOSD should be made with caution, however, since the interval between the first and the second attack varies widely, ranging from just 1 month to more than a decade (median 9 months in a large European cohort [87]). Patients positive for AQP4-IgG should be considered to be at risk of relapse indefinitely.
Accrual of disability in NMOSD is driven mostly by acute clinical attacks. Although ON and myelitis attacks tend to be less severe in MS than in NMOSD on average (at least in AQP4-IgG-positive cases), mild attack-related symptoms should not per se be (mis)taken as evidence against a diagnosis of NMOSD. Indeed, attack severity in NMOSD may range from mild (e.g., tingling, subtle paresis, blurred vision, visual deficiency detectable only by means of low-contrast charts) to extremely severe (e.g., blindness, tetraparesis, respiratory insufficiency). Severe visual deficiency (Snellen score < 20/200 for high-contrast visual acuity) during acute attacks is frequent in AQP4-IgG-positive NMOSD (and MOG-EM/MOGAD) but rare in MS. Whereas persistent severe motor impairment after acute myelitis is more suggestive of AQP4-IgG-positive NMOSD than of MOG-EM/MOGAD, persisting sphincter disturbance/sexual dysfunction is more commonly seen in MOG-EM/MOGAD, as the latter disorder more often affects the conus than does NMOSD [36]. While significant chronic progressive deterioration of symptoms independent of or in addition to clinical relapses frequently occurs in MS, such a course of disease is highly atypical and should be considered a red flag in NMOSD [231] (as well as MOG-EM/MOGAD [84]). However, growing evidence from electrophysiological, radiological, optical coherence tomography (OCT), histopathological, and laboratory studies suggests a role for at least subtle relapse-independent CNS (including retinal) damage in NMOSD (best shown for AQP4-IgG-positive patients) [1, 3, 50, 57, 94, 148, 178, 218].
Even after a single attack severe, permanent disability can remain, especially if the attack is not treated immediately and properly (see Part 2 of this article series for details) [112]. To prevent accrual of disability from repeated attacks, long-term immunotherapy is essential (see Part 2 of this article series) [200]. Especially if left untreated, NMOSD may result in considerable disability, reduction in quality of life, and increased risk of death from acute and chronic complications, and may impose a substantial burden on patients and caregivers. NMOSD causes high societal costs [9, 59, 185]. Respiratory insufficiency due to brainstem or high cervical lesions and urosepsis due to bladder dysfunction count among the most common causes of death from NMOSD. However, mortality rates have dropped significantly since the 1980s and 1990s, most likely reflecting earlier diagnosis and improvements in treatment [32, 87, 139, 229].
As contralateral ON may occur in the initially non-affected eye with a delay of some days or a few weeks in NMOSD, repeat ophthalmological examinations should be performed in patients originally presenting with unilateral ON to document the full extent of neurological damage associated with an attack. This is important, since undocumented deficits may later be mistaken for remnants of additional, unrecognized/subclinical attacks and influence treatment decisions. More generally, ON, spinal cord, brainstem, or encephalitis lesions may occur successively during the course of one and the same attack. Repeat neurological and ophthalmological examinations should thus be generally considered and are mandatory in case of new symptoms.
How should NMOSD be diagnosed?
The initial workup in patients with suspected NMOSD must include a detailed medical history and an extended physical and neurological examination. Additional mandatory diagnostic tests include cranial and spinal MRI and serum AQP4-IgG antibody testing. Orbital MRI may be required in selected patients (see section on MRI diagnostics for details). Lumbar puncture is highly recommended for differential diagnostic purposes. Optionally, OCT may be performed and evoked potentials assessed. Fig. 1 shows a proposed diagnostic algorithm.

Proposed algorithm for the diagnosis of NMOSD. AQP4-IgG aquaporin-4 immunoglobulin G antibodies, CSF cerebrospinal fluid, MOG-EM/MOGAD myelin oligodendrocyte glycoprotein (MOG) antibody-associated encephalomyelitis/MOG autoantibody-associated disease, MRI magnetic resonance imaging, OCT optic coherence tomography
We strongly recommend that NMOSD be diagnosed according to the revised diagnostic criteria proposed by the International Panel for NMO Diagnosis (IPND) in 2015 [228]. These criteria take into account clinical and MRI findings and the AQP4-IgG serostatus and allow NMOSD to be diagnosed after only a single clinical event. Use of the 2006 criteria [230] is strictly discouraged, since it may result in significant underreporting of NMOSD [190]. The 1999 criteria did not yet take into account AQP4-IgG serostatus and must not be used [229].
In patients with a positive AQP4-IgG serostatus, at least one of the six core characteristics listed in Box 1 must be present before a diagnosis of definite NMOSD can be made (note that the definition of core characteristics 5 and 6 includes not only clinical but also MRI findings), and alternative diagnoses that might better explain the patient’s symptoms must be excluded. This is different from previous criteria, which required a history of both acute ON and acute myelitis [230].
To make a diagnosis of NMOSD in patients with negative or unknown AQP4-IgG serostatus, at least two of these core characteristics must have occurred at least once, either with a single or, successively, with multiple clinical attacks (i.e., dissemination in space but not dissemination in time is required). In addition, at least one of the core clinical characteristics present must be acute ON, acute myelitis, or APS. Furthermore, the typical MRI criteria (specified in Box 2) must be met. Again, alternative diagnoses that might better explain the patient’s symptoms must be ruled out.
According to the IPND criteria, a diagnosis of NMOSD can be made also in patients with unknown AQP4-IgG serostatus using the same set of criteria as for patients with negative AQP4-IgG serostatus. However, we recommend that such patients should be tested for AQP4-IgG at the earliest possible time, as long-term treatment options may depend on a patient’s serostatus (see Part 2 for details).
In accordance with a proposal made by the IPND [228], we recommend that a diagnosis of relapsing disease should be made in the event that a new attack occurs more than 4 weeks after onset of the initial attack. Additional symptoms that newly emerge within 4 weeks after onset of an attack should be considered part of one and the same attack.
What alternative diagnoses have to be considered?
A number of differential diagnoses (Box 3) and red flags (Box 4), i.e., clinical, laboratory, or imaging findings that should prompt thorough investigation for competing diagnoses, have to be considered before a definite diagnosis of NMOSD can be made.
The most important antibody-related differential diagnosis of NMOSD is MOG-EM/ MOGAD [84, 91, 132, 134]. A first set of diagnostic criteria for MOG-EM/MOGAD has recently been proposed by an international panel of experts [77] (Box 5), together with a list of red flags (Supplementary Box 1), which we recommend be checked in all patients with signs and symptoms suggestive of that disease. Of note, some patients may meet both the diagnostic criteria for NMOSD and those for MOG-EM/MOGAD, due to the substantial clinical and radiological overlap between the two diseases. However, NEMOS strongly recommends that AQP4-IgG-negative patients who meet the criteria for MOG-EM/MOGAD [77, 78] should no longer be assigned the diagnosis of NMOSD, as convincing evidence now suggests that MOG-EM/MOGAD is a pathogenetically, clinically, and prognostically distinct disorder in its own right, possibly with different treatment needs [75, 77, 78, 84, 86, 154]. The recommended indications for MOG-IgG testing can be found in Supplementary Box 2.
As coexistence of AQP4-IgG-positive NMOSD and MOG-EM/MOGAD is considered to be extremely rare [86, 121] and false-positive serological results still occur, patients meeting the diagnostic criteria for both of these two conditions should be reviewed at a specialized center and their "double-positive" antibody status reconfirmed.
The most important differential diagnosis in AQP4-IgG- and MOG-IgG-negative (“double-negative”) patients with NMOSD is MS. However, several other rare autoimmune, paraneoplastic, neoplastic, infectious, metabolic, and vascular diseases that can mimic NMOSD have to be taken into consideration as well (Box 3). In all patients with double-negative NMOSD, neurosarcoidosis should be considered, especially (but not exclusively) in those presenting with longitudinally extensive transverse myelitis (LETM) [40]. Myelitis in NMOSD may also mimic spinal cancer or lymphoma and vice versa; whenever possible, AQP4-IgG (and MOG-IgG) should be tested and NMOSD excluded before a decision is made to perform spinal cord biopsy—a procedure that can leave patients severely disabled—for suspected spinal neoplasia [180]. For a comprehensive overview of relevant differential diagnoses, see also [105, 110, 116, 214].
Of note, coexistence of NMOSD with any of these differential diagnoses may rarely occur and has to be taken into consideration, both in AQP4-IgG-positive and AQP4-IgG-negative patients. Most importantly, coexisting connective tissue disorders (CTD) and other rheumatic disorders, such as systemic lupus erythematosus, antiphospholipid syndrome, Sjögren’s syndrome, or Sharp syndrome, are not uncommon in patients with (usually AQP4-IgG-positive [87]) NMOSD, suggesting a shared autoimmune predisposition, and must per se not be taken as sufficient reason to reject the diagnosis of NMOSD [7, 65, 74, 87, 167, 208]. AQP4-IgG-positive NMOSD has also been reported in association with other autoimmune disorders such as myasthenia gravis (MG) [80, 97, 123, 138], celiac disease [13, 73], and autoimmune thyroid disease. An interdisciplinary approach is recommended in such cases. Co-existing MG should be considered not only in patients with limb paresis that becoms progressively worse during periods of physical activity but also in patients with ocular weakness, respiratory insufficiency, dysphagia, dysarthria, or dysphonia. Overall, signs of co-existing autoimmunity have been reported in 25–40% of patients with AQP4-IgG-positive NMOSD in some cohorts [74, 167, 208].
Although a number of patients with AQP4-IgG-positive NMOSD and co-existing cancer (with AQP4 expression within the tumor tissue in some) have been reported [166], the disorder is not tumor-associated in the vast majority of cases [87, 192]. While tumor screening may in fact be indicated in individual patients, depending on the presence of risk factors or symptoms suggestive of an underlying or co-existing tumor, general screening of all patients with newly diagnosed AQP4-IgG-positive NMOSD for cancer—especially by means of invasive or otherwise potentially harmful methods—is currently not recommended. Male sex, age > 45 years, and presentation with nausea and vomiting have been identified as risk factors for tumor association in European AQP4-IgG-positive patients [192].
All patients with suspected NMOSD attacks should be examined for signs and symptoms of infection. However, it should be taken into account that the onset of NMOSD (and MOG-EM/MOGAD [85]) is not uncommonly preceded by a common cold or other infection [87]. This can lead to false suspicion of CNS infection. Very rarely, NMOSD (as well as MOG-EM/MOGAD [67, 85]) first occurs following vaccinations [4, 53]. It is unclear whether infection or vaccination induces a de-novo autoimmune reaction in these patients (e.g., by molecular mimicry) or rather acts as a non-specific inflammatory trigger causing clinical exacerbation in patients with pre-existing but previously clinically latent NMOSD. Infections also frequently precede relapses in both NMOSD and MOG-EM/MOGAD; taking into account both the first attack and subsequent attacks, NMOSD attacks were preceded by infections in around 30% of AQP4-IgG-positive and 20% of AQP4-IgG-negative cases in a large European study [87].
Practical recommendations—antibody testing
AQP4-IgG testing is recommended in all patients with clinical or clinico-radiological findings (either present or previous) that would permit the diagnosis of “NMOSD with positive AQP4-IgG serostatus” in the case of a positive test result according to the IPND criteria, i.e., in all patients with one of the core characteristics listed in Box 1, and in all patients diagnosed with “NMOSD with unknown AQP4-IgG serostatus” according to the IPND criteria. In all other cases, the decision on whether or not to test for AQP4-IgG testing should be made individually; however, broad, unselected screening of patients with MS that do not meet the above-mentioned criteria is discouraged, especially in regions of the world in which NMOSD accounts only for a small proportion of IDD cases, since this practice may result in an unfavorable ratio of false-positive to true-positive results. Typical indications for MOG-IgG testing are listed in Supplementary Box 2. Both AQP4-IgG and MOG-IgG (to rule out MOG-EM/MOGAD in patients meeting the IPND criteria for “NMOSD with AQP4-IgG-negative serostatus”) must be tested by means of cell-based assays (CBA). The assay must employ either fixed or live cells expressing conformationally intact, full-length recombinant human AQP4 or MOG protein, respectively, as antigenic substrate and mock-transfected (or, at minimum, non-transfected) cells as control substrate [77, 78, 82, 90, 175, 228].
Formalin-fixed CBA are easier to standardize, are commercially available and thus widely accessible, and can be utilized at all routine laboratories familiar with basic immunofluorescence techniques; in contrast, the technically more demanding live CBA are offered only by a few specialized laboratories. However, live CBA have been found to be slightly more sensitive in some studies, especially for detecting MOG-IgG [44, 175, 223, 224]. Both types of assays may be used.
Immunohistochemistry (IHC) using rodent or primate CNS tissue sections as substrate, enzyme-linked immunosorbent assays (ELISA), fluorescence-based immunoprecipitation assays (FIPA), Western blot assays, and radioimmunoprecipitation assays (RIA) have all been used in the past but are thought to be less sensitive and/or specific than CBA and should therefore no longer be employed for routine clinical testing [71, 77, 78, 90, 160, 228]. IHC-based assays for detecting AQP4-IgG [70, 90, 125] may be used exceptionally if testing by CBA is strictly not available, neither locally nor by shipment to specialized centers, as is still the case in some regions of the world; in that event, confirmatory testing by means of a CBA should be striven for and should be performed as soon as it becomes available. The use of IHC assays for detecting MOG-IgG is strongly discouraged. Assays using peptides instead of full-length protein as antigens lack specificity and thus are not suitable to make a diagnosis of NMOSD or MOG-EM/MOGAD and must no longer be used [71, 77, 78, 90, 228].
The timing of antibody testing is important, since procedures given for attack treatment (glucocorticosteroids, plasma exchange [PLEX], immunoadsorption [IA]), immunosuppressants, intravenous immunoglobulins) or attack prevention may rarely [48, 66] cause false-negative or ambiguous results. Therefore, samples for antibody testing should ideally be taken before treatment is commenced. As AQP4-IgG and MOG-IgG are produced mainly extrathecally, serum should be used for AQP4-IgG determination as biomaterial of choice [72, 90, 228].
Testing of CSF for AQP4-IgG or MOG-IgG may theoretically be helpful in the very rare cases in which non-specific, coexisting serum antibodies cause a strong background signal that may potentially mask a weak AQP4-IgG- or MOG-IgG-specific signal or if testing is performed shortly after PLEX or IA. In fact, a few reports exist on patients positive for AQP4-IgG or MOG-IgG only in the CSF but not in serum [122, 131]. In most of these cases, however, the results were not independently confirmed and/or no sufficiently large control cohorts were included, so the data remain controversial. Importantly, the current diagnostic criteria recommend the use of serum assays [77, 228]. If, deviating from this recommendation, in rare selected cases a provisional diagnosis is made after consultation with a specialized center based on CSF-only positivity, retesting of the CSF and serum sample in a methodologically different assay as well as confirmatory testing of follow-up serum samples is highly advisable.
The specimen analyzed (serum, CSF), the assay type (e.g., live CBA, fixed CBA, ELISA, tissue-based assay) and the assay manufacturer or performing laboratory, as well as the test result (positive, negative, grey area) and, if available, the titer and cut-off, should be documented in the discharge letter (e.g., “Serum AQP4-IgG positive (12-Sept-2022; titer 1:320, cut-off ≥ 1:10; fixed cell-based assay [Euroimmun])”. In addition, the disease status (acute attack, remission) and treatment status at the time of testing should be documented, especially in the event of a negative test result, since samples obtained during remission or under treatment may occasionally be falsely negative or unequivocal and warrant retesting, as mentioned above [66, 223, 234].
The current diagnostic criteria for seropositive NMOSD strictly require the presence of AQP4-specific antibodies of the IgG isotype [77, 90, 228]. Seropositivity only for IgM and/or IgA antibodies to AQP4 must not be considered sufficient, as the pathogenetic and clinical relevance of such findings is unknown. Among other factors, assay specificity depends on the method used to detect patient IgG bound to the test substrate. As detecting patient IgG by use of so-called H + L-chain-specific secondary antibodies carries the risk of occasional false-positive results due to cross-binding of this type of antibodies to IgM and IgA, so-called Fcγ-specific (or, alternatively, IgG1-specific) secondary antibodies should preferentially be used[77, 90]. Information about the exact method used in a particular assay can be obtained from the manufacturer or the performing laboratory. A summary of methodological recommendations can be found in Supplementary Box 3.
As AQP4-IgG and MOG-IgG titers may vary significantly over time, depending on disease activity and treatment [66, 90, 108], and because of the potential differences in sensitivity between assays, retesting (preferentially by means of a standardized live CBA, if available) should be considered in seronegative patients if NMOSD or MOG-EM/MOGAD is still suspected, ideally during a treatment-free interval or during an acute attack. Given the fact that no serological assay is 100% specific and taking into account the important and often life-long diagnostic and therapeutic consequences of a positive test result, positive results should ideally be confirmed, either in a second, methodologically different cell-based assay or, if such an assay is not available, at least in a second sample, especially in the case of low-titer results (e.g., at cut-off or only one dilution step above cut-off) or if a non-approved (in-house) assay was used. Confirming a positive test result is highly recommended if one or more red flags (Box 4 and Supplementary Box 1) are present [77, 219, 228]. As the currently available serological tests for MOG-IgG (especially in patients with low titers) are of lower specificity than those for AQP4-IgG, this is particularly important before a diagnosis of MOG-EM/MOGAD is made and seronegative NMOSD or MS consequently dismissed [77].
Practical recommendation—CSF analysis
Analysis of cerebrospinal fluid (CSF) is not formally required to make a diagnosis of NMOSD according to the current diagnostic criteria; however, we strongly recommend lumbar puncture in patients with suspected NMOSD, since CSF analysis can support a diagnosis of NMOSD and help to distinguish the disorder from MS and other differential diagnoses.
Basic CSF examination should include determining oligoclonal bands, CSF white cell counts (WCC), CSF WCC differentiation (cytology), CSF erythrocyte counts, CSF total protein levels, CSF L-lactate levels, and CSF and serum glucose levels. However, we recommend to assess also IgG, IgM, IgA and albumin CSF/serum ratios (in combination with Reiber’s diagrams [173, 174, 226]), IgG/IgM/IgA fractions, and, if available, the so-called measles, rubella, and zoster virus (MRZ) reaction [68, 69, 76, 83]. The use of Link’s IgG index is discouraged due to limited specificity, especially in patients with moderate to severe blood‒CSF barrier dysfunction). While CSF-specific oligoclonal IgG bands are a diagnostic mainstay of MS (present in > 95% of continental European patients, lower frequency in Asian patients) and persist over time, irrespective of clinical disease activity, they are absent in the majority of patients with AQP4-IgG-positive NMOSD (present in around 25% at first LP in [87] and in only ~20% of acute samples and 10% of remission samples in [79]), AQP4-IgG-negative NMOSD (present in around 35% at first LP in [87]), or MOG-EM/MOGAD (~ 10%) and may be present only transiently [76, 79, 83, 87]. CSF WCC ≥ 50/µl, CSF neutrophils (occasionally also eosinophils), and an albumin CSF/serum ratio (QAlb) > 12 × 10–3 are all atypical for MS but not uncommon in AQP4-IgG-positive NMOSD (WCC ≥ 50/µl in ~ 15%; neutrophils present in ~ 50%; and QAlb > 12 × 10–3 in ~ 30% of all patients with acute attacks and in as many as 50% of those with blood–CSF barrier dysfunction) and MOG-EM/MOGAD (WCC ≥ 50/µl in 46% with acute myelitis, but only very rarely during acute ON; neutrophils in ~ 60% during acute myelitis and ~ 25% during acute ON; QAlb > 12 × 10–3 in 24% of all patients and 43% of those with elevated QAlb during acute myelitis, but only rarely during acute ON) [76, 79, 83]. A positive MRZ reaction (as defined by a positive antibody index against at least two of these three viral antigens), which is present in ~ 67% of patients with MS and is considered the most specific laboratory marker of MS known so far, was absent in virtually all patients with AQP4-IgG-positive or -negative NMOSD tested so far (but also in those with MOG-EM/MOGAD) [68, 69, 76, 83]. In contrast to MS, CSF L-lactate levels were mildly increased in an AQP4-IgG-positive NMOSD cohort (in ~ 45% during acute myelitis and ~ 20% during acute ON; 0% during remission; median 2.9 mmol/l during acute attacks, range 2.1–6.8, in a European cohort[79]), similar to what is seen in MOG-EM/MOGAD (~ 25% in patients with acute myelitis, median 2.5 mmol/l, range 1.9–4.43, > 3 mmol/l in around 10%; rarely in acute ON) [76, 79, 83]. It is important to note (a) that the frequency and extent of CSF abnormalities, as detected by lumbar puncture, depends strongly on the lesion site (spinal cord > brain > optic nerve), reflecting both the rostrocaudal CSF gradient and lesion volume, and (b) that CSF findings may be normal, even during attacks, particularly often in patients with acute ON [76, 79, 83]. AQP4-IgG (as well as MOG-IgG) is often detectable also in the CSF, but antibody indices are usually negative, indicating an extrathecal origin [72, 86]. As mentioned above, the current diagnostic criteria for NMOSD (as well as those for MOG-EM/MOGAD) require serum AQP4-IgG positivity; CSF AQP4-IgG positivity alone is currently not considered sufficient.
Practical recommendation—other laboratory tests
Further laboratory tests can help to identify alternative diagnoses and/or concomitant autoimmune diseases. Besides basic examinations of blood (differential blood count, serum chemistry, and coagulation) and urine, this should include serological examinations for CTD (e.g., ANA, ENA, anti-ds-DNA-antibodies, c/pANCA, cardiolipin antibodies). Further assessments should be considered to exclude hypovitaminosis, other metabolic disorders, infections, autoimmune encephalitis, and neoplastic and paraneoplastic disorders (e.g., anti-collapsin response mediator protein-5 [CRMP5/CV2] antibody-associated optic neuropathy and myelopathy or anti-Ma-associated diencephalic syndrome) (see Box 3 for details).
Practical recommendations on imaging—MRI
MRI examinations are an essential part of the clinical workup for diagnosis and monitoring of NMOSD [199]. The initial MRI serves as an important baseline examination and shall cover both the brain and the spinal cord. MRI is also essential for distinguishing NMOSD from other CNS conditions.
If available, (fat-suppressed) MRI of the orbits is highly recommended, since it allows more precise depiction of the optic nerve than routine brain MR imaging. It may be required in patients with negative or unknown serostatus if making a diagnosis of NMOSD depends on the presence of longitudinally extensive optic neuritis (LEON) or an optic nerve lesion involving the optic chiasm (see Box 2) and conventional brain MRI does not allow reliable assessment of lesion extension. Particular notice should be paid to lesion location and extension, the presence or absence of optic head swelling, perineural gadolinium (Gd) enhancement, chiasmal lesions, and lesions of the optic tracts. While NMOSD more often affects the posterior (intracranial) part of the optic nerve, lesions in MS and MOG-EM/MOGAD are more frequently located in the anterior portion [196]. Swelling of the optic nerve disk/head on orbital MRI is typically seen in MOG-EM/MOGAD, may infrequently occur in AQP4-IgG-positive NMOSD, and is very rare in MS [171], which typically manifests with retrobulbar ON. LEON is frequent in both NMOSD and MOG-EM/MOGAD but typically absent in MS [140, 171]. The optic chiasm and, more rarely, the optic tracts (often bilaterally [171]) may be affected as well in AQP4-IgG-positive NMOSD, whereas these structures are mostly spared in MS [171]; according to recent data, chiasmal lesions are also not uncommon in MOG-EM/MOGAD and then often extend from the anterior part of the optic nerve to the chiasm [84, 203]. Simultaneous bilateral optic nerve involvement is rather infrequent at onset in AQP4-IgG-positive NMOSD (~ 15% in a European cohort [87]) but may occur later in the disease course. It is more common in seronegative NMOSD and in MOG-EM/MOGAD [84, 87] and relatively rare in MS. Gd-enhanced imaging of the optic nerve is recommended for differential diagnostic purposes. Enhancement of the perineural optic nerve sheath strongly favors a diagnosis of MOG-EM/MOGAD over NMOSD or MS. Demonstration of optic nerve Gd enhancement may also facilitate the discrimination between true relapses and pseudorelapses (defined as worsening or re-occurrence of pre-existing neurological symptoms caused by systemic metabolic changes rather than immune-mediated mechanisms) by providing supportive information [199]. Of note, recent studies found Gd-enhancing lesions of the optic nerve in 20–50% of inter-attack MRI scans, i.e., in asymptomatic patients [159, 193]. In one study, these lesions were mainly located to sites of previous ON attacks, whereas new asymptomatic lesions as detected by Gd-enhanced imaging at sites not previously affected were very rare [193]. Asymptomatic lesions were significantly shorter in length (in mm) than attack-related lesions and were not associated with an increased risk for site-specific attacks in the following year in another study [159].
Spinal MRI should, whenever possible, cover the entire spinal cord, including the conus, since lesion site and longitudinal lesion extension can be diagnostically informative. While myelitis is typically longitudinally extensive—extending over three or more complete vertebral segments—in both AQP4-IgG-positive and AQP4-IgG-negative NMOSD (and MOG-EM/MOGAD), as mentioned above, long lesions are extremely rare in MS and, if present, usually result from the coalescence of adjacent lesions. A finding of exclusively new short lesions during an acute attack of myelitis is more common in MOG-EM/MOGAD (being found at least once in up to 50% of cases over the course of disease in a European cohort [84]) than in NMOSD (10–15% [41, 87]). Short lesions may be present also in combination with LETM lesions [87]. When assessing lesion length, it must be taken into account that lesion extension depends to some extent on timing, with short lesions sometimes representing still evolving or already resolving (if MRI is performed late) LETM. If only short spinal cord lesions are detected but the MRI was performed early during an attack and NMOSD is still suspected, a second MRI during the same attack may be considered; this may be particularly helpful in those patients with suspected NMOSD without AQP4-IgG or NMOSD with unknown AQP4-IgG serostatus according to the IPND criteria in whom the diagnosis depends on the presence of a longitudinally extensive spinal cord lesion [228]. Inflammation of the entire spinal cord has mostly been observed in AQP4-IgG-positive NMOSD cases [87], occasionally occurs in MOG-EM/MOGAD [84], and is not seen in MS. Complete resolution of T2 lesions (and especially of spinal lesions) was shown to be significantly less common and the reduction in T2 lesion area on axial spinal cord imaging to be significantly smaller in AQP4-IgG-positive NMOSD (and MS) than in MOGAD [187]. After severe and/or recurrent myelitis attacks, MRI may also show (longitudinally extensive) spinal cord atrophy in NMOSD (but also in MOG-EM and in long-standing MS). The mean upper cervical cord area may be reduced (AQP4-IgG-positive NMOSD > MOG-EM/MOGAD) [25]. Involvement of the lumbar spinal cord and the conus is less common in AQP4-IgG-positive NMOSD and MS, so its presence favors MOG-EM/MOGAD [36].
Spinal MRI should, whenever possible, include both sagittal and axial imaging. AQP4-IgG-positive NMOSD (as well as MOG-EM/MOGAD) predominantly affects the central portion of the spinal cord (with a preserved peripheral dark rim), whereas MS lesions are mostly located in the peripheral portion [99, 161], often involving the lateral and dorsal columns. Sometimes, only the gray matter is affected, resulting in H-shaped T2 hyperintensity on axial images, termed the “H-sign” [33]; however, this occurs more frequently in MOG-EM/MOGAD and is virtually absent in MS. In contrast, lesions in MS often appear cigar-shaped/cylindrical on sagittal images and wedge-shaped on axial images and are typically sharply demarcated [38].
Spinal MRI should also be examined for swelling, gadolinium enhancement, cavitations, and bright spotty lesions (BSL). Spinal cord swelling during acute myelitis attacks is common in NMOSD, but may also be present in MOG-EM [84] and MS. In severe cases of AQP4-IgG-positive NMOSD, T1-hypointense spinal cord lesions and cavitations may occur [99, 161]. Recently, so-called BSL, defined as hyperintense lesions on axial T2-weighted images and sometimes associated with T1 low signal, have been reported to strongly favor a diagnosis of (especially AQP4-IgG-positive) NMOSD in patients presenting with myelitis [27, 161, 169, 181]. Gd enhancement of spinal cord lesions is common in all three diseases, may be ring-shaped in AQP4-IgG-positive NMOSD (as well as in MS) [235], but may also be patchy and irregular [99, 161]. Contrast-enhanced MRI of the spinal cord is not formally required for making a diagnosis of AQP4-IgG-positive NMOSD according to the IPND criteria [228], but we recommend its use for differential diagnostic purposes [145]. Moreover, by increasing the sensitivity of MRI for detecting active lesions, it may be helpful in improving the discrimination of true relapses and pseudorelapses when relapses cannot be adjudicated based on clinical presentation or T2 imaging alone. This said, it should be taken into account as a limitation, however, that Gd enhancement may persist for more than 1 month after an attack in some patients with NMOSD (but Gd enhancement persisting for more than 3 months should be considered a red flag prompting search for alternative causes [227]) and true myelitis attacks with no new Gd-enhancing lesions may occasionally occur[233].
Spinal cord remission MRI scans, if available, should be examined for new subclinical lesions. While clinically silent spinal cord lesions may be present in all three conditions during acute attacks, an accumulation of silent spinal cord lesions on T2-weighted imaging/fluid-attenuated inverse recovery MRI performed during remission (i.e., outside of a relapse and at least 3 months from the last attack), a characteristic feature used in the diagnosis of MS [207], was mostly absent in NMOSD (4%) in a large, mixed adult and pediatric AQP4-IgG-positive cohort in the UK) [21], as well as in MOG-EM/MOGAD (0%) in the same study [21]. New subclinical lesions during remission, although rare, indicated a high risk of imminent relapse in this study [21]. As a caveat, it should be noted that Gd-enhancing spinal cord lesions may be more commonly present during remission. In the N-MOmentum trial, contrast-enhancing lesions occurred independent of clinical disease activity in 15% of patients, probably representing blood–brain barrier damage or subclinical attacks, and were associated with a significantly increased risk of domain-specific attacks in the following year [30, 159]. Moreover, follow-up spinal cord imaging after acute attacks may be justified to assess the efficacy of attack treatment and the presence or absence of spinal cord atrophy.
Spinal nerve root enhancement or myeloradiculitis has been observed in a few patients with AQP4-IgG-positive NMOSD [47, 101, 102, 204, 211], but was recently reported also in MOG-EM/MOGAD [176, 202, 206, 217] and even in individual patients with (pediatric) MS [206]. Importantly, root enhancement should also prompt radiologists to consider spinal cord sarcoidosis as an alternative diagnosis (which may be associated with subpial, leptomeningeal enhancement; LETM; and a “trident sign” on axial imaging).
Brain and/or spinal MRI should always cover the cranio-cervical junction, as upper cervical spinal cord lesions in AQP4-IgG-positive NMOSD (and MOG-EM/MOGAD) frequently extend into the brainstem [5, 161]. MRI should also include contrast-enhanced, thin-section imaging of the brainstem in case signs of symptoms suggestive of brainstem encephalitis are present. Brainstem lesions, which are more common in AQP4-IgG-positive than in AQP4-IgG-negative patients [5, 87], may also occur independently of spinal cord lesions. They often involve the dorsal medulla oblongata (area postrema) in (typically AQP4-IgG-positive) NMOSD and predominantly the pons in MOG-EM/MOGAD. Lesions affecting the middle cerebellar peduncle are more suggestive of MOG-EM/MOGAD (or MS) than of AQP4-IgG-positive NMOSD [136]. Brainstem lesions are often bilateral in AQP4-IgG-positive NMOSD. Diencephalic lesions are suggestive of (especially AQP4-IgG-positive) NMOSD, whereas the deep gray matter (thalami, basal ganglia) is more often affected in MOG-EM/MOGAD.
Importantly, neither a normal brain MRI nor clinically silent lesions should per se be taken as evidence against NMOSD. While a normal brain MRI is not unusual in NMOSD (and, less frequently, may occur also in MOG-EM/MOGAD), especially at onset (both in AQP4-IgG-positive and in AQP4-IgG-negative patients [87]), brain lesions are mandatory to make a diagnosis of MS according to current criteria. Clinically silent brain lesions may accompany acute ON or myelitis attacks (most frequently seen in the corpus callosum, followed by the internal capsule, and the cerebral peduncles in AQP4-IgG-positive patients) [103, 168], but large tumefactive (even bilateral) lesions (as occasionally seen also in MOG-EM/MOGAD and MS), cavitary lesions, and residual T1 lesions (rare in MOG-EM/MOGAD but frequent in MS) may occur as well. As with silent spinal cord lesions, accumulation of silent brain lesions during remission, as frequently seen in MS, is widely absent in AQP4-IgG-positive NMOSD (5%) (and also rare in MOG-EM/MOGAD [4% in a mixed adult and pediatric cohort, 12% in an exclusively pediatric cohort]) [21, 37].
Particular attention must be paid to brain lesion extension and distribution. Like spinal cord and optic nerve lesions, brain lesions tend to be longitudinally extensive in NMOSD (with the best evidence available for AQP4-IgG-positive patients), including corticospinal tract lesions and corpus callosum lesions [45, 99]. An ADEM-like lesion pattern on MRI is suggestive of MOG-EM/MOGAD, not NMOSD, as is a ‘leukodystrophy-like’ MRI pattern (characterized by confluent, largely symmetrical lesions) [51], which mainly occurs in children. MOG-EM/MOGAD is further characterized by so-called fluffy, poorly demarcated/ill-defined T2 lesions (as opposed to well-demarcated lesions in MS) [109]. Periependymal lesions around the lateral, third, and/or fourth ventricle are a common finding in (typically AQP4-IgG-positive) NMOSD and should not be (mis)taken as evidence of MS. The temporal lobe is rarely affected in AQP4-IgG-positive NMOSD [136] but frequently involved in MS (typically the inferior temporal lobe [95, 96, 137]) and in MOG-EM/MOGAD [136].
Contrast-enhanced MRI of the brain is not formally required to make a diagnosis of NMOSD but is recommended, especially at onset, for differential diagnostic purposes. Brain lesions in NMOSD often show patchy, cloud-like Gd enhancement. Gd enhancement may also be seen on T1 imaging alongside extensive periependymal T2 brain lesions [228]. Finally, pencil-thin ependymal Gd enhancement is considered to be relatively specific for AQP4-IgG-positive NMOSD [11]. Leptomeningeal brainstem enhancement and leptomeningeal enhancement accompanying cortical encephalitis on fluid-attenuated inversion recovery (FLAIR) imaging, on the other hand, argue rather against NMOSD (although rare cortical involvement has been reported in patients with positive or unknown AQP4-IgG [107, 201]) and more in favor of MOG-EM/MOGAD [18, 20]. As with spinal MRI, brain MRI examinations conducted very early during an attack may underestimate the peak lesion load, which may not occur until a few days later; follow-up MRI during acute attacks may therefore be advisable, especially in the event of newly emerging symptoms or worsening of attack-related symptoms.
Further MRI findings considered typical for NMOSD according to the current criteria are shown in Box 2. As a caveat, CNS lesions in both NMOSD (~ 15%) and MOG-EM/MOGAD (~ 40% at least once in the course of disease in a European cohort [84]) may formally meet the Barkhof or Swanton criteria for MS in some cases. We strongly recommend consideration of the MRI features listed in Box 4 as ‘red flags’, the presence of which should prompt physicians to critically challenge a diagnosis of NMOSD [99, 228].
Imaging protocols and sequences should be chosen according to the clinical situation (diagnosis vs. monitoring of disease course and/or treatment responses) and with respect to the region of interest (brain, optic nerve/orbits, spinal cord). Generally, 3 T systems should be preferred to those of 1.5 T because of their better resolution. 3D T1 and FLAIR sequences (e.g., 1 mm isotropic) should be preferred over 2D acquisitions. In the case of 2D acquisitions, the slice thickness should not exceed 3 mm. As said before, spinal cord imaging should, if possible, include complete coverage with axial T2 acquisition to increase sensitivity for short lesions [41, 43]. MRI reports should ideally state the total number of lesions and the number of new lesions, together with their dimensions and location in detail (e.g., centralized versus lateral lesions in the spinal cord) [228]. The MRI at onset should include contrast-enhanced T1 images and ideally a diffusion-weighted sequence and a susceptibility-weighted sequence or conventional gradient echo T2* sequence, to separate inflammatory from malignant and vascular processes. Macrocyclic rather than linear Gd agents should be used and unnecessary contrast-enhanced MRI studies avoided to mitigate potential health risks associated with accumulation of Gd in the CNS [49]. Reports on attack-related and chronic-progressive brain atrophy in AQP4-IgG-positive NMOSD exist, but data on the structured involved and the severity vary among studies [50, 63, 135, 209, 221]; currently, monitoring of brain atrophy is not generally recommended outside of clinical studies. Beyond this, the clinical utility of non-conventional MRI (e.g., 7-T MRI, central vein sign, diffusion tensor imaging) for diagnosis, differential diagnosis, and disease monitoring remains to be evaluated [29, 118, 120, 198].
Practical recommendations—visual acuity, OCT, fundoscopy, and perimetry
Habitually corrected visual acuity (VA) must be assessed in both eyes in all patients presenting with a suspected attack of NMOSD. As subclinical ON damage has been observed in patients with clinical myelitis, brainstem encephalitis, or encephalitis, VA should be assessed irrespective of a patient’s clinical presentation. VA assessment should be performed in a standardized manner by an experienced examiner, ideally by an ophthalmologist or optometrist. Especially if VA appears normal using standard Snellen charts, low-contrast VA, which is exclusively altered in some patients, should be tested in addition, e.g., with the ETDRS (Early Treatment Diabetic Retinopathy Study) contrast chart, SLOAN letters 2.5%. In patients with acute ON, VA should be assessed at presentation, shortly after attack treatment (to inform about the need for potential treatment escalation; see Part 2), and before discharge and should be documented in the discharge letter. VA should be assessed also at all follow-up visits.
OCT is a non-invasive technique to generate high-resolution 2D and 3D images of the neural and vascular components of the retina [46, 151]. OCT is an optional procedure not formally needed to reach a diagnosis of NMOSD. However, as it can provide useful supportive information of differential diagnostic relevance, its use is recommended. If OCT is carried out, a high-resolution macular volume scan should be performed to allow analysis of all macular layers and semi-automatic segmentation of all layers. The peripapillary retinal nerve fiber layer (pRNFL) thickness should be assessed as the most informative parameter by performing a ring scan around the optic nerve. In addition, the combined ganglion cell/inner plexiform layer (GCIPL) thickness, the presence or absence of microcystic macular edema (MME) in the inner nuclear layer (INL), the size and shape of the fovea, and the retinal vessel density (using OCT angiography) may be assessed. All scans must be checked for sufficient quality and for segmentation errors before analysis [184].
In NMOSD, most studies have consistently shown thinning of the RNFL and the GCIPL after ON attacks that is, on average, more severe in AQP4-IgG-positive NMOSD than in MS [12, 150, 172, 186, 216]. Based on retrospective analysis of 11 studies (not all of which distinguished between AQP4-IgG-positive and AQP4-IgG-negative patients), OCT shows average RNFL thinning after ON to 55–83 µm in NMOSD and 74–95 µm in MS, compared to an average RNFL thickness of 93–108 µm in healthy individuals without a history of ON [12]; more profound RNFL atrophy (especially in non-temporal quadrants) than in MS has also been demonstrated in MOGAD in a recent pediatric cohort after the first ON attack [155]. A correlation between RNFL thinning and atrophy of the visual pericalcarine cortex has been found in an almost exclusively AQP4-IgG-positive cohort [221]. OCT may be used as a supportive diagnostic tool already early in the disease course. Notably, 88% of NMOSD eyes displayed RNFL thickness < 60 µm even after just one episode of ON in a mixed AQP4-IgG-positive and -negative cohort [216].
Looking into cross-sectional images in more detail, up to 26% of eyes after ON featured MME in the INL in mixed AQP4-IgG-positive and negative NMOSD cohorts. INL-MME rarely appears after MS-ON (5–6%) and is not observed in healthy controls [12, 186]. Moreover, foveal morphometry revealed a wider and flatter fovea in AQP4-IgG-positive NMOSD than in MS and healthy controls [144].
Fundoscopy may facilitate the differential diagnosis of NMOSD and is thus recommended. Demonstration of papillitis/papilledema and/or retinal hemorrhages renders MOG-EM/MOGAD more likely than NMOSD and MS, in which optic disk edema is both less frequent and, usually, less severe. However, a number of differential diagnoses exist that need to be excluded (see Ref. [14, 82]).
Visual field testing is also recommended, since it can provide supportive differential diagnostic information, help to assess attack severity, and may be important for monitoring the efficacy and extent of treatment-driven or spontaneous attack resolution in patients with acute NMOSD-ON. Perimetric studies have demonstrated non-central scotomas (altitudinal, quadrant, three-quadrant, hemianopsia, and bitemporal hemianopsia) in around 25% of patients with AQP4-IgG-positive NMOSD-ON, whereas MS patients typically exhibit central scotoma [64, 141, 146]. Altitudinal hemianopia is the most common type of non-central scotoma in NMOSD and is rare in MOG-EM/MOGAD but may occur also in double-seronegative ON [64]. Complete visual field loss during acute attacks is more frequent in NMOSD and MOG-EM/MOGAD than in MS.
Practical recommendations on electrophysiology—evoked potentials
Visual-evoked potentials (VEP) and, to a lesser degree, somatosensory-evoked potentials (SSEP) examinations are helpful in the differential diagnosis of NMOSD and are thus recommended, although neither is formally required to make a diagnosis of NMOSD according to the IPND criteria. As in MS and MOG-EM/MOGAD, ON in NMOSD was associated with prolonged P100 latencies caused by demyelination both in AQP4-IgG-positive and in mixed, AQP4-IgG-positive and AQP4-IgG-negative cohorts [84, 178, 179]. Reduced amplitudes, suggesting axonal damage, or even a complete lack of response are seen more commonly in AQP4-IgG-positive NMOSD and MOG-EM/MOGAD than in MS [84, 178, 179]. VEP evidence of demyelination is considered a sufficient substitute for MRI evidence of CNS demyelination in patients with isolated ON in the current diagnostic criteria for MOG-EM/MOGAD [78].
What other biomarkers are of interest? New developments
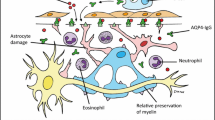
Pathophysiologically, AQP4-IgG-positive NMOSD is characterized by direct damage of astrocytes induced by antibodies against the water channel protein AQP4 and secondary damage to oligodendrocytes and neurons, leading to demyelination and axonal loss [82, 89, 126]. Biomarkers for astrocytic damage and neuroaxonal degeneration may thus potentially be of diagnostic and differential diagnostic impact. Neurofilament light chain (NfL), glial fibrillary acidic protein (GFAP), chitinase 3-like 1 protein (CHI3L1), and glutamine synthetase (GS) have all been found to be increased in the CSF of AQP4-IgG-positive NMOSD patients (and NfL and GFAP also in the serum[1, 61, 98, 183, 222]), especially during acute attacks [1, 31, 111, 205]. Elevated CSF GFAP levels have been occasionally observed also in AQP4-IgG-negative patients [111, 222]). Similarly, elevated CSF levels of the neutrophil-related chemokines CXCL1, CXCL5, and CXCL7, the B-cell chemoattractant CXCL13, the T-cell activation marker soluble CD27 (sCD27) [128, 130, 236], and the proinflammatory cytokine interleukin-6 (IL-6), a key mediator of the immune response in AQP4-IgG-positive NMOSD [26, 42, 215], have been described. Finally, a role for autoantibodies to glucose-regulated protein 78 (GRP78) in the pathogenesis of blood–brain barrier breakdown in both AQP4-IgG-positive NMOSD and MOG-EM/MOGAD has been suggested [194, 195]. However, none of these markers is yet part of the standard diagnostic workup of patients with suspected NMOSD, and we recommend (in the absence of gold standard assays and/or generally accepted cut-offs) that these markers should currently be used mainly in the context of scientific studies.
Unmet needs and outlook
The 2015 IPND diagnostic criteria for “NMOSD without AQP4-IgG” or “NMOSD with unknown AQP4-IgG serostatus” are based mainly on clinical and MRI features typically found in AQP4-IgG-positive NMO. This is useful in enabling a diagnosis of NMOSD also in patients with a false-negative AQP4-IgG test result and in AQP4-IgG-positive patients in whom AQP4-IgG testing has not yet been performed or who do not have access to AQP4-IgG testing. However, patients with genuinely negative AQP4-IgG serostatus may also meet these criteria, which renders the subgroup heterogenous. Revised criteria should thus stress the need for strictly distinguishing “NMOSD with unknown AQP4-IgG serostatus” and “NMOSD without AQP4-IgG” as different diagnostic categories and should define strategies aiming at identifying patients with potentially false-negative AQP4-IgG results who may be included in the latter category. Moreover, more studies addressing the pathogenesis of seronegative NMOSD and the question of potential pathogenetic, prognostic and/or therapeutic heterogeneity within that category are highly desirable. Demonstration of such heterogeneity might have important implications for nosology and future diagnostic criteria, as it might bring about a need for further diagnostic stratification. Most importantly, future diagnostic criteria for NMOSD should pay particular attention to improving the discrimination of NMOSD and MOG-EM/MOGAD, should explicitly exclude patients with MOG-EM/MOGAD from the category of “NMOSD without AQP4-IgG”, and, accordingly, should make MOG-IgG testing mandatory before a diagnosis of “NMOSD without AQP4-IgG” can be assigned.
Although OCT is of differential diagnostic value in general (and may, according to recent studies [24, 155], help discriminate MOG-EM/MOGAD and MS after a first ON attack), its utility for reliably distinguishing NMOSD from MOG-EM/MOGAD and MS is still limited. Combined scores taking into account structural OCT/OCT angiography parameters (such as, e.g., peripapillary RNFL, combined GCIPL thickness, and retinal vessel density), spatial distribution of OCT alterations, and functional parameters such as high- and low-contrast VA and the application of artificial intelligence and deep learning algorithms may have the potential to increase the discriminatory power of OCT in the future [100, 164]. However, inclusion of OCT in future criteria will be hampered by the limited availability of this technique in many parts of the world. The same is true for the implementation of 7 T MRI, novel advanced MRI sequences, or extended CSF analysis (including MRZ testing). Revised future criteria may yet well exploit these new developments by providing a list of (weigthed) supportive criteria, which could help to increase diagnostic certainty in patients with low-titer AQP4-IgG test results. Combined MRI scores may also help to improve both diagnostic specificity and sensitivity.
Conclusions
The diagnosis of NMOSD should be made according to the 2015 IPND criteria. The use of up-to-date, standardized CBA is of crucial importance. Both fixed and live CBA have been shown to be highly specific and sensitive for detecting AQP4-IgG; the use of other methods is currently discouraged. Confirmation of positive test results is generally recommended and is mandatory if red flags are present. MRI, CSF, electrophysiological, and OCT analyses can support the diagnosis. For NMOSD with negative or unknown AQP4-IgG status, obligatory MRI criteria exist. Particularly careful attention must be paid to ruling out differential diagnoses, especially in patients with seronegative NMOSD. The most important differential diagnoses include MOG-EM/MOGAD, MS, neurosarcoidosis, paraneoplastic neurological syndromes, and infectious diseases. In part 2 of this article series, we will provide detailed recommendations regarding the treatment of NMOSD.
Abbreviations
- ACE:
-
Angiotensin-converting enzyme
- ADEM:
-
Acute disseminated encephalomyelitis
- APS:
-
Area postrema syndrome
- AQP4:
-
Aquaporin-4
- CADASIL:
-
Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy
- CBA:
-
Cell-based assay
- CHI3LI:
-
Chitinase 3-like 1 protein
- CLIPPERS:
-
Chronic lymphocytic inflammation with pontine perivascular enhancement
- CNS:
-
Central nervous system
- CSF:
-
Cerebrospinal fluid
- CTD:
-
Connective tissue disorder
- ELISA:
-
Enzyme-linked immunosorbent assay
- ETDRS:
-
Early Treatment Diabetic Retinopathy Study
- FIPA:
-
Fluorescence-based immunoprecipitation assay
- FLAIR:
-
Fluid-attenuated inversion recovery
- GCIPL:
-
Ganglion cell/inner plexiform layer
- GAD:
-
Glutamic acid decarboxylase
- Gd:
-
Gadolinium
- GFAP:
-
Glial fibrillary acidic protein
- GRP78:
-
Glucose-regulated protein 78
- GS:
-
Glutamine synthetase
- HIV:
-
Human immunodeficiency virus
- HLA:
-
Human leukocyte antigen
- HTLV:
-
Human T-lymphotropic virus
- IDD:
-
Inflammatory demyelinating disorders
- IgG:
-
Immunoglobulin G
- IL-6:
-
Interleukin-6
- IL2R:
-
Interleukin-2 receptor
- INL:
-
Inner nuclear layer
- IPND:
-
International Panel for NMO Diagnosis
- LEON:
-
Longitudinaly extensive optic neuritis
- LETM:
-
Longitudinally extensive transverse myelitis
- MOG:
-
Myelin oligodendrocyte glycoprotein
- MOG-EM:
-
MOG encephalomyelitis
- MOGAD:
-
MOG antibody-associated disease
- MRI:
-
Magnetic resonance imaging
- MRZ:
-
Measles/rubella/zoster virus reaction
- MS:
-
Multiple sclerosis
- NEMOS:
-
Neuromyelitis optica study group
- NfL:
-
Neurofilament light chains
- NMDA:
-
N-Methyl-d-aspartate
- NMO:
-
Neuromyelitis optica
- NMOSD:
-
Neuromyelitis optica spectrum disorders
- OCB:
-
Oligoclonal bands
- OCT:
-
Optical coherence tomography
- ON:
-
Optic neuritis
- PLEX:
-
Plasma exchange
- QAlb:
-
Albumin CSF/serum quotient
- RIA:
-
Radioimmunoprecipitation assay
- RNFL:
-
Retinal nerve fiber layer
- sCD27:
-
Soluble CD27
- SIADH:
-
Syndrome of inadequate antidiuretic hormone secretion
- Tb:
-
Tuberculosis
- TM:
-
Transverse myelitis
- VEP:
-
Visual evoked potentials
- VS:
-
Vertebral segments
References
Aktas O, Smith MA, Rees WA, Bennett JL, She D, Katz E, Cree BAC, group NMs, the NMsi (2021) Serum glial fibrillary acidic protein: a neuromyelitis optica spectrum disorder biomarker. Ann Neurol 89:895–910
Alvarenga MP, do Carmo LF, Vasconcelos CCF, Alvarenga MP, Alvarenga-Filho H, de Melo Bento CA, Paiva CLA, Leyva-Fernandez L, Fernandez O, Papais-Alvarenga RM (2021) Neuromyelitis optica is an HLA associated disease different from Multiple Sclerosis: a systematic review with meta-analysis. Sci Rep 11:152
Aly L, Strauss EM, Feucht N, Weiss I, Berthele A, Mitsdoerffer M, Haass C, Hemmer B, Maier M, Korn T, Knier B (2022) Optical coherence tomography angiography indicates subclinical retinal disease in neuromyelitis optica spectrum disorders. Mult Scler 28:522–531
Anamnart C, Tisavipat N, Owattanapanich W, Apiwattanakul M, Savangned P, Prayoonwiwat N, Siritho S, Rattanathamsakul N, Jitprapaikulsan J (2022) Newly diagnosed neuromyelitis optica spectrum disorders following vaccination: case report and systematic review. Mult Scler Relat Disord 58:103414
Asgari N, Skejoe HP, Lillevang ST, Steenstrup T, Stenager E, Kyvik KO (2013) Modifications of longitudinally extensive transverse myelitis and brainstem lesions in the course of neuromyelitis optica (NMO): a population-based, descriptive study. BMC Neurol 13:33
Asseyer S, Henke E, Trebst C, Hümmert MW, Wildemann B, Jarius S, Ringelstein M, Aktas O, Pawlitzki M, Korsen M, Klotz L, Siebert N, Ruprecht K, Bellmann-Strobl J, Wernecke KD, Haussler V, Havla J, Gahlen A, Gold R, Paul F, Kleiter I, Ayzenberg I, Neuromyelitis Optica Study Group (2021) Pain, depression, and quality of life in adults with MOG-antibody-associated disease. Eur J Neurol 28:1645–1658
Asseyer S, Masuda H, Mori M, Bellmann-Strobl J, Ruprecht K, Siebert N, Cooper G, Chien C, Duchow A, Schliesseit J, Liu J, Sugimoto K, Uzawa A, Ohtani R, Paul F, Brandt AU, Kuwabara S, Zimmermann HG (2021) AQP4-IgG autoimmunity in Japan and Germany: differences in clinical profiles and prognosis in seropositive neuromyelitis optica spectrum disorders. Mult Scler J Exp Transl Clin 7:20552173211006864
Asseyer S, Schmidt F, Chien C, Scheel M, Ruprecht K, Bellmann-Strobl J, Brandt AU, Paul F (2018) Pain in AQP4-IgG-positive and MOG-IgG-positive neuromyelitis optica spectrum disorders. Mult Scler J Exp Transl Clin 4:2055217318796684
Ayzenberg I, Richter D, Henke E, Asseyer S, Paul F, Trebst C, Hümmert MW, Havla J, Kumpfel T, Ringelstein M, Aktas O, Wildemann B, Jarius S, Haussler V, Stellmann JP, Senel M, Klotz L, Pellkofer HL, Weber MS, Pawlitzki M, Rommer PS, Berthele A, Wernecke KD, Hellwig K, Gold R, Kleiter I, on behalf of the NEMOS (Neuromyelitis Optica Study Group) (2021) Pain, depression, and quality of life in neuromyelitis optica spectrum disorder: a cross-sectional study of 166 AQP4 antibody-seropositive patients. Neurol Neuroimmunol Neuroinflamm 8:e985
Ayzenberg I, Schollhammer J, Hoepner R, Hellwig K, Ringelstein M, Aktas O, Kumpfel T, Krumbholz M, Trebst C, Paul F, Pache F, Obermann M, Zeltner L, Schwab M, Berthele A, Jarius S, Kleiter I, on behalf of the Neuromyelitis Optica Study Group (NEMOS) (2016) Efficacy of glatiramer acetate in neuromyelitis optica spectrum disorder: a multicenter retrospective study. J Neurol 263:575–582
Banker P, Sonni S, Kister I, Loh JP, Lui YW (2012) Pencil-thin ependymal enhancement in neuromyelitis optica spectrum disorders. Mult Scler 18:1050–1053
Bennett JL, de Seze J, Lana-Peixoto M, Palace J, Waldman A, Schippling S, Tenembaum S, Banwell B, Greenberg B, Levy M, Fujihara K, Chan KH, Kim HJ, Asgari N, Sato DK, Saiz A, Wuerfel J, Zimmermann H, Green A, Villoslada P, Paul F, Gjcf ICC (2015) Neuromyelitis optica and multiple sclerosis: Seeing differences through optical coherence tomography. Mult Scler 21:678–688
Bergamaschi R, Jarius S, Robotti M, Pichiecchio A, Wildemann B, Meola G (2009) Two cases of benign neuromyelitis optica in patients with celiac disease. J Neurol 256:2097–2099
Biotti D, Bonneville F, Tournaire E, Ayrignac X, Dalliere CC, Mahieu L, Vignal C, Dulau C, Brochet B, Ruet A, Ouallet JC, Gout O, Heran F, Menjot de Champfleur N, Tourdias T, Deneve M, Labauge P, Deschamps R (2017) Optic neuritis in patients with anti-MOG antibodies spectrum disorder: MRI and clinical features from a large multicentric cohort in France. J Neurol 264:2173–2175
Boesen MS, Jensen PEH, Born AP, Magyari M, Nilsson AC, Hoei-Hansen C, Blinkenberg M, Sellebjerg F (2019) Incidence of pediatric neuromyelitis optica spectrum disorder and myelin oligodendrocyte glycoprotein antibody-associated disease in Denmark 20082018: a nationwide, population-based cohort study. Mult Scler Relat Disord 33:162–167
Borisow N, Kleiter I, Gahlen A, Fischer K, Wernecke KD, Pache F, Ruprecht K, Havla J, Krumbholz M, Kumpfel T, Aktas O, Ringelstein M, Geis C, Kleinschnitz C, Berthele A, Hemmer B, Angstwurm K, Weissert R, Stellmann JP, Schuster S, Stangel M, Lauda F, Tumani H, Mayer C, Zeltner L, Ziemann U, Linker RA, Schwab M, Marziniak M, Then Bergh F, Hofstadt-van Oy U, Neuhaus O, Winkelmann A, Marouf W, Ruckriem L, Faiss J, Wildemann B, Paul F, Jarius S, Trebst C, Hellwig K, on behalf of NEMOS (Neuromyelitis Optica Study Group) (2017) Influence of female sex and fertile age on neuromyelitis optica spectrum disorders. Mult Scler 23:1092–1103
Bradl M, Kanamori Y, Nakashima I, Misu T, Fujihara K, Lassmann H, Sandkuhler J (2014) Pain in neuromyelitis optica–prevalence, pathogenesis and therapy. Nat Rev Neurol 10:529–536
Budhram A, Mirian A, Le C, Hosseini-Moghaddam SM, Sharma M, Nicolle MW (2019) Unilateral cortical FLAIR-hyperintense Lesions in Anti-MOG-associated Encephalitis with Seizures (FLAMES): characterization of a distinct clinico-radiographic syndrome. J Neurol 266:2481–2487
Bukhari W, Prain KM, Waters P, Woodhall M, O’Gorman CM, Clarke L, Silvestrini RA, Bundell CS, Abernethy D, Bhuta S, Blum S, Boggild M, Boundy K, Brew BJ, Brown M, Brownlee WJ, Butzkueven H, Carroll WM, Chen C, Coulthard A, Dale RC, Das C, Dear K, Fabis-Pedrini MJ, Fulcher D, Gillis D, Hawke S, Heard R, Henderson APD, Heshmat S, Hodgkinson S, Jimenez-Sanchez S, Killpatrick T, King J, Kneebone C, Kornberg AJ, Lechner-Scott J, Lin MW, Lynch C, Macdonell R, Mason DF, McCombe PA, Pender MP, Pereira JA, Pollard JD, Reddel SW, Shaw C, Spies J, Stankovich J, Sutton I, Vucic S, Walsh M, Wong RC, Yiu EM, Barnett MH, Kermode AG, Marriott MP, Parratt JDE, Slee M, Taylor BV, Willoughby E, Wilson RJ, Vincent A, Broadley SA (2017) Incidence and prevalence of NMOSD in Australia and New Zealand. J Neurol Neurosurg Psychiatry 88:632–638
Calabrese M, Oh MS, Favaretto A, Rinaldi F, Poretto V, Alessio S, Lee BC, Yu KH, Ma HI, Perini P, Gallo P (2012) No MRI evidence of cortical lesions in neuromyelitis optica. Neurology 79:1671–1676
Camera V, Holm-Mercer L, Ali AAH, Messina S, Horvat T, Kuker W, Leite MI, Palace J (2021) Frequency of new silent MRI lesions in myelin oligodendrocyte glycoprotein antibody disease and aquaporin-4 antibody neuromyelitis optica spectrum disorder. JAMA Netw Open 4:e2137833
Carnero Contentti E, Leguizamon F, Hryb JP, Celso J, Pace JL, Ferrari J, Knorre E, Perassolo MB (2016) Neuromyelitis optica: association with paroxysmal painful tonic spasms. Neurologia 31:511–515
Chavarro VS, Mealy MA, Simpson A, Lacheta A, Pache F, Ruprecht K, Gold SM, Paul F, Brandt AU, Levy M (2016) Insufficient treatment of severe depression in neuromyelitis optica spectrum disorder. Neurol Neuroimmunol Neuroinflamm 3:e286
Chen JJ, Sotirchos ES, Henderson AD, Vasileiou ES, Flanagan EP, Bhatti MT, Jamali S, Eggenberger ER, Dinome M, Frohman LP, Arnold AC, Bonelli L, Seleme N, Mejia-Vergara AJ, Moss HE, Padungkiatsagul T, Stiebel-Kalish H, Lotan I, Hellmann MA, Hodge D, Oertel FC, Paul F, Saidha S, Calabresi PA, Pittock SJ (2022) OCT retinal nerve fiber layer thickness differentiates acute optic neuritis from MOG antibody-associated disease and Multiple Sclerosis: RNFL thickening in acute optic neuritis from MOGAD vs MS. Mult Scler Relat Disord 58:103525
Chien C, Scheel M, Schmitz-Hubsch T, Borisow N, Ruprecht K, Bellmann-Strobl J, Paul F, Brandt AU (2019) Spinal cord lesions and atrophy in NMOSD with AQP4-IgG and MOG-IgG associated autoimmunity. Mult Scler 25:1926–1936
Chihara N, Aranami T, Sato W, Miyazaki Y, Miyake S, Okamoto T, Ogawa M, Toda T, Yamamura T (2011) Interleukin 6 signaling promotes anti-aquaporin 4 autoantibody production from plasmablasts in neuromyelitis optica. Proc Natl Acad Sci USA 108:3701–3706
Clarke L, Arnett S, Bukhari W, Khalilidehkordi E, Jimenez Sanchez S, O’Gorman C, Sun J, Prain KM, Woodhall M, Silvestrini R, Bundell CS, Abernethy DA, Bhuta S, Blum S, Boggild M, Boundy K, Brew BJ, Brownlee W, Butzkueven H, Carroll WM, Chen C, Coulthard A, Dale RC, Das C, Fabis-Pedrini MJ, Gillis D, Hawke S, Heard R, Henderson APD, Heshmat S, Hodgkinson S, Kilpatrick TJ, King J, Kneebone C, Kornberg AJ, Lechner-Scott J, Lin MW, Lynch C, Macdonell RAL, Mason DF, McCombe PA, Pereira J, Pollard JD, Ramanathan S, Reddel SW, Shaw CP, Spies JM, Stankovich J, Sutton I, Vucic S, Walsh M, Wong RC, Yiu EM, Barnett MH, Kermode AGK, Marriott MP, Parratt JDE, Slee M, Taylor BV, Willoughby E, Brilot F, Vincent A, Waters P, Broadley SA (2021) MRI patterns distinguish AQP4 antibody positive neuromyelitis optica spectrum disorder from multiple sclerosis. Front Neurol 12:722237
Collongues N, Marignier R, Jacob A, Leite MI, Siva A, Paul F, Zephir H, Akman-Demir G, Elsone L, Jarius S, Papeix C, Mutch K, Saip S, Wildemann B, Kitley J, Karabudak R, Aktas O, Kuscu D, Altintas A, Palace J, Confavreux C, De Seze J (2014) Characterization of neuromyelitis optica and neuromyelitis optica spectrum disorder patients with a late onset. Mult Scler 20:1086–1094
Cortese R, Magnollay L, Tur C, Abdel-Aziz K, Jacob A, De Angelis F, Yiannakas MC, Prados F, Ourselin S, Yousry TA, Barkhof F, Ciccarelli O (2018) Value of the central vein sign at 3T to differentiate MS from seropositive NMOSD. Neurology 90:e1183–e1190
Cree BAC, Bennett JL, Kim HJ, Weinshenker BG, Pittock SJ, Wingerchuk DM, Fujihara K, Paul F, Cutter GR, Marignier R, Green AJ, Aktas O, Hartung HP, Lublin FD, Drappa J, Barron G, Madani S, Ratchford JN, She D, Cimbora D, Katz E, investigators NMs (2019) Inebilizumab for the treatment of neuromyelitis optica spectrum disorder (N-MOmentum): a double-blind, randomised placebo-controlled phase 2/3 trial. Lancet 394:1352–1363
Cubas-Nunez L, Gil-Perotin S, Castillo-Villalba J, Lopez V, Solis Tarazona L, Gasque-Rubio R, Carratala-Bosca S, Alcala-Vicente C, Perez-Miralles F, Lassmann H, Casanova B (2021) Potential role of CHI3L1+ astrocytes in progression in MS. Neurol Neuroimmunol Neuroinflamm 8:e972
Du Q, Shi Z, Chen H, Zhang Y, Wang J, Qiu Y, Zhao Z, Zhang Q, Zhou H (2021) Mortality of neuromyelitis optica spectrum disorders in a Chinese population. Annals Clin Transl Neurol 8:1471–1479
Dubey D, Pittock SJ, Krecke KN, Morris PP, Sechi E, Zalewski NL, Weinshenker BG, Shosha E, Lucchinetti CF, Fryer JP, Lopez-Chiriboga AS, Chen JC, Jitprapaikulsan J, McKeon A, Gadoth A, Keegan BM, Tillema JM, Naddaf E, Patterson MC, Messacar K, Tyler KL, Flanagan EP (2019) Clinical, radiologic, and prognostic features of myelitis associated with myelin oligodendrocyte glycoprotein autoantibody. JAMA Neurol 76:301–309
Duignan S, Wright S, Rossor T, Cazabon J, Gilmour K, Ciccarelli O, Wassmer E, Lim M, Hemingway C, Hacohen Y (2018) Myelin oligodendrocyte glycoprotein and aquaporin-4 antibodies are highly specific in children with acquired demyelinating syndromes. Dev Med Child Neurol 60:958–962
Elsone L, Townsend T, Mutch K, Das K, Boggild M, Nurmikko T, Jacob A (2013) Neuropathic pruritus (itch) in neuromyelitis optica. Mult Scler 19:475–479
Etemadifar M, Salari M, Kargaran PK, Sigari AA, Nouri H, Etemadifar F, Ebrahimi S, Sayahi N, Sedaghat N (2021) Conus medullaris involvement in demyelinating disorders of the CNS: a comparative study. Mult Scler Relat Disord 54:103127
Fadda G, Banwell B, Waters P, Marrie RA, Yeh EA, O’Mahony J, Arnold DA, Bar-Or A, Canadian Pediatric Demyelinating Disease N (2021) Silent new brain MRI lesions in children with MOG-antibody associated disease. Ann Neurol 89:408–413
Filippi M, Preziosa P, Banwell BL, Barkhof F, Ciccarelli O, De Stefano N, Geurts JJG, Paul F, Reich DS, Toosy AT, Traboulsee A, Wattjes MP, Yousry TA, Gass A, Lubetzki C, Weinshenker BG, Rocca MA (2019) Assessment of lesions on magnetic resonance imaging in multiple sclerosis: practical guidelines. Brain 142:1858–1875
Flanagan EP, Cabre P, Weinshenker BG, Sauver JS, Jacobson DJ, Majed M, Lennon VA, Lucchinetti CF, McKeon A, Matiello M, Kale N, Wingerchuk DM, Mandrekar J, Sagen JA, Fryer JP, Robinson AB, Pittock SJ (2016) Epidemiology of aquaporin-4 autoimmunity and neuromyelitis optica spectrum. Ann Neurol 79:775–783
Flanagan EP, Kaufmann TJ, Krecke KN, Aksamit AJ, Pittock SJ, Keegan BM, Giannini C, Weinshenker BG (2016) Discriminating long myelitis of neuromyelitis optica from sarcoidosis. Ann Neurol 79:437–447
Flanagan EP, Weinshenker BG, Krecke KN, Lennon VA, Lucchinetti CF, McKeon A, Wingerchuk DM, Shuster EA, Jiao Y, Horta ES, Pittock SJ (2015) Short myelitis lesions in aquaporin-4-IgG-positive neuromyelitis optica spectrum disorders. JAMA Neurol 72:81–87
Fujihara K, Bennett JL, de Seze J, Haramura M, Kleiter I, Weinshenker BG, Kang D, Mughal T, Yamamura T (2020) Interleukin-6 in neuromyelitis optica spectrum disorder pathophysiology. Neurol Neuroimmunol Neuroinflamm 7:e841
Galler S, Stellmann JP, Young KL, Kutzner D, Heesen C, Fiehler J, Siemonsen S (2016) Improved lesion detection by using axial T2-weighted MRI with full spinal cord coverage in multiple sclerosis. AJNR Am J Neuroradiol 37:963–969
Gastaldi M, Scaranzin S, Jarius S, Wildeman B, Zardini E, Mallucci G, Rigoni E, Vegezzi E, Foiadelli T, Savasta S, Banfi P, Versino M, Benedetti L, Novi G, Mancardi MM, Giacomini T, Annovazzi P, Baroncini D, Ferraro D, Lampasona V, Reindl M, Waters P, Franciotta D (2020) Cell-based assays for the detection of MOG antibodies: a comparative study. J Neurol 267:3555–3564
Geraldes R, Ciccarelli O, Barkhof F, De Stefano N, Enzinger C, Filippi M, Hofer M, Paul F, Preziosa P, Rovira A, DeLuca GC, Kappos L, Yousry T, Fazekas F, Frederiksen J, Gasperini C, Sastre-Garriga J, Evangelou N, Palace J, group Ms (2018) The current role of MRI in differentiating multiple sclerosis from its imaging mimics. Nat Rev Neurol 14:199–213
Graves JS, Oertel FC, Van der Walt A, Collorone S, Sotirchos ES, Pihl-Jensen G, Albrecht P, Yeh EA, Saidha S, Frederiksen J, Newsome SD, Paul F, Imsvisual, (2022) Leveraging visual outcome measures to advance therapy development in neuroimmunologic disorders. Neurol Neuroimmunol Neuroinflamm 9:e1126
Grüter T, Ayzenberg I, Gahlen A, Kneiphof J, Gold R, Kleiter I (2018) Flaccid paralysis in neuromyelitis optica: an atypical presentation with possible involvement of the peripheral nervous system. Mult Scler Relat Disord 25:83–86
Grüter T, Ott A, Meyer W, Jarius S, Kinner M, Motte J, Pitarokoili K, Gold R, Komorowski L, Ayzenberg I (2020) Effects of IVIg treatment on autoantibody testing in neurological patients: marked reduction in sensitivity but reliable specificity. J Neurol 267:715–720
Guo BJ, Yang ZL, Zhang LJ (2018) Gadolinium deposition in brain: current scientific evidence and future perspectives. Front Mol Neurosci 11:335
Guo Y, Lennon VA, Parisi JE, Popescu B, Vasquez C, Pittock SJ, Howe CL, Lucchinetti CF (2022) Spectrum of sublytic astrocytopathy in neuromyelitis optica. Brain 145:1379–1390
Hacohen Y, Rossor T, Mankad K, Chong W, Lux A, Wassmer E, Lim M, Barkhof F, Ciccarelli O, Hemingway C (2018) “Leukodystrophy-like” phenotype in children with myelin oligodendrocyte glycoprotein antibody-associated disease. Dev Med Child Neurol 60:417–423
Hamid SHM, Whittam D, Saviour M, Alorainy A, Mutch K, Linaker S, Solomon T, Bhojak M, Woodhall M, Waters P, Appleton R, Duddy M, Jacob A (2018) Seizures and encephalitis in myelin oligodendrocyte glycoprotein IgG disease vs aquaporin 4 IgG disease. JAMA Neurol 75:65–71
Harahsheh E, Callister M, Hasan S, Gritsch D, Valencia-Sanchez C (2022) Aquaporin-4 IgG neuromyelitis optica spectrum disorder onset after Covid-19 vaccination: systematic review. J Neuroimmunol 373:577994
He Z, Ren M, Wang X, Guo Q, Qi X (2017) Pruritus may be a common symptom related to neuromyelitis optica spectrum disorders. Mult Scler Relat Disord 13:1–3
Hoftberger R, Sepulveda M, Armangue T, Blanco Y, Rostasy K, Cobo Calvo A, Olascoaga J, Ramio-Torrenta L, Reindl M, Benito-Leon J, Casanova B, Arrambide G, Sabater L, Graus F, Dalmau J, Saiz A (2015) Antibodies to MOG and AQP4 in adults with neuromyelitis optica and suspected limited forms of the disease. Mult Scler 21:866–874
Hor JY, Asgari N, Nakashima I, Broadley SA, Leite MI, Kissani N, Jacob A, Marignier R, Weinshenker BG, Paul F, Pittock SJ, Palace J, Wingerchuk DM, Behne JM, Yeaman MR, Fujihara K (2020) Epidemiology of neuromyelitis optica spectrum disorder and its prevalence and incidence worldwide. Front Neurol 11:501
Huang Y, Zhou L, ZhangBao J, Cai T, Wang B, Li X, Wang L, Lu C, Zhao C, Lu J, Quan C, Wang M (2019) Peripapillary and parafoveal vascular network assessment by optical coherence tomography angiography in aquaporin-4 antibody-positive neuromyelitis optica spectrum disorders. Br J Ophthalmol 103:789–796
Hümmert MW, Bütow F, Tkachenko D, Ayzenberg I, Pakeerathan T, Hellwig K, Klotz L, Häußler V, Stellmann JP, Warnke C, Goereci Y, Etgen T, Luessi F, Bronzlik P, Gingele S, Lauenstein AS, Kleiter I, Rommer P, Paul F, Bellmann-Strobl J, Duchow AS, Then Bergh F, Pul R, Walter A, Pellkofer H, Kümpfel T, Pompsch M, Kraemer M, Albrecht P, Aktas O, Ringelstein M, Senel M, Giglhuber K, Berthele A, Jarius S, Wildemann B, Trebst C; on behalf of the Neuromyelitis Optica Study Group (NEMOS) (2023) Effects of the COVID-19 pandemic on patients with NMO spectrum disorders and MOG-antibody associated diseases (COPANMO(G)-Study). Neurol Neuroimmunol Neuroinflamm 10:e200082
Hümmert MW, Schoppe LM, Bellmann-Strobl J, Siebert N, Paul F, Duchow A, Pellkofer H, Kümpfel T, Havla J, Jarius S, Wildemann B, Berthele A, Bergh FT, Pawlitzki M, Klotz L, Kleiter I, Stangel M, Gingele S, Weber MS, Faiss JH, Pul R, Walter A, Zettl UK, Senel M, Stellmann JP, Haussler V, Hellwig K, Ayzenberg I, Aktas O, Ringelstein M, Schreiber-Katz O, Trebst C, and on behalf of the Neuromyelitis Optica Study Group (NEMOS) (2022) Costs and health-related quality of life in patients with NMO spectrum disorders and MOG-antibody-associated disease: CHANCE(NMO) study. Neurology 98:e1184–e1196
Hümmert MW, Stern C, Paul F, Duchow A, Bellmann-Strobl J, Ayzenberg I, Schwake C, Kleiter I, Hellwig K, Jarius S, Wildemann B, Senel M, Berthele A, Giglhuber K, Luessi F, Grothe M, Klotz L, Schülke R, Gingele S, Faiss JH, Walter A, Warnke C, Then Bergh F, Aktas O, Ringelstein M, Stellmann JP, Häußler V, Havla J, Pellkofer H, Kümpfel T, Kopp B, Trebst C (2023) Cognition in patients with neuromyelitis optica spectrum disorders: a prospective multicentre study of 217 patients (CogniNMO study). Mult Scler. https://doi.org/10.1177/13524585231151212
Hyun JW, Kim Y, Kim SY, Lee MY, Kim SH, Kim HJ (2021) Investigating the presence of interattack astrocyte damage in neuromyelitis optica spectrum disorder: longitudinal analysis of serum glial fibrillary acidic protein. Neurol Neuroimmunol Neuroinflamm 8:e965
Hyun JW, Kwon YN, Kim SM, Lee HL, Jeong WK, Lee HJ, Kim BJ, Kim SW, Shin HY, Shin HJ, Oh SY, Huh SY, Kim W, Park MS, Oh J, Jang H, Park NY, Lee MY, Kim SH, Kim HJ (2020) Value of area postrema syndrome in differentiating adults with AQP4 vs. MOG antibodies. Front Neurol 11:396
Hyun JW, Park G, Kwak K, Jo HJ, Joung A, Kim JH, Lee SH, Kim S, Lee JM, Kim SH, Kim HJ (2017) Deep gray matter atrophy in neuromyelitis optica spectrum disorder and multiple sclerosis. Eur J Neurol 24:437–445
Ishikawa H, Kezuka T, Shikishima K, Yamagami A, Hiraoka M, Chuman H, Nakamura M, Hoshi K, Goseki T, Mashimo K, Mimura O, Yoshitomi T, Tanaka K, Working Group on Diagnostic Criteria for Refractory Optic Neuritis Based on Neuroimmunological P (2019) Epidemiologic and clinical characteristics of optic neuritis in Japan. Ophthalmology 126:1385–1398
Iyer A, Elsone L, Appleton R, Jacob A (2014) A review of the current literature and a guide to the early diagnosis of autoimmune disorders associated with neuromyelitis optica. Autoimmunity 47:154–161
Jarius S, Aboul-Enein F, Waters P, Kuenz B, Hauser A, Berger T, Lang W, Reindl M, Vincent A, Kristoferitsch W (2008) Antibody to aquaporin-4 in the long-term course of neuromyelitis optica. Brain 131:3072–3080
Jarius S, Bieber N, Haas J, Wildemann B (2022) MOG encephalomyelitis after vaccination against severe acute respiratory syndrome coronavirus type 2 (SARS-CoV-2): case report and comprehensive review of the literature. J Neurol 269:5198–5212
Jarius S, Eichhorn P, Franciotta D, Petereit HF, Akman-Demir G, Wick M, Wildemann B (2017) The MRZ reaction as a highly specific marker of multiple sclerosis: re-evaluation and structured review of the literature. J Neurol 264:453–466
Jarius S, Franciotta D, Bergamaschi R, Rauer S, Wandinger KP, Petereit HF, Maurer M, Tumani H, Vincent A, Eichhorn P, Wildemann B, Wick M, Voltz R (2008) Polyspecific, antiviral immune response distinguishes multiple sclerosis and neuromyelitis optica. J Neurol Neurosurg Psychiatry 79:1134–1136
Jarius S, Franciotta D, Bergamaschi R, Wright H, Littleton E, Palace J, Hohlfeld R, Vincent A (2007) NMO-IgG in the diagnosis of neuromyelitis optica. Neurology 68:1076–1077
Jarius S, Franciotta D, Paul F, Bergamaschi R, Rommer PS, Ruprecht K, Ringelstein M, Aktas O, Kristoferitsch W, Wildemann B (2012) Testing for antibodies to human aquaporin-4 by ELISA: sensitivity, specificity, and direct comparison with immunohistochemistry. J Neurol Sci 320:32–37
Jarius S, Franciotta D, Paul F, Ruprecht K, Bergamaschi R, Rommer PS, Reuss R, Probst C, Kristoferitsch W, Wandinger KP, Wildemann B (2010) Cerebrospinal fluid antibodies to aquaporin-4 in neuromyelitis optica and related disorders: frequency, origin, and diagnostic relevance. J Neuroinflammation 7:52
Jarius S, Jacob S, Waters P, Jacob A, Littleton E, Vincent A (2008) Neuromyelitis optica in patients with gluten sensitivity associated with antibodies to aquaporin-4. J Neurol Neurosurg Psychiatry 79:1084
Jarius S, Jacobi C, de Seze J, Zephir H, Paul F, Franciotta D, Rommer P, Mader S, Kleiter I, Reindl M, Akman-Demir G, Seifert-Held T, Kristoferitsch W, Melms A, Wandinger KP, Wildemann B (2011) Frequency and syndrome specificity of antibodies to aquaporin-4 in neurological patients with rheumatic disorders. Mult Scler 17:1067–1073
Jarius S, Kleiter I, Ruprecht K, Asgari N, Pitarokoili K, Borisow N, Hümmert MW, Trebst C, Pache F, Winkelmann A, Beume LA, Ringelstein M, Stich O, Aktas O, Korporal-Kuhnke M, Schwarz A, Lukas C, Haas J, Fechner K, Buttmann M, Bellmann-Strobl J, Zimmermann H, Brandt AU, Franciotta D, Schanda K, Paul F, Reindl M, Wildemann B (2016) MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 3: Brainstem involvement—frequency, presentation and outcome. J Neuroinflamm 13:281
Jarius S, Lechner C, Wendel EM, Baumann M, Breu M, Schimmel M, Karenfort M, Marina AD, Merkenschlager A, Thiels C, Blaschek A, Salandin M, Leiz S, Leypoldt F, Pschibul A, Hackenberg A, Hahn A, Syrbe S, Strautmanis J, Hausler M, Krieg P, Eisenkolbl A, Stoffels J, Eckenweiler M, Ayzenberg I, Haas J, Hoftberger R, Kleiter I, Korporal-Kuhnke M, Ringelstein M, Ruprecht K, Siebert N, Schanda K, Aktas O, Paul F, Reindl M, Wildemann B, Rostasy K, in cooperation with the BIOMARKER study group and the Neuromyelitis optica Study Group (NEMOS) (2020) Cerebrospinal fluid findings in patients with myelin oligodendrocyte glycoprotein (MOG) antibodies Part 2: Results from 108 lumbar punctures in 80 pediatric patients. J Neuroinflammation 17:262
Jarius S, Paul F, Aktas O, Asgari N, Dale RC, de Seze J, Franciotta D, Fujihara K, Jacob A, Kim HJ, Kleiter I, Kumpfel T, Levy M, Palace J, Ruprecht K, Saiz A, Trebst C, Weinshenker BG, Wildemann B (2018) MOG encephalomyelitis: international recommendations on diagnosis and antibody testing. J Neuroinflamm 15:134
Jarius S, Paul F, Aktas O, Asgari N, Dale RC, de Seze J, Franciotta D, Fujihara K, Jacob A, Kim HJ, Kleiter I, Kumpfel T, Levy M, Palace J, Ruprecht K, Saiz A, Trebst C, Weinshenker BG, Wildemann B (2018) MOG encephalomyelitis: international recommendations on diagnosis and antibody testing. Nervenarzt 89:1388–1399
Jarius S, Paul F, Franciotta D, Ruprecht K, Ringelstein M, Bergamaschi R, Rommer P, Kleiter I, Stich O, Reuss R, Rauer S, Zettl UK, Wandinger KP, Melms A, Aktas O, Kristoferitsch W, Wildemann B (2011) Cerebrospinal fluid findings in aquaporin-4 antibody positive neuromyelitis optica: results from 211 lumbar punctures. J Neurol Sci 306:82–90
Jarius S, Paul F, Martins da Silva A, Leite MI, Waters P, Littleton E, Franciotta D, Palace J, Zipp F, Vincent A (2007) Neuromyelitis optica and longitudinally extensive transverse myelitis following thymectomy for myasthenia gravis. Mult Scler 13:P534
Jarius S, Paul F, Ruprecht K, Wildemann B (2012) Low vitamin B12 levels and gastric parietal cell antibodies in patients with aquaporin-4 antibody-positive neuromyelitis optica spectrum disorders. J Neurol 259:2743–2745
Jarius S, Paul F, Weinshenker BG, Levy M, Kim HJ, Wildemann B (2020) Neuromyelitis optica. Nat Rev Dis Primers 6:85
Jarius S, Pellkofer H, Siebert N, Korporal-Kuhnke M, Hümmert MW, Ringelstein M, Rommer PS, Ayzenberg I, Ruprecht K, Klotz L, Asgari N, Zrzavy T, Hoftberger R, Tobia R, Buttmann M, Fechner K, Schanda K, Weber M, Asseyer S, Haas J, Lechner C, Kleiter I, Aktas O, Trebst C, Rostasy K, Reindl M, Kumpfel T, Paul F, Wildemann B, in cooperation with the Neuromyelitis Optica Study Group (NEMOS) (2020) Cerebrospinal fluid findings in patients with myelin oligodendrocyte glycoprotein (MOG) antibodies. Part 1: Results from 163 lumbar punctures in 100 adult patients. J Neuroinflammation 17:261
Jarius S, Ruprecht K, Kleiter I, Borisow N, Asgari N, Pitarokoili K, Pache F, Stich O, Beume LA, Hümmert MW, Ringelstein M, Trebst C, Winkelmann A, Schwarz A, Buttmann M, Zimmermann H, Kuchling J, Franciotta D, Capobianco M, Siebert E, Lukas C, Korporal-Kuhnke M, Haas J, Fechner K, Brandt AU, Schanda K, Aktas O, Paul F, Reindl M, Wildemann B (2016) MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 2: Epidemiology, clinical presentation, radiological and laboratory features, treatment responses, and long-term outcome. J Neuroinflamm 13:280
Jarius S, Ruprecht K, Kleiter I, Borisow N, Asgari N, Pitarokoili K, Pache F, Stich O, Beume LA, Hümmert MW, Ringelstein M, Trebst C, Winkelmann A, Schwarz A, Buttmann M, Zimmermann H, Kuchling J, Franciotta D, Capobianco M, Siebert E, Lukas C, Korporal-Kuhnke M, Haas J, Fechner K, Brandt AU, Schanda K, Aktas O, Paul F, Reindl M, Wildemann B, in cooperation with the Neuromyelitis Optica Study Group (NEMOS) (2016) MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 2: epidemiology, clinical presentation, radiological and laboratory features, treatment responses, and long-term outcome. J Neuroinflamm 13:280
Jarius S, Ruprecht K, Kleiter I, Borisow N, Asgari N, Pitarokoili K, Pache F, Stich O, Beume LA, Hümmert MW, Trebst C, Ringelstein M, Aktas O, Winkelmann A, Buttmann M, Schwarz A, Zimmermann H, Brandt AU, Franciotta D, Capobianco M, Kuchling J, Haas J, Korporal-Kuhnke M, Lillevang ST, Fechner K, Schanda K, Paul F, Wildemann B, Reindl M, in cooperation with the Neuromyelitis Optica Study Group (NEMOS) (2016) MOG-IgG in NMO and related disorders: a multicenter study of 50 patients Part 1: Frequency, syndrome specificity, influence of disease activity, long-term course, association with AQP4-IgG, and origin. J Neuroinflamm 13:279
Jarius S, Ruprecht K, Wildemann B, Kuempfel T, Ringelstein M, Geis C, Kleiter I, Kleinschnitz C, Berthele A, Brettschneider J, Hellwig K, Hemmer B, Linker RA, Lauda F, Mayer CA, Tumani H, Melms A, Trebst C, Stangel M, Marziniak M, Hoffmann F, Schippling S, Faiss JH, Neuhaus O, Ettrich B, Zentner C, Guthke K, Hofstadt-van Oy U, Reuss R, Pellkofer H, Ziemann U, Kern P, Wandinger KP, Then Bergh F, Boettcher T, Langel S, Liebetrau M, Rommer PS, Niehaus S, Munch C, Winkelmann A, Zettl UK, Metz I, Veauthier C, Sieb JP, Wilke C, Hartung HP, Aktas O, Paul F (2012) Contrasting disease patterns in seropositive and seronegative neuromyelitis optica: a multicentre study of 175 patients. J Neuroinflammation 9:14
Jarius S, Wandinger KP, Borowski K, Stoecker W, Wildemann B (2012) Antibodies to CV2/CRMP5 in neuromyelitis optica-like disease: case report and review of the literature. Clin Neurol Neurosurg 114:331–335
Jarius S, Wildemann B (2010) AQP4 antibodies in neuromyelitis optica: diagnostic and pathogenetic relevance. Nat Rev Neurol 6:383–392
Jarius S, Wildemann B (2013) Aquaporin-4 antibodies (NMO-IgG) as a serological marker of neuromyelitis optica: a critical review of the literature. Brain Pathol 23:661–683
Jarius S, Wildemann B (2019) Devic’s index case: a critical reappraisal—AQP4-IgG-mediated neuromyelitis optica spectrum disorder, or rather MOG encephalomyelitis? J Neurol Sci 407:116396
Jarius S, Wildemann B (2013) The history of neuromyelitis optica. J Neuroinflamm 10:8
Jarius S, Wildemann B (2019) The history of neuromyelitis optica. Part 2: “Spinal amaurosis”, or how it all began. J Neuroinflamm 16:280
Jeong IH, Kim HJ, Kim NH, Jeong KS, Park CY (2016) Subclinical primary retinal pathology in neuromyelitis optica spectrum disorder. J Neurol 263:1343–1348
Jurynczyk M, Geraldes R, Probert F, Woodhall MR, Waters P, Tackley G, DeLuca G, Chandratre S, Leite MI, Vincent A, Palace J (2017) Distinct brain imaging characteristics of autoantibody-mediated CNS conditions and multiple sclerosis. Brain 140:617–627
Jurynczyk M, Tackley G, Kong Y, Geraldes R, Matthews L, Woodhall M, Waters P, Kuker W, Craner M, Weir A, DeLuca GC, Kremer S, Leite MI, Vincent A, Jacob A, de Seze J, Palace J (2017) Brain lesion distribution criteria distinguish MS from AQP4-antibody NMOSD and MOG-antibody disease. J Neurol Neurosurg Psychiatry 88:132–136
Kay CS, Scola RH, Lorenzoni PJ, Jarius S, Arruda WO, Werneck LC (2008) NMO-IgG positive neuromyelitis optica in a patient with myasthenia gravis but no thymectomy. J Neurol Sci 275:148–150
Kim H, Lee EJ, Kim S, Choi LK, Kim K, Kim HW, Kim KK, Lim YM (2020) Serum biomarkers in myelin oligodendrocyte glycoprotein antibody-associated disease. Neurol Neuroimmunol Neuroinflamm 7:e708
Kim HJ, Paul F, Lana-Peixoto MA, Tenembaum S, Asgari N, Palace J, Klawiter EC, Sato DK, de Seze J, Wuerfel J, Banwell BL, Villoslada P, Saiz A, Fujihara K, Kim SH, Guthy-Jackson Charitable Foundation NMOICC, Biorepository (2015) MRI characteristics of neuromyelitis optica spectrum disorder: an international update. Neurology 84:1165–1173
Kim NH, Kim HJ, Park CY, Jeong KS (2019) Retinal Degeneration after first-ever optic neuritis helps differentiate multiple sclerosis and neuromyelitis optica spectrum disorder. Front Neurol 10:1076
Kim S, Kwon B, Park J, Lee H, Kim H, Park D, Nam K (2016) Neuromyelitis optica masquerading as lumbosacral radiculopathy: a case report. Ann Rehabil Med 40:943–948
Kim S, Park J, Kwon BS, Park JW, Lee HJ, Choi JH, Nam K (2017) Radiculopathy in neuromyelitis optica. How does anti-AQP4 Ab involve PNS? Mult Scler Relat Disord 18:77–81
Kim SH, Hyun JW, Joung A, Lee SH, Kim HJ (2016) Occurrence of asymptomatic acute neuromyelitis optica spectrum disorder-typical brain lesions during an attack of optic neuritis or myelitis. PLoS ONE 11:e0167783
Kim SM, Go MJ, Sung JJ, Park KS, Lee KW (2012) Painful tonic spasm in neuromyelitis optica: incidence, diagnostic utility, and clinical characteristics. Arch Neurol 69:1026–1031
Kim SM, Kim SJ, Lee HJ, Kuroda H, Palace J, Fujihara K (2017) Differential diagnosis of neuromyelitis optica spectrum disorders. Ther Adv Neurol Disord 10:265–289
Kim SM, Woodhall MR, Kim JS, Kim SJ, Park KS, Vincent A, Lee KW, Waters P (2015) Antibodies to MOG in adults with inflammatory demyelinating disease of the CNS. Neurol Neuroimmunol Neuroinflamm 2:e163
Kim W, Lee JE, Kim SH, Huh SY, Hyun JW, Jeong IH, Park MS, Cho JY, Lee SH, Lee KS, Kim HJ (2016) Cerebral cortex involvement in neuromyelitis optica spectrum disorder. J Clin Neurol 12:188–193
Kim W, Lee JE, Li XF, Kim SH, Han BG, Lee BI, Kim JK, Choi K, Kim HJ (2012) Quantitative measurement of anti-aquaporin-4 antibodies by enzyme-linked immunosorbent assay using purified recombinant human aquaporin-4. Mult Scler 18:578–586
Kitley J, Waters P, Woodhall M, Leite MI, Murchison A, George J, Kuker W, Chandratre S, Vincent A, Palace J (2014) Neuromyelitis optica spectrum disorders with aquaporin-4 and myelin-oligodendrocyte glycoprotein antibodies: a comparative study. JAMA Neurol 71:276–283
Kitley JL, Leite MI, George JS, Palace JA (2012) The differential diagnosis of longitudinally extensive transverse myelitis. Mult Scler 18:271–285
Kleerekooper I, Herbert MK, Kuiperij HB, Sato DK, Fujihara K, Callegaro D, Marignier R, Saiz A, Senel M, Tumani H, De Jong BA, Trip SA, Nakashima I, Verbeek MM, Petzold A (2020) CSF levels of glutamine synthetase and GFAP to explore astrocytic damage in seronegative NMOSD. J Neurol Neurosurg Psychiatry 91:605–611
Kleiter I, Gahlen A, Borisow N, Fischer K, Wernecke KD, Hellwig K, Pache F, Ruprecht K, Havla J, Kumpfel T, Aktas O, Hartung HP, Ringelstein M, Geis C, Kleinschnitz C, Berthele A, Hemmer B, Angstwurm K, Stellmann JP, Schuster S, Stangel M, Lauda F, Tumani H, Mayer C, Krumbholz M, Zeltner L, Ziemann U, Linker R, Schwab M, Marziniak M, Then Bergh F, Hofstadt-van Oy U, Neuhaus O, Zettl UK, Faiss J, Wildemann B, Paul F, Jarius S, Trebst C, on behalf of NEMOS (Neuromyelitis Optica Study Group) (2018) Apheresis therapies for NMOSD attacks: a retrospective study of 207 therapeutic interventions. Neurol Neuroimmunol Neuroinflamm 5:e504
Kleiter I, Gahlen A, Borisow N, Fischer K, Wernecke KD, Wegner B, Hellwig K, Pache F, Ruprecht K, Havla J, Krumbholz M, Kumpfel T, Aktas O, Hartung HP, Ringelstein M, Geis C, Kleinschnitz C, Berthele A, Hemmer B, Angstwurm K, Stellmann JP, Schuster S, Stangel M, Lauda F, Tumani H, Mayer C, Zeltner L, Ziemann U, Linker R, Schwab M, Marziniak M, Then Bergh F, Hofstadt-van Oy U, Neuhaus O, Winkelmann A, Marouf W, Faiss J, Wildemann B, Paul F, Jarius S, Trebst C, on behalf of the Neuromyelitis Optica Study Group (2016) Neuromyelitis optica: evaluation of 871 attacks and 1,153 treatment courses. Ann Neurol 79:206–216
Kleiter I, Hellwig K, Berthele A, Kumpfel T, Linker RA, Hartung HP, Paul F, Aktas O, for the Neuromyelitis Optica Study Group (2012) Failure of natalizumab to prevent relapses in neuromyelitis optica. Arch Neurol 69:239–245
Kleiter I, Hellwig K, Berthele A, Kumpfel T, Linker RA, Hartung HP, Paul F, Aktas O, for the Neuromyelitis Optica Study Group (NEMOS) (2012) Is it too early to predict the failure of natalizumab in NMO?-Reply. Arch Neurol 69:1085–1086
Kleiter I, Schmutzhard E, Trebst C (2017) Infectious, autoimmune and other immune-mediated causes of myelitis. In: Weidner N, Rupp R, Tansey KE (eds) Neurological aspects of spinal cord injury. Springer International Publishing, Cham, pp 123–160
Kremer L, Mealy M, Jacob A, Nakashima I, Cabre P, Bigi S, Paul F, Jarius S, Aktas O, Elsone L, Mutch K, Levy M, Takai Y, Collongues N, Banwell B, Fujihara K, de Seze J (2014) Brainstem manifestations in neuromyelitis optica: a multicenter study of 258 patients. Mult Scler 20:843–847
Kremer S, Renard F, Achard S, Lana-Peixoto MA, Palace J, Asgari N, Klawiter EC, Tenembaum SN, Banwell B, Greenberg BM, Bennett JL, Levy M, Villoslada P, Saiz A, Fujihara K, Chan KH, Schippling S, Paul F, Kim HJ, de Seze J, Wuerfel JT, Guthy-Jackson Charitable Foundation Neuromyelitis Optica International Clinical C, Biorepository, Cabre P, Marignier R, Tedder T, van Pelt D, Broadley S, Chitnis T, Wingerchuk D, Pandit L, Leite MI, Apiwattanakul M, Kleiter I, Prayoonwiwat N, Han M, Hellwig K, van Herle K, John G, Hooper DC, Nakashima I, Sato D, Yeaman MR, Waubant E, Zamvil S, Stuve O, Aktas O, Smith TJ, Jacob A, O’Connor K (2015) Use of advanced magnetic resonance imaging techniques in neuromyelitis optica spectrum disorder. JAMA Neurol 72:815–822
Krumbholz M, Hofstadt-van Oy U, Angstwurm K, Kleiter I, Jarius S, Paul F, Aktas O, Buchholz G, Kern P, Straube A, Kumpfel T (2015) Very late-onset neuromyelitis optica spectrum disorder beyond the age of 75. J Neurol 262:1379–1384
Kuchling J, Paul F (2020) Visualizing the central nervous system: imaging tools for multiple sclerosis and neuromyelitis optica spectrum disorders. Front Neurol 11:450
Kunchok A, Chen JJ, McKeon A, Mills JR, Flanagan EP, Pittock SJ (2020) Coexistence of myelin oligodendrocyte glycoprotein and aquaporin-4 antibodies in adult and pediatric patients. JAMA Neurol 77:257–259
Kwon YN, Kim B, Kim JS, Mo H, Choi K, Oh SI, Kim JE, Nam TS, Sohn EH, Heo SH, Kim SB, Park KC, Yoon SS, Oh J, Baek SH, Kim BJ, Park KS, Sung JJ, Jung JH, Kim SJ, Park SH, Waters P, Kim SM (2022) Myelin oligodendrocyte glycoprotein-immunoglobulin G in the CSF: clinical implication of testing and association with disability. Neurol Neuroimmunol Neuroinflamm 9:e1095
Leite MI, Coutinho E, Lana-Peixoto M, Apostolos S, Waters P, Sato D, Melamud L, Marta M, Graham A, Spillane J, Villa AM, Callegaro D, Santos E, da Silva AM, Jarius S, Howard R, Nakashima I, Giovannoni G, Buckley C, Hilton-Jones D, Vincent A, Palace J (2012) Myasthenia gravis and neuromyelitis optica spectrum disorder: a multicenter study of 16 patients. Neurology 78:1601–1607
Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SR (2005) IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med 202:473–477
Lennon VA, Wingerchuk DM, Kryzer TJ, Pittock SJ, Lucchinetti CF, Fujihara K, Nakashima I, Weinshenker BG (2004) A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet 364:2106–2112
Levy M, Wildemann B, Jarius S, Orellano B, Sasidharan S, Weber MS, Stuve O (2014) Immunopathogenesis of neuromyelitis optica. Adv Immunol 121:213–242
Li R, Lu D, Li H, Wang Y, Shu Y, Chang Y, Sun X, Lu Z, Qiu W, Yang Z (2021) Neuromyelitis optica spectrum disorders with non opticospinal manifestations as initial symptoms: a long-term observational study. BMC Neurol 21:35
Liu B, Zhong X, Lu Z, Qiu W, Hu X, Wang H (2018) Cerebrospinal fluid level of soluble CD27 is associated with disease severity in neuromyelitis optica spectrum disorder. NeuroImmunoModulation 25:185–192
Liu J, Zhang Q, Lian Z, Chen H, Shi Z, Feng H, Miao X, Du Q, Zhou H (2017) Painful tonic spasm in neuromyelitis optica spectrum disorders: prevalence, clinical implications and treatment options. Mult Scler Relat Disord 17:99–102
Liu Z, Chen J, Wang Z, Wang Y, Zheng D, Wang H, Peng Y (2020) The CSF levels of neutrophil-related chemokines in patients with neuromyelitis optica. Ann Clin Transl Neurol 7:1245–1251
Long Y, Qiu W, Lu Z, Bao J, Wu A, Wang Y, Wang H, Hu X (2012) Aquaporin 4 antibodies in the cerebrospinal fluid are helpful in diagnosing Chinese patients with neuromyelitis optica. NeuroImmunoModulation 19:96–102
Mader S, Gredler V, Schanda K, Rostasy K, Dujmovic I, Pfaller K, Lutterotti A, Jarius S, Di Pauli F, Kuenz B, Ehling R, Hegen H, Deisenhammer F, Aboul-Enein F, Storch MK, Koson P, Drulovic J, Kristoferitsch W, Berger T, Reindl M (2011) Complement activating antibodies to myelin oligodendrocyte glycoprotein in neuromyelitis optica and related disorders. J Neuroinflamm 8:184
Marelli C, Salsano E, Politi LS, Labauge P (2019) Spinal cord involvement in adult-onset metabolic and genetic diseases. J Neurol Neurosurg Psychiatry 90:211–218
Marignier R, Hacohen Y, Cobo-Calvo A, Probstel AK, Aktas O, Alexopoulos H, Amato MP, Asgari N, Banwell B, Bennett J, Brilot F, Capobianco M, Chitnis T, Ciccarelli O, Deiva K, De Seze J, Fujihara K, Jacob A, Kim HJ, Kleiter I, Lassmann H, Leite MI, Linington C, Meinl E, Palace J, Paul F, Petzold A, Pittock S, Reindl M, Sato DK, Selmaj K, Siva A, Stankoff B, Tintore M, Traboulsee A, Waters P, Waubant E, Weinshenker B, Derfuss T, Vukusic S, Hemmer B (2021) Myelin-oligodendrocyte glycoprotein antibody-associated disease. Lancet Neurol 20:762–772
Masuda H, Mori M, Hirano S, Uzawa A, Uchida T, Muto M, Ohtani R, Aoki R, Kuwabara S (2022) Silent progression of brain atrophy in aquaporin-4 antibody-positive neuromyelitis optica spectrum disorder. J Neurol Neurosurg Psychiatry 93:32–40
Matsumoto Y, Misu T, Mugikura S, Takai Y, Nishiyama S, Kuroda H, Takahashi T, Fujimori J, Nakashima I, Fujihara K, Aoki M (2020) Distinctive lesions of brain MRI between MOG-antibody-associated and AQP4-antibody-associated diseases. J Neurol Neurosurg Psychiatry. https://doi.org/10.1136/jnnp-2020-324818
Matthews L, Marasco R, Jenkinson M, Kuker W, Luppe S, Leite MI, Giorgio A, De Stefano N, Robertson N, Johansen-Berg H, Evangelou N, Palace J (2013) Distinction of seropositive NMO spectrum disorder and MS brain lesion distribution. Neurology 80:1330–1337
McKeon A, Lennon VA, Jacob A, Matiello M, Lucchinetti CF, Kale N, Chan KH, Weinshenker BG, Apiwattinakul M, Wingerchuk DM, Pittock SJ (2009) Coexistence of myasthenia gravis and serological markers of neurological autoimmunity in neuromyelitis optica. Muscle Nerve 39:87–90
Mealy MA, Kessler RA, Rimler Z, Reid A, Totonis L, Cutter G, Kister I, Levy M (2018) Mortality in neuromyelitis optica is strongly associated with African ancestry. Neurol Neuroimmunol Neuroinflamm 5:e468
Mealy MA, Whetstone A, Orman G, Izbudak I, Calabresi PA, Levy M (2015) Longitudinally extensive optic neuritis as an MRI biomarker distinguishes neuromyelitis optica from multiple sclerosis. J Neurol Sci 355:59–63
Merle H, Olindo S, Jeannin S, Hage R, Donnio A, Richer R, Cabre P (2013) Visual field characteristics in neuromyelitis optica in absence of and after one episode of optic neuritis. Clin Ophthalmol 7:1145–1153
Miyamoto K, Fujihara K, Kira JI, Kuriyama N, Matsui M, Tamakoshi A, Kusunoki S (2018) Nationwide epidemiological study of neuromyelitis optica in Japan. J Neurol Neurosurg Psychiatry 89:667–668
Mori M, Kuwabara S, Paul F (2018) Worldwide prevalence of neuromyelitis optica spectrum disorders. J Neurol Neurosurg Psychiatry 89:555–556
Motamedi S, Oertel FC, Yadav SK, Kadas EM, Weise M, Havla J, Ringelstein M, Aktas O, Albrecht P, Ruprecht K, Bellmann-Strobl J, Zimmermann HG, Paul F, Brandt AU (2020) Altered fovea in AQP4-IgG-seropositive neuromyelitis optica spectrum disorders. Neurol Neuroimmunol Neuroinflamm 7:e805
Mustafa R, Passe TJ, Lopez-Chiriboga AS, Weinshenker BG, Krecke KN, Zalewski NL, Diehn FE, Sechi E, Mandrekar J, Kaufmann TJ, Morris PP, Pittock SJ, Toledano M, Lanzino G, Aksamit AJ, Kumar N, Lucchinetti CF, Flanagan EP (2021) Utility of MRI enhancement pattern in myelopathies with longitudinally extensive T2 lesions. Neurol Clin Pract 11:e601–e611
Nakajima H, Hosokawa T, Sugino M, Kimura F, Sugasawa J, Hanafusa T, Takahashi T (2010) Visual field defects of optic neuritis in neuromyelitis optica compared with multiple sclerosis. BMC Neurol 10:45
Ochi H, Fujihara K (2016) Demyelinating diseases in Asia. Curr Opin Neurol 29:222–228
Oertel FC, Havla J, Roca-Fernandez A, Lizak N, Zimmermann H, Motamedi S, Borisow N, White OB, Bellmann-Strobl J, Albrecht P, Ruprecht K, Jarius S, Palace J, Leite MI, Kuempfel T, Paul F, Brandt AU (2018) Retinal ganglion cell loss in neuromyelitis optica: a longitudinal study. J Neurol Neurosurg Psychiatry 89:1259–1265
Oertel FC, Schliesseit J, Brandt AU, Paul F (2019) Cognitive impairment in neuromyelitis optica spectrum disorders: a review of clinical and neuroradiological features. Front Neurol 10:608
Oertel FC, Specovius S, Zimmermann HG, Chien C, Motamedi S, Bereuter C, Cook L, Lana Peixoto MA, Fontanelle MA, Kim HJ, Hyun JW, Palace J, Roca-Fernandez A, Leite MI, Sharma S, Ashtari F, Kafieh R, Dehghani A, Pourazizi M, Pandit L, D’Cunha A, Aktas O, Ringelstein M, Albrecht P, May E, Tongco C, Leocani L, Pisa M, Radaelli M, Martinez-Lapiscina EH, Stiebel-Kalish H, Siritho S, de Seze J, Senger T, Havla J, Marignier R, Cobo-Calvo A, Bichuetti D, Tavares IM, Asgari N, Soelberg K, Altintas A, Yildirim R, Tanriverdi U, Jacob A, Huda S, Rimler Z, Reid A, Mao-Draayer Y, Soto de Castillo I, Petzold A, Green AJ, Yeaman MR, Smith T, Brandt AU, Paul F (2021) Retinal optical coherence tomography in neuromyelitis optica. Neurol Neuroimmunol Neuroinflamm 8:e1068
Oertel FC, Zimmermann H, Paul F, Brandt AU (2018) Optical coherence tomography in neuromyelitis optica spectrum disorders: potential advantages for individualized monitoring of progression and therapy. EPMA J 9:21–33
Ogawa R, Nakashima I, Takahashi T, Kaneko K, Akaishi T, Takai Y, Sato DK, Nishiyama S, Misu T, Kuroda H, Aoki M, Fujihara K (2017) MOG antibody-positive, benign, unilateral, cerebral cortical encephalitis with epilepsy. Neurol Neuroimmunol Neuroinflamm 4:e322
Otto C, Wengert O, Unterwalder N, Meisel C, Ruprecht K (2020) Analysis of soluble interleukin-2 receptor as CSF biomarker for neurosarcoidosis. Neurol Neuroimmunol Neuroinflamm 7(4):e725
Pache F, Zimmermann H, Mikolajczak J, Schumacher S, Lacheta A, Oertel FC, Bellmann-Strobl J, Jarius S, Wildemann B, Reindl M, Waldman A, Soelberg K, Asgari N, Ringelstein M, Aktas O, Gross N, Buttmann M, Ach T, Ruprecht K, Paul F, Brandt AU, in cooperation with the Neuromyelitis Optica Study Group (NEMOS) (2016) MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 4: afferent visual system damage after optic neuritis in MOG-IgG-seropositive versus AQP4-IgG-seropositive patients. J Neuroinflamm 13:282
Pakeerathan T, Havla J, Schwake C, Salmen A, Bigi S, Abegg M, Brügger D, Ferrazzini T, Runge A-K, Breu M, Kornek B, Bsteh G, Felipe-Rucián A, Ringelstein M, Aktas O, Karenfort M, Wendel E, Kleiter I, Hellwig K, Kümpfel T, Thiels C, Lücke T, Gold R, Rostasy K, Ayzenberg I (2022) Characteristic retinal atrophy pattern allows differentiation between pediatric MOGAD and MS after a single optic neuritis episode. J Neurol 269:6366–6376
Papp V, Iljicsov A, Rajda C, Magyari M, Koch-Henriksen N, Petersen T, Jakab G, Deme I, Nagy F, Imre P, Lohner Z, Kovacs K, Birkas AJ, Koves A, Rum G, Nagy Z, Kerenyi L, Vecsei L, Bencsik K, Jobbagy Z, Dioszeghy P, Horvath L, Galantai G, Kasza J, Molnar G, Simo M, Satori M, Rozsa C, Acs P, Berki T, Lovas G, Komoly S, Illes Z (2019) A population-based epidemiological study of neuromyelitis optica spectrum disorder in Hungary. Eur J Neurol 27(2):308-317
Papp V, Illes Z, Magyari M, Koch-Henriksen N, Kant M, Pfleger CC, Roemer SF, Jensen MB, Petersen AE, Nielsen HH, Rosendahl L, Mezei Z, Christensen T, Svendsen K, Hyldgaard Jensen PE, Lydolph MC, Heegaard N, Frederiksen JL, Sellebjerg F, Stenager E, Petersen T (2018) Nationwide prevalence and incidence study of neuromyelitis optica spectrum disorder in Denmark. Neurology 91:e2265–e2275
Papp V, Magyari M, Aktas O, Berger T, Broadley SA, Cabre P, Jacob A, Kira JI, Leite MI, Marignier R, Miyamoto K, Palace J, Saiz A, Sepulveda M, Sveinsson O, Illes Z (2021) Worldwide incidence and prevalence of neuromyelitis optica: a systematic review. Neurology 96:59–77
Paul F, Bennett JL, Chen JJ, Smith MA, Cimbora DM, Patterson KR, Hartung HP, Marignier R, Kim HJ, Fujihara K, Aktas O, Cree BAC (2023) Characterization of subclinical spinal cord and optic nerve MRI lesions in the N-MOmentum trial. EAN 2023 (abstract)
Paul F, Jarius S, Aktas O, Bluthner M, Bauer O, Appelhans H, Franciotta D, Bergamaschi R, Littleton E, Palace J, Seelig HP, Hohlfeld R, Vincent A, Zipp F (2007) Antibody to aquaporin 4 in the diagnosis of neuromyelitis optica. PLoS Med 4:e133
Pekcevik Y, Mitchell CH, Mealy MA, Orman G, Lee IH, Newsome SD, Thompson CB, Pardo CA, Calabresi PA, Levy M, Izbudak I (2016) Differentiating neuromyelitis optica from other causes of longitudinally extensive transverse myelitis on spinal magnetic resonance imaging. Mult Scler 22:302–311
Pellkofer HL, Havla J, Hauer D, Schelling G, Azad SC, Kuempfel T, Magerl W, Huge V (2013) The major brain endocannabinoid 2-AG controls neuropathic pain and mechanical hyperalgesia in patients with neuromyelitis optica. PLoS ONE 8:e71500
Petereit HF, Reske D, Tumani H, Jarius S, Markus Leweke F, Woitalla D, Pfister HW, Rubbert A (2010) Soluble CSF interleukin 2 receptor as indicator of neurosarcoidosis. J Neurol 257:1855–1863
Petzold A, Albrecht P, Balcer L, Bekkers E, Brandt AU, Calabresi PA, Deborah OG, Graves JS, Green A, Keane PA, Nij Bijvank JA, Sander JW, Paul F, Saidha S, Villoslada P, Wagner SK, Yeh EA, Imsvisual ERNEYEC (2021) Artificial intelligence extension of the OSCAR-IB criteria. Ann Clin Transla Neurol 8:1528–1542
Petzold A, Fraser CL, Abegg M, Alroughani R, Alshowaeir D, Alvarenga R, Andris C, Asgari N, Barnett Y, Battistella R, Behbehani R, Berger T, Bikbov MM, Biotti D, Biousse V, Boschi A, Brazdil M, Brezhnev A, Calabresi PA, Cordonnier M, Costello F, Cruz FM, Cunha LP, Daoudi S, Deschamps R, de Seze J, Diem R, Etemadifar M, Flores-Rivera J, Fonseca P, Frederiksen J, Frohman E, Frohman T, Tilikete CF, Fujihara K, Galvez A, Gouider R, Gracia F, Grigoriadis N, Guajardo JM, Habek M, Hawlina M, Martinez-Lapiscina EH, Hooker J, Hor JY, Howlett W, Huang-Link Y, Idrissova Z, Illes Z, Jancic J, Jindahra P, Karussis D, Kerty E, Kim HJ, Lagreze W, Leocani L, Levin N, Liskova P, Liu Y, Maiga Y, Marignier R, McGuigan C, Meira D, Merle H, Monteiro MLR, Moodley A, Moura F, Munoz S, Mustafa S, Nakashima I, Noval S, Oehninger C, Ogun O, Omoti A, Pandit L, Paul F, Rebolleda G, Reddel S, Rejdak K, Rejdak R, Rodriguez-Morales AJ, Rougier MB, Sa MJ, Sanchez-Dalmau B, Saylor D, Shatriah I, Siva A, Stiebel-Kalish H, Szatmary G, Ta L, Tenembaum S, Tran H, Trufanov Y, van Pesch V, Wang AG, Wattjes MP, Willoughby E, Zakaria M, Zvornicanin J, Balcer L, Plant GT (2022) Diagnosis and classification of optic neuritis. Lancet Neurol 21:1120-1134
Pittock SJ, Lennon VA (2008) Aquaporin-4 autoantibodies in a paraneoplastic context. Arch Neurol 65:629–632
Pittock SJ, Lennon VA, de Seze J, Vermersch P, Homburger HA, Wingerchuk DM, Lucchinetti CF, Zephir H, Moder K, Weinshenker BG (2008) Neuromyelitis optica and non organ-specific autoimmunity. Arch Neurol 65:78–83
Pittock SJ, Lennon VA, Krecke K, Wingerchuk DM, Lucchinetti CF, Weinshenker BG (2006) Brain abnormalities in neuromyelitis optica. Arch Neurol 63:390–396
Rabaste S, Cobo-Calvo A, Nistiriuc-Muntean V, Vukusic S, Marignier R, Cotton F, Ofsep NSG (2021) Diagnostic value of bright spotty lesions on MRI after a first episode of acute myelopathy. J Neuroradiol 48:28–36
Ramanathan S, Mohammad S, Tantsis E, Nguyen TK, Merheb V, Fung VSC, White OB, Broadley S, Lechner-Scott J, Vucic S, Henderson APD, Barnett MH, Reddel SW, Brilot F, Dale RC, Australasian, New Zealand MOGSG (2018) Clinical course, therapeutic responses and outcomes in relapsing MOG antibody-associated demyelination. J Neurol Neurosurg Psychiatry 89:127–137
Ramanathan S, Prelog K, Barnes EH, Tantsis EM, Reddel SW, Henderson AP, Vucic S, Gorman MP, Benson LA, Alper G, Riney CJ, Barnett M, Parratt JD, Hardy TA, Leventer RJ, Merheb V, Nosadini M, Fung VS, Brilot F, Dale RC (2016) Radiological differentiation of optic neuritis with myelin oligodendrocyte glycoprotein antibodies, aquaporin-4 antibodies, and multiple sclerosis. Mult Scler 22:470–482
Ratchford JN, Quigg ME, Conger A, Frohman T, Frohman E, Balcer LJ, Calabresi PA, Kerr DA (2009) Optical coherence tomography helps differentiate neuromyelitis optica and MS optic neuropathies. Neurology 73:302–308
Reiber H (1998) Cerebrospinal fluid–physiology, analysis and interpretation of protein patterns for diagnosis of neurological diseases. Mult Scler 4:99–107
Reiber H, Peter JB (2001) Cerebrospinal fluid analysis: disease-related data patterns and evaluation programs. J Neurol Sci 184:101–122
Reindl M, Schanda K, Woodhall M, Tea F, Ramanathan S, Sagen J, Fryer JP, Mills J, Teegen B, Mindorf S, Ritter N, Krummrei U, Stocker W, Eggert J, Flanagan EP, Ramberger M, Hegen H, Rostasy K, Berger T, Leite MI, Palace J, Irani SR, Dale RC, Probst C, Probst M, Brilot F, Pittock SJ, Waters P (2020) International multicenter examination of MOG antibody assays. Neurol Neuroimmunol Neuroinflamm 7:e674
Rinaldi S, Davies A, Fehmi J, Beadnall HN, Wang J, Hardy TA, Barnett MH, Broadley SA, Waters P, Reddel SW, Irani SR, Brilot F, Dale RC, Ramanathan S, Australian NZ, MOGSG, (2021) Overlapping central and peripheral nervous system syndromes in MOG antibody-associated disorders. Neurol Neuroimmunol Neuroinflamm 8:e924
Ringelstein M, Ayzenberg I, Lindenblatt G, Fischer K, Gahlen A, Novi G, Hayward-Konnecke H, Schippling S, Rommer PS, Kornek B, Zrzavy T, Biotti D, Ciron J, Audoin B, Berthele A, Giglhuber K, Zephir H, Kümpfel T, Berger R, Rother J, Haussler V, Stellmann JP, Whittam D, Jacob A, Kraemer M, Gueguen A, Deschamps R, Bayas A, Hümmert MW, Trebst C, Haarmann A, Jarius S, Wildemann B, Grothe M, Siebert N, Ruprecht K, Paul F, Collongues N, Marignier R, Levy M, Karenfort M, Deppe M, Albrecht P, Hellwig K, Gold R, Hartung HP, Meuth SG, Kleiter I, Aktas O, on behalf of the Neuromyelitis Optica Study Group (NEMOS) (2022) Interleukin-6 receptor blockade in treatment-refractory MOG-IgG-associated disease and neuromyelitis optica spectrum disorders. Neurol Neuroimmunol Neuroinflamm 9:e1100
Ringelstein M, Harmel J, Zimmermann H, Brandt AU, Paul F, Haarmann A, Buttmann M, Hümmert MW, Trebst C, Schroeder C, Ayzenberg I, Kleiter I, Hellwig K, Havla J, Kumpfel T, Jarius S, Wildemann B, Rommer P, Weber MS, Pellkofer H, Ropke L, Geis C, Retzlaff N, Zettl U, Deppe M, Klotz L, Young K, Stellmann JP, Kaste M, Kermer P, Marouf W, Lauda F, Tumani H, Graf J, Klistorner A, Hartung HP, Aktas O, Albrecht P, on behalf of the Neuromyelitis Optica Study Group (NEMOS) (2020) Longitudinal optic neuritis-unrelated visual evoked potential changes in NMO spectrum disorders. Neurology 94:e407–e418
Ringelstein M, Kleiter I, Ayzenberg I, Borisow N, Paul F, Ruprecht K, Kraemer M, Cohn E, Wildemann B, Jarius S, Hartung HP, Aktas O, Albrecht P (2014) Visual evoked potentials in neuromyelitis optica and its spectrum disorders. Mult Scler 20:617–620
Ringelstein M, Metz I, Ruprecht K, Koch A, Rappold J, Ingwersen J, Mathys C, Jarius S, Bruck W, Hartung HP, Paul F, Aktas O, for the Neuromyelitis Optica Study Group (NEMOS) (2014) Contribution of spinal cord biopsy to diagnosis of aquaporin-4 antibody positive neuromyelitis optica spectrum disorder. Mult Scler 20:882–888
Salama S, Levy M (2021) Bright spotty lesions as an imaging marker for neuromyelitis optica spectrum disorder. Mult Scler 28:1663–1666
Samartzis D, Gillis CC, Shih P, O’Toole JE, Fessler RG (2015) Intramedullary spinal cord tumors: part I-epidemiology, pathophysiology, and diagnosis. Global Spine J 5:425–435
Schindler P, Grittner U, Oechtering J, Leppert D, Siebert N, Duchow AS, Oertel FC, Asseyer S, Kuchling J, Zimmermann HG, Brandt AU, Benkert P, Reindl M, Jarius S, Paul F, Bellmann-Strobl J, Kuhle J, Ruprecht K (2021) Serum GFAP and NfL as disease severity and prognostic biomarkers in patients with aquaporin-4 antibody-positive neuromyelitis optica spectrum disorder. J Neuroinflamm 18:105
Schippling S, Balk LJ, Costello F, Albrecht P, Balcer L, Calabresi PA, Frederiksen JL, Frohman E, Green AJ, Klistorner A, Outteryck O, Paul F, Plant GT, Traber G, Vermersch P, Villoslada P, Wolf S, Petzold A (2015) Quality control for retinal OCT in multiple sclerosis: validation of the OSCAR-IB criteria. Mult Scler 21:163–170
Schmidt F, Zimmermann H, Mikolajczak J, Oertel FC, Pache F, Weinhold M, Schinzel J, Bellmann-Strobl J, Ruprecht K, Paul F, Brandt AU (2017) Severe structural and functional visual system damage leads to profound loss of vision-related quality of life in patients with neuromyelitis optica spectrum disorders. Mult Scler Relat Disord 11:45–50
Schneider E, Zimmermann H, Oberwahrenbrock T, Kaufhold F, Kadas EM, Petzold A, Bilger F, Borisow N, Jarius S, Wildemann B, Ruprecht K, Brandt AU, Paul F (2013) Optical coherence tomography reveals distinct patterns of retinal damage in neuromyelitis optica and multiple sclerosis. PLoS ONE 8:e66151
Sechi E, Krecke KN, Messina SA, Buciuc M, Pittock SJ, Chen JJ, Weinshenker BG, Lopez-Chiriboga AS, Lucchinetti CF, Zalewski NL, Tillema JM, Kunchok A, Monaco S, Morris PP, Fryer JP, Nguyen A, Greenwood T, Syc-Mazurek SB, Keegan BM, Flanagan EP (2021) Comparison of MRI lesion evolution in different central nervous system demyelinating disorders. Neurology 97:e1097–e1109
Senanayake B, Jitprapaikulsan J, Aravinthan M, Wijesekera JC, Ranawaka UK, Riffsy MT, Paramanathan T, Sagen J, Fryer JP, Schmeling J, Majed M, Flanagan EP, Pittock SJ (2019) Seroprevalence and clinical phenotype of MOG-IgG-associated disorders in Sri Lanka. J Neurol Neurosurg Psychiatry 90:1381–1383
Seok JM, Cho HJ, Ahn SW, Cho EB, Park MS, Joo IS, Shin HY, Kim SY, Kim BJ, Kim JK, Cho JY, Huh SY, Kwon O, Lee KH, Kim BJ, Min JH (2017) Clinical characteristics of late-onset neuromyelitis optica spectrum disorder: a multicenter retrospective study in Korea. Mult Scler 23:1748–1756
Sepulveda M, Aldea M, Escudero D, Llufriu S, Arrambide G, Otero-Romero S, Sastre-Garriga J, Romero-Pinel L, Martinez-Yelamos S, Sola-Valls N, Armangue T, Sotoca J, Escartin A, Robles-Cedeno R, Ramio-Torrenta L, Presas-Rodriguez S, Ramo-Tello C, Munteis E, Pelayo R, Gubieras L, Brieva L, Ortiz N, Hervas M, Mane-Martinez MA, Cano A, Vela E, Tintore M, Blanco Y, Montalban X, Graus F, Saiz A (2017) Epidemiology of NMOSD in Catalonia: influence of the new 2015 criteria in incidence and prevalence estimates. Mult Scler 24:1843–1851
Sepulveda M, Delgado-Garcia G, Blanco Y, Sola-Valls N, Martinez-Lapiscina EH, Armangue T, Montejo C, Pulido-Valdeolivas I, Martinez-Hernandez E, Arino H, Escudero D, Ruiz-Garcia R, Llufriu S, Dalmau J, Graus F, Saiz A (2019) Late-onset neuromyelitis optica spectrum disorder: the importance of autoantibody serostatus. Neurol Neuroimmunol Neuroinflamm 6:e607
Sepulveda M, Sola-Valls N, Escudero D, Rojc B, Baron M, Hernandez-Echebarria L, Gomez B, Dalmau J, Saiz A, Graus F (2018) Clinical profile of patients with paraneoplastic neuromyelitis optica spectrum disorder and aquaporin-4 antibodies. Mult Scler 24:1753–1759
Shah SS, Morris P, Buciuc M, Tajfirouz D, Wingerchuk DM, Weinshenker BG, Eggenberger ER, Di Nome M, Pittock SJ, Flanagan EP, Bhatti MT, Chen JJ (2022) Frequency of asymptomatic optic nerve enhancement in a large retrospective cohort of patients with aquaporin-4+ NMOSD. Neurology 99:e851–e857
Shimizu F, Ogawa R, Mizukami Y, Watanabe K, Hara K, Kadono C, Takahashi T, Misu T, Takeshita Y, Sano Y, Fujisawa M, Maeda T, Nakashima I, Fujihara K, Kanda T (2022) GRP78 antibodies are associated with blood-brain barrier breakdown in anti-myelin oligodendrocyte glycoprotein antibody-associated disorder. Neurol Neuroimmunol Neuroinflamm 9:e1038
Shimizu F, Schaller KL, Owens GP, Cotleur AC, Kellner D, Takeshita Y, Obermeier B, Kryzer TJ, Sano Y, Kanda T, Lennon VA, Ransohoff RM, Bennett JL (2017) Glucose-regulated protein 78 autoantibody associates with blood-brain barrier disruption in neuromyelitis optica. Sci Transl Med 9:eaai9111
Shor N, Deschamps R, Cobo Calvo A, Maillart E, Zephir H, Ciron J, Papeix C, Durand-Dubief F, Ruet A, Ayrignac X, Cohen M, Deiva K, Laplaud D, Bourre B, Audoin B, Collongues N, Vukusic S, Cotton F, Marignier R, group Ns (2021) MRI characteristics of MOG-Ab associated disease in adults: an update. Rev Neurol (Paris) 177:39–50
Shosha E, Dubey D, Palace J, Nakashima I, Jacob A, Fujihara K, Takahashi T, Whittam D, Leite MI, Misu T, Yoshiki T, Messina S, Elsone L, Majed M, Flanagan E, Gadoth A, Huebert C, Sagen J, Greenberg BM, Levy M, Banerjee A, Weinshenker B, Pittock SJ (2018) Area postrema syndrome: frequency, criteria, and severity in AQP4-IgG-positive NMOSD. Neurology 91:e1642–e1651
Sinnecker T, Clarke MA, Meier D, Enzinger C, Calabrese M, De Stefano N, Pitiot A, Giorgio A, Schoonheim MM, Paul F, Pawlak MA, Schmidt R, Kappos L, Montalban X, Rovira A, Evangelou N, Wuerfel J, Group MS (2019) Evaluation of the central vein sign as a diagnostic imaging biomarker in multiple sclerosis. JAMA Neurol 76:1446–1456
Solomon JM, Paul F, Chien C, Oh J, Rotstein DL (2021) A window into the future? MRI for evaluation of neuromyelitis optica spectrum disorder throughout the disease course. Ther Adv Neurol Disord 14:17562864211014388
Stellmann JP, Krumbholz M, Friede T, Gahlen A, Borisow N, Fischer K, Hellwig K, Pache F, Ruprecht K, Havla J, Kumpfel T, Aktas O, Hartung HP, Ringelstein M, Geis C, Kleinschnitz C, Berthele A, Hemmer B, Angstwurm K, Young KL, Schuster S, Stangel M, Lauda F, Tumani H, Mayer C, Zeltner L, Ziemann U, Linker RA, Schwab M, Marziniak M, Then Bergh F, Hofstadt-van Oy U, Neuhaus O, Zettl U, Faiss J, Wildemann B, Paul F, Jarius S, Trebst C, Kleiter I, on behalf of NEMOS (Neuromyelitis Optica Study Group) (2017) Immunotherapies in neuromyelitis optica spectrum disorder: efficacy and predictors of response. J Neurol Neurosurg Psychiatry 88:639–647
Sun H, Sun X, Huang D, Wu L, Yu S (2019) Cerebral cortex impairment in neuromyelitis optica spectrum disorder: a case report and literature review. Mult Scler Relat Disord 32:9–12
Sundaram S, Nair SS, Jaganmohan D, Unnikrishnan G, Nair M (2020) Relapsing lumbosacral myeloradiculitis: an unusual presentation of MOG antibody disease. Mult Scler 26:509–511
Tajfirouz D, Padungkiatsagul T, Beres S, Moss HE, Pittock S, Flanagan E, Kunchok A, Shah S, Bhatti MT, Chen JJ (2022) Optic chiasm involvement in AQP-4 antibody-positive NMO and MOG antibody-associated disorder. Mult Scler 28:149–153
Takai Y, Misu T, Nakashima I, Takahashi T, Itoyama Y, Fujihara K, Aoki M (2012) Two cases of lumbosacral myeloradiculitis with anti-aquaporin-4 antibody. Neurology 79:1826–1828
Takano R, Misu T, Takahashi T, Sato S, Fujihara K, Itoyama Y (2010) Astrocytic damage is far more severe than demyelination in NMO: a clinical CSF biomarker study. Neurology 75:208–216
Tantsis EM, Prelog K, Alper G, Benson L, Gorman M, Lim M, Mohammad SS, Ramanathan S, Brilot F, Dale RC, Group PMMS (2019) Magnetic resonance imaging in enterovirus-71, myelin oligodendrocyte glycoprotein antibody, aquaporin-4 antibody, and multiple sclerosis-associated myelitis in children. Dev Med Child Neurol 61:1108–1116
Thompson AJ, Banwell BL, Barkhof F, Carroll WM, Coetzee T, Comi G, Correale J, Fazekas F, Filippi M, Freedman MS, Fujihara K, Galetta SL, Hartung HP, Kappos L, Lublin FD, Marrie RA, Miller AE, Miller DH, Montalban X, Mowry EM, Sorensen PS, Tintore M, Traboulsee AL, Trojano M, Uitdehaag BMJ, Vukusic S, Waubant E, Weinshenker BG, Reingold SC, Cohen JA (2018) Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol 17:162–173
Tian DC, Li Z, Yuan M, Zhang C, Gu H, Wang Y, Shi FD (2020) Incidence of neuromyelitis optica spectrum disorder (NMOSD) in China: a national population-based study. Lancet Reg Health West Pac 2:100021
Tian DC, Xiu Y, Wang X, Shi K, Fan M, Li T, Li H, Su L, Ma Y, Xu W, Song T, Liu Y, Shi FD, Zhang X (2020) Cortical thinning and ventricle enlargement in neuromyelitis optica spectrum disorders. Front Neurol 11:872
Tian DC, Zhang C, Yuan M, Yang X, Gu H, Li Z, Wang Y, Shi FD (2020) Incidence of multiple sclerosis in China: a nationwide hospital-based study. Lancet Reg Health West Pac 1:100010
Toru S, Soejima I, Katayama Y, Saito K, Yokote H (2020) A case of anti-AQP4 antibody-positive neuromyelitis optica spectrum disorder with MRI-proven lesions in lumbar nerve roots. Mult Scler Relat Disord 46:102557
Trebst C, Berthele A, Jarius S, Kumpfel T, Schippling S, Wildemann B, Wilke C, Neuromyelitis Optica Study Group (NEMOS) (2011) Diagnosis and treatment of neuromyelitis optica. Consensus recommendations of the Neuromyelitis Optica Study Group. Nervenarzt 82:768–777
Trebst C, Jarius S, Berthele A, Paul F, Schippling S, Wildemann B, Borisow N, Kleiter I, Aktas O, Kumpfel T, Neuromyelitis Optica Study Group (NEMOS) (2014) Update on the diagnosis and treatment of neuromyelitis optica: recommendations of the Neuromyelitis Optica Study Group (NEMOS). J Neurol 261:1–16
Trebst C, Raab P, Voss EV, Rommer P, Abu-Mugheisib M, Zettl UK, Stangel M (2011) Longitudinal extensive transverse myelitis–it’s not all neuromyelitis optica. Nat Rev Neurol 7:688–698
Uzawa A, Mori M, Ito M, Uchida T, Hayakawa S, Masuda S, Kuwabara S (2009) Markedly increased CSF interleukin-6 levels in neuromyelitis optica, but not in multiple sclerosis. J Neurol 256:2082–2084
Vabanesi M, Pisa M, Guerrieri S, Moiola L, Radaelli M, Medaglini S, Martinelli V, Comi G, Leocani L (2019) In vivo structural and functional assessment of optic nerve damage in neuromyelitis optica spectrum disorders and multiple sclerosis. Sci Rep 9:10371
Vazquez Do Campo R, Stephens A, Marin Collazo IV, Rubin DI (2018) MOG antibodies in combined central and peripheral demyelination syndromes. Neurol Neuroimmunol Neuroinflamm 5:e503
Ventura RE, Kister I, Chung S, Babb JS, Shepherd TM (2016) Cervical spinal cord atrophy in NMOSD without a history of myelitis or MRI-visible lesions. Neurol Neuroimmunol Neuroinflamm 3:e224
Veselaj K, Kamber N, Briner M, Friedli C, Diem L, Guse K, Miclea A, Wiest R, Wagner F, Grabe H, Abegg M, Horn MP, Bigi S, Chan A, Hoepner R, Salmen A (2021) Evaluation of diagnostic criteria and red flags of myelin oligodendrocyte glycoprotein encephalomyelitis in a clinical routine cohort. CNS Neurosci Ther 27:426–438
Viswanathan S, Wah LM (2019) A nationwide epidemiological study on the prevalence of multiple sclerosis and neuromyelitis optica spectrum disorder with important multi-ethnic differences in Malaysia. Mult Scler 25:1452–1461
von Glehn F, Jarius S, Cavalcanti Lira RP, Alves Ferreira MC, von Glehn FH, Costa ECSM, Beltramini GC, Bergo FP, Farias AS, Brandao CO, Wildemann B, Damasceno BP, Cendes F, Santos LM, Yasuda CL (2014) Structural brain abnormalities are related to retinal nerve fiber layer thinning and disease duration in neuromyelitis optica spectrum disorders. Mult Scler 20:1189–1197
Watanabe M, Nakamura Y, Michalak Z, Isobe N, Barro C, Leppert D, Matsushita T, Hayashi F, Yamasaki R, Kuhle J, Kira JI (2019) Serum GFAP and neurofilament light as biomarkers of disease activity and disability in NMOSD. Neurology 93:e1299–e1311
Waters P, Reindl M, Saiz A, Schanda K, Tuller F, Kral V, Nytrova P, Sobek O, Nielsen HH, Barington T, Lillevang ST, Illes Z, Rentzsch K, Berthele A, Berki T, Granieri L, Bertolotto A, Giometto B, Zuliani L, Hamann D, van Pelt ED, Hintzen R, Hoftberger R, Costa C, Comabella M, Montalban X, Tintore M, Siva A, Altintas A, Deniz G, Woodhall M, Palace J, Paul F, Hartung HP, Aktas O, Jarius S, Wildemann B, Vedeler C, Ruiz A, Leite MI, Trillenberg P, Probst M, Saschenbrecker S, Vincent A, Marignier R (2016) Multicentre comparison of a diagnostic assay: aquaporin-4 antibodies in neuromyelitis optica. J Neurol Neurosurg Psychiatry 87:1005–1015
Waters PJ, Komorowski L, Woodhall M, Lederer S, Majed M, Fryer J, Mills J, Flanagan EP, Irani SR, Kunchok AC, McKeon A, Pittock SJ (2019) A multicenter comparison of MOG-IgG cell-based assays. Neurology 92:e1250–e1255
West TW, Hess C, Cree BA (2012) Acute transverse myelitis: demyelinating, inflammatory, and infectious myelopathies. Semin Neurol 32:97–113
Wildemann B, Oschmann P, Reiber H (2011) Laboratory diagnosis in neurology. Thieme, Stuttgart
Wingerchuk D, Banwell B, Bennett JL, Cabre P, Carroll W, Chitnis T, de Seze J, Fujihara K, Greenberg B, Jacob A, Jarius S, Lana-Peicoto M, Levy M, Simon JH, Tenembaum S, Traboulsee AL, Waters P, Wellik K (2015) Weinshenker B (2015) International Consensus Diagnostic Criteria for Neuromyelitis Optica Spectrum Disorders. Neurology 85:177–189
Wingerchuk DM, Banwell B, Bennett JL, Cabre P, Carroll W, Chitnis T, de Seze J, Fujihara K, Greenberg B, Jacob A, Jarius S, Lana-Peixoto M, Levy M, Simon JH, Tenembaum S, Traboulsee AL, Waters P, Wellik KE, Weinshenker BG, International Panel for NMOD (2015) International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology 85:177–189
Wingerchuk DM, Hogancamp WF, O’Brien PC, Weinshenker BG (1999) The clinical course of neuromyelitis optica (Devic’s syndrome). Neurology 53:1107–1114
Wingerchuk DM, Lennon VA, Pittock SJ, Lucchinetti CF, Weinshenker BG (2006) Revised diagnostic criteria for neuromyelitis optica. Neurology 66:1485–1489
Wingerchuk DM, Pittock SJ, Lucchinetti CF, Lennon VA, Weinshenker BG (2007) A secondary progressive clinical course is uncommon in neuromyelitis optica. Neurology 68:603–605
Wu Y, Yang M, Gao P, Wang Z, Wu J, Wang J, Xu Q, Zhou H, Wu T, Wu W, Wei S, Hu YH (2022) Incidence of neuromyelitis optica spectrum disorders in China: a large cohort study using claim data. BMJ Open 12:e048942
Xu Y, Ren Y, Li X, Xu W, Wang X, Duan Y, Liu Y, Zhang X, Tian DC (2021) Persistently gadolinium-enhancing lesion is a predictor of poor prognosis in NMOSD attack: a clinical trial. Neurotherapeutics 18:868–877
Yang J, Kim SM, Kim YJ, Cheon SY, Kim B, Jung KC, Park KS (2016) Accuracy of the fluorescence-activated cell sorting assay for the aquaporin-4 antibody (AQP4-Ab): comparison with the commercial AQP4-Ab assay kit. PLoS ONE 11:e0162900
Zalewski NL, Morris PP, Weinshenker BG, Lucchinetti CF, Guo Y, Pittock SJ, Krecke KN, Kaufmann TJ, Wingerchuk DM, Kumar N, Flanagan EP (2017) Ring-enhancing spinal cord lesions in neuromyelitis optica spectrum disorders. J Neurol Neurosurg Psychiatry 88:218–225
Zhong X, Wang H, Dai Y, Wu A, Bao J, Xu W, Cheng C, Lu Z, Qiu W, Hu X (2011) Cerebrospinal fluid levels of CXCL13 are elevated in neuromyelitis optica. J Neuroimmunol 240–241:104–108
Acknowledgements
The authors are grateful to Kathleen Ingenhoven for excellent organizational support. B.W. is grateful to the Dietmar Hopp Stiftung for funding research on MOG-EM/MOGAD and NMOSD.
Members of the Neuromyelitis Optica Study Group (NEMOS) in alphabetical order: Orhan Aktas, Heinrich Heine University Düsseldorf, Düsseldorf, Germany; Philipp Albrecht, Heinrich Heine University Düsseldorf, Germany; Klemens Angstwurm, University Hospital, Regensburg, Germany; Susanna Asseyer, Charité—Universitätsmedizin, corporate member of Freie Universität Berlin and Humboldt-Universität zu Berlin, Berlin, Germany; Ilya Ayzenberg, St. Josef Hospital, Ruhr University Bochum, Bochum, Germany; Antonios Bayas, University Hospital, Augsburg, Germany; Stefanie Behnke, Knappschaftsklinikum Saar GmbH, Sulzbach; Judith Bellmann-Strobl, Charité—Universitätsmedizin Berlin, corporate member of Freie Universität Berlin and Humboldt-Universität zu Berlin, Berlin, Germany, and Experimental and Clinical Research Center, Berlin, Germany, and Max Delbrück Center for Molecular Medicine in the Helmholtz Association (MDC), Berlin, Germany; Achim Berthele, Klinikum rechts der Isar, School of Medicine, Technical University Munich, Munich, Germany; Stefan Bittner, University Medical Center of the Johannes Gutenberg University, Mainz, Germany; Tobias Böttcher, MVZ Johanna-Odebrecht- Stiftung, Greifswald, Germany; Mathias Buttmann, Caritas Krankenhaus Bad Mergentheim, Germany; Ankelien Duchow, Charité —Universitätsmedizin Berlin, Germany; Daniel Engels, Ludwig-Maximilians University, Munich, Germany; Thorleif Etgen, Kliniken Südostbayern-Klinikum Traunstein, Germany; Barbara Ettrich, University Hospital, Leipzig, Germany; Katinka Fischer, MSc, Heinrich Heine University, Düsseldorf, Germany; Benedikt Frank, University Hospital, Essen, Germany; Frank Freitag, Nervenzentrum Potsdam, Potsdam, Germany; Anna Gahlen, MVZ für Neurologie, Psychiatrie und Physiotherapie, Herne, Germany; Achim Gass, University Hospital, Mannheim, Germany; Christian Geis, Department of Neurology, Jena University Hospital, Germany; Katrin Giglhuber, Klinikum rechts der Isar, School of Medicine, Technical University Munich, Munich, Germany; Ralf Gold, Ruhr University of Bochum, Germany; Yasemin Göreci, Faculty of Medicine and University Hospital Cologne, Germany; Matthias Grothe, University Hospital, Greifswald, Germany; Julia Gutbrod, University Hospital, Augsburg, Germany; Kersten Guthke, Klinikum Görlitz, Germany; Axel Haarmann, University of Würzburg, Germany; Jürgen Haas, University of Heidelberg, Germany; Vivien Häußler, University Medical Center Hamburg-Eppendorf, Hamburg, Germany; Joachim Havla, Ludwig-Maximilians-Universität München, Munich, Germany; Kerstin Hellwig, St. Josef Hospital, Ruhr University Bochum, Germany; Bernhard Hemmer, Klinikum rechts der Isar, School of Medicine, Technical University Munich, Germany; Frank Hoffmann, Krankenhaus Martha-Maria, Halle, Germany; Olaf Hoffmann, St. Josefs-Krankenhaus, Potsdam, Germany; Ulrich Hofstadt-van Oy, Klinikum Westfalen, Dortmund, Germany; Martin W. Hümmert, Hannover Medical School, Hannover, Germany; Sven Jarius, University of Heidelberg, Heidelberg, Germany; Jutta Junghans, Krankenhaus Martha-Maria, Halle, Germany; Matthias Kaste, Nordwest Hospital Sanderbusch, Sande, Germany; Barbara Kaulen, University Hospital, Hamburg, Germany; Peter Kern, Asklepios Klinik, Teupitz, Germany; Karsten Kern, Knappschaftsklinikum Saar GmbH, Sulzbach, Germany; Pawel Kermer, Nordwest Hospital Sanderbusch, Sande, Germany; Christoph Kleinschnitz, University Hospital, Essen, Germany; Ingo Kleiter, Marianne-Strauß-Klinik, Berg, Germany; Luisa Klotz, University of Münster, Münster, Germany; Wolfgang Köhler, University Hospital Leipzig, Germany; Mirjam Korporal-Kuhnke, University of Heidelberg, Heidelberg, Germany; Markus Kowarik, University Hospital, Tübingen, Germany; Markus Kraemer, Alfried-Krupp-Krankenhaus Essen, Germany; Markus Krumbholz, Brandenburg Medical School, Rüdersdorf bei Berlin, Germany; Tania Kümpfel, LMU Hospital, Ludwig-Maximilians- Universität München, Munich, Germany; Stefan Langel, Landeskrankenhaus Rheinhessen, Germany; Ann-Sophie Lauenstein, German Diagnostic Clinic, DKD Helios Clinic Wiesbaden, Germany; Frank Leypoldt, University Medical Center Schleswig Holstein —Kiel, Germany; Martin Liebetrau, St. Josefs-Hospital, Wiesbaden, Germany; Ralf Linker, University Hospital, Regensburg, Germany; De-Hyung Lee, University Hospital, Regensburg, Germany; Lisa Lohmann, University of Münster, Münster, Germany; Peter Luedemann, Agaplesion ev. Bathildiskrankenhaus Bad Pyrmont, Germany; Felix Lüsse, University Medical Center of the Johannes Gutenberg University Mainz, Germany; Martin Marziniak, Isar-Amper Klinik Ost, Munich, Germany ; Christoph Mayer, Neurologischen Gemeinschaftspraxis am Kaiserplatz, Frankfurt, Germany; Stefanie Meister, University Hospital, Rostock, Germany; Arthur Melms, Facharztpraxis für Neurologie und Psychiatrie, Stuttgart, Germany; Mathias von Mering, Klinikum Bremen Nord, Germany; Imke Metz, University Hospital, Göttingen, Germany; Sven Meuth, Heinrich Heine University, Düsseldorf, Germany; Jasmin Naumann, Knappschaftsklinikum Saar GmbH, Sulzbach, Germany; Oliver Neuhaus, SRH Krankenhaus Sigmaringen, Germany; Moritz Niederschweiberer, Charité Universitätsmedizin, Berlin, Germany; Sabine Niehaus, Klinikum Dortmund, Germany; Carolin Otto, Charité Universitätsmedizin, Berlin, Germany; Florence Pache, Charité Universitätsmedizin, Berlin, Germany; Sandra Paryjas, University Medical Center of the Johannes Gutenberg University Mainz, Germany; Friedemann Paul, Charité—Universitätsmedizin Berlin, corporate member of Freie Universität Berlin and Humboldt- Universität zu Berlin, Berlin, Germany, and Experimental and Clinical Research Center, a cooperation between the Max Delbrück Center for Molecular Medicine in the Helmholtz Association and Charité—Universitätsmedizin Berlin, Berlin, Germany, and Max Delbrück Center for Molecular Medicine in the Helmholtz Association (MDC), Berlin, Germany, and NeuroCure Clinical Research Center, Charité Universitätsmedizin Berlin, corporate member of Freie Universität Berlin and Humboldt-Universität zu Berlin, and Berlin Institute of Health and Max Delbrück Center for Molecular Medicine, Berlin, Germany; Hannah Pellkofer, Ludwig- Maximilians University, Munich, Germany; Mosche Pompsch, Alfried-Krupp-Krankenhaus, Essen, Germany; Hans-Ulrich Puhlmann, Schlosspark-Klinik, Berlin, Germany; Refik Pul, University of Essen, Germany; Sebastian Rauer, University Medical Center Freiburg, Germany; Thorsten Rehfeldt, Dietrich-Bonhoeffer Clinic Neubrandenburg, Germany; Nele Retzlaff, University Hospital, Rostock, Germany; Marius Ringelstein, Heinrich Heine University Düsseldorf, Düsseldorf, Germany; Paulus Rommer, Medical University of Vienna, Austria; Kevin Rostásy, Vestische Caritas-Kliniken GmbH, Datteln, Germany; Lioba Rückriem, MediClin Hedon-Klinik, Lingen (Ems), Germany; Klemens Ruprecht, Charité—Universitätsmedizin Berlin, corporate member of Freie Universität Berlin and Humboldt-Universität zu Berlin, Berlin, Germany; Christoph Ruschil, University Hospital, Tübingen, Germany; Patrick Schindler, Charité—Universitätsmedizin Berlin, corporate member of Freie Universität Berlin and Humboldt- Universität zu Berlin, Berlin, Germany; Matthias Schwab, Jena University Hospital, Jena, Germany; Makbule Senel, University of Ulm, Ulm, Germany; Jörn Peter Sieb, Helios Hanseklinikum, Stralsund, Germany; Claudia Sommer, University Hospital, Würzburg, Germany; Till Sprenger, German Diagnostic Clinic, DKD Helios Clinic Wiesbaden, Germany; Jan-Patrick Stellmann, CEMEREM, Marseille, France, and Aix Marseille Univ, Marseille, France; Heike Stephanik, University Hospital, Magdeburg, Germany; Muriel Stoppe, University Hospital, Leipzig, Germany; Klarissa Stürner, University Medical Center Schleswig Holstein— Kiel, Germany; Marie Süße, University Hospital, Greifswald, Germany; Florian Then Bergh, University of Leipzig, Leipzig, Germany; Daria Tkachenko, Hannover Medical School, Hannover Germany; Corinna Trebst, Hannover Medical School, Hannover, Germany; Johannes Tünnerhoff, University Hospital, Tübingen, Germany; Hayrettin Tumani, University of Ulm, Ulm, Germany; Annette Walter, Herford Hospital, Germany; Clemens Warnke, Faculty of Medicine and University Hospital Cologne, Germany; Klaus- Peter Wandinger, University Medical Center Schleswig-Holstein Campus, Lübeck, Germany; Martin Weber, University Hospital, Göttingen, Germany; Jens Weise, Helios Vogtland-Klinikum Plauen, Germany; Robert Weissert, University Hospital, Regensburg, Germany; Heinz Wiendl, University Hospital, Münster, Germany; Brigitte Wildemann, University of Heidelberg, Heidelberg, Germany; Alexander Winkelmann, University Hospital, Rostock, Germany; Yavor Yalachkov, University Hospital, Frankfurt, Germany; Lena Zeltner, University Hospital, Tübingen, Germany; Uwe Zettl, University Hospital, Rostock, Germany; Ulf Ziemann, University Hospital, Tübingen, Germany; Frauke Zipp, University Medical Center of the Johannes Gutenberg University, Mainz, Germany.
Funding
Open Access funding enabled and organized by Projekt DEAL. There was no specific funding for these recommendations.
Author information
Authors and Affiliations
Consortia
Contributions
The initial draft was prepared by SJ and CT with contributions from all authors. All authors were involved in critical revision of the manuscript. All authors have read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
SJ reports no conflicts of interest. OA has received speaking honoraria and travel grants from Alexion, Almirall, Biogen, Celgene, Merck, Novartis, Roche, and VielaBio. IA has received speaking honoraria and scientific advisory board compensation from Alexion, Roche, Merck Serono, Santhera, and Sanofi Genzyme and research support from Chugai and Diamed. JBS has received speaking honoraria and travel grants from Bayer Healthcare, sanofi-aventis/Genzyme, and Biogen and compensation for serving on a scientific advisory board from Roche. AB has received consulting and/or speaking fees from Alexion, Bayer Healthcare, Biogen, Celgene, and Roche. His institution has received compensations for clinical trials from Alexion, Biogen, Merck, Novartis, Roche, and Sanofi. VH reports no conflicts of interest. JH reports a grant for OCT research from the Friedrich-Baur-Stiftung, personal fees and non-financial support from Merck, Alexion, Novartis, Roche, Santhera, Biogen, Heidelberg Engineering, and Sanofi Genzyme, and non-financial support from the Guthy-Jackson Charitable Foundation; he is partially funded by the German Federal Ministry of Education and Research [grant numbers 01ZZ1603[A-D] and 01ZZ1804[A-H] (DIFUTURE)]. KH reports no conflicts of interest. MH reports no conflicts of interest. IK has received personal compensation for consulting, serving on a scientific advisory board, speaking, or other activities from Alexion, Almirall, Bayer, Biogen, Hexal, Horizon, Merck, Neuraxpharm, Sanofi, and Roche/Chugai. LK has received compensation for serving on scientific advisory boards from Alexion, Biogen, BMS, Celgene GmbH, Genzyme, Horizon, Janssen, Merck Serono, Novartis, and Roche. She has received speaker honoraria and travel support from Bayer, Biogen, Celgene GmbH, Genzyme, Grifols, Merck Serono, Novartis, Roche, Santhera, and Teva; she receives research support from German Research Foundation, IZKF Münster, IMF Münster, Biogen, Immunic AG, Novartis, and Merck Serono. MK has received travel funding, speaker honoraria, and research support from Bristol Myers Squibb, Merck, Novartis, and Roche. TK has received speaker honoraria and/or personal fees for advisory boards from Novartis Pharma, Roche Pharma, Alexion/Astra Zeneca and Biogen; the Institution she works for has received grant support for her research from Bayer-Schering AG, Novartis and Chugai Pharma. FP has received grants from Guthy Jackson Charitable Foundation, German Research Foundation, German Federal Ministry of Education and Research, Novartis, Bayer, Biogen Idec, Teva, Alexion, Roche, Horizon, Sanofi-Aventis/Genzyme, Merck Serono, Chugai, German Research Council, Werth Stiftung of the City of Cologne, Arthur Arnstein Stiftung Berlin, EU FP7 Framework Program, National Multiple Sclerosis (USA), MedImmune, Shire, and Alexion, and has a patent on Foveal Morphometry pending to Nocturne GmbH; he is Academic Editor at PLoS ONE and Associate Editor of Neurology® Neuroimmunology & Neuroinflammation. MR has received speaker honoraria from Novartis, Bayer Vital GmbH, Roche, Alexion, and Ipsen and travel reimbursement from Bayer Schering, Biogen Idec, Merz, Genzyme, Teva, Roche, and Merck. KR has received research support from Novartis Pharma, Merck Serono, German Ministry of Education and Research, European Union (821283-2), Stiftung Charité (BIH Clinical Fellow Program) and Arthur Arnstein Foundation, speaker honoraria from Bayer, and travel grants from Guthy Jackson Charitable Foundation. MS has received consulting and/or speaker honoraria from Alexion, Bayer, Biogen, Bristol-Myers-Squibb, Merck, Roche, and Sanofi Genzyme. She has received research funding from the Hertha-Nathorff-Program. PS reports no conflict of interest. FTB has received personal compensation for advisory board participation or speaking, and through his institution has received research support for investigator-initiated studies or for attending scientific meetings, from Actelion, Alexion, Bayer-Schering, Biogen, Merck, Novartis, Roche, Sanofi-Genzyme, Teva, UCB, German Research Foundation (DFG), and German Federal Ministry of Education and Research (BMBF). HT has received honoraria for consultation and expert testimony from Alexion Pharma, Bayer, Biogen, Janssen, MERCK, Novartis, Roche, Sanofi Genzyme, and Teva. BW has received grants from the German Ministry of Education and Research, German Research Foundation, Dietmar Hopp Stiftung, Klaus Tschira Stiftung, and Merck, and personal fees from Alexion, Bayer, Biogen, Roche. CT has received honoraria for consultation and expert testimony from Alexion Pharma Germany GmbH, Chugai Pharma Germany GmbH, and Roche Pharma GmbH.
Ethical approval
The manuscript does not contain patient or animal data.
Additional information
The members of the Neuromyelitis Optica Study Group (NEMOS) are listed in acknowledgements section.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jarius, S., Aktas, O., Ayzenberg, I. et al. Update on the diagnosis and treatment of neuromyelits optica spectrum disorders (NMOSD) – revised recommendations of the Neuromyelitis Optica Study Group (NEMOS). Part I: Diagnosis and differential diagnosis. J Neurol 270, 3341–3368 (2023). https://doi.org/10.1007/s00415-023-11634-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-023-11634-0
Keywords
- Neuromyelitis optica spectrum disorders (NMOSD)
- Neuromyelitis optica (NMO)
- Optic neuritis
- Myelitis
- Diagnostic criteria
- Diagnosis
- Differential diagnosis
- MOG antibody-associated encephalomyelitis (MOG-EM)
- MOG antibody-associated disease (MOGAD)
- Magnetic resonance imaging (MRI)
- Serology
- Cerebrospinal fluid (CSF)
- Optic coherence tomography (OCT)
- Clinical presentation
- Aquaporin-4 (AQP4)
- Myelin oligodendrocyte glycoprotein (MOG)
- Autoantibodies