
Abstract
Introduction
The absence of nigrosome 1 on brain MRI and the hyperechogenicity of substantia nigra (SNh) by transcranial sonography are two useful biomarkers in the diagnosis of parkinsonisms. We aimed to evaluate the absence of nigrosome 1 in amyotrophic lateral sclerosis (ALS) and to address its meaning.
Methods
136 ALS patients were recruited, including 16 progressive muscular atrophy (PMA) and 22 primary lateral sclerosis (PLS) patients. The SNh area was measured planimetrically by standard protocols. The nigrosome 1 status was qualitatively assessed by two blind evaluators in susceptibility weight images of 3T MRI. Demographic and clinical data were collected and the C9ORF72 expansion was tested in all patients.
Results
Nigrosome 1 was absent in 30% of ALS patients (36% of PLS, 29% of classical ALS and 19% of PMA patients). There was no relationship between radiological and clinical laterality, nor between nigrosome 1 and SNh area. Male sex (OR = 3.63 [1.51, 9.38], p = 0.005) and a higher upper motor neuron (UMN) score (OR = 1.10 [1.02, 1.2], p = 0.022) were independently associated to nigrosome 1 absence, which also was an independent marker of poor survival (HR = 1.79 [1.3, 2.8], p = 0.013).
Conclusion
In ALS patients, the absence of nigrosome 1 is associated with male sex, UMN impairment and shorter survival. This suggests that constitutional factors and the degree of pyramidal involvement are related to the substantia nigra involvement in ALS. Thus, nigrosome 1 could be a marker of a multisystem degeneration, which in turn associates to poor prognosis.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease clinically characterized by a progressive weakness and signs of upper (UMN) and lower motor neuron (LMN) impairment. According to the degree of UMN and LMN involvement, three phenotypes can be distinguished: classical ALS (cALS), primary lateral sclerosis (PLS) and progressive muscular atrophy (PMA) [1].
The hallmark of ALS is the presence of TDP-43 aggregates in degenerating UMN and LMN [1]. However, these aggregates can be found far beyond motor neurons in many ALS patients, including substantia nigra (SN) [2,3,4], probably accounting for the extra-motor features seen in ALS patients [1].
By means of transcranial sonography (TCS), a larger area of hyperechogenicity in SN (SNh) has been described in patients with several neurodegenerative diseases, including Parkinson’s disease (PD), atypical parkinsonisms, and ALS [5,6,7,8,9,10,11,12,13,14]. In them, the SNh has been proposed to be a marker of vulnerability for neuronal degeneration, probably caused by a disturbance in iron metabolism [10, 12, 15, 16].
Two different parts can be distinguished in the SN in terms of anatomy and function: the pars reticulata (SNpr) and the pars compacta (SNpc). The latter contains dopaminergic neurons, some of which are packed in clusters called nigrosomes [17,18,19]. In PD patients, postmortem studies have shown that dopaminergic neuronal loss typically affects nigrosomes, especially the largest one, called nigrosome 1 [20]. Nigrosome 1 is located along the rostro-caudal axis of the SNpc, in its dorsal part, and is visible as hyper-intense ovoid sub-structure within the dorsal hypo-intense midbrain region, medial to the cerebral peduncles in high-resolution susceptibility weight (SW) images of 3T and 7T brain magnetic resonance imaging (MRI) [20, 21].
Interestingly, the loss of nigrosome 1 has been demonstrated in 7T and 3T brain MRI of PD patients with postmortem confirmation and has been suggested to be a consequence of neuromelanin loss, an increase in iron content and/or a change in iron oxidation state, associated to the degeneration of dopaminergic neurons of the SNpc [20]. Moreover, the absence of nigrosome 1 differentiates with high-specificity and -sensitivity healthy controls from PD patients [22], where it has been associated with the disease severity and clinical laterality of symptoms [23].
Despite clinical, imaging and neuropathological data suggesting the impairment of SNpc, at least in a subset of ALS patients [2,3,4], no previous study has assessed nigrosome 1 status in ALS patients.
Therefore, the aims of this study were: (1) to evaluate the nigrosome 1 status in patients with ALS; (2) to evaluate the association between nigrosome 1 status and SNh; (3) to address the contribution of demographical, clinical and genetic factors to the absence or presence of nigrosome 1 in patients with ALS and finally; (4) to evaluate the prognostic value of the absence of nigrosome 1 in the disease.
Methods
Subjects and definitions
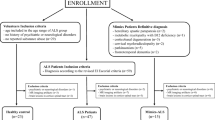
For this cross-sectional study, patients diagnosed with classical ALS (cALS), PMA or PLS who came to our ALS Unit between February 2014 and June 2019 and gave written informed consent, were recruited. Patients are routinely evaluated by the same neurologist (JFVC), and demographical and clinical data are prospectively recorded in a database. The cALS patients met the El Escorial revised criteria of possible, probable or definitive ALS [24]. PMA was defined as a progressive isolated impairment of LMN in at least two regions [25], and PLS as a progressive isolated impairment of UMN in at least one region other than the lumbar region [26].
Patients were followed up until October 2020 and the date of death or tracheostomy were recorded.
Genetic analysis
All patients were screened for C9ORF72 with repeat primed PCR, as previously reported [27].
Clinical and analytical variables
Age, gender, time of symptom onset, region of onset (bulbar vs. spinal) and King’s clinical staging were recorded for all patients.
ALS patients were examined at the time of recruitment by the same expert neurologist (JFVC). The disability (measured with the ALSFRS-R score) [28] and degree of UMN impairment (measured with the UMN score, for a maximum of 16) [29], were recorded. The laterality of symptoms was established as the side of onset or, in bulba onset patients, of clinical predominance. Parkinsonian features (such as bradykinesia and cogwheel rigidity) were not systematically recorded, since they may be masked by and/or confounded with UMN and LMN signs [30].
The presence of behavioral and language disturbances was systematically assessed and the diagnosis of ALS with frontotemporal dementia (ALSFTD) was established based on the current criteria [31].
Nigrosome 1 examination
MR imaging examinations were performed in a 3T scanner (Signa HDxt, GE Healthcare, Milwaukee, WI, USA) using transmit–receive head coil array with eight elements. SW images were obtained with a 3D multi-echo gradient echo T2*-weighted sequence acquired on the transverse plane (TR = 43 ms; 9 echoes with center TE around 26 ms; TE = 15.1–35.8 ms with 5.1 echo spacing; FOV = 220 × 220 mm; matrix = 256 × 256 mm; slice thickness = 2 mm, flip angle = 15°; bandwidth = 62.5 Hz/px).
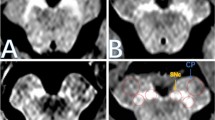
Nigrosome 1 status on brain MRI was visually assessed by a neurologist (MIMG) with 4 years of experience and one expert neuroradiologist (MMM), blinded to each other and to the patients’ data. For the nigrosome 1 evaluation, axial brain MRI SWI sequences at the midbrain level were selected. Healthy nigrosome 1 is a comma-shaped structure located in the posterior area of the SN. It has a high signal on SWI sequences compared to the surrounding elements: the SNpr, anterior and laterally; and the medial lemniscus, medially. Healthy nigrosome 1 and its surrounding structures has been described to resemble a “swallow tail” (Fig. 1) [21].

Anatomic representation of mesencephalon including nigrosome 1 and its surrounding structures (a, asterisk = substantia nigra; arrowhead = medial lemniscus; arrow = nigrosome 1; circle = red nucleus). Nigrosome 1 present on susceptibility weight images of MRI (b, arrow) and area of the SNh (c). Nigrosome 1 absent on susceptibility weight images of MRI (d) and larger area of SNh (e). ALS: amyotrophic lateral sclerosis; SNh: substantia nigra hyperechogenicity
Nigrosome 1 status was qualitatively assessed as (Fig. 1): present (“swallow tail” identified), absent (“swallow tail” not identified) or doubtful (when the quality of the images did not allow to define the nigrosome 1 status). Discrepancies between the two readers were resolved by consensus. Patients with doubtful nigrosomes (3 of all patients), in whom a consensus on nigrosome status was not reached, were excluded from further analysis.
TCS examination
In most patients, a TCS examination was performed within 3 months from MRI, by an expert neurologist sonographer (JIT), who was blind to the clinical data, with the same ultrasound system (Toshiba Aplio XG, Tokyo, Japan 2008) equipped with a 2.5 MHz phased-array transducer to obtain B-mode images through a temporal acoustic bone window. Right and left SNh were obtained and measured by this examiner, as previously reported [32] (Fig. 1).
Statistical analysis
Data were summarized by the mean, standard deviation, median and the first and third quartiles for the continuous variables, and by the relative and absolute frequencies for the categorical variables. For the statistical analysis, the nigrosome was considered to be present in a patient when both nigrosomes were visible, and absent, when either of both nigrosomes was absent. Inter- and intra-rater agreements were assessed using weighted κ test for the ratings (present, absent, doubtful). To assess the agreement between the side of disease onset and the absence of contralateral nigrosome, a Cohen κ test was performed. A mixed logistic regression model, including the subject and side as a random effect, was performed to compare the SNh area according to the nigrosome status. The association of demographical and clinical variables of ALS patients with the absence of nigrosome was assessed by a logistic regression model. The effect of the absence of nigrosome in survival was assessed with a Cox regression model and the results are displayed in a forest plot. Expected survival curves for subpopulations according to nigrosome absence, were calculated based on Cox model and are represented in a Kaplan–Meier curve (Fig. 3).
In both models, co-variables were selected based on previous literature and pre-specified hypothesis. P values of < 0.05 were considered statistically significant. All the statistical analyses and graphs were performed using version 4.3.0 of the R software.
Results
Population characteristics
One hundred and thirty six ALS patients (72% cALS; 12% PMA and 16% PLS) were recruited for this study. Briefly, mean age of patients was 62 years, and 56% were male. Most patients had a spinal onset of symptoms (70%), 15% were diagnosed with concomitant FTD and 12% carried a C9ORF72 expansion. At the time of examination, median disease duration was 17 months, and patients presented a mean ALSFRS-R of 37 and a median progression rate of 0.6. The demographic, clinical, and genetic characteristics of ALS patients, as per phenotype, are summarized in Table 1.
Contribution of demographic, clinical and genetic variables to nigrosome in the ALS patients
The nigrosome 1 status was assessable in 133 (97.7%) patients. The intra- and inter-rater agreement of the nigrosome 1 status was acceptable (weighted κ of 0.6 and 0.69, respectively).
At least one nigrosome 1 was absent in 30% (n = 39) of ALS patients, being more frequent in PLS patients (36%), followed by cALS patients (29%) and PMA patients (19%). Nigrosome 1 was absent in both sides in most patients (63%), only in the left side in 14% and in the right side in 20% of patients. There was no agreement between radiological laterality and the side of symptoms onset (Cohen κ = 0.03). Table 2 summarizes patients’ characteristics according to the nigrosome 1 status. Overall, patients with absent nigrosome 1 were more frequently male, had higher UMN score, and were more frequently diagnosed with FTD and with a C9ORF72 expansion. Interestingly, they showed similar ALSFRS-R score than patients with both nigrosomes 1, despite presenting shorter disease duration. Accordingly, they showed somewhat faster disease progression rate.
The multivariable model showed an independent association between the absence of nigrosome 1 and male gender (estimate = 3.637, p = 0.005) and higher UMN-score (estimate = 1.101, p = 0.022). Conversely, no statistically significant differences in ALSFS-R score, FTD diagnosis, C9ORF72 status and disease duration were found (see Table 3 and Fig. 2).

Graphical representation of the estimated effect of male sex (a) and upper motor neuron score (b) in the absence of nigrosome 1, as per the multivariable model
Nigrosome and prognosis
In the Cox model, the absence of nigrosome 1 was found to be an independent predictor of poor prognosis in ALS patients (HR = 1.79; p value = 0.013) together with age (HR = 1.02; p value = 0.025) and progression rate (HR = 2.35; p value < 0.001), while spinal onset (HR = 0.50; p value = 0.005) and PLS phenotype (HR = 0.12; p value < 0.001) were protective factors (Fig. 3).

Forest plot displaying the effect (Hazard Ratio) of co-variables in the survival of ALS patients (a). Kaplan–Meier curve, based on the Cox model, representing the independent effect of nigrosome 1 absence in the survival of ALS patients (b). Red = Nigrosome 1 present; Blue = Nigrosome 1 absent. Time is measured in months. FTD frontotemporal dementia, PLS primary lateral sclerosis, PMA progressive muscular atrophy
Nigrosome and SNh area
TCS was performed in 111 patients within 3 months from MRI (median delay of 1 day), and it was possible to measure the SNh area through the temporal acoustic bone window in 95 (81.9%) of them.
No differences in the SNh area were found according to the nigrosome status (OR = 0.46, p = 0.63, Fig. 4).

Box plots representing the SNh area according to the nigrosome status. SNh: substantia nigra hyperechogenicity
Discussion
The SNpc is a small midbrain region comprising neuro-melanin-containing dopaminergic neurons (some of them packed in nigrosomes), which becomes frequently affected in neurodegenerative diseases with motor involvement, such as parkinsonisms and motor neuron diseases [2, 4, 10]. Up till now, several imaging biomarkers have been described in the SN in these diseases. The SNh, measured by TCS, is thought to reflect iron deposits and has been shown to be a marker of SN vulnerability rather than neuronal loss [10, 16]. Accordingly, the SNh is not related with motor symptoms, but with genetic and constitutional factors that increase the risk of those neurodegenerative diseases [10, 16]. Unlike the SNh, the absence of nigrosome 1, detected in high SWI of brain MRI, is thought to represent a neuro-melanin/iron imbalance due to neuronal loss, and has been associated with motor symptoms in the contralateral brain hemisphere in PD patients [20, 23]. Despite the profuse literature about both biomarkers, to the best of our knowledge, no previous study has compared the absence of nigrosome 1 with the SNh in any neurodegenerative disease. Here, we demonstrate that there is not an association between the SNh and the absence of nigrosome 1, reinforcing the idea that they measure different pathophysiological processes. In short, the SNh probably measures an increase in iron content in the SNpc acting as a marker of neuronal vulnerability, while the nigrosome 1 absence is rather a marker of neuro-melanin loss in the SNpc. Considering this, it is not surprising that, in some diseases specifically causing SNpc degeneration (such as PD), the SNh is as frequently found as the nigrosome 1 absence [22]. Conversely, in other diseases, such as ALS, the SNh is much more frequently found than the absence nigrosome 1 on MRI or on the neuropathological assessment.
Thus, in postmortem studies, the SNpc becomes involved in about 20–30% of patients [2, 4, 33], a similar proportion to the nigrosome 1 loss found in our study. Different neuropathological patterns of involvement of the SNpc can be distinguished among ALS cases. On the one hand, the SNpc involvement is a feature of a subset of cALS patients being in an advanced neuropathological stage (2–4) [2, 4]. On the other hand, the SNpc involvement is a hallmark of ALS with pallidonigroluysian degeneration [2, 4, 33]. Similarly, different clinical patterns of patients combining motor neuron disease and parkinsonism have been described (e.g. ALS-parkinsonism, PLS-parkinsonism, ALS-FTD-parkinsonism…) [34]. Moreover, parkinsonian features and a dopaminergic deficit have also been described in the striatum of a variable proportion of cALS patients [30, 35,36,37]. Intriguingly, no association between extrapyramidal features and dopaminergic striatal deficit in ALS patients has been found [30, 35, 36], suggesting that other circuits are responsible of these symptoms. Accordingly, the SNh was not associated with clinical features in ALS patients, but with genetic and constitutional factors [16]. However, SNh is not a marker of neuronal loss and, up till now, no previous study has assessed nigrosome 1 in ALS patients.
In this study, we show for the first time that nigrosome 1 is absent, on at least one side of the SNpc, in 30% of ALS patients with different phenotypes, compared to about 95% of PD patients and 10% of healthy controls [22]. Interestingly, the neuropathologic involvement of the SNpc has been found in a similar percentage of patients in postmortem studies [2, 4, 33].
Unlike in PD patients [23], in most ALS patients, the nigrosome 1 was absent in both sides and, in those with an asymmetric involvement, no relationship with the clinical side of onset was found. Moreover, no associations between nigrosome 1 absence and age, disease duration or disability were found. Furthermore, although the frequency of FTD and C9ORF72 mutations was proportionally higher in patients with absent nigrosome 1, no independent association could be found in the multivariable model (perhaps due to the small sample size). However, male sex and the UMN score (a proxy of the degree of UMN impairment) were independently associated with the nigrosome 1 absence. Moreover, the nigrosome 1 absence was most frequent in PLS patients and least frequent in PMA patients. These findings are not surprising, considering previous literature. Firstly, male sex is a well-known risk factor for ALS, Parkinson’s disease and larger area of hyperechogenicity of substantia nigra [16]. Second, both parkinsonian features and dopaminergic dysfunction have been more frequently described in male and UMN-predominant ALS patients [30, 35]. Third, extrapyramidal features are much more frequent in PLS than in ALS patients and can respond to levodopa [38]. All these suggest a relationship between male sex, pyramidal and extrapyramidal involvement. The distinction between extrapyramidal and pyramidal symptoms and signs is complex and it has been hypothesized that parkinsonian traits in UMN-predominant ALS patients could actually be attributed to spasticity [30]. However, our data showing an association between UMN signs and the absence of nigrosome 1, rather suggest a direct relationship between the degree of pyramidal and extrapyramidal involvement in ALS patients. This fits well with the proposed model of corticofugal axonal spread of the disease [39] and the existence of a direct cortico-nigral pathway [40]. Thus, the absence of nigrosome 1 in ALS patients could be a marker of the cerebral neuropathological expansion of the disease to the second neuropathological stage, as proposed by Brettschneider et al. [2]. In this respect, the lack of association between the nigrosome absence and disability or disease duration is not surprising, since the neuropathological extension in ALS is not related with none of these variables [2, 3, 33].
Finally, we found that the nigrosome 1 absence is an independent prognostic factor, when adjusted for other well-known risk factors. As commented above, the absence of nigrosome 1 could be a marker of neuropathological multisystem degeneration, which ultimately associates to poor prognosis [3], as found in ALS patients with concomitant FTD or other extramotor symptoms [31, 41].
Strengths and limitations
Our study represents the first study assessing the status of nigrosome 1 and SNh in a large cohort of thoroughly characterized ALS patients, which allowed the use of multivariable analysis.
The main limitation is that parkinsonian features were not assessed systematically and consequently have not been analyzed. However, interpreting parkinsonian features in ALS patients may be challenging, since they can be masked by UMN and LMN signs [30]. Moreover, previous studies have not found relationship between extrapyramidal traits and SN dysfunction [30, 35, 36]. Another limitation is that the assessment of the nigrosome 1 is observer-dependent. To minimize this limitation, the nigrosome 1 was assessed by two experienced readers and discrepancies between the two were resolved by consensus. The use of 123 I-FP-CIT scintigraphy could have help to validate our results of nigrosome 1 absence, but was not available in this study. Although we studied a large cohort of ALS patients, some characteristics (FTD, C9ORF72 expansion) are infrequent, which could have limited the power to detect an association between nigrosome 1 and those variables. Finally, healthy controls or ALS mimics were not included to assess its role as a diagnostic biomarker. However, given its frequency in ALS patients, it does not appear to be useful in the diagnostic work up of ALS patients.
Conclusion
The absence of nigrosome 1 is found in up to a third of ALS patients and is not related with the SNh area. Moreover, it is independently associated with male sex, UMN impairment and shorter survival. This suggests that constitutional factors and the degree of pyramidal impairment influence the neuropathological involvement of the SNpc in ALS patients. Thus, nigrosome 1 could be a marker of a multisystem degeneration, which in turn associates to poor prognosis. Neuropathological studies are warranted to confirm our findings.
Data availability
The data that support the findings of this study are available from the corresponding author, (JFVC), upon reasonable request. There are no additional unpublished data from the study.
Code availability
Not applicable.
Change history
12 November 2021
Funding note is missing and funding note should be Open Access funding provided thanks to the CRUE-CSIC agreement with Springer Nature.
References
Al-Chalabi A, Hardiman O, Kiernan MC et al (2016) Amyotrophic lateral sclerosis: moving towards a new classification system. Lancet Neurol 15:1182–1194. https://doi.org/10.1016/S1474-4422(16)30199-5
Brettschneider J, Del Tredici K, Toledo JB et al (2013) Stages of pTDP-43 pathology in amyotrophic lateral sclerosis. Ann Neurol 74:20–38. https://doi.org/10.1002/ana.23937
Nishihira Y, Tan CF, Onodera O et al (2008) Sporadic amyotrophic lateral sclerosis: Two pathological patterns shown by analysis of distribution of TDP-43-immunoreactive neuronal and glial cytoplasmic inclusions. Acta Neuropathol 116:169–182. https://doi.org/10.1007/s00401-008-0385-z
Geser F, Brandmeir NJ, Kwong LK et al (2008) Evidence of multisystem disorder in whole-brain map of pathological TDP-43 in amyotrophic lateral sclerosis. Arch Neurol 65:636–641. https://doi.org/10.1001/archneur.65.5.636
Pilotto A, Yilmaz R, Berg D (2015) Developments in the role of transcranial sonography for the differential diagnosis of Parkinsonism. Curr Neurol Neurosci Rep. https://doi.org/10.1007/s11910-015-0566-9
Berg D, Godau J, Walter U (2008) Transcranial sonography in movement disorders. Lancet Neurol 7:1044–1055
Berg D, Siefker C, Becker G (2001) Echogenicity of the substantia nigra in Parkinson’s disease and its relation to clinical findings. J Neurol 248:684–689. https://doi.org/10.1007/s004150170114
Zhou HY, Sun Q, Tan YY et al (2016) Substantia nigra echogenicity correlated with clinical features of Parkinson’s disease. Parkinsonism Relat Disord 24:28–33. https://doi.org/10.1016/j.parkreldis.2016.01.021
Walter U, Dressler D, Probst T et al (2007) Transcranial brain sonography findings in discriminating between Parkinsonism and idiopathic Parkinson disease. Arch Neurol 64:1635–1640. https://doi.org/10.1001/archneur.64.11.1635
Berg D (2011) Substantia nigra hyperechogenicity is a risk marker of Parkinson’s disease: yes. J Neural Transm 118:613–619
Hermann A, Reuner U, Schaefer J et al (2015) The diagnostic value of midbrain hyperechogenicity in ALS is limited for discriminating key ALS differential diagnoses. BMC Neurol 15:33. https://doi.org/10.1186/s12883-015-0280-x
Fathinia P, Hermann A, Reuner U et al (2013) Parkinson’s disease-like midbrain hyperechogenicity is frequent in amyotrophic lateral sclerosis. J Neurol 260:454–457. https://doi.org/10.1007/s00415-012-6654-8
Prell T, Schenk A, Witte OW et al (2014) Transcranial brainstem sonography as a diagnostic tool for amyotrophic lateral sclerosis. Amyotroph Lateral Scler Front Degener 15:244–249. https://doi.org/10.3109/21678421.2014.881499
Pavlovic AM, Stevic Z, Pekmezovic T et al (2015) Increased frequency of pathologic findings on transcranial b-mode parenchymal sonography in patients with sporadic amyotrophic lateral sclerosis. Ultrasound Med Biol 41:982–988. https://doi.org/10.1016/j.ultrasmedbio.2014.12.005
Schweitzer KJ, Behnke S, Liepelt I et al (2007) Cross-sectional study discloses a positive family history for Parkinson’s disease and male gender as epidemiological risk factors for substantia nigra hyperechogenicity. J Neural Transm 114:1167–1171. https://doi.org/10.1007/s00702-007-0725-5
Vázquez-Costa JF, Tembl JI, Fornés-Ferrer V et al (2017) Genetic and constitutional factors are major contributors to substantia nigra hyperechogenicity. Sci Rep 7:7119. https://doi.org/10.1038/s41598-017-07835-z
Massey LA, Yousry TA (2010) Anatomy of the substantia Nigra and Subthalamic nucleus on MR imaging. Neuroimaging Clin N Am 20:7–27. https://doi.org/10.1016/j.nic.2009.10.001
Damier P, Hirsch EC, Agid Y, Graybiel AM (1999) The substantia nigra of the human brain: I. Nigrosomes and the nigral matrix, a compartmental organization based on calbindin D(28K) immunohistochemistry. Brain 122:1421–1436. https://doi.org/10.1093/brain/122.8.1421
Damier P, Hirsch EC, Agid Y, Graybiel AM (1999) The substantia nigra of the human brain: II. Patterns of loss of dopamine-containing neurons in Parkinson’s disease. Brain 122:1437–1448. https://doi.org/10.1093/brain/122.8.1437
Blazejewska AI, Schwarz ST, Pitiot A et al (2013) Visualization of nigrosome 1 and its loss in PD. Neurology 81:534–540. https://doi.org/10.1212/WNL.0b013e31829e6fd2
Schwarz ST, Afzal M, Morgan PS et al (2014) The “swallow tail” appearance of the healthy nigrosome—a new accurate test of Parkinson’s disease: a case-control and retrospective cross-sectional MRI study at 3T. PLoS ONE. https://doi.org/10.1371/journal.pone.0093814
Chau MT, Todd G, Wilcox R et al (2020) Diagnostic accuracy of the appearance of Nigrosome-1 on magnetic resonance imaging in Parkinson’s disease: a systematic review and meta-analysis. Park Relat Disord 78:12–20
Noh Y, Sung YH, Lee J, Kim EY (2015) Nigrosome 1 detection at 3T MRI for the diagnosis of early-stage idiopathic Parkinson disease: assessment of diagnostic accuracy and agreement on imaging asymmetry and clinical laterality. Am J Neuroradiol 36:2010–2016. https://doi.org/10.3174/ajnr.A4412
Brooks BR, Miller RG, Swash M, Munsat TL (2000) El escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler 1:293–299. https://doi.org/10.1080/146608200300079536
Visser J, de Jong JMBV, de Visser M (2008) The history of progressive muscular atrophy: syndrome or disease? Neurology 70:723–727. https://doi.org/10.1212/01.wnl.0000302187.20239.93
Gordon PH, Cheng B, Katz IB et al (2006) The natural history of primary lateral sclerosis. Neurology 66:647–653. https://doi.org/10.1212/01.wnl.0000200962.94777.71
Dejesus-Hernandez M, Mackenzie IR, Boeve BF et al (2011) Chromosome 9p-linked FTD and ALS. Neuron 72:245–256
Cedarbaum JM, Stambler N, Malta E et al (1999) The ALSFRS-R: a revised ALS functional rating scale that incorporates assessments of respiratory function. J Neurol Sci 169:13–21. https://doi.org/10.1016/S0022-510X(99)00210-5
Ellis CM, Simmons A, Jones DK et al (1999) Diffusion tensor MRI assesses corticospinal tract damage in ALS. Neurology 53:1051–1058. https://doi.org/10.1212/wnl.53.5.1051
D’Ascenzo C, Cecchin D, Santelli L et al (2012) Parkinson-like features in ALS with predominant upper motor neuron involvement. Amyotroph Lateral Scler 13:137–143. https://doi.org/10.3109/17482968.2011.603732
Strong MJ, Abrahams S, Goldstein LH et al (2017) Amyotrophic lateral sclerosis—frontotemporal spectrum disorder (ALS-FTSD): Revised diagnostic criteria. Amyotroph Lateral Scler Front Degener 18:153–174. https://doi.org/10.1080/21678421.2016.1267768
Vivo-Orti MN, Tembl JIJJI, Sastre-Bataller I et al (2013) Evaluation of the substantia nigra by means of transcranial ultrasound imaging: evaluación de la sustancia negra mediante ultrasonografía transcraneal. Rev Neurol 56:268–274
Ito J, Shimizu H, Ohta K et al (2019) amyotrophic lateral sclerosis with pallidonigroluysian degeneration: a clinicopathological study. Ann Neurol. https://doi.org/10.1002/ana.25652
Wolf Gilbert RM, Fahn S, Mitsumoto H, Rowland LP (2010) Parkinsonism and motor neuron diseases: twenty-seven patients with diverse overlap syndromes. Mov Disord 25:1868–1875. https://doi.org/10.1002/mds.23200
Calvo A, Chiò A, Pagani M et al (2019) Parkinsonian traits in amyotrophic lateral sclerosis (ALS): a prospective population-based study. J Neurol 266:1633–1642. https://doi.org/10.1007/s00415-019-09305-0
Borasio GD, Linke R, Schwarz J et al (1998) Dopaminergic deficit in amyotrophic lateral sclerosis assessed with [I-123] IPT single photon emission computed tomography. J Neurol Neurosurg Psychiatry 65:263–265. https://doi.org/10.1136/jnnp.65.2.263
Pradat PF, Bruneteau G, Munerati E et al (2009) Extrapyramidal stiffness in patients with amyotrophic lateral sclerosis. Mov Disord 24:2143–2148. https://doi.org/10.1002/mds.22762
Wais V, Rosenbohm A, Petri S et al (2016) The concept and diagnostic criteria of primary lateral sclerosis. Acta Neurol Scand. https://doi.org/10.1111/ane.12713
Braak H, Brettschneider J, Ludolph AC et al (2013) Amyotrophic lateral sclerosis—a model of corticofugal axonal spread. Nat Rev Neurol 9:708–714. https://doi.org/10.1038/nrneurol.2013.221
Cacciola A, Milardi D, Anastasi GP et al (2016) A direct cortico-nigral pathway as revealed by constrained spherical deconvolution tractography in humans. Front Hum Neurosci 10:374. https://doi.org/10.3389/fnhum.2016.00374
Vázquez-Costa JF, Arlandis S, Hervas D et al (2017) Clinical profile of motor neuron disease patients with lower urinary tract symptoms and neurogenic bladder. J Neurol Sci 378:130–136. https://doi.org/10.1016/j.jns.2017.04.053
Acknowledgements
We would like to thank patients for their participation in this study. Samples were processed, preserved, and delivered by the Biobank La Fe (PT13/0010/0026).
Funding
This research has received funding from Instituto de Salud Carlos III (JR19/00030 PI JFVC, 19/01178 PI TS), and from Generalitat Valenciana (PROMETEO/ 2018/135, PI TS). The Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER) is initiative from the ISCIII. TS and JFVC are members of the European Reference Network for Rare Neuromuscular Diseases (ERN EURO-NMD). Open Access funding provided thanks to the CRUE-CSIC agreement with Springer Nature.
Author information
Authors and Affiliations
Contributions
JFVC designed and supervised the study, participated in clinical data acquisition and interpretation, and wrote and edited the manuscript. MIMG participated in clinical and imaging data acquisition and interpretation and wrote and edited the manuscript. JIT and MMM participated in imaging data acquisition and critically revised the manuscript. LMB and TS supervised the study and critically revised the manuscript. JFVC had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. All authors have approved the submitted version of the paper.
Corresponding author
Ethics declarations
Conflicts of interest
Dr. Vázquez-Costa served on advisory boards for Biogen and Roche and received travel and speaker honoraria from Biogen and Roche.
Ethical approval
The study was approved by the Ethics Committee for Biomedical Research of La Fe Hospital (Valencia, Spain).
Consent to participate
All participants gave their informed consent prior to their inclusion in the study.
Consent for publication
Not applicable.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Moreno-Gambín, M.I., Tembl, J.I., Mazón, M. et al. Role of the nigrosome 1 absence as a biomarker in amyotrophic lateral sclerosis. J Neurol 269, 1631–1640 (2022). https://doi.org/10.1007/s00415-021-10729-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-021-10729-w