
Abstract
Background
The optimal treatment strategy with disease-modifying therapies (DMTs) in relapsing–remitting multiple sclerosis (RRMS) remains uncertain.
Objective
To compare outcomes of initial treatment with infusion therapies and starting therapy with medium efficacy therapy in a propensity-matched cohort of Finnish RRMS patients.
Methods
A total of 154 RRMS patients initiating natalizumab, alemtuzumab, ocrelizumab or rituximab as first DMT (high efficacy DMT, heDMT group) and 1771 patients initially treated with injectable therapies, teriflunomide or dimethylfumarate and escalated based on disease activity (moderate efficacy DMT, meDMT group) were identified from the Finnish MS registry. Nearest neighbor propensity matching (1:1, caliper 0.1) was performed for age, sex, baseline Expanded Disability Status Scale (EDSS), annual relapse rate (ARR) one year prior DMT and time since MS symptom onset. Primary outcome was time to 6-month confirmed EDSS progression and the secondary outcome time to first relapse.
Results
In the propensity-matched group comparisons, the probability of 6-month confirmed disability progression (CDP) at 5 years after DMT start was 28.4% (95% CI 15.7–39.3) in the heDMT group (n = 66) and 47.0% (95% CI 33.1–58.1) in meDMT group (n = 66), p = 0.013. Probability of relapse at 5 years was 34.6% (95% CI 24.1–43.6) for heDMT (n = 105) and 47.2% (95% CI 36.6–56.1) for meDMT (n = 105), p = 0.019.
Conclusions
Initiating MS-therapy with heDMT significantly reduced the risk of 5-year disability progression and relapse compared to using meDMT as first DMT choice in propensity-matched groups of Finnish MS-patients.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The development of disease-modifying therapies (DMTs) has enabled significant advances in the treatment of multiple sclerosis (MS) during the past decades. DMTs have shown to delay the transition from clinically isolated syndrome to confirmed MS and from relapsing–remitting MS (RRMS) to secondary progressive MS (SPMS) and to reduce the rate and severity of relapses, new lesion formation and brain volume loss [1,2,3,4,5,6]. However, the optimal treatment strategy with DMTs in RRMS remains elusive [2, 7,8,9]. Current treatment approaches may miss a window of opportunity for achieving the highest effectiveness of DMTs [9,10,11,12].
In a real-world clinical setting, the most common treatment algorithm of newly diagnosed RRMS is starting therapy with a low-risk, moderate efficacy DMT and escalating treatment in the presence of continued disease activity. Initial treatment with high-efficacy DMTs is an approach often reserved for a minority of patients with high disease activity. This is mainly due to the potential risks, often more complex monitoring requirements or higher cost of high-efficacy DMTs, but also lack of established guidelines and comparative studies on optimal treatment strategies [13,14,15,16,17,18,19,20].
Previously, a propensity-matched nationwide register study from Denmark and an observational clinical cohort study from UK have shown better clinical outcomes in patients initiating MS therapy with high-efficacy DMTs compared to the escalation approach [10, 11]. In this study, we compared the risk of disability progression and relapse in treatment naïve Finnish MS patients initiating MS therapy with high-efficacy infusion therapies: natalizumab, alemtuzumab, ocrelizumab or rituximab, to a propensity-matched cohort of patients initiating therapy with medium efficacy DMTs.
Methods
Study design
We performed a population-based, propensity-matched register study of Finnish RRMS patients from four Finnish hospital districts: Helsinki and Uusimaa (HUS), Southwest Finland (SwF), Kanta-Häme and Pirkanmaa, jointly covering a population of 2.8 million inhabitants. Data collection was conducted from 1st Jan 2006 to 31st Dec 2020. The demographic and clinical data were collected using the Finnish MS register (www.neurorekisteri.fi), that holds a full disease history data of approximately 7000 and diagnosis data of 11 094 (at 9th Mar 2020) Finnish MS patients. The register coverage is over 90% of the estimated Finnish MS population [21, 22].
Patients
Patients were eligible for inclusion if they had confirmed RRMS diagnosis recorded after 1st Jan 2006, had started first-line treatment within 3 years after MS diagnosis and after 1st Jan 2006, and had at least 2.5 years of follow-up. The exclusion criteria were: follow-up less than 2.5 years, time from RRMS diagnosis to first DMT more than 3 years or disease course at first DMT either SPMS, primary progressive MS (PPMS) or undefined. Patients receiving azathioprine, mitoxantrone, fingolimod or cladribine as first-line treatment were excluded because azatiophrine and mitoxantrone are not used as first-line DMTs for MS in Finland [23], fingolimod is reimbursed in Finland in treatment naïve patients in highly active disease but was not categorized as high efficacy therapy in this study on the basis of comparative data [24], and cladribine use did not begin in Finland until 2018. However, we also performed additional analyses with fingolimod included in the heDMT group. A flowchart of the study patients is presented in Fig. 1.

Flowchart of Participants. Of the eligible 1925 patients entering the study, 154 (8.0%) had been prescribed a high-efficacy DMT as first-line treatment (heDMT), while 1771 (92.0%) initiated treatment with a moderate-efficacy DMT (meDMT)
Study treatments
Eligible treatment naïve patients were categorized as initiating high-efficacy infusion therapy (heDMT) or moderate efficacy therapies (meDMT). Alemtuzumab, natalizumab, ocrelizumab and rituximab were categorized as high-efficacy and dimethyl fumarate, glatiramer acetate, interferon beta and teriflunomide as moderate efficacy DMTs [18]. All DMTs were administered according to published protocols. Rituximab was administered as one dose of 1.0 g intravenously followed by 0.5 g every 6 months.
Statistical analysis
Primary outcome was time to 6-month confirmed disability progression (CDP) and secondary outcome time to first relapse in the propensity-matched cohorts. As an exploratory outcome, we assessed safety. As an additional outcome, we studied odds for disability progression at 3 and 5 years in the unmatched and matched cohorts using conditional logistic regression.
Disability was assessed with EDSS [25]. Disability progression was defined by 3-strata progression in EDSS: ≥ 1.5 point increase from baseline EDSS of 0; ≥ 1 point increase for baseline 1 to 5.5 and ≥ 0.5 increase for baseline ≥ 6 [26] The 6-month CDP was defined by the same EDSS criteria and a 6-month confirmation period for verification of progression [9]. As baseline EDSS, the closest date to DMT onset (12 months prior to or 6 months after) was used, prioritizing EDSS assessments within 6 months prior. EDSS assessments within 1 month after relapse were excluded at baseline and at follow-up unless verified by the 6-month confirmation period.
Group comparisons for continuous variables were performed using the Wilcoxon rank-sum test or Student’s t test depending on the normality of the groups, and for categorical variables using Fisher’s exact test. For controlling and checking the False Discovery Rate, Benjamini–Hochberg procedure was used as a correction for multiple comparisons. All significant raw p values remained under 0.05 after adjustment.
Separate complete case matching was performed for all outcome analyses. Propensity scores were matched using Nearest Neighbor matching with 1:1 ratio and a caliper of 0.1 SDs controlling adequate pair similarities. Matching variables for outcome analyses were age, sex, baseline EDSS, Annual Relapse Rate (ARR) 1 year prior DMT onset and time since MS onset. In addition, time difference between baseline EDSS and DMT onset was added to matching for the logistic regression analyses.
Hazard ratios and corresponding confidence intervals were analyzed using semiparametric Cox proportional hazard regression model. Significance testing for rate differences between heDMT and meDMT was based on Wald test. Probabilities of 6-month CDP and first relapse at specific timepoints were analyzed using cumulated events analysis based on 1-Kaplan–Meier estimates and curves. Log-rank test was utilized to assess differences between overall event probabilities.
In the time to 6-month CDP analysis time origin for patients with baseline EDSS before DMT onset was fixed to DMT onset date. Group balances were checked with standardized differences before and after matching. In the 6-month CDP analysis, standardized mean difference for sex falling over 0.2 was considered acceptable based on non-significant omnibus test using Chi-squared test. Diagnostics for the Cox regression model included testing the proportional-hazards (PH) assumptions and visual residual checks. In addition, univariate and multivariate analyses for unmatched data were performed to detect the matching effect and raw data bias.
Results
Patient characteristics and disability and relapse outcomes in the unmatched cohort
The clinical and demographic characteristics of all the study patients are presented in Table 1. The mean ARR prior treatment was higher in the heDMT group vs meDMT (1.6 vs 1.1, p < 0.001), as well as baseline EDSS (median 2.0 vs. 1.0, p < 0.001). A conditional regression analysis with raw data indicated that the odds for disability progression at 3 years in the heDMT patients (n = 100) was significantly lower than in the meDMT patients (n = 308, OR 0.51 95% CI 0.31–0.76, p = 0.002) in a univariate model. In a multivariate model, the OR was 0.53 (95% CI 0.32–0.84, p = 0.010). At 5 years, the odds for disability progression in the heDMT patients (n = 72) compared to 233 meDMT patients was 0.46 (95% CI 0.29–0.79, p < 0.001) in a univariate model, and 0.58 (95% CI 0.34–0.95, p = 0.034) in a multivariate model.
In the heDMT group, ARR reduction from baseline was 69% at 3 years and 75% at 5 years, and in the meDMT group 45% at 3 years and 64% at 5 years, respectively. The patients in the heDMT group had more Gd + lesions (p < 0.001) and T2 lesions (p < 0.001) on brain MRI at study onset, however, MRI data was not available in all patients (see Tables 1, 2, 3). Therefore, propensity matching based on MRI characteristics was not feasible.
Patient characteristics and disability and relapse outcomes in the propensity-matched cohorts
Time to 6-month confirmed disability progression
A total of 66 heDMT patients had frequent EDSS data enabling time to 6-month CDP analysis (a mean of 5.5 [SD 1.97] EDSS evaluations during the follow-up). They were propensity-matched to 66 meDMT patients with a mean of 5.1 (SD 2.22) EDSS evaluations. (Table 2). The median (Q1, Q3) follow-up time to an event or censoring was 4.7 (3.1, 5.8) years in the heDMT group and 4.0 (2.3, 5.7) years in the meDMT group. Patients were censored at death, data cut-off or at 6-year follow-up mark.
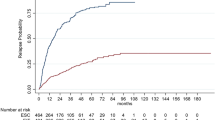
The probability of 6-month CDP at 3 years after initiating DMT was 15.2% (95% CI 6.1–23.4) in the heDMT group and 35.0% (95% CI 22.4–45.6) in the meDMT group and at 5 years, 28.4% (95% CI 15.7–39.3) vs 47.0% (95% CI 33.1–58.1), respectively. The absolute risk reduction for CDP was 19.8% at 3 years and 18.6% at 5 years. The event probabilities between the groups differed significantly (p = 0.013). The heDMT group had a 40% lower rate of 6-month CDP compared to meDMT (HR 0.60, 95% CI 0.39–0.91, p = 0.015). The results are depicted as a Cumulated events (1-KM) curve displayed in Fig. 2. When fingolimod was included in the heDMT-group, the rate of 6-month CDP compared to meDMT was no more significant (n = 73; HR 0.74 95% CI 0.49–1.11, p = 0.143.)

Probability of 6-month CDP in the propensity-matched heDMT vs meDMT groups
Time to first relapse
A total of 105 heDMT patients were propensity matched with 105 meDMT patients (Table 3). The probability of the first relapse at 3 years was 27.6% (95% CI 18.5–35.7) in the heDMT group and 43.9% (95% CI 33.5–52.6) in the meDMT group. Probability of the first relapse at 5 years was 34.6% (95% CI 24.1–43.6) in the heDMT group and 47.2% (95% CI 36.6–56.1) in the meDMT group. The absolute risk reduction for relapse was 16.3% at 3 years and 12.6% at 5 years. The mean (SD) number of relapses during the follow-up was 0.7 (1.54) in the heDMT group and 1.4 (2.5) in the meDMT group. The event probabilities between the groups differed significantly (p = 0.019, Fig. 3). The heDMT group had a 30% lower rate of the first relapse compared to meDMT (HR 0.70, 95% CI 0.52–0.94, p = 0.020).

Probability of the first relapse in the propensity-matched heDMT vs meDMT groups
The probability of the first relapse at 3 years was 27.6% (95% CI 18.5–35.7) in the heDMT group and 43.9% (95% CI 33.5–52.6) in the meDMT group. Probability of the first relapse at 5 years was 34.6% (95% CI 24.1–43.6) in the heDMT group and 47.2% (95% CI 36.6–56.1) in the meDMT group. The absolute risk reduction for relapse was 16.3% at 3 years and 12.6% at 5 years. The mean (SD) number of relapses during the follow-up was 0.7 (1.54) in the heDMT group and 1.4 (2.5) in the meDMT group. The event probabilities between the groups differed significantly (p = 0.019, Fig. 3). The heDMT group had a 30% lower rate of the first relapse compared to meDMT (HR 0.70, 95% CI 0.52–0.94, p = 0.020). The results were similar with fingolimod included in the heDMT group (n = 115; HR 0.64 95% CI 0.52–0.94, p = 0.020, p = 0.003).
Disability progression at 3 and 5 years
A conditional regression analysis was performed to assess the difference in disability progression in the propensity-matched cohorts of heDMT and meDMT at 3 and 5 years after the first DMT. The results were in line with the raw data results; at 3 years, the OR (95% CI) for disability progression in a univariate model comparing heDMT (n = 76) and meDMT (n = 76) was 0.43 (95% CI 0.24–0.75, p = 0.004), and at 5 years, the OR was 0.54 (95% CI 0.30–0.94, p = 0.032), (n = 57 for both groups). When fingolimod was included in the heDMT group, the results remained significant for the 3-year disability progression (n = 76; OR 0.47, 95% CI 0.26–0.80, p = 0.006), but not for 5-year disability progression (n = 56; OR 0.60, 95% CI 0.34–1.05, p = 0.076).
Treatment escalations
A total of 219 out of 1771 (12.4%) patients in the whole meDMT group escalated into natalizumab, alemtuzumab, rituximab or ocrelizumab at a median of 2.4 years after first DMT initiation. Of these 219 patients, 95 escalated during the first 2 years of follow-up. In most of the patients going through treatment escalation (80.8%), the reason for escalation was recorded to be lack of efficacy. A relapse within one year before the switch was observed in 74.2% of the patients switching because of a lack of efficacy. In the matched subcohort of 105 meDMT patients in the time to first relapse analysis, a total of 26 patients (24.8%) escalated at a median of 2.5 years and in the matched subcohort of 66 patients in the meDMT group in the time to 6-month CDP analysis, a total of 24 patients (36.3%) escalated at a median of 2.5 years. A total of 20 patients had a relapse within a year before the escalation in both of the propensity-matched meDMT groups (76.9% and 83.3%, respectively).
Safety
The numbers of adverse events between the groups did not significantly differ. In the heDMT group, adverse events were recorded for 13 patients (8.4%) and in the meDMT group for 252 (14.2%) patients. The adverse events recorded are shown in Supplemental Table 1. There was one case of Progressive Multifocal Leucoencephalopathy (PML) in the meDMT group after escalation from interferon to natalizumab; the patient was not included in the propensity-matched group comparisons. There were 7 deaths in the meDMT group among 1771 patients during the 6-year follow-up and no deaths among the 154 patients in the heDMT group. The mean age at death was 48.4 years (range 29 to 58 years) and the causes of death were subarachnoid hemorrhage, stroke, breast cancer, lung cancer, amyotrophic lateral sclerosis (patient not included in the matched comparisons), advanced MS and aspiration pneumonia and one unknown.
Discussion
In this propensity-matched retrospective register study, we compared disability and relapse outcomes in Finnish MS patients receiving high-efficacy infusion therapies as first treatment versus those initially treated with moderate efficacy therapies. We found that early high-efficacy infusion therapy resulted in a lower probability of 5-year confirmed disability progression and first relapse than starting first treatment with moderate efficacy therapies.
These findings are in line with previous studies [10, 11]. In a propensity-matched study with 388 patients from the Danish MS register, Buron et al. found a lower probability for 6-month confirmed EDSS worsening and first relapse in patients starting a high-efficacy DMT as first therapy compared to a matched sample starting moderate-efficacy DMT [11]. In a population-based observational study of 592 patients, Harding et al. discovered that the 5-year change in EDSS was lower and time to sustained accumulation of disability was longer in the group receiving early intensive vs escalation treatment [10]. No propensity matching was performed in this study.
To capture both progressions independent of relapses (PIRA) and relapse-associated worsening [27], we included relapse-associated follow-up EDSS-values that were confirmed similarly as in the Danish register study [11]. We used a confirmatory period of a minimum of 6 months to catch consistent disability [28] and excluded EDSS scores determined within 4 weeks after relapse onset at baseline and for confirmation [27].
In a large international observational study, He et al. showed that high-efficacy therapy commenced within 2 years of disease onset in comparison to 4–6 years after disease onset is associated with less disability after 6–10 years [29]. The high-efficacy therapies used in our study were the same as in the study by He et al. and the mean time since MS onset to treatment was 1.7 years in the heDMT group. In the propensity-matched meDMT groups in our study, a total of 25–36% of the patients escalated into infusion therapies at a median of 2.5 years after DMT start. Therefore, the start of the heDMT in the patients initiating with meDMT and escalating later was delayed beyond 4 years in the majority of the patients escalating therapy.
Inclusion of fingolimod into the heDMT group in our study changed the results such that 5-year disability progression and time to 6-month CDP no longer significantly differed between the heDMT and meDMT groups. However, the risk of 3-year disability progression and time to first relapse remained significantly lower in the heDMT group also when fingolimod was included.
The criteria of the treatment escalation in Finland are guided by the national MS Current Care Guidelines and combine clinical relapses and MRI active lesions while on treatment [23]. The guidelines include rebaseline MRI within 6 months of DMT start and annual MRI monitoring thereafter to monitor MRI activity [23]. Majority of the patients in the meDMT group in our study used beta interferons or glatiramer acetate as the first DMT. Prosperini et al. recently showed that marginal MRI activity in the absence of both relapses and Gd-enhancing lesions after the first year of treatment was associated with a minor risk of future disability after starting injectables [30]. Majority of the patients escalating therapy in our study had experienced a relapse within the year preceding treatment escalation, but in approximately one in five patients with initial meDMT was escalated based on lack of efficacy but without relapse activity, likely based on MRI activity. Median time for escalation was 2.5 years. Almost half of the propensity-matched meDMT patients escalated into heDMTs within 2 years of DMT start. Subgroup analyses of patients escalating before or after 2 years did not significantly change our results (data not shown). The numbers of patients escalating within the first year after DMT start and without relapse activity was too small for statistical analyses. It is thence possible, that early escalation within the first year after disease onset, using minimal or no evidence of disease activity as a treatment goal, could have led to similarly good outcomes in the meDMT group than in the heDMT group.
We only included patients diagnosed and treated with first DMTs after the year 2006, since the first high-efficacy therapy natalizumab became available for clinical use in Finland then. Majority of the patients in the heDMT group of our study used natalizumab, similarly as in the Danish register study [11] that included patients from 2001 to 2018. Therefore our study results cannot be generalized and extended to all current high efficacy treatments. Our study period was from the beginning of the year 2006 to the end of the year 2020. There was a discrepancy in the DMTs used in the Danish register study and our study; we did not include fingolimod in the heDMT group but included rituximab similarly as in the study by He et al. [29]. Further, in our study a higher proportion of patients treated with alemtuzumab were included in the heDMT group and a smaller proportion were treated with teriflunomide and a larger proportion with dimethylfumarate in the meDMT group. The majority of meDMT patients included in both studies had received interferons as the first DMT. In our study, the mean time from the disease onset in the matched groups was 2 years, in comparison to 4 years in the Danish study. The number of patients after propensity matching was smaller in our study, but we took two separate statistical approaches to assess the difference in the risk of disability progression between the propensity-matched groups: logistic regression analysis to determine odds for disability progression at 3- and 5-year milestones, and Cox proportional hazard regression to study the time to CDP. However, the primary endpoint of time to 6-month CDP and the secondary endpoint of time to first relapse were the same. Both studies yielded similar primary and secondary endpoint results, supporting the generalizability of the results across MS populations in different countries.
In a prospective study with a follow-up up to 10 years, it was found that rates of worsening and evolution to SPMS during the treatment era have become substantially lower as compared with earlier natural history studies [8]. Therefore, longer follow-up than in our or previous studies comparing treatment strategies is necessary to establish whether early high efficacy treatment results in a lower risk of SPMS evolution.
The limitations of this study include retrospective study design, small sample size, possible imbalance of MRI parameters and selection bias attributable to exclusion from the analysis of a greater proportion of patients in the meDMT group than in the heDMT group. MRI data is not a mandatory element in the Finnish MS register. Among the patients with MRI data, there were more patients with Gd + -lesions and a higher number of T2 lesions in the heDMT group at baseline. However, half of the meDMT patients in the propensity-matched groups had missing baseline MRI results. Inclusion of MRI into the matching would have increased selection bias and resulted in too small subgroups for statistical comparisons. Therefore, matching for MRI parameters was not feasible. Since Gd + -lesions and a higher number of T2 lesions are established prognostic brain MRI biomarkers associated with worse prognosis [31], the possible imbalance in the MRI parameters would rather have favored the meDMT group both in the relapse and disability outcomes.
The secondary exploratory outcome of safety did not indicate a higher risk of heDMT vs meDMT approach in our patient cohort, but adverse events reporting it is not a mandatory register element and reporting was likely incomplete. Further, assessing long-term risks of potent immunosuppression, such as malignancies, needs decades of follow-up and studies comparing the risks against the background population. However, the paradigm that patients need to fail a first-line therapy before being offered more potent therapies risks irreversible neural tissue injury during the process of changing treatments to find an appropriate medication. A major unmet need in MS is to find biomarkers to aid the selection of optimal personalized therapy from the start.
In conclusion, we showed in a propensity-matched cohort of Finnish MS patients that early high efficacy infusion therapy in RRMS patients reduces the probability of disability progression and relapses compared to initiating treatment with moderate efficacy therapies. No randomized controlled trials have yet directly compared the effects of these different treatment strategies, but such studies are needed and currently recruiting patients [16, 32].
Data availability
Data are available upon request from the corresponding author.
Code availability
Not applicable.
References
Jokubaitis VG et al (2016) Predictors of long-term disability accrual in relapse-onset multiple sclerosis. Ann Neurol 80(1):89–100. https://doi.org/10.1002/ana.24682
Palace J et al (2015) Effectiveness and cost-effectiveness of interferon beta and glatiramer acetate in the UK Multiple Sclerosis Risk Sharing Scheme at 6 years: a clinical cohort study with natural history comparator. Lancet Neurol 14(5):497–505. https://doi.org/10.1016/S1474-4422(15)00018-6
Sotirchos ES et al (2020) Effect of disease-modifying therapies on subcortical gray matter atrophy in multiple sclerosis. Mult Scler J 26(3):312–321. https://doi.org/10.1177/1352458519826364
Armoiry X et al (2018) Short- and long-term clinical outcomes of use of beta-interferon or glatiramer acetate for people with clinically isolated syndrome: a systematic review of randomised controlled trials and network meta-analysis. J Neurol 265(5):999–1009. https://doi.org/10.1007/s00415-018-8752-8
Tedeholm H et al (2013) Time to secondary progression in patients with multiple sclerosis who were treated with first generation immunomodulating drugs. Mult Scler J 19(6):765–774. https://doi.org/10.1177/1352458512463764
Freedman MS (2014) Evidence for the efficacy of interferon beta-1b in delaying the onset of clinically definite multiple sclerosis in individuals with clinically isolated syndrome. Ther Adv Neurol Disord 7(6):279–288. https://doi.org/10.1177/1756285614549554
Shirani A et al (2012) Association between use of interferon beta and progression of disability in patients with relapsing-remitting multiple sclerosis. JAMA 308(3):247–256. https://doi.org/10.1001/jama.2012.7625
Cree BAC et al (2016) Long-term evolution of multiple sclerosis disability in the treatment era. Ann Neurol 80(4):499–510. https://doi.org/10.1002/ana.24747
Kalincik T et al (2017) Towards personalized therapy for multiple sclerosis: prediction of individual treatment response. Brain 140(9):2426–2443. https://doi.org/10.1093/brain/awx185
Harding K et al (2019) Clinical outcomes of escalation vs early intensive disease-modifying therapy in patients with multiple sclerosis. JAMA Neurol 76(5):536–541. https://doi.org/10.1001/jamaneurol.2018.4905
Buron MD et al (2020) Initial high-efficacy disease-modifying therapy in multiple sclerosis: a nationwide cohort study. Neurology 95(8):e1041–e1051. https://doi.org/10.1212/WNL.0000000000010135
Rush CA, Maclean HJ, Freedman MS (2015) Aggressive multiple sclerosis: proposed definition and treatment algorithm. Nat Rev Neurol 11(7):379–389. https://doi.org/10.1038/nrneurol.2015.85
Montalban X et al (2018) ECTRIMS/EAN Guideline on the pharmacological treatment of people with multiple sclerosis. Mult Scler 24(2):96–120. https://doi.org/10.1177/1352458517751049
Freedman MS (2008) Induction vs. escalation of therapy for relapsing multiple sclerosis: the evidence. Neurol Sci 29(SUPPL. 2):250–253. https://doi.org/10.1007/s10072-008-0953-y
Soelberg Sorensen P (2017) Safety concerns and risk management of multiple sclerosis therapies. Acta Neurol Scand 136(3):168–186. https://doi.org/10.1111/ane.12712
Ontaneda D, Tallantyre E, Kalincik T, Planchon SM, Evangelou N (2019) Early highly effective versus escalation treatment approaches in relapsing multiple sclerosis. Lancet Neurol 18(10):973–980. https://doi.org/10.1016/S1474-4422(19)30151-6
Faissner S, Gold R (2018) Efficacy and safety of the newer multiple sclerosis drugs approved since 2010. CNS Drugs 32(3):269–287. https://doi.org/10.1007/s40263-018-0488-6
Scolding N et al (2015) Association of British Neurologists: revised (2015) guidelines for prescribing disease-modifying treatments in multiple sclerosis. Pract Neurol 15(4):273–279. https://doi.org/10.1136/practneurol-2015-001139
Wattjes MP et al (2015) Evidence-based guidelines: MAGNIMS consensus guidelines on the use of MRI in multiple sclerosis—establishing disease prognosis and monitoring patients. Nat Rev Neurol 11(10):597–606. https://doi.org/10.1038/nrneurol.2015.157
Marziniak M et al (2016) Variations in multiple sclerosis practice within Europe—is it time for a new treatment guideline? Mult Scler Relat Disord 8:35–44. https://doi.org/10.1016/j.msard.2016.04.004
Laakso SM et al (2019) Multiple sclerosis in Finland 2018—Data from the national register. Acta Neurol Scand 140(5):303–311. https://doi.org/10.1111/ane.13145
Glaser A et al (2019) Multiple sclerosis registries in Europe—an updated mapping survey. Mult Scler Relat Disord 27:171–178. https://doi.org/10.1016/j.msard.2018.09.032
W. G. A. by the F. M. S. D. and the F. N. Society. Multiple Sclerosis Current Care Guidelines 2020. https://www.kaypahoito.fi/hoi36070. Accessed Sep. 01 2020
Kalincik T et al (2017) Treatment effectiveness of alemtuzumab compared with natalizumab, fingolimod, and interferon beta in relapsing-remitting multiple sclerosis: a cohort study. Lancet Neurol. https://doi.org/10.1016/S1474-4422(17)30007-8
Kurtzke JF (1983) Rating neurologic impairment in multiple sclerosis: an expanded disability status scale (EDSS). Neurology 33(11):1444–1444. https://doi.org/10.1212/WNL.33.11.1444
Alasdair DA, Coles J, Compston KS, Selmaj SLLW, Moran DHS, Norris Margolin K, Tandon PK (2008) Alemtuzumab vs Interferon Beta-1a in Early Multiple Sclerosis. N Engl J Med 359(17):1786–1801. https://doi.org/10.1056/NEJMoa0802670
Kappos L et al (2020) Contribution of relapse-independent progression vs relapse-associated worsening to overall confirmed disability accumulation in typical relapsing multiple sclerosis in a pooled analysis of 2 Randomized Clinical Trials. JAMA Neurol. https://doi.org/10.1001/jamaneurol.2020.1568
Kalincik T et al (2015) Defining reliable disability outcomes in multiple sclerosis. Brain 138(11):3287–3298. https://doi.org/10.1093/brain/awv258
He A, Merkel B, Brown JWL, Zhovits Ryerson L, Kister I, Malpas CB, Sharmin S, Horakova D, Kubala Havrdova E, Spelman T, Izquierdo G, Eichau S, Trojano M, Lugaresi A, Hupperts R, Sola P, Ferraro D, Lycke J, Grand’Maison F, Prat A, Girard M, Duquette P, Lar (2020) Timing of high-efficacy therapy for multiple sclerosis: a retrospective observational cohort study. Lancet Neurol 19(4):307–316. https://doi.org/10.1016/S1474-4422(20)30067-3
Prosperini L et al (2020) Minimal evidence of disease activity (MEDA) in relapsing-remitting multiple sclerosis. J Neurol Neurosurg Psychiatry. https://doi.org/10.1136/jnnp-2019-322348
Comabella M, Sastre-Garriga J, Montalban X (2016) Precision medicine in multiple sclerosis: biomarkers for diagnosis, prognosis, and treatment response. Curr Opin Neurol. https://doi.org/10.1097/WCO.0000000000000336
Ontaneda D et al (2020) Determining the effectiveness of early intensive versus escalation approaches for the treatment of relapsing-remitting multiple sclerosis: the DELIVER-MS study protocol. Contemp Clin Trials. https://doi.org/10.1016/j.cct.2020.106009
Funding
Open access funding provided by University of Turku (UTU) including Turku University Central Hospital. K Hänninen received personal research grants from the Finnish Multiple Sclerosis Foundation, Turku University Foundation and Turku University State Research Funding.
Author information
Authors and Affiliations
Contributions
MS-H conceptualized and designed the study and edited the manuscript. MV performed statistical analyses and contributed to designing the study and editing the manuscript. KH gathered data, drafted and edited the manuscript. SA, SML and HK contributed data, interpreted the results and participated in editing the manuscript.
Corresponding author
Ethics declarations
Conflicts of interest
S Atula has received honoraria for lectures, advisory boards or for serving as an investigator for clinical trials from Biogen, Novartis, Merck and Roche. H Kuusisto has received honoraria for lectures, advisory boards or for serving as an investigator for clinical trials from Biogen, Celgene, Genzyme, Novartis, Merck, Roche and Sanofi. M Soilu-Hänninen has received honoraria for lectures, advisory boards or for serving as an investigator for clinical trials from Biogen, Celgene, Genzyme, Novartis, Merck, Roche, Sanofi and Teva. SM Laakso has received honoraria for lectures from Merck and congress expenses covered by Roche and Merck. K Hänninen and M Viitala have no disclosures.
Ethical approval
According to Finnish law, ethics committee approval was not required since the study was based on administrative register data and did not involve any contact with patients. The study was approved by the Turku, Helsinki and Tampere University Hospital and Kanta-Häme Central Hospital Research Services. The data processing practices followed the EU Data Protection Directive rules (permission numbers T16/2017 for SwF, HUS/163/2019 for Helsinki, R19613S for Pirkanmaa and KHSHP/1571/13.00.01/2018 for Kanta-Häme).
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hänninen, K., Viitala, M., Atula, S. et al. Initial treatment strategy and clinical outcomes in Finnish MS patients: a propensity-matched study. J Neurol 269, 913–922 (2022). https://doi.org/10.1007/s00415-021-10673-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-021-10673-9