
Abstract
Objective
To describe adult-onset limb-girdle-type muscular dystrophy caused by biallelic variants in the PYROXD1 gene, which has been recently linked to early-onset congenital myofibrillar myopathy.
Methods
Whole exome sequencing was performed for adult-onset neuromuscular disease patients with no molecular diagnosis. Patients with PYROXD1 variants underwent clinical characterization, lower limb muscle MRI, muscle biopsy and spirometry. A yeast complementation assay was used to determine the biochemical consequences of the genetic variants.
Results
We identified four patients with biallelic PYROXD1 variants. Three patients, who had symptom onset in their 20s or 30s, were homozygous for the previously described p.Asn155Ser. The fourth patient, with symptom onset at age 49, was compound heterozygous for p.Asn155Ser variant and previously unknown p.Tyr354Cys. All patients presented with a LGMD-type phenotype of symmetric muscle weakness and wasting. Symptoms started in proximal muscles of the lower limbs, and progressed slowly to involve also upper limbs in a proximal-predominant fashion. All patients remained ambulant past the age of 60. They had restrictive lung disease but no cardiac impairment. Muscle MRI showed strong involvement of anterolateral thigh muscles. Muscle biopsy displayed chronic myopathic changes. Yeast complementation assay demonstrated the p.Tyr354Cys mutation to impair PYROXD1 oxidoreductase ability.
Conclusion
PYROXD1 variants can cause an adult-onset slowly progressive LGMD-type phenotype.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Inherited disorders of the skeletal muscles are classified based on clinical, histopathological and genetic features. Limb-girdle muscular dystrophy (LGMD) is a genetically heterogeneous group of muscular dystrophies with autosomal inheritance characterized by slowly progressive degeneration of proximal limb muscles. In many types of LGMD also other muscles, e.g., distal muscles, respiratory muscle and the heart may be affected. Inheritance can be either dominant (LGMD1) or recessive (LGMD2), and to date at least 34 LGMD disease genes are known [1,2,3]. Distribution of muscle involvement, age at onset and rate of progression show great variability, which are underscored by the extensive genetic diversity.
Biallelic PYROXD1 gene mutations were identified to underlie congenital myopathy in nine patients from five different families [4]. These patients had the onset of muscle weakness between birth and 8 years with slow disease progression, and muscle pathology consistent with myofibrillar myopathy. All patients were still ambulant at the time of study, the oldest patient being 31 years of age. PYROXD1 encodes a nuclear-cytoplasmic oxidoreductase, with a currently unknown exact function in cellular redox regulation [4]. Patients from four families had a mutation causing p.Asn155Ser amino acid change, which was shown to impair reductase activity in a complementation assay of yeast lacking glutathione reductase [4]. Another patient with homozygous p.Asn155Ser variant was recently reported, presenting with progressive muscle weakness starting at the age of 9 years and leading to loss of ambulation at the age of 37 years [5].
Here we describe additional four patients from three families with recessive PYROXD1 variants. In contrast to the previous report, our patients had the disease onset in adulthood and have now reached 60 years of age, allowing evaluation of the natural history of PYROXD1 associated disease.
Materials and methods
Patients
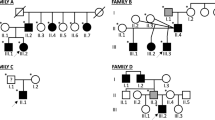
The patients in this study are from three families of Finnish origin with non-consanguineous parents (Fig.1). Patients; patient 1 (P1, Family 1), patient 2 (P2, Family 2), and siblings patient 3 and 4 (P3 and P4, Family 3), were studied in cohorts of undiagnosed neuromuscular disease patients. In addition to detailed clinical neurological examinations, all patients underwent electroneuromyography (ENMG) investigations, muscle biopsy, lower limb muscle MRI, spirometry/respiratory assessment and measurement of serum creatine kinase (CK) values (Table1). The MR images of patient P4 had been performed a long time ago at age 45 years and therefore only the written radiology report was available for review. Whole-body MRI had been performed in two patients (P1 and P2).

Family pedigrees and the identified PYROXD1 variants
Muscle biopsies were histochemically stained with haematoxylin & eosin (H&E), Gomöri trichrome, nicotinamide adenine dinucleotide tetrazolium reductase (NADH-TR) and combined cytochrome oxidase (COX)/succinate dehydrogenase (SDH). The following immunohistochemical stainings were performed for P1 and P3: MyHC double staining [Myosin Heavy Chain, Slow, Myosin Heavy Chain, A4.74 (fast)], p62, myotilin and desmin. Furthermore, biopsies from P1 and P2 were stained for sarcolemmal proteins dystrophin 1–3, dysferlin, sarcoglycan-alpha, dystroglycan-alpha and caveolin-3 in addition to merosin and emerin. For P2, only archival histological and histochemical stainings were available, and thus MyHC, p62, myotilin or desmin immunohistochemical stainings were not possible.
All subjects in this study have provided written consent for the use of clinical data and material. The study was approved by Helsinki University Hospital and Tampere University Hospital ethics boards.
DNA sequencing
For P1 and P2, whole exome sequencing (WES) was performed at the Finnish Institute of Molecular Medicine (FIMM). Briefly, 150ng of gDNA was fragmented with a Covaris E220 evolution instrument (Covaris). Sample libraries were processed according to SeqCapEZ Library SR (Roche Nimblegen) manual. NimbleGen capture was performed according to NimbleGen SeqCap EZ Exome Library SR User’s Guide. Sequencing was performed with Illumina HiSeq2500 system in Rapid mode using HiSeq Rapid v2 kits (Illumina). Reads were then aligned to the GRCh37 reference genome with the BWA (0.6.2), and the mpileup from the SAMTOOLS (1.4) package was used for variant calling.
For family 3 (P3, P4 and their unaffected brother), WES was performed essentially by the same method, but library preparation was done using KAPA Hyper library preparation Kit (Kapa Biosystems, Wilmington, Ma, USA) and SeqCap EZ MedExome assay (Roche Nimblegen) was used for target enrichment.
Sanger sequencing was performed with primers specific for PYROXD1 exon 5 (CAGTGGGAAAGTGAGATTCATTT and ATTACGGATTCCACAAGAGCT) and exon 10 (CCATGGAAATTCAGCTCAGGT and AACAACTGTGCTAGCTTCCT).
Plasmids, strains, media, and methods for yeast cells
The human PYROXD1 and PYROXD1-p.Tyr354Cys cDNAs were cloned by the Gateway® (Invitrogen) method into pDONR221 entry vector and then recombined into yeast destination vectors (Addgene;[6]) to obtain pAG415-promGPD-PYROXD1 (pSF371) and pAG415-promGPD-PYROXD1-p.Tyr354Cys (pSF501) plasmids. The pAG415 is a low-copy number CEN plasmid bearing the constitutive GPD (glyceraldehyde-3-phosphate dehydrogenase) promoter and the LEU2 auxotrophic marker for selection of the transformants on SC-Leu medium. Plasmid sequences were verified (GATC Biotech). The Saccharomyces cerevisiae wild-type BY4742 (MATα leu2Δ0 ura3Δ0 his3Δ0 lys2Δ0) reference strain and the glr1∆ (MATα leu2Δ0 ura3Δ0 his3Δ0 lys2Δ0 glr1::KanMX) mutant strain were used. Yeast cells were transformed using the modified lithium acetate method [7]. The indicated yeast strains were grown at 30°C to mid-exponential growth phase in synthetic complete (SC) medium SC-Leu: 0.67% yeast nitrogen base (YNB) without amino acids, 2% glucose and the appropriate—Leu dropout mix, to maintain the plasmid. These precultures were used to inoculate the rich medium YPD: 1% yeast extract, 2% peptone, 2% glucose, or the oxidative stress medium YPD + H2O2: 1% yeast extract, 2% peptone, 2% glucose, 3mM H2O2, and the growth of the yeast cells was analysed at 30°C under agitation in liquid medium by measuring the OD at 600nm over time, with measurements every 10min over 17h.
For western blot analysis, total yeast protein extracts were prepared by NaOH lysis of 1.5 OD600nm unit of yeast cells, followed by trichloroacetic acid (TCA) precipitation and the pellet was resuspended in 50µl of 2X Laemmli buffer plus Tris Base. Samples were incubated 5 min at 37°C prior western-blot analysis by 8% SDS-PAGE, followed by transfer on a nitrocellulose blotting membrane (Amersham™ Protran™ 0.45µm NC) and immunoblotting with rabbit polyclonal anti-PYROXD1 (1/500, R3500) antibodies [4] using standard procedures. Images were acquired with the ChemiDoc Touch Imaging System (Bio-Rad).
Results
Clinical features of P1
Patient 1 (Family 1) is a male who had been previously diagnosed with Ménière’s disease, but had been otherwise healthy. He had slowly progressive proximal muscle weakness combined with back pain starting at the age of 49 years (Table1). Before this his strengths had been normal, and he reported no problems with doing sports as a child or young adult. A lumbar disk herniation had been operated at the age of 51 years. At the age of 60 years, he was referred for neurologic consultation because of an incidental MRI finding of atrophy and fat replacement symmetrically in the paravertebral lower back muscles. The family history was negative for neuromuscular disease.
On clinical examination at the age of 63 years, he was ambulant without aids, but needed to use both hands when rising from a chair or climbing stairs. Moderate volume loss was noted in the upper paraspinal muscles, but otherwise there was no evident atrophy. Distally upper limb strengths were normal, proximally slightly reduced on both sides. In the lower limbs, hip flexion and knee flexion/extension strengths were decreased on both sides. Distal lower limb strengths were within normal limits. There was no facial or bulbar weakness.
Electromyography (EMG) of both proximal and distal muscles in all limbs showed abnormal motor unit potentials (MUPs), either small polyphasic or large and partially polyphasic. Nerve conduction studies showed normal conduction velocities and sensory and motor amplitudes.
Plasma creatine kinase (CK), acylcarnitine profile and cardiac ultrasound were normal. Spirometry showed evidence of moderate restrictive lung disease. High resolution CT of the thorax showed nodular parenchymal change consistent with pulmonary sarcoidosis. He was placed on an inhaled glucocorticoid.
Clinical features of P2
In his late teens before the age of 20 years, patient 2 (Family 2) needed support from his hands to climb stairs and he had trouble running. Nevertheless, he played football at the age of 35 years, and started requiring walking aids only at the age of 54 years. On examination at the age of 64 years he walked with two sticks, and short distances without aids. He had kyphotic posture and upper back muscles were noted as atrophic.
There was slight ptosis and facial weakness bilaterally. Limb strength was decreased predominantly proximally in a symmetric fashion. Arm elevation was less than 90° and elbow extension was severely decreased. Distal hand muscle strength was better preserved. In the lower limbs, hip flexion, knee extension and ankle dorsiflexion/plantar flexion were severely affected, while knee flexion was moderately reduced.
EMG showed myopathic changes. Plasma CK was slightly increased. Cardiac ultrasound at age 61 years was normal. He had episodic dyspnea and reduced respiratory function (age 64 years), but so far has not required ventilator support.
The patient’s brother had also been diagnosed with muscle disease and died of pneumonia at the age of 43 years. Both parents and two older sisters were healthy.
Clinical features of patients P3 and P4
The proband P3 and his sister P4 (Family 3) were asymptomatic until early thirties when they noticed weakness in their proximal lower limbs. They had difficulties rising from squat or chairs. Few years later muscle weakness was also reported in proximal upper limbs. The progression was very slow. At the age of 70 years the proband was ambulant with sticks for 200 m but rising from low chair was impossible. Gait was waddling with hyperextended knees. He could elevate his arms less than 90°. His posture was normal although spinal and abdominal muscles were weak with mild neck flexor weakness. Also distal lower limb muscles were weak (ankle plantar and dorsal flexion grade 3). Scapular winging or facial weakness was not observed. He had hoarseness in his voice but no other bulbar symptoms. He had had two strokes (left hemisphere aged 61 years and right hemisphere 66 years), which might contribute to the symptoms although acute hemiplegic symptoms had resolved. There was severe symmetrical muscle atrophy in the anterior part of the thigh, proximal upper limbs and shoulder girdle. He underwent echocardiography with normal results. Mild respiratory insufficiency and weak cough strength was observed in pulmonary function tests.
The sister, P4, started to use rollator at the age of 65 years and wheelchair aged 66 years. She could not extend her knees and hip flexion was weak as well as neck flexors and abdominal muscles. She had asymmetrical upper limb weakness both proximally and distally with left side more severely affected. No bulbar symptoms were present. She had severe progressive respiratory insufficiency and she died from pneumonia aged 70 years. There were no cardiac symptoms; echocardiography was not performed. Both siblings had normal CK levels. EMG showed myopathic changes especially in the proximal upper and lower limbs.
Muscle MRI and biopsy
Muscle MRI in all patients (P1, P2, P3 and P4) showed an unusual pattern of fatty dystrophic changes with total or near-total replacement of gluteus maximus, sartorius, gracilis and quadriceps (Fig.2a–c). Similar to the original report on PYROXD1-related myopathy [4], rectus femoris was less affected than the vasti muscles. In the distal lower limbs, gastrocnemii were the most affected muscles in patients P2, P3 and P4, while all lower leg muscles were only mildly and diffusely affected in patient P1. In two patients (P2 and P3), a very peculiar pattern of fatty degeneration was observed, where the outer parts of the anterior and lateral compartment muscles were replaced by fat, but not the inner portions (Fig.2b, c). No significant STIR edema was detected in any muscle, which is consistent with a relatively inactive and chronic degenerative process. Muscles of the upper girdle and the torso displayed diffuse fatty degenerative changes to a mild-moderate degree, while the muscles of the forearm and hand were minimally affected or entirely spared (not shown).

Muscle MRI and biopsies. MRI was performed for a individual P1 at age 60, b individual P2 at age 59, and c individual P3 at age 65. Gluteus maximus, quadriceps, sartorius and gracilis muscles were most severely affected in all patients, although the most proximal parts of the rectus femoris muscles were relatively spared or even hypertrophic. Anterior thigh muscles were always more severely affected than the hamstrings and adductor compartments. A very peculiar pattern of fatty degeneration was observed in patients P2 and P3, where only the outer parts of the anterior and lateral compartment muscles were replaced by fat. In patient P1, there was an unusual crescent-shaped fatty-degeneration involving the inner parts of vastus lateralis and medialis muscles. Muscle biopsy (vastus lateralis) from d individual P2 and e individual P1 showed a dystrophic pattern, where individual P2 had an almost end-stage pathology with severe atrophy, fibrosis and fatty infiltration. Scale bars 100µm
Muscle biopsies from the vastus lateralis of P2 (Fig.2d) and P1 (Fig.2e) showed chronic myopathic changes with atrophy, fat infiltration and strong fiber size variability, multiple internalized nuclei, and without significant myofibrillar pathology. Immunohistochemistry of sarcolemmal membrane-associated proteins, merosin and emerin was normal. Stainings for p62, myotilin and desmin in P1 showed no myofibrillar features.
The first biopsies of the siblings, P3 and P4, were performed 30 years ago and were not available for further analysis. The morphological changes were reported to be dystrophic without specific features. Patient 3 underwent a new biopsy from tibialis anterior muscle at the age of 70. The muscle pathology showed extensive end-stage dystrophic changes. No fiber type predominance was noted. Immunohistochemical staining for myotilin revealed a few positive cytoplasmic inclusions in scattered atrophic muscle fibers (Supplementary Fig.1). However, no desmin- or p62-positive protein aggregates were found.
Genetic findings
The patients’ pedigrees suggested recessively inherited disease (Fig.1). Thus, the exome sequencing data were filtered for homozygous or compound heterozygous variants that induce damaging changes to amino acid sequence, have population frequency of less than 0.001 in the total population and Finnish sub-population in ExAC variant database, CADD-scores of 20 or greater, and are present in less than 1% of an in-house database with 429 samples (P1 and P2) or less than 4% of a separate in-house database of 63 samples (P3 and P4). For P2, P3 and P4, the homozygous variant c.464A > G p.Asn155Ser in PYROXD1 (ENST00000240651.9) caught our attention because of its recent association with myopathy [4]. In the previous study, this variant was identified in four out of five studied families as homozygous (two families) or compound heterozygous (two families), and it is present in variant databases as a low frequency European variant always in heterozygous state. According to the GnomAD database, its allele frequency is slightly higher in the Finnish population (1.186 × 10−4) than elsewhere. The unaffected brother in the family 3 was a heterozygous carrier of the p.Asn155Ser variant.
Patient 1 was compound heterozygous for the p.Asn155Ser and a previously unknown variant c.1061A > G p.Tyr354Cys. The latter variant is found in four heterozygous individuals in the GnomAD database with a frequency of 1.444 × 10−5. The mutated tyrosine is located in the N-terminal end of the pyridine nucleotide oxidoreductase domain of PYROXD1 (Fig.3a), and the amino acid is evolutionarily conserved (Fig.3b). Segregation of the variants was investigated in the family of patient 1, and the affected individual was the only one carrying both mutations. The p.Asn155Ser allele was inherited maternally and the p.Tyr354Cys paternally.

PYROXD1 variants identified in this study and the functional characterization of p.Tyr354Cys in a yeast complementation assay. a A schematic of PYROXD1 with functional domains and the missense variants of patients of this study. b Evolutionary conservation of Tyr354. c Western blot of glr1Δ yeast cells transformed with yeast expression vectors empty (pAG415) or bearing the PYROXD1 wild-type (WT) or p.Tyr354Cys (Y354C) under the control of a constitutive promoter, two different clones (cl1 and cl2) were analyzed. The black arrows indicate the PYROXD1 protein and the loading control. d The indicated yeast cells were grown at 30 °C in rich medium (YPD) containing H2O2 (3 mM) to induce an oxidative stress. The growth curves of the different strains were determined by measuring the cell growth (OD at 600 nm) over time (h)
The PYROXD1-p.Tyr354Cys is defective in oxidative stress resistance in yeast cells
Humanization of Saccharomyces cerevisiae yeast cells can be used to better understand and/or to identify the cellular role of a human protein [8, 9]. Indeed, the human PYROXD1 cDNA was shown to rescue the oxidative stress defect associated with the yeast glutathione oxidoreductase glr1∆ deletion mutant strain [4]. Furthermore, the p.Asn155Ser variant was shown to have impaired rescue ability in the yeast complementation assay [4]. We tested here the effect of the new p.Tyr354Cys variant using the same yeast glr1∆ strain. The glr1∆ mutant cells were transformed by empty plasmid (pAG415) or by plasmids bearing either PYROXD1 wild-type cDNA, or the mutant allele p.Tyr354Cys. Expression of the human cDNAs was controlled by western blot analysis with anti-PYROXD1 antibody, showing that the wild-type and the mutant form of PYROXD1 were expressed in two different clones (cl1 and cl2) of yeast cells (Fig.3c).
We observed that the yeast glutathione reductase glr1∆ mutant had a strong growth defect when grown in the presence of hydrogen peroxide (Fig.3d). As shown before, the expression of the human PYROXD1 cDNA was able to complement the growth defect of the glr1∆ mutant cells [4], indicating that PYROXD1 has an oxidoreductase activity in vivo in yeast cells. Compared to wild-type PYROXD1, the p.Tyr354Cys variant had a lower growth rate in the presence of H2O2, showing that this new patient mutation impairs the oxidoreductase activity of PYROXD1 in vivo in yeast cells (Fig.3d).
Discussion
We report here new patients with recessive PYROXD1 variants underlying myopathy phenotypes. In contrast to the congenital myopathy cases described previously [4, 5], our patients had a considerably later disease onset and lacked significant myofibrillar pathology. In fact, a few myotilin-positive inclusions were only observed in the end-stage degenerated muscle of one of our patients. The clinical picture of our patients was consistent with adult-onset, slowly progressive LGMD type disease. Reasons for the clear difference in disease onset between the patients of Turkish [4] and Sudanese of Arab [5] ancestry, and the Finnish patients of this study, who were homozygous for the p.Asn155Ser variant, are speculative at this point but may involve the population-specific genetic background or some environmental factors that are relevant for the regulation of PYROXD1 oxidoreductase activity.
Muscle MRI may help to distinguish PYROXD1 myopathy from other myopathies, as our patients showed an unusual preference for anterolateral thigh muscles. Overall, the clinical course of the disease appears relatively benign, with preservation of ambulation past the age of 60. Respiratory impairment is a concern, however, as all our patients had developed respiratory muscle weakness and the brother of P2, who had been affected by a very similar kind of progressive muscle weakness, died from a respiratory infection at age 43 years. Also a 21-year-old patient described by O’Grady et al. had restrictive lung disease [4]. Regular monitoring of pulmonary function is therefore mandated in PYROXD1 myopathy. Fortunately, there appears to be no cardiac involvement.
It is important to note that the aged individuals in this study did not have evidence of neuropathy. While two affected individuals aged 26 and 29 years described by O’Grady et al. had an axonal neuropathy [4], our finding suggests that neuropathy is not a universal consequence of PYROXD1 variants even with advancing age. An important feature to note is axial myopathy, which was prominent in P1, and also present in 8/9 of O’Grady’s patients. It may be of interest to look for PYROXD1 variants in individuals presenting with pure axial myopathy, the genetics of which remains incompletely studied [10].
Three of our four patients were homozygous for the previously described p.Asn155Ser, whereas one was compound heterozygous for the same variant and the previously undescribed p.Tyr354Cys. Both of these variants are found in different populations according to exome and genome databases, suggesting that additional patients will probably be identified. The p.Asn155Ser change appears to be a common pathogenic PYROXD1 variant, as most described patients carry it in one or both alleles. However, the associated myopathy can have highly variable ages of onset. Our patient, P1, who was compound heterozygous for the p.Tyr354Cys variant together with p.Asn155Ser had the highest age of onset of all patients, suggesting that p.Tyr354Cys might impair the function of PYROXD1 less than the p.Asn155Ser variant.
In conclusion, our study shows that PYROXD1 should be considered as a causative gene also in later-onset unsolved myopathies, which resemble LGMD.
References
Nigro V, Savarese M (2014) Genetic basis of limb-girdle muscular dystrophies: the 2014 update. Acta Myol 33:1–12
Liewluck T, Milone M (2018) Untangling the complexity of limb-girdle muscular dystrophies. Muscle Nerve 58(2):167–177
Vissing J (2016) Limb girdle muscular dystrophies: classification, clinical spectrum and emerging therapies. Curr Opin Neurol 29:635–641
O’Grady GL, Best HA, Sztal TE et al (2016) Variants in the oxidoreductase PYROXD1 cause early-onset myopathy with internalized nuclei and myofibrillar disorganization. Am J Hum Genet 99:1086–1105
Saha M, Reddy HM, Salih MA et al (2018) Impact of PYROXD1 deficiency on cellular respiration and correlations with genetic analyses of limb-girdle muscular dystrophy in Saudi Arabia and Sudan. Physiol Genom 50:929–939
Alberti S, Gitler AD, Lindquist S (2007) A suite of gateway cloning vectors for high-throughput genetic analysis in saccharomyces cerevisiae. Yeast 24:913–919
Gietz D, St Jean A, Woods RA et al (1992) Improved method for high efficiency transformation of intact yeast cells. Nucleic Acids Res 20:1425
Amoasii L, Bertazzi DL, Tronchere H et al (2012) Phosphatase-dead myotubularin ameliorates X-linked centronuclear myopathy phenotypes in mice. PLoS Genet 8:e1002965
Kachroo AH, Laurent JM, Yellman CM et al (2015) Systematic humanization of yeast genes reveals conserved functions and genetic modularity. Science 348:921–925
Witting N, Andersen LK, Vissing J (2016) Axial myopathy: an overlooked feature of muscle diseases. Brain 139:13–22
Acknowledgements
Open access funding provided by University of Helsinki including Helsinki University Central Hospital. We thank the families in participation on this study. Riitta Lehtinen is acknowledged for technical assistance. We thank Sergio Buenestado Serrano (Erasmus student, Université de Strasbourg), Tom Schelcher and Sylvain Debard for help with the yeast experiments, and Jocelyn Laporte (IGBMC, Illkirch, France) for sharing the rabbit polyclonal anti-PYROXD1 antibody. We acknowledge the exome capture and sequencing performed by the Institute for Molecular Medicine Finland FIMM, Technology Centre, and University of Helsinki, and CSC—IT Center for Science, Finland, for computational resources.
Funding
This study was funded by the Academy of Finland pHealth project ‘Neurogenomics in routine diagnostics of children and adults: impact on patient care and cost-effectiveness’ for HT and MA, and by CNRS and Université de Strasbourg for BR and SF.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare that they have no conflict of interest.
Ethical Standards
All subjects in this study have provided written consent for the use of patient data and material. The study was approved by Helsinki University Hospital and Tampere University Hospital ethics boards.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Sainio, M.T., Välipakka, S., Rinaldi, B. et al. Recessive PYROXD1 mutations cause adult-onset limb-girdle-type muscular dystrophy. J Neurol 266, 353–360 (2019). https://doi.org/10.1007/s00415-018-9137-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-018-9137-8