
Abstract
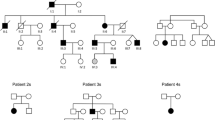
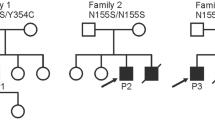
DTNA encodes α-dystrobrevin, a component of the macromolecular dystrophin–glycoprotein complex (DGC) that binds to dystrophin/utrophin and α-syntrophin. Mice lacking α-dystrobrevin have a muscular dystrophy phenotype, but variants in DTNA have not previously been associated with human skeletal muscle disease. We present 12 individuals from four unrelated families with two different monoallelic DTNA variants affecting the coiled-coil domain of α-dystrobrevin. The five affected individuals from family A harbor a c.1585G > A; p.Glu529Lys variant, while the recurrent c.1567_1587del; p.Gln523_Glu529del DTNA variant was identified in the other three families (family B: four affected individuals, family C: one affected individual, and family D: two affected individuals). Myalgia and exercise intolerance, with variable ages of onset, were reported in 10 of 12 affected individuals. Proximal lower limb weakness with onset in the first decade of life was noted in three individuals. Persistent elevations of serum creatine kinase (CK) levels were detected in 11 of 12 affected individuals, 1 of whom had an episode of rhabdomyolysis at 20 years of age. Autism spectrum disorder or learning disabilities were reported in four individuals with the c.1567_1587 deletion. Muscle biopsies in eight affected individuals showed mixed myopathic and dystrophic findings, characterized by fiber size variability, internalized nuclei, and slightly increased extracellular connective tissue and inflammation. Immunofluorescence analysis of biopsies from five affected individuals showed reduced α-dystrobrevin immunoreactivity and variably reduced immunoreactivity of other DGC proteins: dystrophin, α, β, δ and γ-sarcoglycans, and α and β-dystroglycans. The DTNA deletion disrupted an interaction between α-dystrobrevin and syntrophin. Specific variants in the coiled-coil domain of DTNA cause skeletal muscle disease with variable penetrance. Affected individuals show a spectrum of clinical manifestations, with severity ranging from hyperCKemia, myalgias, and exercise intolerance to childhood-onset proximal muscle weakness. Our findings expand the molecular etiologies of both muscular dystrophy and paucisymptomatic hyperCKemia, to now include monoallelic DTNA variants as a novel cause of skeletal muscle disease in humans.






Similar content being viewed by others

Data availability
Any data not published within the article will be shared from the corresponding authors upon reasonable request.
References
Amici DR, Pinal-Fernandez I, Mázala DAG, Lloyd TE, Corse AM, Christopher-Stine L et al (2017) Calcium dysregulation, functional calpainopathy, and endoplasmic reticulum stress in sporadic inclusion body myositis. Acta Neuropathol Commun 5:24. https://doi.org/10.1186/s40478-017-0427-7
Belhasan DC, Akaaboune M (2020) The role of the dystrophin glycoprotein complex on the neuromuscular system. Neurosci Lett 722:134833. https://doi.org/10.1016/j.neulet.2020.134833
Böhm S, Jin H, Hughes SM, Roberts RG, Hinits Y (2008) Dystrobrevin and dystrophin family gene expression in zebrafish. Gene Expr Patterns 8:71–78. https://doi.org/10.1016/j.modgep.2007.10.004
Bruels CC, Li C, Mendoza T, Khan J, Reddy HM, Estrella EA et al (2019) Identification of a pathogenic mutation in ATP2A1 via in silico analysis of exome data for cryptic aberrant splice sites. Mol Genet Genom Med 7:e552. https://doi.org/10.1002/mgg3.552
Cagliani R, Comi GP, Tancredi L, Sironi M, Fortunate F, Giorda R et al (2001) Primary beta-sarcoglycanopathy manifesting as recurrent exercise-induced myoglobinuria. Neuromuscul Disord 11:389–394. https://doi.org/10.1016/S0960-8966(00)00207-8
Chen B, Liu P, Zhan H, Wang ZW (2011) Dystrobrevin controls neurotransmitter release and muscle Ca 2+transients by localizing BK channels in Caenorhabditis elegans. J Neurosci 31:17338–17347. https://doi.org/10.1523/JNEUROSCI.3638-11.2011
Chung W, Campanelli JT (1999) WW and EF hand domains of dystrophin-family proteins mediate dystroglycan binding. Mol Cell Biol Res Commun 2:162–171. https://doi.org/10.1006/mcbr.1999.0168
Davydov EV, Goode DL, Sirota M, Cooper GM, Sidow A, Batzoglou S (2010) Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Computat Biol 6:e1001025. https://doi.org/10.1371/journal.pcbi.1001025
Dekkers LC, van der Plas MC, van Loenen PB, den Dunnen JT, van Ommen GJB, Fradkin LG et al (2004) Embryonic expression patterns of the Drosophila dystrophin-associated glycoprotein complex orthologs. Gene Expr Patterns 4:153–159. https://doi.org/10.1016/j.modgep.2003.09.004
Dubowitz V, Sewry C, Oldfors A (2013) Muscle biopsy: a practical approach, 4th edn. Saunders Elsevier, Philadelphia
Figarella-Branger D, Machado AMB, Putzu GA, Malzac P, Voelckel MA, Pellissier JF (1997) Exertional rhabdomyolysis and exercise intolerance revealing dystrophinopathies. Acta Neuropathol 94:48–53. https://doi.org/10.1007/s004010050671
Gieseler K, Mariol MC, Bessou C, Migaud M, Franks CJ, Holden-Dye L et al (2001) Molecular, genetic and physiological characterisation of dystrobrevin-like (dyb-1) mutants of Caenorhabditis elegans. J Mol Biol 307:107–117. https://doi.org/10.1006/jmbi.2000.4480
Grady RM, Akaaboune M, Cohen AL, Maimone MM, Lichtman JW, Sanes JR (2003) Tyrosine-phosphorylated and nonphosphorylated isoforms of α-dystrobrevin: Roles in skeletal muscle and its neuromuscular and myotendinous junctions. J Cell Biol 160:741–752. https://doi.org/10.1083/jcb.200209045
Grady RM, Grange RW, Lau KS, Maimone MM, Nichol MC, Stull JT et al (1999) Role for α-dystrobrevin in the pathogenesis of dystrophin-dependent muscular dystrophies. Nat Cell Biol 1:215–220. https://doi.org/10.1038/12034
Grady RM, Wozniak DF, Ohlemiller KK, Sanes JR (2006) Cerebellar synaptic defects and abnormal motor behavior in mice lacking α- and β-dystrobrevin. J Neurosci 26:2841–2851. https://doi.org/10.1523/JNEUROSCI.4823-05.2006
Grady RM, Zhou H, Cunningham JM, Henry MD, Campbell KP, Sanes JR (2000) Maturation and maintenance of the neuromuscular synapse: genetic evidence for roles of the dystrophin–glycoprotein complex. Neuron 25:279–293. https://doi.org/10.1016/S0896-6273(00)80894-6
Ichida F, Tsubata S, Bowles KR, Haneda N, Uese K, Miyawaki T et al (2001) Novel gene mutations in patients with left ventricular noncompaction or Barth syndrome. Circulation 103:1256–1263. https://doi.org/10.1161/01.CIR.103.9.1256
Ishikawa-Sakurai M, Yoshida M, Imamura M, Davies KE, Ozawa E (2004) ZZ domain is essentially required for the physiological binding of dystrophin and utrophin to β-dystroglycan. Hum Mol Genet 13:693–702. https://doi.org/10.1093/hmg/ddh087
Jantrapirom S, Nimlamool W, Temviriyanukul P, Ahmadian S, Locke CJ, Davis GW et al (2019) Dystrobrevin is required postsynaptically for homeostatic potentiation at the Drosophila NMJ. Biochim Biophys Acta Mol Basis Dis 1865:1579–1591. https://doi.org/10.1016/j.bbadis.2019.03.008
Jones KJ, Compton AG, Yang N, Mills MA, Peters MF, Mowat D et al (2003) Deficiency of the syntrophins and α-dystrobrevin in patients with inherited myopathy. Neuromuscul Disord 13:456–467. https://doi.org/10.1016/S0960-8966(03)00066-X
Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q et al (2020) The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581:434–443. https://doi.org/10.1038/s41586-020-2308-7
Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJE (2015) The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc 10:845–858. https://doi.org/10.1038/nprot.2015.053
Kircher M, Witten DM, Jain P, O’roak BJ, Cooper GM, Shendure J (2014) A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 46:310–315. https://doi.org/10.1038/ng.2892
Kruglyak L, Daly MJ, Reeve-Daly MP, Lander ES (1996) Parametric and nonparametric linkage analysis: a unified multipoint approach. Am J Hum Genet 58:1347–1363
Li H, Durbin R (2010) Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26:589–595. https://doi.org/10.1093/bioinformatics/btp698
Malakootian M, Jalilian M, Kalayinia S, Hosseini Moghadam M, Heidarali M, Haghjoo M (2022) Whole-exome sequencing reveals a rare missense variant in DTNA in an Iranian pedigree with early-onset atrial fibrillation. BMC Cardiovasc Disord 22:37. https://doi.org/10.1186/s12872-022-02485-0
Mathews KD, Stephan CM, Laubenthal K, Winder TL, Michele DE, Moore SA et al (2011) Myoglobinuria and muscle pain are common in patients with limb-girdle muscular dystrophy 2I. Neurology 76:194–195. https://doi.org/10.1212/WNL.0b013e3182061ad4
Mercuri E, Bönnemann CG, Muntoni F (2019) Muscular dystrophies. The Lancet 394:2025–2038. https://doi.org/10.1016/S0140-6736(19)32910-1
Metzinger L, Blake DJ, Squier MV, Anderson LVB, Deconinck AE, Nawrotzki R et al (1997) Dystrobrevin deficiency at the sarcolemma of patients with muscular dystrophy. Hum Mol Genet 6:1185–1191. https://doi.org/10.1093/hmg/6.7.1185
Nakamori M, Takahashi MP (2011) The role of alpha-dystrobrevin in striated muscle. Int J Mol Sci 12:1660–1671. https://doi.org/10.3390/ijms12031660
Nallamilli BRR, Chakravorty S, Kesari A, Tanner A, Ankala A, Schneider T et al (2018) Genetic landscape and novel disease mechanisms from a large LGMD cohort of 4656 patients. Ann Clin Transl Neurol 5:1574–1587. https://doi.org/10.1002/acn3.649
Nguyen K, Bassez G, Krahn M, Bernard R, Laforêt P, Labelle V et al (2007) Phenotypic study in 40 patients with dysferlin gene mutations: high frequency of atypical phenotypes. Arch Neurol 64:1176–1182. https://doi.org/10.1001/archneur.64.8.1176
Panadés-de Oliveira L, Bermejo-Guerrero L, de Fuenmayor-Fernández de la Hoz CP, Cantero Montenegro D, Hernández Lain A, Martí P et al (2020) Persistent asymptomatic or mild symptomatic hyperCKemia due to mutations in ANO5: the mildest end of the anoctaminopathies spectrum. J Neurol 267:2546–2555. https://doi.org/10.1007/s00415-020-09872-7
Pena L, Kim K, Charrow J (2010) Episodic myoglobinuria in a primary gamma-sarcoglycanopathy. Neuromuscul Disord 20:337–339. https://doi.org/10.1016/j.nmd.2010.02.015
Pilgram GSK, Potikanond S, Baines RA, Fradkin LG, Noordermeer JN (2010) The roles of the dystrophin-associated glycoprotein complex at the synapse. Mol Neurobiol 41:1–21
Pinal-Fernandez I, Amici DR, Parks CA, Derfoul A, Casal-Dominguez M, Pak K et al (2019) Myositis autoantigen expression correlates with muscle regeneration but not autoantibody specificity. Arthritis Rheumatol 71:1371–1376. https://doi.org/10.1002/art.40883
Pinal-Fernandez I, Casal-Dominguez M, Derfoul A, Pak K, Miller FW, Milisenda JC et al (2020) Machine learning algorithms reveal unique gene expression profiles in muscle biopsies from patients with different types of myositis. Ann Rheum Dis 79:1234–1242. https://doi.org/10.1136/annrheumdis-2019-216599
Pinal-Fernandez I, Casal-Dominguez M, Derfoul A, Pak K, Plotz P, Miller FW et al (2019) Identification of distinctive interferon gene signatures in different types of myositis. Neurology 93:e1193–e1204. https://doi.org/10.1212/WNL.0000000000008128
Ponting CP, Blake DJ, Davies KE, Kendrick-Jones J, Winder SJ (1996) ZZ and TAZ: new putative zinc fingers in dystrophin and other proteins. Trends Biochem Sci 21:11–13. https://doi.org/10.1016/S0968-0004(06)80020-4
Quinlivan R, Jungbluth H (2012) Myopathic causes of exercise intolerance with rhabdomyolysis. Dev Med Child Neurol 54:886–891. https://doi.org/10.1111/j.1469-8749.2012.04320.x
Reddy HM, Cho KA, Lek M, Estrella E, Valkanas E, Jones MD et al (2017) The sensitivity of exome sequencing in identifying pathogenic mutations for LGMD in the United States. J Hum Genet 62:243–252. https://doi.org/10.1038/jhg.2016.116
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J et al (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17:405–424. https://doi.org/10.1038/gim.2015.30
Rubegni A, Malandrini A, Dosi C, Astrea G, Baldacci J, Battisti C et al (2019) Next-generation sequencing approach to hyperCKemia: a 2-year cohort study. Neurol Genet 5:e352. https://doi.org/10.1212/NXG.0000000000000352
Scalco RS, Gardiner AR, Pitceathly RDS, Hilton-Jones D, Schapira AH, Turner C et al (2016) CAV3 mutations causing exercise intolerance, myalgia and rhabdomyolysis: Expanding the phenotypic spectrum of caveolinopathies. Neuromuscul Disord 26:504–510. https://doi.org/10.1016/j.nmd.2016.05.006
Simon MJ, Murchison C, Iliff JJ (2018) A transcriptome-based assessment of the astrocytic dystrophin-associated complex in the developing human brain. J Neurosci Res 96:180–193. https://doi.org/10.1002/jnr.24082
Straub V, Murphy A, Udd B (2018) 229th ENMC international workshop: limb girdle muscular dystrophies—nomenclature and reformed classification Naarden, the Netherlands, 17–19 March 2017. Neuromuscul Disord 28:702–710. https://doi.org/10.1016/j.nmd.2018.05.007
Wang K, Li M, Hakonarson H (2010) Analysing biological pathways in genome-wide association studies. Nat Rev Genet 11:843–854
Wiel L, Baakman C, Gilissen D, Veltman JA, Vriend G, Gilissen C (2019) MetaDome: pathogenicity analysis of genetic variants through aggregation of homologous human protein domains. Hum Mutat 40:1030–1038. https://doi.org/10.1002/humu.23798
Zima J, Eaton A, Pál E, Till Á, Ito YA, Warman-Chardon J et al (2020) Intrafamilial variability of limb-girdle muscular dystrophy, LGMD1D type. Eur J Med Genet 63:103655. https://doi.org/10.1016/j.ejmg.2019.04.012
Acknowledgements
We are indebted to the “Biobanc de l’Hospital Infantil Sant Joan de Déu per a la Investigació”, part of the Spanish Biobank Network of ISCIII for the sample and data procurement.
Funding
Daniel Natera-de Benito is supported by the Miguel Servet program from Instituto de Salud Carlos III, Spain (CP22/00141). Jordi Pijuan is supported by a Carmen de Torres fellowship of the SJD Research Institute. Francesc Palau is supported by Fundacion Isabel Gemio and AGAUR (2017 SGR 324). Janet Hoenicka is supported by the Torro Solidari-RAC1 i Torrons Vicens. The work in Peter B. Kang's laboratory was supported in part by NIH R01 NS080929. The work in Louis M. Kunkel's laboratory was supported in part by NIH R01AR064300 and the Bernard F. and Alva B. Gimbel Foundation. Work in Carsten G. Bönneman’s laboratory is supported by intramural funds by the NIH National Institute of Neurological Disorders and Stroke. Sequencing and analysis of families C and D were provided by the Broad Institute of MIT and Harvard Center for Mendelian Genomics (Broad CMG) and was funded by the National Human Genome Research Institute, the National Eye Institute, and the National Heart, Lung and Blood Institute grant UM1HG008900 and in part by National Human Genome Research Institute grant R01HG009141.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
None of the authors has any conflict of interest to disclose.
Ethics/consent to participate
This study was performed in accordance with the Declaration of Helsinki and was approved by the Hospital Sant Joan de Déu Clinical Research Ethics Committee (reference PIC-04-18). Data were collected in accordance with the ethical guidelines of each of the institutions involved. Written informed consent for study participation was obtained by qualified clinicians.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Nascimento, A., Bruels, C.C., Donkervoort, S. et al. Variants in DTNA cause a mild, dominantly inherited muscular dystrophy. Acta Neuropathol 145, 479–496 (2023). https://doi.org/10.1007/s00401-023-02551-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-023-02551-7