
Abstract
Frontotemporal lobar degeneration (FTLD) is a clinically, genetically and pathologically heterogeneous disorder. Within FTLD with ubiquitin-positive inclusions (FTLD-U), a new pathological subtype named FTLD-FUS was recently found with fused in sarcoma (FUS) positive, TDP-43-negative inclusions, and striking atrophy of the caudate nucleus. The aim of this study was to determine the frequency of FTLD-FUS in our pathological FTLD series, and to describe the clinical, neuroimaging and neuropathological features of FTLD-FUS, especially caudate atrophy. Demographic and clinical data collected prospectively from 387 patients with frontotemporal dementia (FTD) yielded 74 brain specimens. Immunostaining was carried out using a panel of antibodies, including AT-8, ubiquitin, p62, FUS, and TDP-43. Cortical and caudate atrophy on MRI (n = 136) was rated as normal, mild-moderate or severe. Of the 37 FTLD-U cases, 33 were reclassified as FTLD-TDP and four (0.11, 95%: 0.00–0.21) as FTLD-FUS, with ubiquitin and FUS-positive, p62 and TDP-43-negative neuronal intranuclear inclusions (NII). All four FTLD-FUS cases had a negative family history, behavioural variant FTD (bvFTD), and three had an age at onset ≤40 years. MRI revealed mild-moderate or severe caudate atrophy in all, with a mean duration from onset till MRI of 63 months (range 16–119 months). In our total clinical FTD cohort, we found 11 patients (0.03; 95% CI: 0.01–0.05) with bvFTD, negative family history, and age at onset ≤40 years. Caudate atrophy was present in 10 out of 136 MRIs, and included all four FUS-cases. The newly identified FTLD-FUS has a frequency of 11% in FTLD-U, and an estimated frequency of three percent in our clinical FTD cohort. The existence of this pathological subtype can be predicted with reasonable certainty by age at onset ≤40 years, negative family history, bvFTD and caudate atrophy on MRI.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Frontotemporal dementia (FTD) is the second most common type of presenile dementia, and a clinically, genetically and pathologically heterogeneous disorder [1, 2]. Behavioural changes and cognitive dysfunctions, especially language, are core clinical features. Four clinical subtypes can be distinguished: the behavioural variant of FTD (bvFTD), semantic dementia (SD), progressive non-fluent aphasia (PNFA), and FTD with motor neuron disease (FTD-MND).
Frontotemporal lobar degeneration (FTLD) is the common underlying pathology of all four clinical variants, and can be divided into FTLD with tau-positive immunoreactive inclusions (FTLD-tau) and FTLD with ubiquitin-positive immunoreactive inclusions (FTLD-U). After the TDP-43 protein was found to be the major constituent of ubiquitin-positive inclusions [3], the latter term was changed to FTLD-TDP. However, the identification of TDP-43-negative inclusions in some FTLD-U cases has resulted in the designation of a new neuropathological subtype, atypical FTLD-U [4–6]. Very recently, the name of this subtype has been changed into FTLD-fused in sarcoma (FTLD-FUS) by the observation of positive staining of these inclusions with antibody against FUS protein [7].
The characteristic features of FTLD-FUS are an early-onset FTD with severe progressive psychobehavioural changes, a negative family history and striking atrophy of the striatum at neuropathological examination [4–7]. Its exact frequency is yet unknown, and neuroimaging features may differentiate it from other subtypes during life. The aim of this study was to determine the frequency of FTLD-FUS in our FTLD-U cases and to describe the clinical, neuroimaging and neuropathological features of FTLD-FUS. Furthermore, we estimated the prevalence of FTLD-FUS in our clinical cohort of FTD on the basis of a combination of specific clinical features.
Methods
Patients with FTD were ascertained in an ongoing genetic-epidemiologic study conducted in The Netherlands since 1994 [1, 8, 9], after referral to the out-clinic department of the Erasmus Medical Center, or after a visit in nursing homes and psychogeriatric hospitals by the research physician. Detailed clinical history was obtained from the spouses and first-degree relatives using a checklist of behavioural and cognitive changes, and motor neuron signs.
The age at onset was defined as the age at which the first symptom compatible with FTD diagnosis was observed by a close relative or caretaker [1, 9]. During the neurological examination carried out in all patients, special attention was paid to the presence of extrapyramidal and motor neuron disease signs. Data on family history were provided by a spouse or first-degree relative using a structured questionnaire. Family history was defined as positive if patients had at least one-first-degree relative with dementia, parkinsonism, or MND, irrespective of their age of onset.
Neuropsychological evaluation consisted of tests for memory functions (Rey figure, 15-Word test [10]), attention and concentration, executive functions (Stroop, Trailmaking A and B, Wisconsin Card Sorting Test, WAIS subtest substitution), language (Boston Naming Test, the Dutch revised version of the Semantic association test [11]), and visuoconstructive and visuospatial skills (Clock drawing, Block design of the WAIS), and was performed in a subset of patients at our out-clinic department. Due to the variation in test batteries, a detailed comparison between subgroups was not possible. Severity of dementia at ascertainment was assessed using the Clinical Dementia Rating scale [12].
MRI scans performed between 1990 and 2008 were available for evaluation in 136 out of 387 patients. Available T1-weighted MR, T2-weighted MR, Proton-density (PD) weighted MR, and fluid attenuated inversion recovery (FLAIR) MR images were used for evaluation.
The severity of cortical and caudate atrophy on MRI was evaluated and semi-quantitatively rated according to a three-point scale: (0) normal caudate nucleus; (1) mild or moderate atrophy of the caudate nucleus, either still bulging into the lateral ventricle or with a flat contour; (2) severe atrophy with no visible caudate nucleus. The presence of caudate atrophy was evaluated by a radiologist and neurologist blinded to clinical and pathological findings. Disagreement was solved by consensus.
Mutation screening of all exons and exon–intron regions of MAPT, GRN and CHMP2B genes was performed in all patients with a positive family history, and in a subset of patients with sporadic FTD (n = 72) as previously described [1, 9].
Two hundred thirty-two patients died during a follow up period of 14 years, of which 74 patients underwent brain autopsy. Brain autopsy was carried out within four hours of death according to the Legal and Ethical Code of Conduct of The Netherlands Brain Bank. Macroscopic inspection of the brain included the frontal, temporal, parietal, and occipital lobes, cerebellum, hippocampus, basal ganglia and substantia nigra.
Tissue blocks were taken from all cortical areas, hippocampus, amygdala, basal ganglia, substantia nigra, pons, medulla oblongata, cerebellum, and cervical spinal cord. They were embedded in paraffin blocks and subjected to routine staining with haematoxylin-eosin, Bodian, methenamine-silver, and Congo red. The severity of neuron loss was scored as absent, mild, or moderate-severe.
Immunohistochemistry was performed with antibodies directed against: hyperphosphorylated tau (AT-8, Innogenetics, Ghent, Belgium; 1:400); ubiquitin (anti-ubiquitin, DAKO, Glostrup, Denmark; 1:500, following 80°C antigen retrieval); β-amyloid protein (anti-beta amyloid, DAKO, Glostrup, Denmark; 1:100, following formic acid pre-treatment); α-synuclein (anti-α-synuclein; Zymed Laboratories, San Francisco, California, USA; undiluted, following formic acid pre-treatment); poli-ubiquitin-binding protein p62 (BD Biosciences Pharmingen, San Diego, CA, USA; 1:200, following pressure cooking); TDP-43 (Biotech, Chicago, IL, USA; 1:100, following pressure cooking); and α–internexin (anti-alpha-internexin, Invitrogen, Camarillo, CA, USA, 1:100, following pressure-cooking), FUS (Sigma-Aldrich anti-FUS; 1:25–1:200 with initial overnight incubation at room temperature, following pressure cooking).
Antigen retrieval was done either for 30 min in 0.1 M sodium citrate buffer at 80°C and pH 7.7 or using pressure cooking in 0.1 M sodium citrate buffer (pH 6) for 5 min. Pre-treatment with 70% formic acid was done for 5 min (α-synuclein and β-amyloid).
Primary antibodies were incubated overnight at 4°C. Endogenous peroxidase activity was inhibited by incubation for 30 min in PBS-hydrogen peroxide-sodiumazide solution (100 ml 0,1 M PBS + 2 ml 30% H2O2 + 1 ml natriumazide). The Histostain-Plus broad-spectrum kit DAB (Zymed, San Francisco, California, USA) was used and slides were counterstained with Mayer’s haematoxylin and mounted in entellan.
Neuropathological examination of brains from the 74 autopsied patients with clinical FTD revealed 70 brains with FTLD (FTLD-U in 37, FTLD-tau in 32 and FTLD with no inclusions (FTLD-ni) in one), whereas familial Creutzfeldt-Jakob disease (fCJD) was diagnosed in two brains, and Alzheimer’s disease (AD) in the remaining two.
The study was approved by the Medical Ethical Committee at the Erasmus MC-University Medical Center Rotterdam. For each patient, a spouse or first-degree relative of the patient gave written informed consent.
Results
Thirty-three out of 37 FTLD-U cases (0.89; 95% CI: 0.79–1.00) showed positive staining with the TDP-43 antibody and were reclassified as FTLD-TDP, whereas four cases had ubiquitin-positive, TDP-43-negative inclusions. All four FTLD-U patients with TDP-43 negative inclusions showed positive staining with the FUS antibody and were designated as FTLD-FUS. Inclusions of FTLD-TDP cases did not stain with the FUS antibody. FTLD-FUS was present in 4 out of all 70 FTLD brains (0.06; 95% CI: 0.00–0.11), and in four out of 37 FTLD-U cases (0.11; 95% CI: 0.00–0.21).
Demographic and clinical data for FTLD-FUS
Demographic and clinical data of the four patients with FTLD-FUS are presented in Table 1. All four patients had clinical bvFTD and a negative family history, and three out of four patients had an age at onset before the age of 40. The presenting symptoms varied among these four cases. Two patients presented with obsessive–compulsive behaviour, in particular showing extremely parsimonious behaviour. Case 1 and Case 3 showed sexually disinhibited behaviour (Case 1 frequently visited prostitutes, while married). Three out of four patients shoplifted several shops, especially the two patients who presented with obsessive–compulsive behaviour. Hyperorality has developed in all four patients, and was the presenting symptom in three patients. Visual hallucinations of deformed paintings were mentioned in the case of one patient, and persecution delusions in another. All patients developed language problems later in the course of the disease, with economy of utterances in all, perseverations (n = 3), echolalia (n = 2), stereotypy (n = 2) and eventually mutism in all. Neurological examination was unremarkable without signs of motor neuron disease, pyramidal or extrapyramidal dysfunction, except for mild left-sided cogwheeling in one patient.
Neuropsychological testing revealed impaired attention and concentration, and executive deficits, with normal orientation, memory and visuoconstructive functions in all four patients, except for evident memory problems in one.
MRI showed severe frontal and/or temporal atrophy in three patients, and mild to moderate frontal atrophy in one. All four patients had mild-moderate (n = 1), or severe caudate atrophy (n = 3), at a mean duration of illness of 63 months (range 16–119 months) (Figure 1). Genetic screening revealed no mutations in MAPT or GRN in all four patients, and in CHMP2B genes in three patients. DNA was not available anymore in Case 1 to screen for CHMP2B gene mutations.

MRI scans of FTLD-FUS cases. a Patient 1 with severe atrophy (PD-weighted MR image). b Patient 2 with severe atrophy (PD-weighted MR image). c Patient 3 with mild-moderate atrophy (FLAIR MR image). d Patient 4 with severe atrophy (T1-weighted MR image)
Age at onset ≤40 years, negative family history, and behavioural variant FTD
The frequency of FTLD-FUS in our clinical cohort was estimated by using negative family history, age at onset ≤40 years and bvFTD as distinctive features for FTLD-FUS patients. We selected patients with a negative family history and bvFTD (n = 126). We stratified our patients according to age at onset and made three subgroups: age at onset ≤40 years (n = 11), >40 to ≤50 years (n = 23), and >50 years (n = 92). FTLD-FUS was found in three out of 11 patients (0.27) in the group with age at onset ≤40 years, one out of 23 (0.04) in the group of patients with age at onset >40 to ≤50 years, and was absent in the group of 92 patients with age at onset >50 years. FTLD-FUS is not more frequently found in patients with an age at onset ≤40, than in patients with an age at onset >40 to ≤50 years (Fisher’s Exact Test, p = 0.089). However, FTLD-FUS is more frequently found in patients with an age at onset ≤40 years than in patients with an age at onset >50 years (Fisher’s Exact Test, p = 0.001).
The estimated prevalence of FTLD-FUS for the total clinical FTD group based on family history, age at onset ≤ 40 years and bvFTD as clinical subtype, is 11 out of 387 patients (0.03; 95% CI: 0.01–0.05).
Caudate atrophy
Severe caudate atrophy is one of the pathological hallmarks of FTLD-FUS. We reviewed all available MRIs of all clinical FTD patients (n = 136) for the presence of caudate atrophy. Caudate atrophy was present in 10 out of 136 MRIs (0.07; 95% CI: 0.03–0.12). Seven of these 10 patients had a negative family history, onset ≤40 years and bvFTD, of whom three had pathological proven FTLD-FUS. Pathology was not available in the other four cases. Of the remaining three cases with caudate atrophy, two had a GRN mutation and had an age at onset >40 years, and the third patient had FTLD-FUS with an age at onset of 49 years (Case 1). The interval between onset and time of MRI scanning between cases with and without caudate atrophy did not differ (36 months in cases with caudate atrophy as well as in cases without caudate atrophy).
Seven of the eleven patients with a negative family history, onset ≤40 years and bvFTD had caudate atrophy. There were no MRIs available of the other four patients.
Pathology of FTLD-FUS
Brain weight, gross atrophy and depigmentation of the substantia nigra are summarized in Table 2. The most characteristic feature was the striking atrophy of the caudate in all four cases.
All four brains showed severe neuronal loss of CA1 and subiculum consistent with hippocampal sclerosis, as well as severe neuronal loss of the caudate nucleus. The frontal and/or temporal cortices showed moderate-to-severe neuronal loss in all, whereas the parietal cortex was mildly affected in only one brain. The substantia nigra was severely affected in all four brains. There was no involvement of the pons, medulla oblongata, cerebellum, or spinal cord.
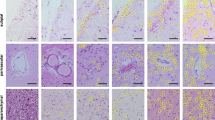
Immunohistochemistry revealed many ubiquitin, p62, and FUS-positive, TDP-43-negative neuronal cytoplasmatic inclusions (NCI) in the granular cells of the dentate gyrus of the hippocampus, whereas the number of NCI and dystrophic neurites (DN) in the frontotemporal cortex and caudate nucleus was low. Several ubiquitin and FUS-positive, p62 and TDP-43-negative neuronal intranuclear inclusions (NII) of variable shapes (straight, worm-like, C-shaped, or ring-like) were found in the granular cells of the dentate gyrus in all four brains (Fig. 2a, c, d); two brains also had NII in the CA4 pyramidal neurons, including one with NII in CA3 (Case 1). Case 3 had NCI and NII in the medulla oblongata. The antibody against the ubiquitin-binding protein p62 gave positive staining of NCI and DN, but not with NII (Fig. 2b). There were no inclusions found in the substantia nigra, pons, medulla, or cerebellum of any of the four brains. Immunostaining with AT-8, α-synuclein, and α-internexin was negative in all cases.

Immunohistochemistry of the FTLD-FUS cases. Ubiquitin-positive neuronal intranuclear inclusions (NII) in the granular cells of the dentate gyrus, which are worm-like (a). Only the ubiquitin-positive neuronal cytoplasmatic inclusions (NCI) stained positive with p62 (b); NII did not. TDP-43-immunohistochemistry stained only normal nuclei, and stained neither ubiquitin-positive NCI nor NII (c). NCI as well as NII stained positive with FUS antibody (d). Scale bar 50 μm
Discussion
This study showed a frequency of FTLD-FUS of six percent of all our pathologically proven FTLD cases, and 11 percent of the FTLD-U cases. We estimated a frequency of FTLD-FUS of three percent in the total series of patients with clinical FTD, based on the clinical variables age at onset, family history, and clinical subtype. All four FTLD-FUS cases presented with severe behavioural changes, young age at onset, negative family history, and caudate atrophy on MRI. FTLD-FUS cases were pathologically characterized by striking atrophy of the caudate nucleus, ubiquitin and FUS-positive, p62 and TDP-43-negative NII with remarkable morphology.
The present observation of three percent of FTLD-FUS in a large clinical cohort of FTD patients gives a frequency estimation of this subtype for the first time. Of the four FTLD-FUS patients, three had an age at onset ≤40 years, negative family history, and clinical presentation of bvFTD. As 11 patients within the age group ≤40 years of our cohort meet the combination of these features, this suggests an even higher frequency in this age group. Our observed age distribution is in line with that of the other reported series of FTLD with TDP-43 negative inclusions (28 to 63 years) [4–6]. Delusions and hallucinations in the present cases may be discriminative features of this type of FTLD, as their occurrence is uncommon for the total FTD group [13]. The initial diagnosis of a primary psychiatric disorder has been considered in one of the present patients [14], as well as in several other recently reported cases [4, 5]. The occurrence of disinhibition and obsessive compulsive behaviour was reported in the present cases, while aggressive behaviour was reported in others [4].
Although we did not have pathological verification of four other patients, the combination of age at onset ≤40 years, negative family history, presence of severe behavioural changes and caudate atrophy is highly suggestive of FTLD-FUS. Our suggested prevalence of three percent could be an underestimate, as ~30 percent of all cases described so far had an age at onset above 40 years, and not all patients in our cohort had an available MRI to semi-quantitatively score the caudate atrophy [4–6].
The observation of caudate atrophy on MRI in our FTLD-FUS cases during life has not been reported so far, but is in line with the presence of caudate atrophy and severe neuron loss at neuropathological examination. Neuroimaging features during life were not explicitly mentioned in three pathological studies on TDP-43-negative FTLD-U cases (FTLD-FUS in the new nomenclature) [15–17]. Although smaller caudate volumes may be seen in the end-stage of FTD [18, 19], severe caudate atrophy was already detectable in the early disease stage of the present FTLD-FUS cases. Prominent caudate atrophy is characteristic for Huntington’s disease, but chorea or other extrapyramidal signs were absent in the present cases.
The occurrence of FTLD-FUS in 11% of our FTLD-U cases lies between the frequencies reported in other series [4–6], and emphasizes the idea that alternative pathophysiological mechanisms exist in the clinicopathological spectrum of FTLD and motor neuron disease. The FUS protein containing 526 amino-acids has structural similarities with TDP-43 and is involved in DNA repair and the regulation of transcription, RNA splicing, and export to cytoplasm [7, 20, 21]. Mutations in the FUS gene located on chromosome 16 have recently been identified in familial ALS with ubiquitin and FUS-positive, TDP-43-negative inclusions [20, 21].
The TDP-43-negative, FUS-positive cytoplasmatic inclusions have also been found in neuronal intermediate filament inclusion disease (NIFID) cases [22], whereas their presence has yet to be investigated in other related disorders: FTLD caused by CHMP2B mutations, basophilic inclusion body disease (BIBD), and ALS due to SOD1 mutations [23–26].
The remarkable vermiform or C-shaped morphology of the NII reported in all cases seems to be pathognomic for FTLD-FUS and differs from the lentiform or cat-eye shaped NII in GRN mutations [4–6, 27–29]. Moreover, NII of GRN mutations shows a different immunohistochemical pattern with TDP-43 positive and FUS negative staining.
The relevance of exclusive p62-positive staining of the cytoplasmatic inclusions (and not of NII) still has to be determined. The question is whether the p62 protein is merely entrapped in these inclusions or plays a pathophysiological role in these disorders. The recent finding of accumulation of hyperphosphorylated tau and neurodegeneration in mice with genetic inactivation of the p62 gene supports the latter hypothesis [30].
A major drawback of the present study is the absence of a quantitative assessment of caudate atrophy on MRI [18]. Future prospective follow-up studies with voxel-based morphometry of the caudate nucleus on MRI are needed to determine the progression of caudate atrophy over time in FTLD-FUS.
In conclusion, FTLD-FUS is estimated to account for three percent of our series of clinical FTD. Age at onset ≤40 years, negative family history, bvFTD and caudate atrophy on MRI could be useful clinical predictors of FTLD-FUS as an underlying pathology in patients with clinical FTD. Larger samples of FTLD-FUS cases are needed to verify and to add new predictors for this clinicopathological subtype.
Abbreviations
- FTD:
-
Frontotemporal dementia
- FTLD:
-
Frontotemporal lobar degeneration
- FTLD-U:
-
FTLD with ubiquitinated inclusions
- TDP-43:
-
TAR DNA binding protein of 43 kDa
- FUS:
-
Fused in sarcoma
- FTLD-TDP:
-
FTLD-TDP-43 positive inclusions
- FTLD-FUS:
-
FTLD with FUS-positive inclusions
- GRN:
-
Progranulin
References
Seelaar H et al (2008) Distinct genetic forms of frontotemporal dementia. Neurology 71(16):1220–1226
van Swieten JC, Heutink P (2008) Mutations in progranulin (GRN) within the spectrum of clinical and pathological phenotypes of frontotemporal dementia. Lancet Neurol 7(10):965–974
Neumann M et al (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314(5796):130–133
Mackenzie IR et al (2008) Atypical frontotemporal lobar degeneration with ubiquitin-positive, TDP-43-negative neuronal inclusions. Brain 131(Pt 5):1282–1293
Roeber S et al (2008) TDP-43-negative FTLD-U is a significant new clinico-pathological subtype of FTLD. Acta Neuropathol 116(2):147–157
Josephs KA et al (2008) Frontotemporal lobar degeneration with ubiquitin-positive, but TDP-43-negative inclusions. Acta Neuropathol 116(2):159–167
Neumann M et al (2009) Frontotemporal lobar degeneration with FUS pathology. Brain
Rosso SM et al (2003) Frontotemporal dementia in The Netherlands: patient characteristics and prevalence estimates from a population-based study. Brain. 126(Pt 9): 2016–201622. (Epub 2003 Jul 22)
Seelaar H et al (2007) TDP-43 pathology in familial frontotemporal dementia and motor neuron disease without Progranulin mutations. Brain 130(Pt 5):1375–1385
Saan RJ, Deelman BG (1986) Dutch version of the Rey auditory verbal learning test. Groningen: Department of Neuropsychology, RUG, provisional manual
Visch-Brink EG, Stroks DL (2005) The semantic association test (the Dutch revised version of the Pyramid and Palmtrees). Harcourt Test Publishers, Lisse
Morris JC (1993) The clinical dementia rating (CDR): current version and scoring rules. Neurology 43(11):2412–2414
Mendez MF et al (2008) Psychotic symptoms in frontotemporal dementia: prevalence and review. Dement Geriatr Cogn Disord 25(3):206–211
Lamote H, Tan KL, Verhoeven WM (1998) Frontotemporal dementia in a young woman with apparent schizophrenia. Ned Tijdschr Geneeskd 142(35):1962–1965
Pikkarainen M, Hartikainen P, Alafuzoff I (2008) Neuropathologic features of frontotemporal lobar degeneration with ubiquitin-positive inclusions visualized with ubiquitin-binding protein p62 immunohistochemistry. J Neuropathol Exp Neurol 67(4):280–298
Hatanpaa KJ et al (2008) TAR DNA-binding protein 43 immunohistochemistry reveals extensive neuritic pathology in FTLD-U: a midwest-southwest consortium for FTLD study. J Neuropathol Exp Neurol 67(4):271–279
Cairns NJ et al (2007) TDP-43 in familial and sporadic frontotemporal lobar degeneration with ubiquitin inclusions. Am J Pathol
Looi JC et al (2008) Caudate nucleus volumes in frontotemporal lobar degeneration: differential atrophy in subtypes. AJNR Am J Neuroradiol 29(8):1537–1543
Broe M et al (2003) Staging disease severity in pathologically confirmed cases of frontotemporal dementia. Neurology 60(6):1005–1011
Kwiatkowski TJ Jr et al (2009) Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323(5918):1205–1208
Vance C et al (2009) Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323(5918):1208–1211
Neumann M et al (2009) Abundant FUS-immunoreactive pathology in neuronal intermediate filament inclusion disease. Acta Neuropathol
Holm IE et al (2007) A reassessment of the neuropathology of frontotemporal dementia linked to chromosome 3. J Neuropathol Exp Neurol 66(10):884–891
Cairns NJ et al (2004) Clinical and neuropathologic variation in neuronal intermediate filament inclusion disease. Neurology 63(8):1376–1384
Yokota O et al (2008) Basophilic inclusion body disease and neuronal intermediate filament inclusion disease: a comparative clinicopathological study. Acta Neuropathol 115(5):561–575
Mackenzie IR et al (2007) Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann Neurol 61(5):427–434
Mackenzie IR (2007) The neuropathology and clinical phenotype of FTD with progranulin mutations. Acta Neuropathol 114(1):49–54
Bronner IF et al (2007) Progranulin mutations in Dutch familial frontotemporal lobar degeneration. Eur J Hum Genet 15(3):369–374
Sampathu DM et al (2006) Pathological heterogeneity of frontotemporal lobar degeneration with ubiquitin-positive inclusions delineated by ubiquitin immunohistochemistry and novel monoclonal antibodies. Am J Pathol 169(4):1343–1352
Ramesh Babu J et al (2008) Genetic inactivation of p62 leads to accumulation of hyperphosphorylated tau and neurodegeneration. J Neurochem 106(1):107–120
Acknowledgments
The authors would like to thank Michiel Kooreman and Afra van den Berg of the Netherlands Brain Bank for technical assistance, and Ruud Koppenol and Tom de Vries Lentsch for photography. The authors would also like to thank the patients and their families for participating in this study. This study received financial support from the Hersenstichting (project. 13F05(2).14) and Stichting Dioraphte (grantnumber 07 01 05).
Conflict of interest statement
None.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Seelaar, H., Klijnsma, K.Y., de Koning, I. et al. Frequency of ubiquitin and FUS-positive, TDP-43-negative frontotemporal lobar degeneration. J Neurol 257, 747–753 (2010). https://doi.org/10.1007/s00415-009-5404-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-009-5404-z