
Abstract
Sudden unexplained death (SUD) takes up a considerable part in overall sudden death cases, especially in adolescents and young adults. During the past decade, many channelopathy- and cardiomyopathy-associated single nucleotide variants (SNVs) have been identified in SUD studies by means of postmortem molecular autopsy, yet the number of cases that remain inconclusive is still high. Recent studies had suggested that structural variants (SVs) might play an important role in SUD, but there is no consensus on the impact of SVs on inherited cardiac diseases. In this study, we searched for potentially pathogenic SVs in 244 genes associated with cardiac diseases. Whole-exome sequencing and appropriate data analysis were performed in 45 SUD cases. Re-analysis of the exome data according to the current ACMG guidelines identified 14 pathogenic or likely pathogenic variants in 10 (22.2%) out of the 45 SUD cases, whereof 2 (4.4%) individuals had variants with likely functional effects in the channelopathy-associated genes SCN5A and TRDN and 1 (2.2%) individual in the cardiomyopathy-associated gene DTNA. In addition, 18 structural variants (SVs) were identified in 15 out of the 45 individuals. Two SVs with likely functional impairment were found in the coding regions of PDSS2 and TRPM4 in 2 SUD cases (4.4%). Both were identified as heterozygous deletions, which were confirmed by multiplex ligation-dependent probe amplification. In conclusion, our findings support that SVs could contribute to the pathology of the sudden death event in some of the cases and therefore should be investigated on a routine basis in suspected SUD cases.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Sudden death events in young individuals often represent the first manifestation of an undetected genetic disease, which remained without any symptoms during lifetime. Although some of the deceased show cardiac structural abnormalities and can be explained by autopsy, approximately 30% of sudden death cases still remain elusive and are therefore termed as sudden unexplained death (SUD) [1, 2]. In the past years, postmortem molecular autopsy enabled the identification of hidden cardiac diseases in a series of SUD cases. These diseases include mainly channelopathies or cardiomyopathies associated with single nucleotide variants (SNVs) located in the coding regions of functional genes [3,4,5,6]. In addition, some variants located in noncoding regions, such as regulatory elements and splice sites, might be accountable for serious functional effects [7,8,9,10]. Recent studies revealed that structural variants (SVs), including copy number variations (CNVs), might play an important role in SUD and could explain some of the unresolved cases [11,12,13,14].
SVs include diverse genomic alterations like deletions, duplications, insertions, inversions, translocations, or complex rearrangements of relatively large segments such as tandem exon duplications, or complex gains or losses of homologues sequences at multiple sites in the genome, commonly referred to as CNVs [15, 16]. While SVs are considerably less common than SNVs, they have greater functional potential due to their larger size, and they are more likely to alter gene structure [17]. SVs and CNVs can influence gene expression, phenotypic variation, and adaptation, by disrupting genes and altering gene dosage. Many SVs are described to confer risk to complex disease traits or increase disease susceptibility to e.g. HIV-1 infection [18] or neurodevelopmental disorders including autism [19], schizophrenia [20], and depression [21]. Besides, pathogenic CNVs have been reported in cardiac diseases such as Brugada syndrome (BrS) [11], arrhythmogenic right ventricular cardiomyopathy (ARVC) [12], or sudden cardiac death (SCD)-related cases [13, 14]. One previous study examined CNVs in a large cohort of SUD cases and patients that had an inherited cardiac disease and discovered that the frequency of identified CNVs varied from 1.4 to 5.1% among cases with different underlying diseases [22]. Another study investigated a small cohort of 13 sudden arrhythmic death syndrome (SADS) and sudden unexplained death in infancy (SUDI) cases with whole genome sequencing and whole transcriptome sequencing. However, they did not find any SVs in the coding and regulatory regions of 100 cardiac genes [23]. A recent study focused on 2 large Amish families with multiple sudden deaths and sudden cardiac arrests in young individuals and identified a homozygous multi-exon duplication in RYR2 [24]. Another study investigated a Tunisian family with high incidence of SCD in young family members, but did not find any pathological relevant CNVs [25]. The lack of a consensus of the impact of SVs on inherited cardiac diseases emphasized the need of more evidence on this topic.
The aim of this study was to screen for potentially pathogenic SVs in 244 genes associated with cardiac diseases in a cohort of 45 SUD cases. Whole-exome sequencing (WES) in 35 out of these 45 cases has already been performed earlier [4], whereas 10 additional SUD cases were analyzed as part of this study. Due to updates in the recommendations for rare variant classification, the exome sequencing data of all 45 SUD cases was analyzed and classified according to the current ACMG guidelines [26].
Materials and methods
SUD cohort
All SUD cases had been autopsied at the Zurich Institute of Forensic Medicine (ZIFM) in Switzerland between 2012 and 2019. The sudden death victims were examined according to the respective European and Swiss guidelines for the management of young SUD cases [27,28,29], including a thorough death scene investigation, a complete autopsy with pharmacological-toxicological and histopathological screening, and a review of the clinical history. The SUD cohort consisted of 45 cases with a mean age of 30.2 ± 14.5 years (range: 1–63 years of age) (Table S1). Seventy-five percent of the deceased were males and most of them were of European origin (89%). Another 5 cases (11.1%) were of African (3 cases), Indian (1 case), and Chinese (1 case) origin. Exome sequencing data was already available for 35 of the SUD cases [4]. Additional 10 SUD cases were analyzed according to the workflow described in our previous study [4].
Exome sequencing and bioinformatics
Genomic DNA of the 10 new SUD cases was obtained from shock-frozen kidney tissue. DNA extraction was performed using the QIAamp DNA Mini Kit (Qiagen, Hombrechtikon, Switzerland) according to the manufacturer’s protocol. DNA quantities were determined with a Quantus™ Fluorometer (Promega, Dübendorf, Switzerland). The SureSelectXT target enrichment and SureSelectXT All Exon V5 + UTR kits (Agilent Technologies AG, Basel, Switzerland) were used for DNA library preparation and exome capture, followed by quality and quantity assessment with the TapeStation system using DNA 1000 ScreenTapes (Agilent Technologies AG). DNA libraries were sequenced on an Illumina NovaSeq 6000 instrument (Illumina Inc., San Diego, CA, USA) with 150 paired-end reads and a mean coverage of 65 × per sample. Sequences were aligned to the reference genome (GRCh37/hg19) using BWA [30] and samples were required to have at least 80% of the exome covered at ≥ 20 × read depth. Variant discovery was performed by means of GATK [31], following the GATK best practice workflow [32].
SNV identification
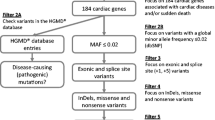
Data analysis was confined to a target gene panel consisting of 244 genes associated with cardiac diseases (Table S2). The annotation of the VCF files was performed with the software Alamut Batch, version 1.11 (Interactive Biosoftware, Rouen, France) and the visualization of the coverage of SNVs was done with the software IGV, version 2.4.16 [33]. SNV screening was performed according to an updated version of our previously published in-house filtering strategy [4] and to the ACMG standards and guidelines for the interpretation of sequence variants [26]. Our filter strategy selects SNVs with a stringent global minor allele frequency value (MAF) of less than or equal to 0.005% derived from the genome aggregation database (gnomAD) which is the largest available human database, including 125,748 exomes and 15,708 whole genomes from unrelated individuals of different ethnicities, sequenced as part of various population genetic studies [34]. In addition, synonymous and intronic variants were excluded, focusing only on exonic and splice site SNVs. Pathogenicity of the SNVs was assigned based on the evaluation of variant types (null variants, frameshift variants, splice site variants, or missense variants) and on the genome interpretation database VarSome [35]. Identified SNVs have been submitted to the Leiden Open Variation Database (Individuals Nr. 00,315,499–00,315,504 and 00,324,655–00,324,658) (http://databases.lovd.nl).
SV identification
SVs were called using LUMPY [15]. Briefly, discordant paired-end alignments were retrieved using samtools [36] (samtools view -b -F 1294) and the split-read alignments were extracted using the LUMPY auxiliary script extractSplitReads_BwaMem. The discordant paired-end and the split-read file were then parsed, together with the alignment (bam) file to lumpyexpress with default settings. The resulting VCF files were annotated using ANNOVAR [37] using the UCSC human genome version GRCh37/hg19 as reference and the Database of Genomic Variants (DGV) as variants database [38]. Among the SVs within our target genes, those identified in more than one sample were excluded from subsequent analysis, since pathogenic SVs that contribute to SUD should be rare variants according to the ACMG guidelines [26]. Furthermore, SVs longer than 1 Mbp were excluded because in these cases, the expression levels of too many genes were considered abnormal, which makes them difficult to validate. The remaining SVs were manually checked using the IGV software; only those with a characteristic gene expression alteration were considered eligible SV candidates. Allele frequencies of the candidate SVs were checked in the gnomAD-SV database [45].
MLPA validation
The identified candidate SVs were confirmed by multiplex ligation-dependent probe amplification (MLPA) according to the manufacturer’s instructions. Self-designed oligos for the detection of our target regions were synthesized by Microsynth (Microsynth AG, Balgach, Switzerland). In brief, 100 ng of DNA and a customized probemix consisting of synthetic oligos (Table S3) and the P200 reference probemix (MRC Holland, Amsterdam, Netherlands) were used in the hybridization reaction. Fragment separation was performed on a 3130xl Genetic Analyzer (Thermo Fisher Scientific, Reinach, Switzerland). Fragment analysis and comparative analysis results were generated using Coffalyser.Net software (MRC Holland).
Results
SNV identification
Following the current ACMG guidelines for variant interpretation [26], 14 pathogenic or likely pathogenic SNVs were identified in 10 (22.2%) out of the 45 SUD cases (Table 1 and Table S4). These variants were located in genes that are linked to cardiomyopathies (1 SUD case), ion channelopathies (2 SUD cases), connective tissue diseases and/or congenital malformation syndromes (3 SUD cases), and metabolic diseases (4 SUD cases). Out of these 14 SNVs, 2 heterozygous stop-gain variants (DTNA, NM_001390.4, c.2224C > T, p.(Gln742*) and LZTR1, NM_006767.4, c.2440C > T, p.(Gln814*)), 1 heterozygous two-base duplication (CALR3, NM_145046.4, c.387dup, p.(Ile130Tyrfs*11)), 1 homozygous one-base deletion (LZTR1, NM_006767.4, c.604_605del, p.(Met202Valfs*57)), and 1 heterozygous missense variant (SCN5A, NM_001099404.1, c.2204C > A, p.(Ala735Glu)) were predicted as pathogenic. In addition, 2 heterozygous stop-gain variants (ALMS1, ENST00000264448.6, c.54_55insTAG, p.(Glu19*) and MLYCD, NM_012213.3, c.1073C > A, p.(Ser358*)), 2 heterozygous deletions (SLC37A4, ENST00000357590.5, c.528del, p.(Val177Trpfs*35) and TRDN, NM_006073.4, c.1923_1924del, p.(Leu643Serfs*19)), and 5 heterozygous missense variants (ACADS, NM_000017.4, c.320G > A, p.(Arg107His); ACADS, NM_000017.4, c.814C > T, p.(Arg272Cys); SOS1, NM_005633.3, c.2728G > T, p.(Asp910Tyr); FBN2, NM_001999.4, c.3437A > G, p.(Tyr1146Cys) and ANK2, NM_001148.5, c.4158C > G, p.(Phe1386Leu)) were predicted as likely pathogenic.
In general, the re-analysis and re-classification of our recently published exome data of the 35 SUD cases [4] caused a change in the classification of the pathogenicity in the previously reported 11 variants (Table S5). Six of the 11 previously reported variants in the genes ACAD9 (p.(Arg420Cys)), AKAP9 (p.(Asn2045Ser)), FBN2 (p.(Gln2432His)), MYLK (p.(Arg1250His)), SEMA3A (p.(Arg66Trp)), and RYR2 (p.(Glu1127Gly)) had a MAF greater than 0.005% and were therefore filtered out. The pathogenicity of the remaining 5 variants were down-classified from probably pathogenic (BMPR2 (p.(Pro864Leu)) and KCN5E (p.(Tyr62Asn))) or likely pathogenic (EFEMP2 (p.(Arg185His)), RYR2 (p.(Ala3814Val)), and RYR2 (p.(Gln4164Glu)) to uncertain significance (Table S5).
SV identification
After pre-screening and IGV check, a total of 18 SVs were identified in 15 out of the 45 SUD cases (Table 2), located in 17 different genes (ABCC9, CDH2, DMPK, DPP6, EFEMP2, FXN, GPD1L, KCNJ2, LAMA4, NOS1AP, PDLIM3, PDSS2, PPA2, PRKAG2, PRKG1, PTPN11, TRPM4). The lengths of these SVs varied from 35 to 6079 bp, and most of them were positioned in the intergenic or intronic regions of our target genes. Out of these 18 SVs, the most promising 2 were heterozygous deletions located in the coding regions of PDSS2 and TRPM4, respectively (Fig. S1).
The 71-bp deletion of PDSS2 (NM_020381.4: c.1009-4103_1009-4033del), which can only be identified in exon 3 of one gene isoform (transcript ID: ENST00000449027), was predicted to be a variant with uncertain significance. This SV was identified in an 11-year-old previously healthy girl, who was swimming in the lake when she suddenly disappeared in the water. A lifeguard observed the incident and rescued her and immediately started resuscitation. The emergency team diagnosed a ventricular fibrillation and she became defibrillated for a single event. In the children’s hospital, she had a serious derailment of the acid–base balance and the sugar metabolism and died shortly after. According to the forensic investigations, the girl was already unconscious when she disappeared under water. Histological examination of the heart tissue revealed pre-existing changes in the excitation conduction system of the heart in the area of the secondary pacemaker center of the heart, which could have triggered cardiac arrhythmia and unconsciousness leading to a cardiovascular arrest.
The 35-bp deletion in exon 11 (transcript ID: ENST00000252826) of TRPM4 (NM_017636.4: c.1459_1494del, p.(Lys487_Leu498del)) was predicted as pathogenic. This SV was detected in a 38-year-old male. He complained of discomfort after drinking some alcohol and was found dead some hours later in his bed. He did not have any medical history; however, his wife reported that he felt unusually tired and exhausted in the last 2 months prior to death. Autopsy investigation revealed an enlarged heart (520 g, 56% enlarged according to Zeek [40]) with thickening of the heart chamber wall muscle.
MLPA validation
MLPA validation was performed on the 2 most promising SVs in the coding regions of PDSS2 and TRPM4, respectively. According to the comparative analysis results of Coffalyser.Net, both SVs were confirmed to be heterozygous deletions (Fig. 1).

SVs confirmed by MLPA. Blue/green bars represent 95% confidence interval over the reference samples (N = 3), and dots with lines represent 95% confidence interval estimate for each probe. In our case, the 95% confidence intervals of a PDSS2 in SUD058 and b TRPM4 in SUD075 did not overlap, which suggests a heterozygous deletion
Discussion
Sudden unexplained death (SUD) takes up a considerable part in overall sudden death cases, especially in adolescents and young adults. During the past decade, many channelopathy- and cardiomyopathy-associated SNVs have been identified in SUD studies by means of postmortem molecular autopsy [3,4,5,6], yet the number of cases that remain inconclusive is still high. The aim of this study was to re-analyze the exome data of 45 SUD cases according to the current ACMG guidelines and to search for potentially pathogenic SNVs and SVs.
Exome sequencing data was already available for 35 SUD cases, whereas additional 10 SUD cases were sequenced and analyzed for rare SNVs within the scope of this study. Following the current ACMG guidelines for variant interpretation [26], we re-analyzed and re-classified the SNVs in the 35 SUD cases in addition to the 10 newly sequenced SUD cases. A total of 14 pathogenic or likely pathogenic variants were identified in 10 (22.2%) of the 45 SUD cases. Out of these 10 cases, 1 individual (2.2%) carried a pathogenic variant in the cardiomyopathy-associated gene DTNA and 2 individuals (4.4%) carried 2 (1 pathogenic and 1 likely pathogenic) variants in the channelopathy-associated genes SCN5A and TRDN, respectively. Variants in the alpha-dystrobrevin encoding gene DTNA have been reported in patients with congenital heart disease and left ventricular non-compaction (LVNC) [41]. The SCN5A gene encodes the alpha subunit of the main cardiac sodium channel Nav1.5 and variants in this gene have been found to be causatively associated with BrS, long QT syndrome, cardiac conduction system dysfunction, and dilated cardiomyopathy [42]. Triadin-1, encoded by TRDN, is an important component of the calcium release unit in the sarcoplasmic reticulum of cardiac myocytes and a number of variants have been identified in patients with catecholaminergic polymorphic ventricular tachycardia (CPVT) [43].
Other studies already pointed out the importance of re-analysis of sequencing data, especially in patients with complex genetic diseases, as genomic databases are continuously updated and adjusted based on new findings in co-segregation studies and functional analyses [44]. Accordingly, when comparing our previously published data and the results presented in this study, some variants were interpreted differently and some new variants popped up. These changes are caused by the updated candidate gene list, a more stringent MAF as recommend by Tester et al. [45] and the assessment of pathogenicity based on the genome interpretation database VarSome that includes additional interpretation criteria, such as if the variant is located in a mutational hot spot and/or critical and well-established functional domain [35]. Nevertheless, many variants remain with uncertain significance emphasizing the importance of cautious interpretation, especially in cases without a clear phenotype and a complex genetic contribution. In addition, co-segregation analysis and functional assays are recommended to further evaluate the pathogenicity of identified variants.
The focus of this study was a genetic screening for SVs as potential contribution to the sudden unexpected death event. A total of 18 SVs were identified in 15 out of the 45 individuals, but only 2 (11.1%) were located on exons. The 2 exonic SVs were confirmed by MLPA as heterozygous deletions. The functional annotation of the 2 SVs was checked in several databases.
The 71-bp heterozygous deletion in PDSS2 was previously identified as nsv4140011 by whole genome sequencing (WGS) in the gnomAD structural variants study [39]. However, neither validation information nor clinical assertion has been reported for this SV. According to the Database of Genomic Variants (DGV) [38], the deletion type was only observed once within 10,847 samples. Such a low frequency could meet the criteria for a pathogenic SV. The protein encoded by PDSS2 is an enzyme that synthesizes the prenyl side chain of coenzyme Q10, which is one of the key elements in the respiratory chain. Previous research has revealed that individuals with primary coenzyme Q10 deficiency could have manifestations associated with multisystemic diseases, including encephalopathy and hypertrophic cardiomyopathy [46]. Therefore, we have reason to suspect SVs that alter the function or expression of this gene might be pathogenic variants. In addition, it is noteworthy that a missense mutation with uncertain significance in RYR2 (p.(Ala3814Val)) has also been found in this individual. However, it is not clear what the biological effects of these genetic variants are and whether they may have contributed to the cause of death in this young girl.
The other 35 bp heterozygous deletion in TRPM4 was reported to have conflicting interpretations of pathogenicity (benign, likely benign, or uncertain significance) according to the NCBI ClinVar database. As a calcium-activated ion channel encoding gene, TRPM4 had already been extensively studied in a series of channelopathy-related reports, and some uncommon missense SNVs had been identified to be likely pathogenic in 20 out of 248 BrS patients and in 13 out of 330 SUD cases, respectively [47, 48]. One study had demonstrated in-frame deletions in individuals with cardiac conduction disturbances, but the deletions co-existed with other missense variants, which makes it hard to determine their real functional impact [49]. In our case, beside the heterozygous deletion in TRPM4, a pathogenic stop variant in the gene LZTR1 (p.(Gln814*)) was identified. Variants in LZTR1 are associated with Noonan syndrome, which is a genetic disorder that causes multiple congenital abnormalities and characteristic facial features that evolve with age [50]. Furthermore, a small portion of patients with Noonan syndrome were reported to show cardiovascular diseases, including atrial septal defects and hypertrophic cardiomyopathy [51].
It is worth noting that a large proportion of SVs identified in this study were located in the intergenic or intronic regions of our target genes and thus were of unknown significance. The evaluation of variants in these noncoding regions has always been challenging as the knowledge about their contribution to electrophysiological dysfunction is still very limited. Introns are usually considered to contribute to the control of gene expression if regulatory regions and noncoding functional RNA genes are affected [52,53,54]. A recent study combined the most extensive maps of CNVs in human populations and discovered that intronic losses are the most frequent CNVs in protein-coding genes [55]. Therefore, the significance of SVs identified in the intronic regions of our target genes might need to be carefully evaluated by functional studies. Moreover, a recent study has cross-referenced human transcriptome, epigenomic, and chromatin datasets to find causal genetic variants in noncoding regions that alter the functionality of transcription regulatory elements and target gene expression associated with atrial fibrillation (AF) [56]. With an improved ability to identify these genetic variants neglected by most previous studies, the pathogenic mechanism behind SUD might eventually be better explained by routine SNV and SV testing in suspected SUD cases.
There are some limitations in our current study. Since we only focused on 244 cardiac-related genes, variants outside these regions could not be identified. Besides, SVs longer than 1 Mbp were not included in our candidate list due to the difficulties in confirming the MPS result. In addition, functional studies would be required to further investigate the 2 identified SVs in PDSS2 and TRPM4 in order to verify their potential pathogenic role and contribution to the sudden death event of these 2 SUD cases. When several potential pathogenic variants are under consideration, it will be important to find out which of the detected variants contributed most to the sudden unexpected death event. In our study, family members were not available for co-segregation analyses. This would be necessary to determine the mode of inheritance and to identify other family members at risk for sudden cardiac death.
In conclusion, our study supports that SVs in cardiac disease-associated genes might be involved in some SUD cases. However, the functional interpretation of pathogenic SVs is complex and genetic evidence should be used cautiously in molecular diagnosis.
Change history
06 May 2021
In Table 2, the 44 in the header MAF (gnom-AD-SV) should be superscript. This is now correctly presented.
References
Ackerman MJ et al (2011) HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies: this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Europace 13(8):1077–1109. https://doi.org/10.1093/europace/eur245
Markwerth P et al (2020) Sudden cardiac death-update. Int J Legal Med. https://doi.org/10.1007/s00414-020-02481-z
Narula N et al (2014) Post-mortem whole exome sequencing with gene-specific analysis for autopsy-negative sudden unexplained death in the young: a case series. Pediatr Cardiol 36(4):768–778. https://doi.org/10.1007/s00246-014-1082-4
Neubauer J et al (2018) Exome analysis in 34 sudden unexplained death (SUD) victims mainly identified variants in channelopathy-associated genes. Int J Legal Med 132(4):1057–1065. https://doi.org/10.1007/s00414-018-1775-y
Anderson JH et al (2016) Whole-exome molecular autopsy after exertion-related sudden unexplained death in the young. Circ Cardiovasc Genet 9(3):260–265. https://doi.org/10.1161/CIRCGENETICS.115.001370
Christiansen SL et al (2016) Genetic investigation of 100 heart genes in sudden unexplained death victims in a forensic setting. Eur J Hum Genet 24(12):1797–1802. https://doi.org/10.1038/ejhg.2016.118
Bezzina Connie R et al (2006) Common sodium channel promoter haplotype in Asian subjects underlies variability in cardiac conduction. Circulation 113(3):338–344. https://doi.org/10.1161/CIRCULATIONAHA.105.580811
Park JK et al (2012) Genetic variants in SCN5A promoter are associated with arrhythmia phenotype severity in patients with heterozygous loss-of-function mutation. Heart Rhythm 9(7):1090–1096. https://doi.org/10.1016/j.hrthm.2012.02.023
Zhao Y et al (2015) Post-transcriptional regulation of cardiac sodium channel gene SCN5A expression and function by miR-192–5p. Biochim Biophys Acta 1852(10 Pt A):2024–2034
Wang S et al (2017) An insertion/deletion polymorphism within 3′UTR of RYR2 modulates sudden unexplained death risk in Chinese populations. Forensic Sci Int 270:165–172. https://doi.org/10.1016/j.forsciint.2016.12.005
Sonoda K et al (2018) Copy number variations of SCN5A in Brugada syndrome. Heart Rhythm. https://doi.org/10.1016/j.hrthm.2018.03.033
Li Mura IE et al (2013) Identification of a PKP2 gene deletion in a family with arrhythmogenic right ventricular cardiomyopathy. Eur J Hum Genet 21(11):1226–1231. https://doi.org/10.1038/ejhg.2013.39
Mates J et al (2020) Sudden cardiac death and copy number variants: what do we know after 10 years of genetic analysis? FSI:Genetics 47. https://doi.org/10.1016/j.fsigen.2020.102281
Tester DJ et al (2020) Identification of a novel homozygous multi-exon duplication in RYR2 among children with exertion-related unexplained sudden deaths in the Amish community. JAMA Cardiol. https://doi.org/10.1001/jamacardio.2019.5400
Layer RM et al (2014) LUMPY: a probabilistic framework for structural variant discovery. Genome Biol 15(6):R84. https://doi.org/10.1186/gb-2014-15-6-r84
Sharp AJ et al (2005) Segmental duplications and copy-number variation in the human genome. The American Journal of Human Genetics 77(1):78–88. https://doi.org/10.1086/431652
Redon R et al (2006) Global variation in copy number in the human genome. Nature 444(7118):444–454. https://doi.org/10.1038/nature05329
Nakajima T et al (2008) HIV-1/AIDS susceptibility and copy number variation in CCL3L1, a gene encoding a natural ligand for HIV-1 co-receptor CCR5. Cytogenet Genome Res 123(1–4):156–160. https://doi.org/10.1159/000184703
Glessner JT et al (2009) Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature 459(7246):569–573. https://doi.org/10.1038/nature07953
Glessner JT et al (2010) Strong synaptic transmission impact by copy number variations in schizophrenia. Proc Natl Acad Sci U S A 107(23):10584–10589. https://doi.org/10.1073/pnas.1000274107
Glessner JT et al (2010) Duplication of the SLIT3 locus on 5q35.1 predisposes to major depressive disorder. PLoS One 5(12):e15463. https://doi.org/10.1371/journal.pone.0015463
Mates J et al (2018) Role of copy number variants in sudden cardiac death and related diseases: genetic analysis and translation into clinical practice. Eur J Hum Genet. https://doi.org/10.1038/s41431-018-0119-1
Andersen JD et al (2019) Whole genome and transcriptome sequencing of post-mortem cardiac tissues from sudden cardiac death victims identifies a gene regulatory variant in NEXN. Int J Legal Med 133(6):1699–1709. https://doi.org/10.1007/s00414-019-02127-9
Tester DJ et al (2020) Identification of a novel homozygous multi-exon duplication in RYR2 among children with exertion-related unexplained sudden deaths in the Amish community. JAMA Cardiology 5(3):13–18. https://doi.org/10.1001/jamacardio.2019.5400
Jaouadi H et al (2020) Multiallelic rare variants support an oligogenic origin of sudden cardiac death in the young. Herz. https://doi.org/10.1007/s00059-019-04883-1
Richards S et al (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17(5):405–424. https://doi.org/10.1038/gim.2015.30
Sabatasso S et al (2020) Second opinion system for sudden cardiac death cases in forensic practice. Int J Legal Med. https://doi.org/10.1007/s00414-019-02225-8
Medeiros Domingo A et al (2018) Recommendations for genetic testing and counselling after sudden cardiac death: practical aspects for Swiss practice. Swiss Med Wkly 148:w14638. https://doi.org/10.4414/smw.2018.14638
Fellmann F et al (2019) European recommendations integrating genetic testing into multidisciplinary management of sudden cardiac death. Eur J Hum Genet 27(12):1763–1773. https://doi.org/10.1038/s41431-019-0445-y
Li H, Durbin R (2010) Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26(5):589–595. https://doi.org/10.1093/bioinformatics/btp698
McKenna A et al (2010) The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20(9):1297–1303. https://doi.org/10.1101/gr.107524.110
DePristo MA et al (2011) A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43(5):491–498. https://doi.org/10.1038/ng.806
Robinson JT et al (2011) Integrative genomics viewer. Nat Biotechnol 29(1):24–26. https://doi.org/10.1038/nbt.1754
Wang Q et al (2020) Landscape of multi-nucleotide variants in 125,748 human exomes and 15,708 genomes. Nat Commun 11(1):2539. https://doi.org/10.1038/s41467-019-12438-5
Kopanos C et al (2019) VarSome: the human genomic variant search engine. Bioinformatics 35(11):1978–1980. https://doi.org/10.1093/bioinformatics/bty897
Li H et al (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics 25(16):2078–2079. https://doi.org/10.1093/bioinformatics/btp352
Wang K et al (2010) ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38(16):e164. https://doi.org/10.1093/nar/gkq603
MacDonald JR et al (2014) The Database of Genomic Variants: a curated collection of structural variation in the human genome. Nucleic Acids Res 42(Database issue):D986–D992. https://doi.org/10.1093/nar/gkt958
Collins RL et al (2020) A structural variation reference for medical and population genetics. Nature 581(7809):444–451. https://doi.org/10.1038/s41586-020-2287-8
Zeek PM (1942) Heart weight I. The weight of the normal human heart. Arch Pathol 34:820–832
Arbustini E et al (2016) Left ventricular noncompaction: a distinct genetic cardiomyopathy? J Am Coll Cardiol 68(9):949–966. https://doi.org/10.1016/j.jacc.2016.05.096
Li W et al (2018) SCN5A variants: association with cardiac disorders. Front Physiol 9:1372. https://doi.org/10.3389/fphys.2018.01372
Hancox JC et al. (2017) Triadin mutations - a cause of ventricular arrhythmias in children and young adults. J Congenital Cardiol 1(1). https://doi.org/10.1186/s40949-017-0011-9
Campuzano O et al (2020) Reanalysis and reclassification of rare genetic variants associated with inherited arrhythmogenic syndromes. EBio Med 54:102732. https://doi.org/10.1016/j.ebiom.2020.102732
Tester DJ et al (2018) Cardiac Genetic Predisposition in Sudden Infant Death Syndrome. J Am Coll Cardiol 71(11):1217–1227. https://doi.org/10.1016/j.jacc.2018.01.030
Salviati L et al (2017) Primary coenzyme Q10 deficiency. In: Gene reviews [Internet]. Seattle (WA): University of Washington, Seattle, 28125198
Liu H et al (2013) Molecular genetics and functional anomalies in a series of 248 Brugada cases with 11 mutations in the TRPM4 channel. PLoS ONE 8(1):e54131–e54131. https://doi.org/10.1371/journal.pone.0054131
Subbotina E et al (2018) Functional characterization of TRPM4 variants identified in sudden unexpected natural death. Forensic Sci Int 293:37–46. https://doi.org/10.1016/j.forsciint.2018.10.006
Stallmeyer B et al (2012) Mutational spectrum in the Ca2+-activated cation channel gene TRPM4 in patients with cardiac conductance disturbances. Hum Mutat 33(1):109–117. https://doi.org/10.1002/humu.21599
Romano AA et al (2010) Noonan syndrome: clinical features, diagnosis, and management guidelines. Pediatrics 126(4):746–759. https://doi.org/10.1542/peds.2009-3207
Prendiville TW et al (2014) Cardiovascular disease in Noonan syndrome. Arch Dis Child 99(7):629–634. https://doi.org/10.1136/archdischild-2013-305047
Rose AB (2008) Intron-Mediated Regulation of Gene Expression. In: Reddy ASN, Golovkin M (eds) Nuclear pre-mRNA Processing in Plants. Springer Berlin Heidelberg, Berlin, pp 277–290. https://doi.org/10.1007/978-3-540-76776-3_15
Le Hir H et al (2003) How introns influence and enhance eukaryotic gene expression. Trends Biochem Sci 28(4):215–220. https://doi.org/10.1016/S0968-0004(03)00052-5
Jo B-S, Choi SS (2015) Introns: the functional benefits of introns in genomes. Genomics Inform 13(4):112–118. https://doi.org/10.5808/GI.2015.13.4.112
Rigau M et al (2019) Intronic CNVs and gene expression variation in human populations. PLoS Genet 15(1):e1007902–e1007902. https://doi.org/10.1371/journal.pgen.1007902
van Ouwerkerk AF et al (2019) Identification of atrial fibrillation associated genes and functional non-coding variants. Nat Commun 10(1):4755–4755. https://doi.org/10.1038/s41467-019-12721-5
Acknowledgment
The authors would like to thank Dr. Urs Graf and Alessandro Maspoli from the Institute of Medical Molecular Genetics (University of Zurich, Switzerland) for their help in data interpretation of the exome data. The authors express their gratitude to the Emma Louise Kessler Foundation that supported this study.
Funding
Open Access funding provided by University of Zurich. The study was funded by the Emma Louise Kessler Foundation. The exome analysis was supported by the Swiss National Science Foundation (SNF, project-No. 320030-149456, 2013-2017). Shouyu Wang was financially supported by the fund of China Scholarship Council (Nr. 201906240236).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethics approval
Ethical approval for this study was provided by the local ethics committee in Zurich (KEK-ZH-Nr. 2013–0086), and the study was conducted in full conformance with Swiss laws and regulations. The requirements of the local ethics committee included written informed consent of family members. If no family members were available, SUD cases had been irreversibly anonymized.
Conflicts of interest
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Neubauer, J., Wang, S., Russo, G. et al. Re-evaluation of single nucleotide variants and identification of structural variants in a cohort of 45 sudden unexplained death cases. Int J Legal Med 135, 1341–1349 (2021). https://doi.org/10.1007/s00414-021-02580-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00414-021-02580-5