
Abstract
Microhaplotypes have become a new type of forensic marker with a great ability to identify and deconvolute mixtures because massively parallel sequencing (MPS) allows the alleles (haplotypes) of the multi-SNP loci to be determined directly for an individual. As originally defined, a microhaplotype locus is a short segment of DNA with two or more SNPs defining three or more haplotypes. The length is short enough, less than about 300 bp, that the read length of current MPS technology can produce a phase-known sequence of each chromosome of an individual. As part of the discovery phase of our studies, data on 130 microhaplotype loci with estimates of haplotype frequency data on 83 populations have been published. To provide a better picture of global allele frequency variation, we have now tested 13 more populations for 65 of the microhaplotype loci from among those with higher levels of inter-population gene frequency variation, including 8 loci not previously published. These loci provide clear distinctions among 6 biogeographic regions and provide some information distinguishing up to 10 clusters of populations.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Microhaplotypes have great ability to identify and deconvolute mixtures because massively parallel sequencing (MPS) allows the alleles (haplotypes) of the multi-SNP loci to be determined directly for an individual [1]. By 2013 [2], our interest in use of haplotypes focused on very short “microhaplotypes.” We have subsequently published on our developing set of microhaplotypes and the criteria for selecting the most useful microhaplotypes for mixture resolution [3,4,5]. As originally defined, a microhaplotype locus (short form, microhap) is a short segment of DNA with two or more SNPs defining three or more haplotypes at reasonable frequencies in a large part of the world. The loci are designed to be typed with MPS, which can determine, for sequences of up to about 300 bp, the specific combination of SNP alleles on each of the parental chromosomes of an individual. Thus, MPS provides phase-known data, in contrast to conventional Sanger sequencing, at any locus with two or more heterozygous SNPs. Microhaplotype loci have several desirable characteristics including, by definition, multiple alleles. Although most microhaps have fewer alleles than most short tandem repeat polymorphisms (STRPs), microhaps have the advantages over STRPs of very low mutation rates, absence of stutter, and the ability to multiplex large numbers of loci. Sets of microhaplotype loci can be optimized to be useful for individual identification, determining biological relationships, providing information on particular phenotypes, providing information on biogeographic ancestry, or, as noted above, deconvolution of a mixture.
Knowing the haplotype frequency variation around the world is important in determining how useful particular microhaplotypes will be for any one of those five uses in any specific population. Thus, it is important that multiple loci be characterized on as many populations as possible from as many regions of the world as possible. We recently published on 130 microhaplotypes that we have identified and have characterized in 83 populations from around the world [1]. In that paper, we noted the many ways that microhaps can be used but emphasized the value of microhaps for mixture deconvolution. To expand global characterization, we have now collected and analyzed new data on 5667 individuals for 198 SNPs that define 65 microhaplotypes in 13 additional populations, bringing the total from 83 to 96 populations for 65 microhaplotypes. With this broader geographic representation, we now are considering how well microhaps provide information on biogeographic ancestry.
Materials and methods
Populations
Figure 1 shows the geographic locations of the 96 populations (5667 individuals) including 13 new populations. (Two populations that have a cultural-religious basis but no recent single geographic location are omitted from the figure.) The full list of populations is given in Supplemental Table S1. The populations in the table are organized by geographic region. The table also includes the three-character abbreviations used in illustrations, and the unique sample identifier (UID) in the ALFRED database <https://alfred.med.yale.edu> [6, 7] for the description of each sample. The new population samples had only small amounts of DNA available and were chosen, in part, to provide somewhat more uniform sampling of populations around the world. All samples were collected with full informed consent per local law allowing studies such as this.

Geographic locations of 96 population samples
Selection of loci
To characterize the additional populations and emphasize ancestry inference, we chose loci with higher ranks by informativeness (I n) based on the 83 populations already evaluated for all 130 microhaplotypes and additional loci from among those loci subsequently characterized [1]. Eight loci not previously published are noted in Table 1. Availability of TaqMan assays already on hand in the laboratory determined which loci were specifically tested first. The current set of 65 loci involves 198 SNPs and represents an empiric balance of available assays and sufficient DNA. Additional loci may be tested on some of these populations in the future, but the available DNA has been exhausted for several of these “new” populations.
Genotyping
All markers were typed using TaqMan assays obtained from Thermo Fisher. The individuals with large amounts of DNA were typed following manufacturer’s protocols with reaction volumes reduced to 3 μl, run in 384-well plates, and read on an AB9700HT using Applied Biosystems’ SDS (sequence detection system) software. To maximize the number of SNPs that could be typed on the small amounts of DNA available, a pre-amplification protocol was employed as described [8].
Haplotyping
The haplotypes were estimated using phase version 2.1.1. [9, 10] as described previously [1]. This approach provides good allele frequency estimates for these reference populations.
Statistics
The effective number of alleles, A e, was calculated following Kidd and Speed [5]. Informativeness, I n, was calculated using the formula of Rosenberg et al. [11]. STRUCTURE analyses [12] were done for the full set of 96 populations and 65 loci with 10 independent runs at each K value. PCA was calculated with Addinsoft’s XLSTAT 2017.
Results
Table 1 lists the 8 previously unpublished loci and their definitions using the nomenclature for microhaplotypes we previously proposed [13]. The definitions of all 65 loci, including those previously published, are in ALFRED <alfred.med.yale.edu>. The allele frequencies for all 65 loci, including the 8 new loci, are now in ALFRED for all 96 populations along with the data on the rest of the original 130 microhaps [1]. Data on all these loci can be retrieved using the key word microhap on the ALFRED home page or on the drop down Search menu.
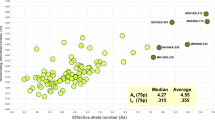
Table 2 lists all 65 microhaplotypes along with their A e and I n values, with the ranks from largest to smallest, using all 96 populations to calculate the statistics. Figure 2 is a scatterplot of the 65 loci by these two statistics. Comparison of the scatterplot in Fig. 2 with that in [1] shows that these 65 loci have proportionately fewer loci with I n less than 0.1 and A e less than 2.0.

Scatterplot of average effective allele number (A e) by informativeness (I n) for 65 microhaplotypes studied on 96 populations based on the values in Table 2
Figure 3 presents scatterplots from the principal components analysis (PCA) of the 96 populations based on their allele frequencies at the 65 loci. The first two PCs account for 39% of the variation (Fig. 3a); the major continental regions––Africa, Southwest Asia, Europe, South Central Asia, East Asia, Americas, and Pacific—clearly separate from one another. Figure 3b shows an enlarged view of the tighter cluster of populations with labels for the individual populations following the labels in Supplemental Table S1. The third PC accounts for an additional 9.28% (Fig. 3c) and separates the Native American populations more distinctly from the East Asian populations. The fourth PC (not shown) accounts for only an additional 4% of the variation and moves the Pacific Island populations away from East Asia.

Figure 4 shows population averages for STRUCTURE analyses at K = 7 and K = 10. At K = 7, except for small amounts of “noise,” the majority of Africans are assigned to a single cluster. The majority of East Asians are assigned to a single cluster, and the majority of the Native Americans are assigned to a single cluster. Three of the “Americas” populations from the 1000 Genomes project [14] are highly admixed in these analyses and are labeled as such. At K = 10, the populations farthest North (Eastern Siberia, Mongolia) in East Asia cluster together apart from other East Asians while the Sub-Saharan African populations subdivide into distinctive patterns for West Africa compared to Central and East African populations.

Estimated cluster membership values in STRUCTURE analyses for 96 populations at K = 7 and K = 10 (highest likelihood run results). See Supplemental Table S1 for population details
Comparison of the STRUCTURE results at K = 10 with Fig. 3b shows, by the two distinct statistics, that information on the finer relationships is present in the dataset.
Discussion
Forensic questions that can be addressed by microhaps include resolution of mixtures, identifying relatives, inferring ancestral origins, estimating phenotype, and individualization. In this study, we have emphasized ancestral origins, but we note that this set of 65 microhaplotypes spread across 21 human autosomes can be very useful for mixture deconvolution, familial inference, and individualization as well. Microhaplotypes can incorporate SNPs useful for estimating aspects of phenotype, but we have not considered that type of information. Our analyses of these 65 loci have focused on biogeographic ancestry. In future papers, we will present analyses of random match probabilities (RMP) and familial inference for sets of microhaps. We note that we have estimated the RMP for the reference CEU population sample for these 65 loci; the estimated RMP is between 10−55 and 10−56.
These 65 loci have different characteristics as measured by A e and I n. Those differences are shown in Fig. 2. We note that the two microhaps with the highest I n values (mh14KK-101 and mh02KK-003) are among the loci with the lowest A e values (ranks 60 and 56, respectively, Table 2). Examination of the haplotype frequencies shows that the global patterns are noticeably different and have large regions of the world with one haplotype at greater than 80% frequencies, albeit different haplotypes in different regions (Supplemental Fig. 2). The other six loci with I n > 0.35 show a range of A e values from nearly three to nearly six. Basically, these loci have multiple alleles everywhere but very different allele frequencies. We also note that some of the loci have low ranks for both measures.
The PCA in Fig. 3 shows that this set of microhaps distinguish among populations from some of the different biogeographic regions. Africa and the Americas are very clearly distinct. The Eastern and Northern of the Eurasian populations also fall into a loose cluster clearly distinct from European populations. What is interesting is how relatively close the European populations cluster in this global set of populations. On the other hand, this is not surprising because Europe is a geographically small area. Figure 3b makes clear how geographic boundaries do not reflect the genetic clustering of populations. The Komi from NW Siberia are clearly “European”; the Khanty from W Siberia are intermediate between European and the Northern Asian populations from Mongolia and Eastern Siberia.
The global dispersion is reflected in the STRUCTURE results in Fig. 4. At K=7, there are quite distinct biogeographic regions with intermediate populations showing partial assignment to flanking clusters. Europe and Southwest Asia are the exception in that a Southwest to Northeast cline of two clusters is seen. This is a pattern that has been seen with many sets of ancestry markers [e.g., 15, 16].
Our previous paper studying 130 loci on 83 populations [1] had 28 loci with A e > 3.0 giving a very high probability of identifying and resolving a mixture of two individuals; the current dataset has 22 loci in this range. By definition, these 22 loci have multiple alleles and relatively low frequencies for each of the alleles. These same attributes make these good for identifying relatives and make the likelihood of two unrelated individuals having the same multi-locus genotype vanishingly small.
Three of the new microhaps are in the region of SLC45A2. The coding SNP at SLC45A2 that is associated with skin color, rs16891982, is located on chromosome 5 at nt 33,951,588 (build 38) and shows a different global pattern of variation from even the closest of these three microhaps which is 17 kb away. The three microhaps also differ in their global patterns (Supplemental Fig. 1). At this stage, we are not choosing which is better since the different microhaps may differ in appropriateness for different purposes. The global variation is not identical, with I n values from a high of 0.299 (rank 15) for mh05KK-122 to 0.198 (rank 49) for mh05KK-123. In the context of 65 loci with multiple alleles, including all three will have little effect other than strengthening whatever pattern in STRUCTURE analyses is favored by that chromosomal region––Europe and SW Asia are distinct from East Asia.
Conclusions
The addition of 13 populations and emphasis on microhaplotype loci with higher I n values on average, compared to the 130 microhaps in [1], has shown that these 65 loci constitute a significant panel for ancestry inference. These results provide additional material for selecting panels of microhaplotypes optimized for different purposes.
That many of the loci also have high A e values argues that most of these loci have value for mixture deconvolution. The overall results support our previous findings [1] that many microhaplotypes have use for both ancestry inference and mixture deconvolution. The markers with A e > 3.0 are particularly good at mixture deconvolution, and the same logic indicates they will be very useful for familial relationships and individualization.
As we have been able to identify and characterize more microhaplotypes as part of our discovery phase, it has become possible to begin the laboratory studies to determine which loci will be the most robust with actual MPS typing. Our objective has been to identify and characterize a large number of loci with useful statistical characteristics such that molecular issues that may exclude some loci from MPS multiplexes will leave sufficient loci. These studies provide the necessary estimates of reference population allele frequencies. When actual multiplex kits are available, the identification of microhaplotype genotypes in individuals using MPS promises to be an important adjunct to forensic casework.
References
Kidd KK, Speed WC, Pakstis AJ, Podini DS, Lagace R, Chang J, Wootton S, Haigh E, Soundararajan U (2017) Evaluating 130 microhaplotypes across a global set of 83 populations. Forensic Sci Int Genet 29:29–37. https://doi.org/10.1016/j.fsigen.2017.03.014
Kidd KK, Pakstis AJ, Speed WC, Lagace R, Chang J, Wootton S, Ihuegbu N (2013) Microhaplotype loci are a powerful new type of forensic marker. Forensic Sci Int Genet Suppl Ser 4(1):e123–e124. https://doi.org/10.1016/j.fsigss.2013.10.063
Kidd KK, Pakstis AJ, Speed WC, Lagace R, Chang J, Wootton S, Haigh E, Kidd JR (2014) Current sequencing technology makes microhaplotypes a powerful new type of genetic marker for forensics. Forensic Sci Int Genet 12:215–224. https://doi.org/10.1016/j.fsigen.2014.06.014
Kidd KK, Speed WC, Wootton S, Lagace R, Langit R, Haigh E, Chang J, Pakstis AJ (2015) Genetic markers for massively parallel sequencing in forensics. Forensic Sci Int Genet Suppl Ser 5:e677–e679. https://doi.org/10.1016/j.fsigss.2015.12.004
Kidd KK, Speed WC (2015) Criteria for selecting microhaplotypes: mixture detection and deconvolution. Investig Genet 6(1):1. https://doi.org/10.1186/s13323-014-0018-3
Rajeevan H, Soundararajan U, Kidd JR, Pakstis AJ, Kidd KK (2012) ALFRED: an allele frequency resource for research and teaching. Nucleic Acids Res 40(D1):D1010–D1015. https://doi.org/10.1093/nar/gkr924
Osier MV, Cheung K-H, Kidd JR, Pakstis AJ, Miller PL, Kidd KK (2002) ALFRED: an allele frequency database for anthropology. Am J Phys Anthropol 119(1):77–83. https://doi.org/10.1002/ajpa.10094
Brissenden JE, Kidd JR, Evsanaa B, Togtokh AJ, Pakstis AJ, Friedlaender F, Kidd KK, Roscoe JM (2015) Mongolians in the genetic landscape of Central Asia: exploring the genetic relations among Mongolians and other world populations. Hum Biol 87(2):5–23
Stephens M, Smith NJ, Donnelly P (2001) A new statistical method for haplotype reconstruction from population data. Am J Hum Genet 68(4):978–989. https://doi.org/10.1086/319501
Stephens M, Scheet P (2005) Accounting for decay of linkage disequilibrium in haplotype inference and missing-data imputation. Am J Hum Genet 76(3):449–462. https://doi.org/10.1086/428594
Rosenberg NA, Li LM, Ward R, Pritchard JK (2003) Informativeness of genetic markers for inference of ancestry. Am J Hum Genet 73(6):1402–1422. https://doi.org/10.1086/380416
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155(2):945–959
Kidd KK (2016) Proposed nomenclature for microhaplotypes. Hum Genomics 10(1):16. https://doi.org/10.1186/s40246-016-0078-y.
1000 Genomes Consortium, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM et al (2015) A global reference for human genetic variation. Nature 526(7571):68–74. https://doi.org/10.1038/nature15393
Bulbul O, Cherni L, El-Khil-Khodjet H, Rajeevan H, Kidd KK (2016) Evaluating a subset of ancestry informative SNPs for discriminating among southwest Asian and circum-Mediterranean populations. Forensic Sci Int Genet 23:153–158. https://doi.org/10.1016/j.fsigen.2016.04.010
Pakstis AJ, Kang L, Liu L, Zhang Z, Jin T, Grigorenko EL, Wendt FR, Budowle B, Hadi S, Salam Al Qahtani M, Morling N, Smidt Morensen H, Themudo GE, Soundararajan U, Rajeevan H, Kidd JR, Kidd KK (2017) Increasing the reference populations for the 55 AISNP panel: the need and benefits. Int J Legal Med 131(4):913–917. https://doi.org/10.1007/s00414-016-1524-z
Acknowledgements
This work was funded primarily by NIJ grants 2013-DN-BX-K023, 2015-DN-BX-K023, and 2014-DN-BX-K030 awarded to KKK by the National Institute of Justice, Office of Justice Programs, U.S. Department of Justice. Funding to ELG is also acknowledged for grant #14.Z50.31.0027 from the government of the Russian Federation. Points of view in this presentation are those of the authors and do not necessarily represent the official position or policies of the U.S. Department of Justice. Special thanks are due to the many hundreds of individuals who volunteered to give blood or saliva samples for studies of gene frequency variation and to the many colleagues who helped collect the samples. In addition, some cell lines were obtained from the National Laboratory for the Genetics of Israeli Populations at Tel Aviv University, and African American samples were obtained from the Coriell Institute for Medical Research, Camden, New Jersey.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
ESM 1
(PDF 665 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Bulbul, O., Pakstis, A.J., Soundararajan, U. et al. Ancestry inference of 96 population samples using microhaplotypes. Int J Legal Med 132, 703–711 (2018). https://doi.org/10.1007/s00414-017-1748-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00414-017-1748-6