
Abstract
Chromosomal aberrations are often associated with incomplete genome duplication, for instance at common fragile sites, or as a consequence of chemical alterations in the DNA template that block replication forks. Studies of the cancer-prone disease Fanconi anaemia (FA) have provided important insights into the resolution of replication problems. The repair of interstrand DNA crosslinks induced by chemotherapy drugs is coupled with DNA replication and controlled by FA proteins. We discuss here the recent discovery of new FA-associated proteins and the development of new tractable repair systems that have dramatically improved our understanding of crosslink repair. We focus also on how FA proteins protect against replication failure in the context of fragile sites and on the identification of reactive metabolites that account for the development of Fanconi anaemia symptoms.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Fanconi anaemia
Fanconi anaemia (FA) was first recognised as a clinical entity by Guido Fanconi in 1927 (Fanconi 1927). The disease manifests with a variety of congenital abnormalities at birth and by a severe depression of all bone marrow-derived haematopoietic cell lineages (pancytopenia) during childhood (Tischkowitz and Hodgson 2003). Patients are at high risk of developing acute myeloid leukaemia and, in adulthood, squamous cell carcinomas of the gastrointestinal and of the female reproductive tracts (Alter et al. 2003). FA cells feature a high frequency of broken and radial chromosomes (Schroeder et al. 1964) and are highly sensitive to interstrand DNA crosslinks (ICLs) (Auerbach and Wolman 1976). Covalent bonds linking the two anti-parallel strands of DNA are extremely toxic, as they prevent DNA unwinding required for DNA replication and transcription. Hence, bi-functional adducts such as nitrogen mustards, mitomycin C or cisplatin are widely used in antitumor therapies (Deans and West 2011).
Fourteen FA genes have been identified to date, plus a 15th one, RAD51C, inactivated in one patient with multiple congenital anomalies (Vaz et al. 2010). The patient, however, had no symptoms of bone marrow failure or malignancies when diagnosed, so bi-allelic inactivation of RAD51C has been provisionally associated with a FA-like disorder. FA genes encode proteins implicated in an ubiquitin signalling pathway, in S phase checkpoint activation, in translesion DNA synthesis and in DNA double-strand break (DSB) repair by homologous recombination (Kee and D’Andrea 2010; Moldovan and D'Andrea 2009; Wang 2007).
The FA pathway
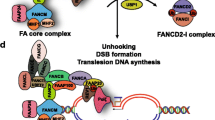
A basic molecular view of the FA pathway is depicted in Fig. 1. FANCA, FANCB, FANCC, FANCE, FANCF, FANCG, the E3 ligase enzyme FANCL and an associated protein of 100 kDa (FAAP100) form a nuclear, ubiquitin ligase FA core complex (Garcia-Higuera et al. 2001; Ling et al. 2007; Meetei et al. 2003a). The FA core complex associates with chromatin and the nuclear matrix during S phase, in a DNA damage-induced and FANCM-dependent manner (Kim et al. 2008; Mi and Kupfer 2004; Qiao et al. 2001). FANCM is a DNA translocase that can bind and move the branch point of three- and four-strand DNA structures akin to replication/repair intermediates (Gari et al. 2008a, b; Meetei et al. 2005; Xue et al. 2008). The DNA translocase forms a conserved DNA remodelling holoenzyme with the histone fold heterodimer MHF (Yan et al. 2010) and is assisted by the associated factor FAAP24 to regulate several independent aspects of the DNA damage response (Ciccia et al. 2007): (1) FANCM/FAAP24 interacts with the checkpoint protein HCLK2 and facilitates DNA damage signalling mediated by ATR (Collis et al. 2008); (2) FANCM associates with the Bloom’s complex (BLM-TopoIIIα-RMI1-RMI2) via direct contacts with the RMI1 and TopoIIIα subunits (Deans and West 2009; Meetei et al. 2003b); (3) FANCM/FAAP24 recruits the FA core complex to chromatin (Kim et al. 2008), through a direct FANCM–FANCF interaction (Deans and West 2009).

Schematic representation of FA proteins during ICL repair. FANCM recognises interstrand DNA crosslinks (in yellow) in association with FAAP24 and MHF and promotes ATR signalling of DNA damage. The FANCJ/BRIP1 helicase also facilitates ATR signalling of stalled replication forks. FANCM recruits the Bloom’s complex and the FA core complex at damaged sites. The FA core complex monoubiquitinates FANCD2/FANCI. Monoubiquitination of FANCD2 and FANCI stabilises the complex and promotes its retention at stalled forks. The FANCD2/FANCI complex contains two sets of dsDNA and ssDNA binding sites, which can bind and stabilise replication intermediates that result from converging replication forks. Ub-FANCD2/Ub-FANCI in turn recruits the structure-specific nuclease FAN1. Ubiquitination mediated by the FA core complex may also promote recruitment of SLX4/FANCP and associated nucleases. In the final stage of crosslink repair, BRCA2/FANCD1 and its partner PALB2/FANCN control double-strand DNA break repair by homologous recombination. For clarity, Fanconi anaemia proteins are in colour and associated factors are filled in grey
Genotoxic stress in S phase activates the FA core complex to monoubiquitinate FANCD2 and FANCI, two key interacting paralogs in the FA pathway (Dorsman et al. 2007; Garcia-Higuera et al. 2001; Sims et al. 2007; Smogorzewska et al. 2007). This signalling event has been proposed to stabilise the ubiquitinated FANCI–FANCD2 complex in chromatin, which in turn allows recruitment of DNA repair factors such as the nuclease FAN1 at damaged sites (Kratz et al. 2010; Liu et al. 2010; MacKay et al. 2010; Shereda et al. 2010; Smogorzewska et al. 2010; Stoepker et al. 2011; Yoshikiyo et al. 2010).
A number of posttranslational modifications regulate FA proteins: Phosphorylation of FANCI by ATR is a prerequisite for FANCD2 monoubiquitination (Ishiai et al. 2008). In mammalian cells, the phosphorylation of FANCD2 by ATR/ATM (T691 and S717) and by Chk1 (S331) is required for potent FANCD2 monoubiquitination and for resistance to DNA cross-linking agents (Ho et al. 2006; Zhi et al. 2009). Several phosphorylation steps also regulate FA core complex proteins. Phosphorylation of FANCA (S1449) by ATR, of FANCG (S7), and of FANCE (T346 and S374) by Chk1 all appears necessary for cellular tolerance to mitomycin C (Collins et al. 2008; Qiao et al. 2004; Wang et al. 2007).
An additional layer of complexity has emerged with the discovery that RAD18 contributes to the regulation of FANCD2 monoubiquitination (Geng et al. 2010; Palle and Vaziri 2011; Park et al. 2010a; Song et al. 2010b; Williams et al. 2011). RAD18 is an E3-ligase that monoubiquitinates proliferating cell nuclear antigen (PCNA) in response to replication fork stalling by bulky DNA damage (Prakash et al. 2005). PCNA functions as a scaffold for the recruitment of translesion DNA polymerases that bypass DNA lesions. In response to bulky DNA adducts, the monoubiquitination of PCNA by RAD18 and the subsequent recruitment of TLS polymerases are necessary for efficient FANCD2 monoubiquitination (Song et al. 2010b). In cells exposed to mitomycin C or camptothecin, however, the monoubiquitination of FANCD2 is independent of PCNA, yet depends on RAD18 (Palle and Vaziri 2011; Williams et al. 2011). How exactly RAD18’s ligase activity participates in the FA pathway remains elusive.
Deubiquitination of FANCD2 is under the control of USP1 (Kim et al. 2009; Nijman et al. 2005), in association with UAF1 (Cohn et al. 2007). USP1 binds FANCI directly via its SUMO-like domain SLD2, which associates with the SUMO-like domain-interacting motif (SIM) on FANCI (Yang et al. 2011). Removal of the ubiquitin moiety on FANCD2 and FANCI is necessary for completion of the DNA repair process and ICL tolerance (Kim et al. 2009).
FANCJ/BRIP1 interacts with BRCA1, unwinds DNA structures that block replication forks and facilitates checkpoint signalling (Bridge et al. 2005; Cantor et al. 2001; Gupta et al. 2005; Hiom 2010; Levitus et al. 2005; Litman et al. 2005). FANCJ/BRIP1 also forms a complex with BLM, and the two helicases can unwind DNA substrates synergistically (Suhasini et al. 2011). Some symptoms in FA-J patients may be linked to a deficiency in BLM protein, as BLM is unstable in the absence of FANCJ (Suhasini et al. 2011). BRCA2/FANCD1 and PALB2/N are key regulators of homologous recombination. BRCA2, the product of the breast cancer susceptibility gene, provides a structural platform for the fine regulation of the strand exchange protein RAD51 (for a recent review, see (Holloman 2011). Additional insights into structural, functional and phenotypic features of Fanconi anaemia are developed below.
FANCD2–FANCI and associated nucleases
Structure of FANCD2–FANCI
A new view of the FA pathway has been provided by the recent crystal structure of the mouse FANCD2–FANCI complex (Joo et al. 2011). Each protein is made of four solenoid segments and two interspersed helical domains that fold into a saxophone-like structure (cartooned in Fig. 1). FANCD2 and FANCI interact in an antiparallel manner along their saxophone body, forming a narrow and shallow platform (Joo et al. 2011). Electron density maps and FANCI-DNA crystals have revealed that the FANCD2/FANCI surface includes two sets of double- (dsDNA) and single- strand DNA (ssDNA) binding sites that could accommodate a replication intermediate comprising two converging forks at a crosslink (Fig. 1). This is consistent with earlier reports on the DNA-binding properties of FANCD2 and FANCI (Longerich et al. 2009; Park et al. 2005; Roques et al. 2009; Yuan et al. 2009). It is noteworthy that the purified FANCI–FANCD2 complex exhibits more affinity for branched DNA structures than either FANCD2 or FANCI proteins alone (Yuan et al. 2009). Three phosphorylation sites in FANCI as well as the FANCI and FANCD2 ubiquitination sites locate at the FANCD2–FANCI interface and are thought to stabilise the complex (Joo et al. 2011). Predictions suggest that the lysine–ubiquitin isopeptide bonds would lie within solvent free accessible tunnels from which the ubiquitin structural domain could emerge, on either side of the FANCD2–FANCI complex (Joo et al. 2011). This molecular view of FANCD2–FANCI shows how the complex may bind and protect stalled replication forks and control the recruitment of structure specific DNA endonucleases for coordinated unhooking of ICLs.
FAN1
The conjugation of FANCD2 with an ubiquitin moiety is not only a signal for the localisation and the retention of FANCD2 on chromatin but also for the recruitment of DNA repair nucleases. One such nuclease is FAN1, discovered independently by five groups that have reported congruent observations (Kratz et al. 2010; Liu et al. 2010; MacKay et al. 2010; Shereda et al. 2010; Smogorzewska et al. 2010): FAN1 stands for FANCD2-associated nuclease. The protein is necessary for chromosome stability and for cellular tolerance to cisplatin and mitomycin C. In cells treated with ICL-inducing agents, FAN1 forms nuclear foci that colocalise with FANCD2 and binds to the monoubiquitinated form of FANCD2. It contains an amino-terminal ubiquitin-binding motif, a RAD18-like CCHC zinc finger known as UBZ domain and a carboxy terminal VRR-nuc domain (virus-type replication-repair nuclease domain). The UBZ domain is both necessary and sufficient for interaction with FANCD2 and for targeting of FAN1 to DNA repair sites (Kratz et al. 2010; Liu et al. 2010; MacKay et al. 2010; Shereda et al. 2010). FAN1 exhibits 5′ flap endonuclease and 5′–3′ exonuclease activities and participates in DNA repair mechanisms dependent on homologous recombination (Kratz et al. 2010; Liu et al. 2010; MacKay et al. 2010; Smogorzewska et al. 2010). Together, these studies provide conclusive evidence of a strong link between Ub-FANCD2 and a structure specific endo/exo nuclease in ICL repair. Some nuances were brought in a follow-up study where the FAN1 locus was disrupted by gene targeting in chicken DT40 cells (Yoshikiyo et al. 2010). First, FA proteins and FAN1 appear to have also non-epistatic functions because DT40 cells lacking both FANCC and FAN1 are more sensitive to cisplatin than either single mutant. Second, unlike FA-deficient cells that exhibit proliferation defects, the growth kinetic of Fan1 −/− cells is similar to wild-type DT40 cells (Yoshikiyo et al. 2010). This may suggest that FAN1 does not function with FA proteins in the resolution of endogenous replication obstacles. Studies using defined repair systems (discussed below) will be necessary to understand precisely how FAN1 acts during repair of DNA damage induced by crosslinking agents.
SLX4
A second connexion with nucleases in the FA network emerged from the discovery of bi-allelic SLX4 mutations in six Fanconi anaemia patients (Kim et al. 2011; Stoepker et al. 2011). Based on this, SLX4 has been renamed Fanconi anaemia complementation group P (FANCP). With FANCM, FANCP is the second FA protein to be conserved in yeast. Inactivation of SLX4 sensitises cells to ICLs-inducing agents and to camptothecin, an inhibitor of topoisomerase 1. The discovery that SLX4 is a FA protein was accompanied with the description of a Slx4 −/− mouse model (Crossan et al. 2011). Mice lacking SLX4 exhibit a number of phenotypes akin to FA symptoms, including multiple developmental defects, reduced fertility as well as genetic instability and deficiencies in the hematopoietic compartment (Crossan et al. 2011). The protein is necessary for homologous recombination measured using a GFP reporter system that involves the repair of a DSB generated by I-SceI endonuclease (Munoz et al. 2009; Svendsen et al. 2009). It has been proposed that SLX4/FANCP may act a regulatory platform associated with multiple structure-specific endonucleases, namely, SLX1, XPF-ERCC1 and MUS81-EME1 (Andersen et al. 2009; Fekairi et al. 2009; Munoz et al. 2009; Svendsen et al. 2009). Whether and how SLX4 coordinates the activities of structure-specific nucleases is a heavily studied topic. SLX4 contains multiple protein–protein interaction modules: two amino terminal C2HC zinc finger (UBZ4) domains; a MEI9XPF-interaction-like region that binds XPF-ERCC1; a BTB/POZ protein–protein interaction domain; a SAP motif necessary for MUS81–EME1 binding; and a conserved C-terminal domain implicated in the interaction with SLX1 (Fekairi et al. 2009; Svendsen et al. 2009). The human SLX1–SLX4 complex exhibits 5′ flap endonuclease activity and resolves Holliday junctions (Fekairi et al. 2009; Munoz et al. 2009; Svendsen et al. 2009). That SLX1–SLX4 is a bona fide Holliday junction resolvase is substantiated by the demonstration that it produces directly “ligatable” nicked duplexes (Fekairi et al. 2009). MUS81–EME1 exhibits structure specificity for 3′ flaps, replication fork-like structures and Holliday junctions, whereas the nucleotide excision repair factor XPF-ERCC1 cleaves bubble and stem loop structures near the 5′ side of the junction between dsDNA and ssDNA, and will also act on 3′ flaps during single-strand annealing (Ciccia et al. 2008). These nucleases are most likely used in specific biological contexts. Expression of a recombinant SLX4 protein bearing a deletion of the SLX1 interaction motif complements the MMC sensitivity of Slx4 −/− mouse embryonic fibroblasts, whereas Slx4 −/− cells expressing a mutant SLX4 protein lacking the XPF-ERCC1 interaction motif remain MMC sensitive (Crossan et al. 2011). Consistently, sub-cellular fractionation studies revealed that, unlike SLX1 and MUS81-EME1, the chromatin association of XPF-ERCC1 was specifically impaired in a FA-P cell line (EUFA1354) (Stoepker et al. 2011). This was confirmed by immunofluorescent studies showing that the formation of ERCC1 nuclear foci was impaired in EUFA1354 cells (Stoepker et al. 2011). These observations show that the association of SLX4 with XPF-ERCC1 is critical for the DNA damage response mediated by FA proteins.
Unlike FAN1, SLX4 is essential for the proliferation of chicken DT40 cells, suggesting that it plays an important role in the repair of DNA lesions that arise spontaneously (Yamamoto et al. 2011). Chicken SLX4 is connected to the Fanconi anaemia network by its UBZ domain, which is necessary for tolerance to ICL-inducing agents. SLX4 forms ICLs-induced foci in DT40 cells and associates with FANCD2 in pull-down experiments. The recruitment of SLX4 at DNA repair sites depends on the integrity of its UBZ domain and on the Fanconi anaemia core complex (Yamamoto et al. 2011). The observation that the function of SLX4 in ICL repair depends on the FA ubiquitination pathway in chicken DT40 cells awaits confirmation from studies based on human cellular systems. Additional work is also necessary to determine whether SLX4 UBZ domain binds Ub-FANCD2 directly. Finally, Fancc −/Slx4 − UBZD double mutants are more sensitive to ICLs inducing agents than either single mutant, suggesting that SLX4 and the FA core complex have also non-epistatic roles in ICL repair (Yamamoto et al. 2011).
FA proteins promote DNA damage signalling
Replication obstacles and stalled forks are signalled by ATR, which detects RPA-covered single-stranded DNA that accumulate as a result of uncoupling of MCM helicases and DNA polymerases (Byun et al. 2005; Zou and Elledge 2003). The replication checkpoint is triggered efficiently by the recognition of primed single-stranded DNA with a free 5′ end (MacDougall et al. 2007). Single-stranded DNA coated by the single-strand DNA-binding protein RPA is bound directly by ATRIP (ATR-interacting protein), which enables the association of the ATRIP–ATR complex at stalled forks (Zou and Elledge 2003). The 5′ junction between ssDNA and dsDNA is recognised by RAD17-RFC, which loads the 9–1–1 checkpoint clamp necessary for efficient ATR activation (Ellison and Stillman 2003; Majka et al. 2006; Zou et al. 2003). FA proteins stimulate DNA damage signalling in multiple ways.
Role of FANCM
Collis et al. (2008) provided initial evidence that FANCM functions in ATR-mediated checkpoint signalling, independently of the Fanconi anaemia core complex. FANCM and FAAP24 associate with the checkpoint protein HCLK2. Depletion of FANCM in mammalian cells results in cellular features shared with ATR-defective cells, including spontaneous increase in DNA damage markers, increased frequency of cells with supernumary centrosomes and inefficient damage-induced Chk1 activation. Importantly, the integrity of the ATPase motif of FANCM is necessary for its role in checkpoint signalling (Collis et al. 2008; Huang et al. 2010). Subsequent studies confirmed that FANCM participates in Chk1 activation and limits replication-associated DNA damage (Luke-Glaser et al. 2010; Schwab et al. 2010). FANCM also promotes the retention of chicken TopBP1 in chromatin (Schwab et al. 2010) and the restart of stalled replication forks (Luke-Glaser et al. 2010; Schwab et al. 2010). Finally, Chk1 and FANCM protect each other from proteosomal degradation during DNA replication stress (Luke-Glaser et al. 2010). Together, these studies have unveiled a tight link between FANCM and S-phase checkpoint signalling.
Role of FANCJ/BRIP1
FANCJ/BRIP1 provides another link between Fanconi anaemia proteins and checkpoint signalling. The BRIP1 helicase interacts with TopBP1 (Gong et al. 2010). This interaction involves S-phase-specific phosphorylation of BRIP1 at Thr 1133 and the TopBP1 BRCT repeats 7 and 8, which undergo important conformational change upon binding with BRIP1 (Leung et al. 2011). In cells exposed to hydroxyurea, the helicase activity of BRIP1 and its interaction with TopBP1 are both required for the accumulation of RPA in chromatin, which in turn promotes the assembly of the ATR signalling complex and the phosphorylation of Chk1 (Gong et al. 2010). Hence, FANCJ/BRIP1 is likely to facilitate ATR activation by unwinding DNA at stalled forks.
Signalling from a DNA crosslink
As crosslinks prevent the separation of DNA strands, and RPA covered single-stranded DNA is a critical determinant of ATR activation, it raises the question of how DNA damage signalling is implemented at ICL-stalled forks. Two reports suggest that Fanconi anaemia proteins sense ICLs directly and promote an ICL-specific mode of ATR activation (Ben-Yehoyada et al. 2009; Huang et al. 2010). Specifically after exposure to ICL-inducing agents, FANCM and FAAP24 proteins are necessary for the accumulation and the phosphorylation of RPA32 at damaged sites (Huang et al. 2010). Interestingly, the formation of RPA foci induced by ICLs occurs in the absence of microscopically detectable single-stranded DNA foci. In vitro, FAAP24 binds ICL-damaged DNA preferentially and promotes the recruitment of RPA (Huang et al. 2010). In a cell-free extract from Xenopus eggs, plasmid DNA containing a single ICL can induce ATR signalling in the absence of DNA replication (Ben-Yehoyada et al. 2009). ICL-induced ATR activation depends on xFANCD2 and xFANCL (Ben-Yehoyada et al. 2009) in contrast to human cells that do not seem to rely on FA core complex proteins for ICL-signalling (Huang et al. 2010). Using episomal-based chromatin IP from HEK293 cells to detect proteins bound to a plasmid bearing a defined interstrand DNA crosslink, Shen et al. (2009) found that the FA core complex proteins and FANCD2 were enriched on the damaged plasmid compared with the unmodified substrate (Shen et al. 2009). The recruitment of FA proteins to the ICL was not dependent on the plasmid undergoing DNA replication (Shen et al. 2009), consistent with the observations that FA proteins bind crosslinked DNA and promote ATR signalling of ICLs independently of DNA replication (Ben-Yehoyada et al. 2009; Huang et al. 2010).
In conclusion, FA proteins seem to bind DNA crosslinks before replication forks reach the lesion and promote an ICL-specific mode of ATR activation that is mechanistically distinct from signal activation induced by uncoupling of MCM helicase and DNA polymerase activities.
Indirect role of FA proteins
More recently, a genome-wide screen for genes necessary to maintain cell cycle arrest after exposure to ionizing radiation revealed that homologous recombination and Fanconi anaemia proteins play a critical role in S-phase checkpoint activation (Cotta-Ramusino et al. 2011). Consistently, the homologous recombination protein RAD51C has been implicated in Chk2 activation and cell cycle arrest in response to DNA damage (Badie et al. 2009). The involvement of homologous recombination (HR) proteins in checkpoint signalling suggests that cells may sense ongoing DNA repair (Cotta-Ramusino et al. 2011). Consequently, the role of Fanconi anaemia protein in DNA damage signalling may also result from their function in channelling DSBs into repair by homologous recombination (Adamo et al. 2010; Pace et al. 2010). Consistent with this, inhibition of the non-homologous end-joining machinery restores most of the IR-induced signalling defects in cells lacking FANCM, FANCL and FANCJ (Cotta-Ramusino et al. 2011).
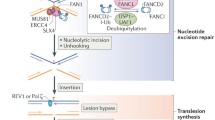
FA proteins promote replication-coupled ICL repair
The removal of interstrand DNA crosslinks mobilises an important part of the cellular DNA repair tool kit, including helicases, nucleases, translesion DNA polymerases, DNA recombinases, and a battery of posttranslational modifications and scaffold proteins, which orchestrate the sequential intervention of DNA processing enzymes. The development of experimentally tractable systems, using DNA plasmids damaged with a single ICL at a defined position, has provided powerful means to tackle ICL repair in Xenopus egg extracts and in mammalian cells. ICLs can be repaired in G0/G1 cells by the combined action of the nucleotide excision repair machinery that unhooks and excises the crosslink and translesion DNA polymerases that ensure DNA repair synthesis (Ben-Yehoyada et al. 2009; Sarkar et al. 2006; Shen et al. 2009). In S/G2, the repair of ICLs is coupled with DNA replication and depends on Fanconi anaemia proteins and homologous recombination (Fig. 2). The Fanconi anaemia pathway has long been thought to promote homologous recombination, but this has been difficult to demonstrate with the widely used GFP reporter mammalian system developed in the laboratory of Maria Jasin, which measures the repair of a double-strand DNA break introduced by I-SceI endonuclease (Moynahan et al. 2001). The Jasin’s laboratory has now modified the reporter system to demonstrate that the Fanconi anaemia pathway facilitates replication-coupled homologous recombination in mammalian cells (Nakanishi et al. 2011). The authors used a triplex-forming oligonucleotide (TFO) conjugate to introduce a psoralen interstrand DNA crosslink into the GFP reporter plasmid at a specific site. After crosslink formation, the TFO was removed by reduction of a disulphide bond that links the oligonucleotide and the psoralen moiety. To measure repair with or without replication, they simply used plasmids containing or not the OriP EBV replication origin. Whereas DNA replication had little impact on recombination repair induced by a DSB, ICL-induced homologous recombination was greatly stimulated when coupled to replication and strongly dependent on the integrity of the FA pathway (Nakanishi et al. 2011).

Replication-coupled, recombination dependent repair of ICLs. Two adjacent replication forks converging at the crosslink (green box) are represented. Single replication forks blocked at a crosslink can also be repaired. DNA synthesis is halted temporarily 20–40 nucleotides away from the lesion, before further extension to within one nucleotide from the ICL. The MCM2-7 accumulates at ICL-stalled forks, and removal of the replicative helicase may be necessary for crosslink repair. The RAD51 recombinase is recruited early during fork stalling, before nucleolytic incisions. The FA core complex and FANCD2–FANCI are recruited at ICLs and coordinate nucleolytic processing of one of the parental DNA strands to “unhook” the crosslink. REV1 may be responsible for the insertion of one nucleotide across the lesion, and DNA polymerase ζ extends the leading strand past the lesion. The restored duplex is used as a template for repair of the broken sister by homologous recombination
Replication-coupled ICL-repair in Xenopus egg extracts
Major advances in our understanding of the mechanism of replication-coupled ICL repair have been made in the laboratory of Johannes Walter, using plasmids containing either nitrogen-mustard like or cisplatin ICL and Xenopus egg extracts (Raschle et al. 2008). Plasmids bearing a defined ICL are replicated by sequential incubation into high-speed supernatant of egg cytoplasm followed by highly concentrated nucleoplasm egg extracts. This procedure promotes synchronous and efficient replication. As the plasmid is small, two replication forks rapidly converge and stall at the ICL after random initiation of bidirectional replication. Fork stalling at the ICL induces the activation of the ATR signalling pathway and the monoubiquitination of FANCD2, suggesting that the system supports the DNA damage response. A detailed examination of plasmid-ICL replication in this system revealed that the leading strand of both forks first pause 20–40 nucleotides from the ICL (Fig. 2). A recent study from the same laboratory has demonstrated that the replicative DNA helicase complex is a 3′–5′ ssDNA translocase that unwinds DNA by steric exclusion (Fu et al. 2011). During replication-coupled repair of crosslinked plasmids, the observed pausing site 20–40 nt from the ICL corresponds to the footprint of the stalled replicative DNA helicase, composed of the hexameric ATPase MCM2-7, CDC45 and GINS. After initial pausing, DNA synthesis proceeds from one of the two leading strands up to one nucleotide from the ICL. The disappearance of MCM7 from the ICL site correlates with the leading strand nearing towards the ICL, suggesting that displacement of the replicative helicase facilitates the resumption of DNA synthesis towards the ICL (Fu et al. 2011).
After DNA replication, nucleolytic digestions occur on one of the parental DNA strands to “unhook” the crosslink (Fig. 2). As a result, one sister chromatid is broken, while the other sister duplex is restored, first by insertion of a nucleotide opposite the adduct and then by DNA repair synthesis dependent on Pol ζ (Raschle et al. 2008).
The “unhooking” of the crosslink and the insertion of a nucleotide opposite the unhooked crosslink are mediated by ubiquitylated FANCD2 and FANCI (Knipscheer et al. 2009). Both events are abolished when the FANCD2–FANCI complex is removed from the extract or when depleted extracts are complemented with recombinant FANCD2 bearing a lysine to arginine substitution in its monoubiquitination site. It is likely that Ub-FANCD2/Ub-FANCI recruits structure-specific nucleases to coordinate incisions in one of the parental DNA strands. A recent study has shown that human SNM1A can digest ICL-containing DNA from a single 5′ nick (Wang et al. 2011). The data suggest that, in principle, an initial incision catalysed by XPF-ERCC1 would be sufficient to prime SNM1A exonucleolytic digestion of DNA past the ICL, yielding a gapped DNA intermediate with a covalently linked mononucleotide.
In DT40 cells, the Fanconi anaemia pathway interacts genetically with REV1 (Niedzwiedz et al. 2004), a dCMP transferase containing an ubiquitin-binding motif (Lehmann et al. 2007). Hence, ub-FANCD2/Ub-FANCI may also recruit REV1 to insert a nucleotide across the unhooked crosslink, followed by DNA polymerase ζ for DNA repair synthesis and restoration of one sister duplex (Raschle et al. 2008).
A recent replication-coupled repair study has used low speed supernatants from Xenopus eggs, which supports a single replication initiation event on plasmid DNA after chromatinization and assembly into pseudo nuclei (Le Breton et al. 2011). Whether both Raschle et al. (2008) and Le Breton et al. (2011) show that concerted incisions occur in one of the parental DNA strands, incisions are observed in the later study whether one or both forks have reached the ICL. Consistent with this, ICL-plasmids containing the origin of replication from Epstein–Barr virus are repaired when introduced in mammalian cells (Nakanishi et al. 2011). Since EBNA1 initiates unidirectional DNA replication at OriP, this shows that crosslink repair can be accomplished from a single stalled fork.
In the final stage of ICL repair, the restored duplex is used as a template for repair of the broken sister by homologous recombination. Remarkably, recombination-dependent ICL repair can be recapitulated in the Xenopus egg extract system: Repair of the broken sister duplex is RAD51 dependent and leads to the formation of a recombination intermediate containing a hemicatenane (Long et al. 2011). As expected, formation of the recombination intermediates requires the presence of FANCD2–FANCI. Chromatin immunoprecipitation analyses showed that RAD51 and FANCD2–FANCI accumulate near the ICL independently of each other. Interestingly, RAD51 is targeted to ICL-stalled fork before the formation of a double-strand DNA break. This is consistent with a role for RAD51 in the protection of nascent DNA strands from degradation at stalled forks (Hashimoto et al. 2010). A similar role has been attributed to BRCA2, which prevents MRE11-mediated degradation of nascent DNA (Schlacher et al. 2011). Fork protection by BRCA2 depends on a RAD51-binding site at the carboxyl terminus of BRCA2, which stabilises RAD51 nucleoprotein filaments. It is therefore possible that BRCA2/FANCD1 targets RAD51 to replication forks stalled at ICLs.
Pathway choice
HR and non-homologous end joining (NHEJ) are competing DSBs repair pathways (Kass and Jasin 2010). Recruitment of DNA-PKcs to DSB sites is observed already two seconds after micro-irradiation (Uematsu et al. 2007). Ku70/Ku80 and DNA-PKcs are very abundant, largely outnumber HR factors, and would easily win in a DNA end binding competition based on the law of mass action (Meek et al. 2008). To prevent this, the repair of DSBs is highly regulated in S phase. Adamo et al. (2010) and Pace et al. (2010) have shown that elimination of NHEJ components in FA-deficient cells suffice to suppress hypersensitivity to DNA crosslinks and accumulation of chromosomal aberration and to correct homologous recombination defects. This suggests that the main function of Fanconi anaemia proteins is to prevent the toxic engagement of the non-homologous end joining machinery, thereby allowing recombination dependent crosslink repair (Adamo et al. 2010; Pace et al. 2010). There is, however, one difference between the two studies: Pace et al. (2010) find that only the knockout of Ku70 eliminates the cisplatin sensitivity of DT40 Fancc −/− cells; suppression of DNA-PKcs or Ligase IV provides no advantages. In contrast, Adamo et al. (2010) observe that deletion of lig-4 in Caenorhabditis elegans fcd-2 mutant strains restores meiotic and ICL repair defects and that the knockdown or drug inhibition of DNA-PKcs suppresses phenotypic features of transformed human cells lacking FA core complex or FANCD2 proteins. Whereas the reason for this difference is not clear yet, the consensus is that an important function of FA proteins is to shield replication-coupled repair intermediates from the promiscuous intervention of NHEJ factors. Inhibitors of DNA-PKcs may relieve the symptoms of Fanconi anaemia, but some questions remain outstanding. Monoubiquitinated FANCD2 targets the crosslink repair nuclease FAN1 to damaged sites (Kratz et al. 2010; MacKay et al. 2010; Smogorzewska et al. 2010). If the FA pathway is necessary to recruit repair endonucleases at ICLs, it is difficult to conceive how crosslink repair can take place without FA core complex proteins or FANCD2. DNA-PKcs may also have hidden functions that are important for the repair of replication-associated double-strand DNA breaks. In response to DNA damage induced by camptothecin, DNA-PKcs phosphorylates RPA32 at Ser4 and Ser8 (Anantha et al. 2007; Shao et al. 1999). Phosphorylation of RPA32 at Ser4 and Ser8 probably has important consequences for DNA repair (Anantha et al. 2007). Interestingly, DNA-PKcs is phosphorylated by ATR at a Thr2609 cluster in response to UV-induced replication stress (Yajima et al. 2006). Mice carrying three alanine substitutions in the Thr2609 cluster of DNA-PKcs die prematurely of severe bone marrow failure (Zhang et al. 2011). MEFs derived from DNA-PK3A/3A cells are hypersensitive to ICLs, exhibit an attenuation of damage-induced monoubiquitination of FANCD2 and RAD51 foci formation and exhibit impaired DSB repair by homologous recombination (Zhang et al. 2011). By contrast, HR activity in DNA-PKcs −/− is enhanced as the competing NHEJ pathway is eliminated. This shows that the mouse Thr2609 phosphorylation cluster promotes FA pathway activation and homologous recombination. Clearly, further work is needed to dissect the complex regulation of replication-coupled and recombination-dependent repair of ICLs.
Role of FA proteins at fragile sites
In general, FA patients are not exposed to chemotherapeutic agents that induce ICLs. Endogenous sources of DNA replication defects may drive the expression of FA symptoms. Some evidence suggests that FA proteins either prevent or resolve replication problems at common fragile sites (Howlett et al. 2005). Fragile loci are intrinsically difficult to replicate and often coincide with chromosomal breakpoints in tumors (Gorgoulis et al. 2005; Yunis 1984). Interestingly, FANCD2 foci that persist in mitosis localize to fragile site loci on metaphase chromosome spreads (Chan et al. 2009). Recent studies have provided fresh insights into the nature of fragile sites, which brings perspective to the role of Fanconi anaemia proteins (Fig. 3). Fragile sites are late-replicating regions that are poor in origins of replication (Letessier et al. 2011). We discuss below why supplementary origins of replication are necessary to prevent replication catastrophes.

Cytological consequences of replication failure at fragile sites. DNA lesions or oncogene activation that results in nucleotide pool imbalance induce the stalling of replication forks. Supplementary origins that remain dormant if replication is unperturbed are mobilised and allow replication restart when forks stall. In the absence of supplementary origins, or at fragile sites, which are intrinsically origin-poor and late replicating, completion of DNA replication is not ensured. The FANCD2–FANCI complex stabilises DNA structures that are partially replicated, forming twin foci detectable by immunofluorescence microscopy in G2. In mitosis, the entanglement of partially replicated sister chromatids induces the formation of ultrafine anaphase bridges (UFBs) flanked on each side by a FANCD2/FANCI focus. UFBs may be resolved by decatenation, yielding damaged duplexes that are transmitted to G1 cells and shielded from nucleolytic degradation in 53BP1 nuclear bodies
Foreword on supplementary replication origins and the rescue of stalled forks
During licensing of DNA replication origins, MCM2-7 complexes are loaded onto chromatin in large excess (Edwards et al. 2002; Hua and Newport 1998; Ritzi et al. 1998). This reserve of chromatin-bound MCM2-7 complexes is much larger than is needed to duplicate the genome, and supplementary origins remain dormant as long as replication forks that proceed from selected origins move freely. Under stressful conditions, however, dormant origins become essential to complete DNA replication and prevent genomic instability (Ge et al. 2007; Ibarra et al. 2008; Woodward et al. 2006). Whereas a reduction of 50% of chromatin-bound MCM2-7 is compatible with cell cycle progression (Ge et al. 2007), it leads to death when cells are exposed to low doses of hydroxyurea or aphidicolin (Ge et al. 2007; Ibarra et al. 2008). Subsequent studies have shown that supplementary origins are also necessary to prevent the pathologic consequences of endogenous replication blocks: Mice with reduced levels of MCM2-7 proteins are cancer prone (Chuang et al. 2010; Kunnev et al. 2010). Cells derived from Mcm4 mutant mice exhibit an increased frequency of stalled replication forks (Kawabata et al. 2011). Together, these studies show that supplementary origins are necessary to rescue stalled replication forks. Letessier et al. (2011) have now demonstrated that chromosomal breaks occur at fragile sites because these loci are late replicating and origin poor. At fragile sites, replication forks have to cover unusually long distances. Therefore, there is a probability that replication forks do not complete the duplication of fragile loci before entry into mitosis, and this probability increases if fork movement is slowed down by nucleotide depletion or polymerase inhibition (Letessier et al. 2011). Another study suggests that breaks at some fragile sites may stem from secondary DNA structures that induce replication fork stalling in a context where no supplementary MCM2-7 complexes are available to rescue stalled forks (Ozeri-Galai et al. 2011). Both studies show that the density of usable replication origins is a key determinant of genome stability in S phase.
Role of FANCD2/I in the response to oncogene-induced DNA replication stress
The activation of oncogenes in an evolving population of tumor cells induces the expression of fragile sites and the formation of double-strand DNA breaks associated with DNA replication (Bartkova et al. 2005; Gorgoulis et al. 2005). One study has shown that inappropriate stimulation of S phase entry by human papillomavirus (HPV) E6/E7 or by cyclin E occurs without concomitant induction of nucleotide biosynthesis pathways to sustain DNA replication (Bester et al. 2011). E7 binds and degrades the retinoblastoma protein, leading to E2F activation, whereas E6 induces p53 degradation. It has been proposed that suboptimal levels of nucleotides in aberrant S phase cells may account for the increased frequency of replication fork stalling and for the formation of replication-associated DSBs (Bester et al. 2011). Individuals infected with HPV are at high risk of developing head and neck squamous cells carcinomas (HNSCCs). Fanconi anaemia patients are also predisposed to HNSCCs. It is noteworthy that FANCD2 counteracts DNA damage induced by (HPV) E7 activation (Park et al. 2010b). In cultured cells, the expression of (HPV) E7 oncoprotein induces FANCD2 foci formation and chromosomal aberrations (Spardy et al. 2007). In vitro, FA deficiency increases hyperplasia of HPV-positive keratinocytes (Hoskins et al. 2009). In vivo, the knockout of Fancd2 predisposes transgenic mice to (HPV) E7-induced HNSCCs (Park et al. 2010b). Conflicting clinical data, however, have been obtained regarding the potential increased susceptibility of Fanconi anaemia patients to HPV-induced carcinogenesis (Kutler et al 2003; van Zeeburg et al 2008). Nevertheless, these studies show strong connections between the FA pathway, the expression of fragile sites induced by (HPV) E7 and carcinogenesis. This is consistent with the notion that the FA pathway either prevents replication fork stalling within origin poor regions or resolves the problem of incomplete DNA replication at fragile sites.
FANCD2/I may coordinate the repair of un-replicated regions flanked by two stalled forks (Kawabata et al. 2011). Alternatively, a recent study has shown that FANCD2 is required for efficient initiation of replication origins in primary human cells (Song et al. 2010a). Hence, the paucity of origins at fragile sites may be further aggravated in FA cells. A third non-exclusive possibility is that FA proteins prevent formation of breaks in mitosis via the resolution of topological entanglements between partially replicated sister chromatids (discussed below).
Role of FA proteins in the resolution of ultrafine anaphase bridges
Abnormal replication intermediates formed at fragile sites have repercussions beyond the S and G2 phases of the cell cycle. Immunofluorescent staining of polo-like kinase 1 interacting checkpoint helicase (PICH) and Bloom’s syndrome protein (BLM) have revealed that ultrafine DNA bridges that connect sister chromatids form frequently in anaphase (Baumann et al. 2007; Chan et al. 2007). Ultrafine DNA structures are not detectable using conventional DNA dyes. Two classes of ultrafine anaphase bridges (UFBs) have been identified: centromeres-associated and non-centromeric/damage-induced, anaphase bridges. In mitosis, FANCD2 and FANCI form sister foci that localise specifically to the termini of UFBs induced by replication blocking agents, such as aphidicolin and mitomycin C (Chan et al 2009; Naim and Rosselli 2009). Most FANCD2/I “sister foci” are already detectable in G2 cells and are located at fragile sites (Chan et al 2009). Thus, it is likely that damage-inducible UFBs in anaphase originate from incomplete DNA replication. Consistent with this, nearly 50% of mice Mcm4 mutant cells that have less potential origins exhibit spontaneous FANCD2 sister foci in prophase (Kawabata et al 2011). Interestingly, only 10% of FANCD2 sister foci detected in metaphase are connected by UFBs in anaphase, suggesting that a significant proportion of abnormal replication intermediates that recruit FANCD2/I are resolved before chromosome migration to the poles of the dividing cell (Chan et al. 2009). Unresolved UFBs would otherwise degenerate into double strand DNA breaks, yield chromosomal abnormalities and cause cytokinesis failure with the production of micronuclei and binucleated cells. Formation of bulky anaphase bridges (stained with DNA dyes) in FA cells has been reported before (Qiao et al. 2004). The recent studies show that FA deficiency is accompanied with increased ultrafine anaphase bridges that derive from fragile sites, consistent with the notion that FA proteins rescue replication failure (Chan et al. 2009; Naim and Rosselli 2009; Vinciguerra et al. 2010).
Bloom’s syndrome protein (BLM) is recruited in anaphase to resolve DNA bridges, in association with its partner proteins RMI1 and the decatenase Topoisomerase IIIα (Chan et al. 2007). Similarly, some FA proteins may also participate in the resolution of topologically intertwined sister chromatids. FANCM decorates UFBs flanked by FANCD2 foci in telophase cells (Vinciguerra et al. 2010). Intriguingly, FANCM UFBs are BLM-dependent, but the two proteins do not colocalise. BLM UBFs staining tend to disappear at the time FANCM bridges form (Vinciguerra et al. 2010). This may reflect a BLM to FANCM “hand off” mechanism for the resolution of UFBs.
Finally, two recent reports show that marks of replication failure can be transmitted to daughter cells. Incomplete DNA replication induced by aphidicolin or unresolved replication intermediates in BLM cells correlate with the formation of 53BP1 nuclear bodies in the subsequent G1 (Harrigan et al. 2011; Lukas et al. 2011). 53BP1 nuclear bodies are enriched at common fragile sites and contain a number of DNA damage response proteins (Harrigan et al. 2011; Lukas et al. 2011). These replication stress structures presumably shield DNA lesions that persist after mitosis until optimal repair conditions are met in the next cell cycle (Lukas et al. 2011). In conclusion, replication failure at fragile sites initiates a cascade of causally linked processes with repercussions up to the next cell cycle.
FA proteins repair DNA lesions induced by reactive aldehydes
The role for FA proteins at fragile sites is not sufficient to explain the severe clinical manifestations of Fanconi anaemia. Recent studies have provided answers to the long-standing question on the identity of endogenous DNA damaging molecules that can promote the appearance of FA symptoms. The Fanconi anaemia pathway appears to limit the toxicity of DNA adducts formed by endogenous aldehydes. Aldehydes are highly reactive products implicated, among other things, in amino acids, carbohydrates, lipids, vitamins and steroids metabolic processes (Voulgaridou et al. 2011). For instance, malondialdehyde and crotonaldehyde are both produced through lipid peroxydation and can induce interstrand crosslinks in DNA (Cho et al. 2006; Niedernhofer et al. 2003; Summerfield and Tappel 1984). Some studies have focused on identifying DNA damage response pathways necessary for tolerance to formaldehyde and acetaldehyde. Formaldehyde is widespread in the environment, in nutriments and in the metabolism of purines and some amino acids. The main alteration induced by formaldehyde is protein–DNA crosslinks (Voulgaridou et al. 2011). DT40 cells lacking Fanconi anaemia or homologous recombination proteins are hypersensitive to endogenous levels of formaldehyde in human plasma (Ridpath et al. 2007). Chinese hamster ovary cell lines bearing inactivating mutations of homologous recombination and Fanconi anaemia genes are also hypersensitive to acetaldehyde (Mechilli et al 2008), the product of ethanol oxidation and an intermediate in carbohydrate metabolism. Acetaldehyde generates mainly mono adducts, and it can also produce ICLs (Brooks and Theruvathu 2005): In a basic environment composed of histones and polyamines, acetaldehyde reacts with deoxyguanine (dG) to form 1,N 2-propano-2′-deoxyguanosine adducts, which can in turn react with dG on the complementary strand to form an ICL (Fig. 4). Cellular exposure to acetaldehyde induces FANCD2 monoubiquitination (Marietta et al 2009). To test the relevance of acetaldehyde-induced DNA damage in vivo, Langevin et al. (2011) have disrupted the main acetaldehyde detoxifying enzymes aldehyde dehydrogenase 2 (ALDH2) in Fancd2 −/− mice. Aldh2 −/− Fancd2 −/− mice were born only from mothers bearing at least one wild-type Aldh2 allele, showing that the catabolism of acetaldehyde in utero was essential for embryonic development (Langevin et al. 2011). Stress overload with intraperitoneal injection of ethanol caused severe developmental defects in double-mutant embryos. After birth, exposure of Aldh2 −/− Fancd2 −/− to ethanol in drinking water induced severe bone marrow failure. Unexposed Aldh2 −/− Fancd2 −/− mice were born with subtle developmental defects and succumbed 3 to 6 months later from a disease similar to acute lymphoblastic leukemia (Langevin et al. 2011). These striking observations demonstrate that in addition to acetaldehyde detoxification pathways, Fanconi anaemia proteins constitute an essential line of defence against the genotoxic consequences of acetaldehyde. Based on the Aldh2 −/− Fancd2 −/− mouse model, a defect in the repair of acetaldehyde-induced lesions seems to account for most of the Fanconi anaemia symptoms. Reduced exposure to exogenous sources of acetaldehyde combined with therapeutic strategies to accelerate the catabolism of acetaldehyde will likely ameliorate the condition of Fanconi anaemia patients. Further studies will be necessary to decipher whether acetaldehyde is the most critical reactive metabolite for Fanconi anaemia or whether exposure to additional aldehyde-derived DNA adducts must be reduced. Limited consumption of alcohol is also advisable for the 36% of East Asians that have an inherited deficiency in ALDH2 (Brooks et al. 2009).

Connections between acetaldehyde metabolism and the Fanconi anaemia pathway. Acetaldehyde is an intermediate in sugar and alcohol metabolism, which is catabolised by aldehyde dehydrogenase enzymes, notably ALDH2, disabled in approximately 36% of East Asians. This reactive metabolite can induce a variety of chemical alterations in DNA, including interstrand DNA crosslinks. The reaction of two aldehydes molecules with deoxyguanosine (dG) forms 1,N 2-propanodeoxyguanosine adducts (1,N 2-PdG) that can react with dG on the opposite strand to form interstrand DNA crosslinks. 1,N 2-PdG can also be formed from crotonaldehyde, a product of lipid peroxidation (Marietta et al 2009). The FA pathway coordinates the repair of DNA lesions induced by acetaldehyde. The accumulation of acetaldehyde-induced lesions may induce congenital malformations, bone marrow failure and leukaemia in Fanconi anaemia patients; foetal damage from excessive alcohol consumption during pregnancy; and oesophageal squamous cell carcinomas in ALDH2-deficient individuals (Brooks et al 2009)
Concluding remarks
Important connections between DNA replication and tumorigenesis have been established. Aberrant entry into S phase may occur without coordination with supportive metabolic programs, such as nucleotide biosynthesis pathways, and lead to chromosomal breaks, primarily at fragile sites, the latest origin-poor region of the genome to be replicated. Replication-blocking lesions induced by reactive metabolites, such as aldehydes, and exogenous DNA damaging agents aggravate difficulties intrinsic to the replication program. FA proteins appear to play a central role in the response to both endogenous and exogenous sources of replication obstacles. The question remains as to why FA-deficient cells appear uniquely hypersensitive to ICL-inducing agents. An important function of FA proteins is to ensure the maintenance of genome stability in cancer prone multicellular organisms. A small number of blocked replication forks do not necessarily induce cell death: In a mouse model that develops spontaneous tumors as a result of insufficient dormant origins, stalled replication forks poorly activate the ATR-Chk1 signalling pathway (Kawabata et al. 2011). One important challenge in the future will be to understand precisely how FA proteins ensure fragile sites stability in the S/G2 and mitotic phases of the cell cycle, which may be relevant for our understanding of the development of solid tumors in FA patients and in the general population. The protection of replication-associated double-strand DNA breaks from toxic repair by NHEJ may be sufficient to explain how the FA network maintains genome integrity. NHEJ components, however, are also required for the maintenance of genome stability. The existence of specific phospho-sites in DNA-PKcs that promote ICL repair suggests that it may be possible to control toxic NHEJ activities without compromising the entire NHEJ pathway in FA patients.
The identification of genotoxic aldehydes that account for disease symptoms in Fanconi anaemia is an important breakthrough that will foster the elaboration of new cancer prevention strategies. Future studies will be necessary to confirm whether acetaldehyde is the most hurtful metabolite in FA patients or whether protection against other aldehydes must be considered. As acetaldehyde induces several types of DNA lesions, mainly monoadducts, the identification of the specific acetaldehyde-induced lesion (s) that depend (s) on FA proteins for repair will shed new light on the FA pathway. After all, the main physiological function of FA proteins may not be to repair ICLs.
Nevertheless, FA proteins are necessary for cellular tolerance to chemotherapeutic, ICL-inducing agents. The complete understanding of the biochemical steps and associated factors involved in FA-mediated crosslink repair will provide new opportunities to improve the use of crosslinking agents in cancer therapy. The development of systems using mammalian or Xenopus extracts to study crosslink repair will greatly accelerate progresses in this field.
It is clear that studies of the rare genetic disease Fanconi anaemia have provided and will continue to provide important knowledge on how cells respond to endogenous replication obstacles, on the nature of these obstacles and on how cells can surmount chemotherapeutic treatments that cause replication failure. This knowledge will not only help improve the care of Fanconi anaemia patients but also contribute to reducing the burden of cancer in the general population.
References
Adamo A, Collis SJ, Adelman CA, Silva N, Horejsi Z, Ward JD, Martinez-Perez E, Boulton SJ, La Volpe A (2010) Preventing nonhomologous end joining suppresses DNA repair defects of Fanconi anemia. Mol Cell 39:25–35
Alter BP, Greene MH, Velazquez I, Rosenberg PS (2003) Cancer in Fanconi anemia. Blood 101:2072
Anantha RW, Vassin VM, Borowiec JA (2007) Sequential and synergistic modification of human RPA stimulates chromosomal DNA repair. J Biol Chem 282:35910–35923
Andersen SL, Bergstralh DT, Kohl KP, LaRocque JR, Moore CB, Sekelsky J (2009) Drosophila MUS312 and the vertebrate ortholog BTBD12 interact with DNA structure-specific endonucleases in DNA repair and recombination. Mol Cell 35:128–135
Auerbach AD, Wolman SR (1976) Susceptibility of Fanconi's anaemia fibroblasts to chromosome damage by carcinogens. Nature 261:494–496
Badie S, Liao C, Thanasoula M, Barber P, Hill MA, Tarsounas M (2009) RAD51C facilitates checkpoint signaling by promoting CHK2 phosphorylation. J Cell Biol 185:587–600
Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K, Guldberg P, Sehested M, Nesland JM, Lukas C, Orntoft T, Lukas J, Bartek J (2005) DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 434:864–870
Baumann C, Korner R, Hofmann K, Nigg EA (2007) PICH, a centromere-associated SNF2 family ATPase, is regulated by Plk1 and required for the spindle checkpoint. Cell 128:101–114
Ben-Yehoyada M, Wang LC, Kozekov ID, Rizzo CJ, Gottesman ME, Gautier J (2009) Checkpoint signaling from a single DNA interstrand crosslink. Mol Cell 35:704–715
Bester AC, Roniger M, Oren YS, Im MM, Sarni D, Chaoat M, Bensimon A, Zamir G, Shewach DS, Kerem B (2011) Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell 145:435–446
Bridge WL, Vandenberg CJ, Franklin RJ, Hiom K (2005) The BRIP1 helicase functions independently of BRCA1 in the Fanconi anemia pathway for DNA crosslink repair. Nat Genet 37:953–957
Brooks PJ, Enoch MA, Goldman D, Li TK, Yokoyama A (2009) The alcohol flushing response: an unrecognized risk factor for esophageal cancer from alcohol consumption. PLoS Med 6:e50
Brooks PJ, Theruvathu JA (2005) DNA adducts from acetaldehyde: implications for alcohol-related carcinogenesis. Alcohol 35:187–193
Byun TS, Pacek M, Yee MC, Walter JC, Cimprich KA (2005) Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev 19:1040–1052
Cantor SB, Bell DW, Ganesan S, Kass EM, Drapkin R, Grossman S, Wahrer DC, Sgroi DC, Lane WS, Haber DA, Livingston DM (2001) BACH1, a novel helicase-like protein, interacts directly with BRCA1 and contributes to its DNA repair function. Cell 105:149–160
Chan KL, North PS, Hickson ID (2007) BLM is required for faithful chromosome segregation and its localization defines a class of ultrafine anaphase bridges. EMBO J 26:3397–3409
Chan KL, Palmai-Pallag T, Ying S, Hickson ID (2009) Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nat Cell Biol 11:753–760
Cho YJ, Wang H, Kozekov ID, Kurtz AJ, Jacob J, Voehler M, Smith J, Harris TM, Lloyd RS, Rizzo CJ, Stone MP (2006) Stereospecific formation of interstrand carbinolamine DNA cross-links by crotonaldehyde- and acetaldehyde-derived alpha-CH3-gamma-OH-1, N2-propano-2′-deoxyguanosine adducts in the 5′-CpG-3′ sequence. Chem Res Toxicol 19:195–208
Chuang CH, Wallace MD, Abratte C, Southard T, Schimenti JC (2010) Incremental genetic perturbations to MCM2-7 expression and subcellular distribution reveal exquisite sensitivity of mice to DNA replication stress. PLoS Genet 6:e1001110
Ciccia A, Ling C, Coulthard R, Yan Z, Xue Y, Meetei AR, el Laghmani H, Joenje H, McDonald N, de Winter JP, Wang W, West SC (2007) Identification of FAAP24, a Fanconi anemia core complex protein that interacts with FANCM. Mol Cell 25:331–343
Ciccia A, McDonald N, West SC (2008) Structural and functional relationships of the XPF/MUS81 family of proteins. Annu Rev Biochem 77:259–287
Cohn MA, Kowal P, Yang K, Haas W, Huang TT, Gygi SP, D'Andrea AD (2007) A UAF1-containing multisubunit protein complex regulates the Fanconi anemia pathway. Mol Cell 28:786–797
Collins NB, Wilson JB, Bush T, Tomashevski A, Roberts KJ, Jones NJ, Kupfer GM (2008) ATR-dependent phosphorylation of FANCA on serine 1449 after DNA damage is important for FA pathway function. Blood 113:2181–2190
Collis SJ, Ciccia A, Deans AJ, Horejsi Z, Martin JS, Maslen SL, Skehel JM, Elledge SJ, West SC, Boulton SJ (2008) FANCM and FAAP24 function in ATR-mediated checkpoint signaling independently of the Fanconi anemia core complex. Mol Cell 32:313–324
Cotta-Ramusino C, McDonald ER 3rd, Hurov K, Sowa ME, Harper JW, Elledge SJ (2011) A DNA damage response screen identifies RHINO, a 9-1-1 and TopBP1 interacting protein required for ATR signaling. Science 332:1313–1317
Crossan GP, van der Weyden L, Rosado IV, Langevin F, Gaillard PH, McIntyre RE, Gallagher F, Kettunen MI, Lewis DY, Brindle K, Arends MJ, Adams DJ, Patel KJ (2011) Disruption of mouse Slx4, a regulator of structure-specific nucleases, phenocopies Fanconi anemia. Nat Genet 43:147–152
Deans AJ, West SC (2009) FANCM connects the genome instability disorders Bloom's syndrome and Fanconi Anemia. Mol Cell 36:943–953
Deans AJ, West SC (2011) DNA interstrand crosslink repair and cancer. Nat Rev Cancer 11:467–480
Dorsman JC, Levitus M, Rockx D, Rooimans MA, Oostra AB, Haitjema A, Bakker ST, Steltenpool J, Schuler D, Mohan S, Schindler D, Arwert F, Pals G, Mathew CG, Waisfisz Q, de Winter JP, Joenje H (2007) Identification of the Fanconi anemia complementation group I gene, FANCI. Cell Oncol 29:211–218
Edwards MC, Tutter AV, Cvetic C, Gilbert CH, Prokhorova TA, Walter JC (2002) MCM2–7 complexes bind chromatin in a distributed pattern surrounding the origin recognition complex in Xenopus egg extracts. J Biol Chem 277:33049–33057
Ellison V, Stillman B (2003) Biochemical characterization of DNA damage checkpoint complexes: clamp loader and clamp complexes with specificity for 5′ recessed DNA. PLoS Biol 1:E33
Fanconi G (1927) Familiäre infantile perniziosartige Anämie (perniziöses Blutbild und Konstitution). Jahrb Kinderh 117:257–280
Fekairi S, Scaglione S, Chahwan C, Taylor ER, Tissier A, Coulon S, Dong MQ, Ruse C, Yates JR 3rd, Russell P, Fuchs RP, McGowan CH, Gaillard PH (2009) Human SLX4 is a Holliday junction resolvase subunit that binds multiple DNA repair/recombination endonucleases. Cell 138:78–89
Fu YV, Yardimci H, Long DT, Guainazzi A, Bermudez VP, Hurwitz J, van Oijen A, Scharer OD, Walter JC (2011) Selective bypass of a lagging strand roadblock by the eukaryotic replicative DNA helicase. Cell 146:931–941
Garcia-Higuera I, Taniguchi T, Ganesan S, Meyn MS, Timmers C, Hejna J, Grompe M, D'Andrea AD (2001) Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Mol Cell 7:249–262
Gari K, Decaillet C, Delannoy M, Wu L, Constantinou A (2008a) Remodeling of DNA replication structures by the branch point translocase FANCM. Proc Natl Acad Sci U S A 105:16107–16112
Gari K, Decaillet C, Stasiak AZ, Stasiak A, Constantinou A (2008b) The Fanconi anemia protein FANCM can promote branch migration of Holliday junctions and replication forks. Mol Cell 29:141–148
Ge XQ, Jackson DA, Blow JJ (2007) Dormant origins licensed by excess Mcm2-7 are required for human cells to survive replicative stress. Genes Dev 21:3331–3341
Geng L, Huntoon CJ, Karnitz LM (2010) RAD18-mediated ubiquitination of PCNA activates the Fanconi anemia DNA repair network. J Cell Biol 191:249–257
Gong Z, Kim JE, Leung CC, Glover JN, Chen J (2010) BACH1/FANCJ acts with TopBP1 and participates early in DNA replication checkpoint control. Mol Cell 37:438–446
Gorgoulis VG, Vassiliou LV, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T, Venere M, Ditullio RA Jr, Kastrinakis NG, Levy B, Kletsas D, Yoneta A, Herlyn M, Kittas C, Halazonetis TD (2005) Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 434:907–913
Gupta R, Sharma S, Sommers JA, Jin Z, Cantor SB, Brosh RM Jr (2005) Analysis of the DNA substrate specificity of the human BACH1 helicase associated with breast cancer. J Biol Chem 280:25450–25460
Harrigan JA, Belotserkovskaya R, Coates J, Dimitrova DS, Polo SE, Bradshaw CR, Fraser P, Jackson SP (2011) Replication stress induces 53BP1-containing OPT domains in G1 cells. J Cell Biol 193:97–108
Hashimoto Y, Chaudhuri AR, Lopes M, Costanzo V (2010) Rad51 protects nascent DNA from Mre11-dependent degradation and promotes continuous DNA synthesis. Nat Struct Mol Biol 17:1305–1311
Hiom K (2010) FANCJ: solving problems in DNA replication. DNA Repair (Amst) 9:250–256
Ho GP, Margossian S, Taniguchi T, D'Andrea AD (2006) Phosphorylation of FANCD2 on two novel sites is required for mitomycin C resistance. Mol Cell Biol 26:7005–7015
Holloman WK (2011) Unraveling the mechanism of BRCA2 in homologous recombination. Nat Struct Mol Biol 18:748–754
Hoskins EE, Morris TA, Higginbotham JM, Spardy N, Cha E, Kelly P, Williams DA, Wikenheiser-Brokamp KA, Duensing S, Wells SI (2009) Fanconi anemia deficiency stimulates HPV-associated hyperplastic growth in organotypic epithelial raft culture. Oncogene 28:674–685
Howlett NG, Taniguchi T, Durkin SG, D'Andrea AD, Glover TW (2005) The Fanconi anemia pathway is required for the DNA replication stress response and for the regulation of common fragile site stability. Hum Mol Genet 14:693–701
Hua XH, Newport J (1998) Identification of a preinitiation step in DNA replication that is independent of origin recognition complex and cdc6, but dependent on cdk2. J Cell Biol 140:271–281
Huang M, Kim JM, Shiotani B, Yang K, Zou L, D'Andrea AD (2010) The FANCM/FAAP24 complex is required for the DNA interstrand crosslink-induced checkpoint response. Mol Cell 39:259–268
Ibarra A, Schwob E, Mendez J (2008) Excess MCM proteins protect human cells from replicative stress by licensing backup origins of replication. Proc Natl Acad Sci U S A 105:8956–8961
Ishiai M, Kitao H, Smogorzewska A, Tomida J, Kinomura A, Uchida E, Saberi A, Kinoshita E, Kinoshita-Kikuta E, Koike T, Tashiro S, Elledge SJ, Takata M (2008) FANCI phosphorylation functions as a molecular switch to turn on the Fanconi anemia pathway. Nat Struct Mol Biol 15:1138–1146
Joo W, Xu G, Persky NS, Smogorzewska A, Rudge DG, Buzovetsky O, Elledge SJ, Pavletich NP (2011) Structure of the FANCI-FANCD2 complex: insights into the Fanconi anemia DNA repair pathway. Science 333:312–316
Kass EM, Jasin M (2010) Collaboration and competition between DNA double-strand break repair pathways. FEBS Lett 584:3703–3708
Kawabata T, Luebben SW, Yamaguchi S, Ilves I, Matise I, Buske T, Botchan MR, Shima N (2011) Stalled fork rescue via dormant replication origins in unchallenged S phase promotes proper chromosome segregation and tumor suppression. Mol Cell 41:543–553
Kee Y, D'Andrea AD (2010) Expanded roles of the Fanconi anemia pathway in preserving genomic stability. Genes Dev 24:1680–1694
Kim JM, Kee Y, Gurtan A, D'Andrea AD (2008) Cell cycle dependent chromatin loading of the fanconi anemia core complex by FANCM/FAAP24. Blood 111:5215–5222
Kim JM, Parmar K, Huang M, Weinstock DM, Ruit CA, Kutok JL, D'Andrea AD (2009) Inactivation of murine Usp1 results in genomic instability and a Fanconi anemia phenotype. Dev Cell 16:314–320
Kim Y, Lach FP, Desetty R, Hanenberg H, Auerbach AD, Smogorzewska A (2011) Mutations of the SLX4 gene in Fanconi anemia. Nat Genet 43:142–146
Knipscheer P, Raschle M, Smogorzewska A, Enoiu M, Ho TV, Scharer OD, Elledge SJ, Walter JC (2009) The Fanconi anemia pathway promotes replication-dependent DNA interstrand cross-link repair. Science 326:1698–1701
Kratz K, Schopf B, Kaden S, Sendoel A, Eberhard R, Lademann C, Cannavo E, Sartori AA, Hengartner MO, Jiricny J (2010) Deficiency of FANCD2-associated nuclease KIAA1018/FAN1 sensitizes cells to interstrand crosslinking agents. Cell 142:77–88
Kunnev D, Rusiniak ME, Kudla A, Freeland A, Cady GK, Pruitt SC (2010) DNA damage response and tumorigenesis in Mcm2-deficient mice. Oncogene 29:3630–3638
Kutler DI, Wreesmann VB, Goberdhan A, Ben-Porat L, Satagopan J, Ngai I, Huvos AG, Giampietro P, Levran O, Pujara K, Diotti R, Carlson D, Huryn LA, Auerbach AD, Singh B (2003) Human papillomavirus DNA and p53 polymorphisms in squamous cell carcinomas from Fanconi anemia patients. J Natl Cancer Inst 95:1718–1721
Langevin F, Crossan GP, Rosado IV, Arends MJ, Patel KJ (2011) Fancd2 counteracts the toxic effects of naturally produced aldehydes in mice. Nature 475:53–58
Le Breton C, Hennion M, Arimondo PB, Hyrien O (2011) Replication-fork stalling and processing at a single psoralen interstrand crosslink in Xenopus egg extracts. PLoS One 6:e18554
Lehmann AR, Niimi A, Ogi T, Brown S, Sabbioneda S, Wing JF, Kannouche PL, Green CM (2007) Translesion synthesis: Y-family polymerases and the polymerase switch. DNA Repair (Amst) 6:891–899
Letessier A, Millot GA, Koundrioukoff S, Lachages AM, Vogt N, Hansen RS, Malfoy B, Brison O, Debatisse M (2011) Cell-type-specific replication initiation programs set fragility of the FRA3B fragile site. Nature 470:120–123
Leung CC, Gong Z, Chen J, Glover JN (2011) Molecular basis of BACH1/FANCJ recognition by TopBP1 in DNA replication checkpoint control. J Biol Chem 286:4292–4301
Levitus M, Waisfisz Q, Godthelp BC, Vries Y, Hussain S, Wiegant WW, Elghalbzouri-Maghrani E, Steltenpool J, Rooimans MA, Pals G, Arwert F, Mathew CG, Zdzienicka MZ, Hiom K, De Winter JP, Joenje H (2005) The DNA helicase BRIP1 is defective in Fanconi anemia complementation group J. Nat Genet 37:934–935
Ling C, Ishiai M, Ali AM, Medhurst AL, Neveling K, Kalb R, Yan Z, Xue Y, Oostra AB, Auerbach AD, Hoatlin ME, Schindler D, Joenje H, de Winter JP, Takata M, Meetei AR, Wang W (2007) FAAP100 is essential for activation of the Fanconi anemia-associated DNA damage response pathway. EMBO J 26:2104–2114
Litman R, Peng M, Jin Z, Zhang F, Zhang J, Powell S, Andreassen PR, Cantor SB (2005) BACH1 is critical for homologous recombination and appears to be the Fanconi anemia gene product FANCJ. Cancer Cell 8:255–265
Liu T, Ghosal G, Yuan J, Chen J, Huang J (2010) FAN1 acts with FANCI-FANCD2 to promote DNA interstrand cross-link repair. Science 329:693–696
Long DT, Raschle M, Joukov V, Walter JC (2011) Mechanism of RAD51-dependent DNA interstrand cross-link repair. Science 333:84–87
Longerich S, San Filippo J, Liu D, Sung P (2009) FANCI binds branched DNA and is monoubiquitinated by UBE2T-FANCL. J Biol Chem 284:23182–23186
Lukas C, Savic V, Bekker-Jensen S, Doil C, Neumann B, Solvhoj Pedersen R, Grofte M, Chan KL, Hickson ID, Bartek J, Lukas J (2011) 53BP1 nuclear bodies form around DNA lesions generated by mitotic transmission of chromosomes under replication stress. Nat Cell Biol 13:243–253
Luke-Glaser S, Luke B, Grossi S, Constantinou A (2010) FANCM regulates DNA chain elongation and is stabilized by S-phase checkpoint signalling. EMBO J 29:795–805
MacDougall CA, Byun TS, Van C, Yee MC, Cimprich KA (2007) The structural determinants of checkpoint activation. Genes Dev 21:898–903
MacKay C, Declais AC, Lundin C, Agostinho A, Deans AJ, MacArtney TJ, Hofmann K, Gartner A, West SC, Helleday T, Lilley DM, Rouse J (2010) Identification of KIAA1018/FAN1, a DNA repair nuclease recruited to DNA damage by monoubiquitinated FANCD2. Cell 142:65–76
Majka J, Binz SK, Wold MS, Burgers PM (2006) Replication protein A directs loading of the DNA damage checkpoint clamp to 5′-DNA junctions. J Biol Chem 281:27855–27861
Marietta C, Thompson LH, Lamerdin JE, Brooks PJ (2009) Acetaldehyde stimulates FANCD2 monoubiquitination, H2AX phosphorylation, and BRCA1 phosphorylation in human cells in vitro: implications for alcohol-related carcinogenesis. Mutat Res 664:77–83
Mechilli M, Schinoppi A, Kobos K, Natarajan AT, Palitti F (2008) DNA repair deficiency and acetaldehyde-induced chromosomal alterations in CHO cells. Mutagenesis 23:51–56
Meek K, Dang V, Lees-Miller SP (2008) DNA-PK: the means to justify the ends? Adv Immunol 99:33–58
Meetei AR, De Winter JP, Medhurst AL, Wallisch M, Waisfisz Q, Van De Vrugt HJ, Oostra AB, Yan Z, Ling C, Bishop CE, Hoatlin ME, Joenje H, Wang W (2003a) A novel ubiquitin ligase is deficient in Fanconi anemia. Nat Genet 35:165–170
Meetei AR, Medhurst AL, Ling C, Xue Y, Singh TR, Bier P, Steltenpool J, Stone S, Dokal I, Mathew CG, Hoatlin M, Joenje H, de Winter JP, Wang W (2005) A human ortholog of archaeal DNA repair protein Hef is defective in Fanconi anemia complementation group M. Nat Genet 37:958–963
Meetei AR, Sechi S, Wallisch M, Yang D, Young MK, Joenje H, Hoatlin ME, Wang W (2003b) A multiprotein nuclear complex connects Fanconi anemia and Bloom syndrome. Mol Cell Biol 23:3417–3426
Mi J, Kupfer GM (2004) The Fanconi anemia core complex associates with chromatin during S phase. Blood 105:759–766
Moldovan GL, D'Andrea AD (2009) How the fanconi anemia pathway guards the genome. Annu Rev Genet 43:223–249
Moynahan ME, Cui TY, Jasin M (2001) Homology-directed DNA repair, mitomycin-c resistance, and chromosome stability is restored with correction of a Brca1 mutation. Cancer Res 61:4842–4850
Munoz IM, Hain K, Declais AC, Gardiner M, Toh GW, Sanchez-Pulido L, Heuckmann JM, Toth R, Macartney T, Eppink B, Kanaar R, Ponting CP, Lilley DM, Rouse J (2009) Coordination of structure-specific nucleases by human SLX4/BTBD12 is required for DNA repair. Mol Cell 35:116–127
Naim V, Rosselli F (2009) The FANC pathway and BLM collaborate during mitosis to prevent micro-nucleation and chromosome abnormalities. Nat Cell Biol 11:761–768
Nakanishi K, Cavallo F, Perrouault L, Giovannangeli C, Moynahan ME, Barchi M, Brunet E, Jasin M (2011) Homology-directed Fanconi anemia pathway cross-link repair is dependent on DNA replication. Nat Struct Mol Biol 18:500–503
Niedernhofer LJ, Daniels JS, Rouzer CA, Greene RE, Marnett LJ (2003) Malondialdehyde, a product of lipid peroxidation, is mutagenic in human cells. J Biol Chem 278:31426–31433
Niedzwiedz W, Mosedale G, Johnson M, Ong CY, Pace P, Patel KJ (2004) The Fanconi anaemia gene FANCC promotes homologous recombination and error-prone DNA repair. Mol Cell 15:607–620
Nijman SM, Huang TT, Dirac AM, Brummelkamp TR, Kerkhoven RM, D'Andrea AD, Bernards R (2005) The deubiquitinating enzyme USP1 regulates the Fanconi anemia pathway. Mol Cell 17:331–339
Ozeri-Galai E, Lebofsky R, Rahat A, Bester AC, Bensimon A, Kerem B (2011) Failure of origin activation in response to fork stalling leads to chromosomal instability at fragile sites. Mol Cell 43:122–131
Pace P, Mosedale G, Hodskinson MR, Rosado IV, Sivasubramaniam M, Patel KJ (2010) Ku70 corrupts DNA repair in the absence of the Fanconi anemia pathway. Science 329:219–223
Palle K, Vaziri C (2011) Rad18 E3 ubiquitin ligase activity mediates Fanconi anemia pathway activation and cell survival following DNA Topoisomerase 1 inhibition. Cell Cycle 10:1625–1638
Park HK, Wang H, Zhang J, Datta S, Fei P (2010a) Convergence of Rad6/Rad18 and Fanconi anemia tumor suppressor pathways upon DNA damage. PLoS One 5:e13313
Park JW, Pitot HC, Strati K, Spardy N, Duensing S, Grompe M, Lambert PF (2010b) Deficiencies in the Fanconi anemia DNA damage response pathway increase sensitivity to HPV-associated head and neck cancer. Cancer Res 70:9959–9968
Park WH, Margossian S, Horwitz AA, Simons AM, D'Andrea AD, Parvin JD (2005) Direct DNA binding activity of the Fanconi anemia D2 protein. J Biol Chem 280:23593–23598
Prakash S, Johnson RE, Prakash L (2005) Eukaryotic translesion synthesis DNA polymerases: specificity of structure and function. Annu Rev Biochem 74:317–353
Qiao F, Mi J, Wilson JB, Zhi G, Bucheimer NR, Jones NJ, Kupfer GM (2004) Phosphorylation of fanconi anemia (FA) complementation group G protein, FANCG, at serine 7 is important for function of the FA pathway. J Biol Chem 279:46035–46045
Qiao F, Moss A, Kupfer GM (2001) Fanconi anemia proteins localize to chromatin and the nuclear matrix in a DNA damage- and cell cycle-regulated manner. J Biol Chem 276:23391–23396
Raschle M, Knipsheer P, Enoiu M, Angelov T, Sun J, Griffith JD, Ellenberger TE, Scharer OD, Walter JC (2008) Mechanism of replication-coupled DNA interstrand crosslink repair. Cell 134:969–980
Ridpath JR, Nakamura A, Tano K, Luke AM, Sonoda E, Arakawa H, Buerstedde JM, Gillespie DA, Sale JE, Yamazoe M, Bishop DK, Takata M, Takeda S, Watanabe M, Swenberg JA, Nakamura J (2007) Cells deficient in the FANC/BRCA pathway are hypersensitive to plasma levels of formaldehyde. Cancer Res 67:11117–11122
Ritzi M, Baack M, Musahl C, Romanowski P, Laskey RA, Knippers R (1998) Human minichromosome maintenance proteins and human origin recognition complex 2 protein on chromatin. J Biol Chem 273:24543–24549
Roques C, Coulombe Y, Delannoy M, Vignard J, Grossi S, Brodeur I, Rodrigue A, Gautier J, Stasiak AZ, Stasiak A, Constantinou A, Masson JY (2009) MRE11-RAD50-NBS1 is a critical regulator of FANCD2 stability and function during DNA double-strand break repair. EMBO J 28:2400–2413
Sarkar S, Davies AA, Ulrich HD, McHugh PJ (2006) DNA interstrand crosslink repair during G1 involves nucleotide excision repair and DNA polymerase zeta. EMBO J 25:1285–1294
Schlacher K, Christ N, Siaud N, Egashira A, Wu H, Jasin M (2011) Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 145:529–542
Schroeder TM, Anschutz F, Knopp A (1964) Spontaneous chromosome aberrations in familial panmyelopathy. Humangenetik 1:194–196
Schwab RA, Blackford AN, Niedzwiedz W (2010) ATR activation and replication fork restart are defective in FANCM-deficient cells. EMBO J 29:806–818
Shao RG, Cao CX, Zhang H, Kohn KW, Wold MS, Pommier Y (1999) Replication-mediated DNA damage by camptothecin induces phosphorylation of RPA by DNA-dependent protein kinase and dissociates RPA:DNA-PK complexes. EMBO J 18:1397–1406
Shen X, Do H, Li Y, Chung WH, Tomasz M, de Winter JP, Xia B, Elledge SJ, Wang W, Li L (2009) Recruitment of fanconi anemia and breast cancer proteins to DNA damage sites is differentially governed by replication. Mol Cell 35:716–723
Shereda RD, Machida Y, Machida YJ (2010) Human KIAA1018/FAN1 localizes to stalled replication forks via its ubiquitin-binding domain. Cell Cycle 9:3977–3983
Sims AE, Spiteri E, Sims RJ 3rd, Arita AG, Lach FP, Landers T, Wurm M, Freund M, Neveling K, Hanenberg H, Auerbach AD, Huang TT (2007) FANCI is a second monoubiquitinated member of the Fanconi anemia pathway. Nat Struct Mol Biol 14:564–567
Smogorzewska A, Desetty R, Saito TT, Schlabach M, Lach FP, Sowa ME, Clark AB, Kunkel TA, Harper JW, Colaiacovo MP, Elledge SJ (2010) A genetic screen identifies FAN1, a Fanconi anemia-associated nuclease necessary for DNA interstrand crosslink repair. Mol Cell 39:36–47
Smogorzewska A, Matsuoka S, Vinciguerra P, McDonald ER 3rd, Hurov KE, Luo J, Ballif BA, Gygi SP, Hofmann K, D'Andrea AD, Elledge SJ (2007) Identification of the FANCI protein, a monoubiquitinated FANCD2 paralog required for DNA repair. Cell 129:289–301
Song IY, Barkley LR, Day TA, Weiss RS, Vaziri C (2010a) A novel role for fanconi anemia (FA) pathway effector protein FANCD2 in cell cycle progression of untransformed primary human cells. Cell Cycle 9:2375–2388
Song IY, Palle K, Gurkar A, Tateishi S, Kupfer GM, Vaziri C (2010b) Rad18-mediated translesion synthesis of bulky DNA adducts is coupled to activation of the Fanconi anemia DNA repair pathway. J Biol Chem 285:31525–31536
Spardy N, Duensing A, Charles D, Haines N, Nakahara T, Lambert PF, Duensing S (2007) The human papillomavirus type 16 E7 oncoprotein activates the Fanconi anemia (FA) pathway and causes accelerated chromosomal instability in FA cells. J Virol 81:13265–13270
Stoepker C, Hain K, Schuster B, Hilhorst-Hofstee Y, Rooimans MA, Steltenpool J, Oostra AB, Eirich K, Korthof ET, Nieuwint AW, Jaspers NG, Bettecken T, Joenje H, Schindler D, Rouse J, de Winter JP (2011) SLX4, a coordinator of structure-specific endonucleases, is mutated in a new Fanconi anemia subtype. Nat Genet 43:138–141
Suhasini AN, Rawtani NA, Wu Y, Sommers JA, Sharma S, Mosedale G, North PS, Cantor SB, Hickson ID, Brosh RM Jr (2011) Interaction between the helicases genetically linked to Fanconi anemia group J and Bloom's syndrome. EMBO J 30:692–705
Summerfield FW, Tappel AL (1984) Detection and measurement by high-performance liquid chromatography of malondialdehyde crosslinks in DNA. Anal Biochem 143:265–271
Svendsen JM, Smogorzewska A, Sowa ME, O'Connell BC, Gygi SP, Elledge SJ, Harper JW (2009) Mammalian BTBD12/SLX4 assembles a Holliday junction resolvase and is required for DNA repair. Cell 138:63–77
Tischkowitz MD, Hodgson SV (2003) Fanconi anaemia. J Med Genet 40:1–10
Uematsu N, Weterings E, Yano K, Morotomi-Yano K, Jakob B, Taucher-Scholz G, Mari PO, van Gent DC, Chen BP, Chen DJ (2007) Autophosphorylation of DNA-PKCS regulates its dynamics at DNA double-strand breaks. J Cell Biol 177:219–229
van Zeeburg HJ, Snijders PJ, Wu T, Gluckman E, Soulier J, Surralles J, Castella M, van der Wal JE, Wennerberg J, Califano J, Velleuer E, Dietrich R, Ebell W, Bloemena E, Joenje H, Leemans CR, Brakenhoff RH (2008) Clinical and molecular characteristics of squamous cell carcinomas from Fanconi anemia patients. J Natl Cancer Inst 100:1649–1653
Vaz F, Hanenberg H, Schuster B, Barker K, Wiek C, Erven V, Neveling K, Endt D, Kesterton I, Autore F, Fraternali F, Freund M, Hartmann L, Grimwade D, Roberts RG, Schaal H, Mohammed S, Rahman N, Schindler D, Mathew CG (2010) Mutation of the RAD51C gene in a Fanconi anemia-like disorder. Nat Genet 42:406–409
Vinciguerra P, Godinho SA, Parmar K, Pellman D, D'Andrea AD (2010) Cytokinesis failure occurs in Fanconi anemia pathway-deficient murine and human bone marrow hematopoietic cells. J Clin Invest 120:3834–3842
Voulgaridou GP, Anestopoulos I, Franco R, Panayiotidis MI, Pappa A (2011) DNA damage induced by endogenous aldehydes: current state of knowledge. Mutat Res 711:13–27
Wang AT, Sengerova B, Cattell E, Inagawa T, Hartley JM, Kiakos K, Burgess-Brown NA, Swift LP, Enzlin JH, Schofield CJ, Gileadi O, Hartley JA, McHugh PJ (2011) Human SNM1A and XPF-ERCC1 collaborate to initiate DNA interstrand cross-link repair. Genes Dev 25:1859–1870
Wang W (2007) Emergence of a DNA-damage response network consisting of Fanconi anaemia and BRCA proteins. Nat Rev Genet 8:735–748
Wang X, Kennedy RD, Ray K, Stuckert P, Ellenberger T, D'Andrea AD (2007) Chk1-mediated phosphorylation of FANCE is required for the Fanconi anemia/BRCA pathway. Mol Cell Biol 27:3098–3108
Williams SA, Longerich S, Sung P, Vaziri C, Kupfer GM (2011) The E3 ubiquitin ligase RAD18 regulates ubiquitylation and chromatin loading of FANCD2 and FANCI. Blood 117:5078–5087
Woodward AM, Gohler T, Luciani MG, Oehlmann M, Ge X, Gartner A, Jackson DA, Blow JJ (2006) Excess Mcm2-7 license dormant origins of replication that can be used under conditions of replicative stress. J Cell Biol 173:673–683
Xue Y, Li Y, Guo R, Ling C, Wang W (2008) FANCM of the Fanconi anemia core complex is required for both monoubiquitination and DNA repair. Hum Mol Genet 17:1641–1652
Yajima H, Lee KJ, Chen BP (2006) ATR-dependent phosphorylation of DNA-dependent protein kinase catalytic subunit in response to UV-induced replication stress. Mol Cell Biol 26:7520–7528
Yamamoto KN, Kobayashi S, Tsuda M, Kurumizaka H, Takata M, Kono K, Jiricny J, Takeda S, Hirota K (2011) Involvement of SLX4 in interstrand cross-link repair is regulated by the Fanconi anemia pathway. Proc Natl Acad Sci U S A 108:6492–6496
Yan Z, Delannoy M, Ling C, Daee D, Osman F, Muniandy PA, Shen X, Oostra AB, Du H, Steltenpool J, Lin T, Schuster B, Decaillet C, Stasiak A, Stasiak AZ, Stone S, Hoatlin ME, Schindler D, Woodcock CL, Joenje H, Sen R, de Winter JP, Li L, Seidman MM, Whitby MC, Myung K, Constantinou A, Wang W (2010) A histone-fold complex and FANCM form a conserved DNA-remodeling complex to maintain genome stability. Mol Cell 37:865–878
Yang K, Moldovan GL, Vinciguerra P, Murai J, Takeda S, D'Andrea AD (2011) Regulation of the Fanconi anemia pathway by a SUMO-like delivery network. Genes Dev 25:1847–1858
Yoshikiyo K, Kratz K, Hirota K, Nishihara K, Takata M, Kurumizaka H, Horimoto S, Takeda S, Jiricny J (2010) KIAA1018/FAN1 nuclease protects cells against genomic instability induced by interstrand cross-linking agents. Proc Natl Acad Sci U S A 107:21553–21557
Yuan F, El Hokayem J, Zhou W, Zhang Y (2009) FANCI protein binds to DNA and interacts with FANCD2 to recognize branched structures. J Biol Chem 284:24443–24452
Yunis JJ (1984) Fragile sites and predisposition to leukemia and lymphoma. Cancer Genet Cytogenet 12:85–88
Zhang S, Yajima H, Huynh H, Zheng J, Callen E, Chen HT, Wong N, Bunting S, Lin YF, Li M, Lee KJ, Story M, Gapud E, Sleckman BP, Nussenzweig A, Zhang CC, Chen DJ, Chen BP (2011) Congenital bone marrow failure in DNA-PKcs mutant mice associated with deficiencies in DNA repair. J Cell Biol 193:295–305
Zhi G, Wilson JB, Chen X, Krause DS, Xiao Y, Jones NJ, Kupfer GM (2009) Fanconi anemia complementation group FANCD2 protein serine 331 phosphorylation is important for fanconi anemia pathway function and BRCA2 interaction. Cancer Res 69:8775–8783
Zou L, Elledge SJ (2003) Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 300:1542–1548
Zou L, Liu D, Elledge SJ (2003) Replication protein A-mediated recruitment and activation of Rad17 complexes. Proc Natl Acad Sci U S A 100:13827–13832
Acknowledgment
We thank Pierre-Henri Gaillard, Philippe Pasero and all of the members of the laboratory for their comments and suggestions on the manuscript. We apologise to any colleagues whose contribution might have been overlooked in this review. This work was supported by ARC Association pour la Recherche sur le Cancer and by ANR Agence Nationale de la Recherche.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Erich Nigg
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Constantinou, A. Rescue of replication failure by Fanconi anaemia proteins. Chromosoma 121, 21–36 (2012). https://doi.org/10.1007/s00412-011-0349-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00412-011-0349-2