
Abstract
This narrative review examines the possible role of microglial cells, first, in neuroinflammation and, second, in schizophrenia, depression, and suicide. Recent research on the interactions between microglia, astrocytes and neurons and their involvement in pathophysiological processes of neuropsychiatric disorders is presented. This review focuses on results from postmortem, positron emission tomography (PET) imaging studies, and animal models of schizophrenia and depression. Third, the effects of antipsychotic and antidepressant drug therapy, and of electroconvulsive therapy on microglial cells are explored and the upcoming development of therapeutic drugs targeting microglia is described. Finally, there is a discussion on the role of microglia in the evolutionary progression of human lineage. This view may contribute to a new understanding of neuropsychiatric disorders.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Microglial cells originate from myeloid precursors in the yolk sac and are regarded as resident macrophages of the central nervous system (CNS) [1]. During the development of the CNS, microglial cells have various functions: they phagocytose apoptotic neurons, and induce neuronal apoptosis, prune weak synapses, form new synapses, promote the survival of pyramidal neurons in the white matter, direct the expression of (S)-2-amino-3-(3-hydroxy-5-methyl-4-isoxazole)-propionic acid—(AMPA) and N-methyl-D-aspartate—(NMDA) receptors at thalamocortical synapses and regulate the production and migration of cortical inhibitory neurons [1]. Activated microglia produce cytokines and nitric oxide (NO). Activated microglial cells are polarized towards the M1 or M2 phenotype. While polarization towards the M1 phenotype, also called the proinflammatory phenotype, is caused by cytokines such as interferon-alpha (IFN-α) and tumor necrosis factor-alpha (TNF-α), polarization towards the M2 phenotype, also termed the anti-inflammatory phenotype, is triggered by various cytokines such as interleukin IL-4, IL-13 and IL-25 [2,3,4,5,6,7,8,9,10,11]. The differences between the M1 and M2 phenotypes have been critically discussed and might be revaluated in the future [10, 12].
Microglial cells and neuroinflammation
Neuroinflammation is inflammation of nervous tissue [13]. The hallmark of neuroinflammation in schizophrenia (SZ) is overactivation of microglial cells [14]. Acute neuroinflammation during fetal development causes neuropathological abnormalities in the cerebellum, insular cortex, fusiform gyrus and deficits in neuronal activation in the right amygdala and fusiform gyrus, and ventrolateral prefrontal cortex (PFC) [15]. Neuroinflammation is mainly facilitated by microglial cells and to same extent by astrocytes and mast cells [16]. Microglial cells are divided into amoeboid and ramified cells. The so-called dark microglial cells, which are highly phagocytic cells under oxidative stress around the vasculature showing an electron-dense, condensed cytoplasm, are either overactivated microglial cells or a novel type of myeloid cell infiltrating the brain [16]. Neuroinflammation is linked to white matter pathology in patients with schizophrenia and is caused by microglial cells [17, 18]. Furthermore, focal neuroinflammation occurs in the hippocampi of patients with schizophrenia with an acute psychosis [19]. Disruption of the interaction between mast cells and microglial cells increases neuroinflammation [20]. However, the term neuroinflammation is not interchangeable with the term inflammation: microarray analyses of postmortem cerebral cortices of patients with Alzheimer’s disease (DAT), Parkinson’s disease (PD), patients with schizophrenia and patients with inflammatory diseases have demonstrated no relationship between these conditions [21].
When microglial cells become activated during neuroinflammation, they become enlarged and phagocytic. Microglial cells contribute to synaptic plasticity by releasing neuroactive molecules such as adenosine triphosphate (ATP), glutamate, D-serine, nitric oxide (NO), brain-derived neurotrophic factor (BDNF), TNF-α, free radicals, prostaglandin E2 (PGE2), and ILs; and microglial cells communicate with astrocytes through glutamatergic neurotransmission [2]. Furthermore, glutamate controls the function of microglial cells via ionotropic and metabotropic receptors located in these cells [22]. Microglial cells also communicate with neurons, promoting neuronal cell death, neurogenesis, and synaptic interactions [7, 23, 24]. Activated microglia control inhibitory inputs from parvalbumin containing interneurons onto the basilar dendritic spines of deep layer 3 pyramidal neurons in the PFC in patients with schizophrenia [25]. Microglia–neuronal interactions involve various signals such as cytokines, neurotransmitters, and neuron-microglia inhibitory factors, such as fractalkine (CXCL1) and cluster of differentiation (CD200) [26, 27]. Microglial cells are also connected with astrocytes and oligodendrocytes, and these interactions might also cause neuropathic pain [28]. Dystrophic microglial cells contribute to substantial dystrophies in adjacent oligodendrocytes in the PFC (layer 5) in patients with schizophrenia with predominately negative symptoms and to a lesser extent in patients with schizophrenia with mainly positive symptoms but not in healthy control subjects [29]. Disruption of the balance between microglial cells and astrocytes (e.g., type-1/type-2 imbalance) causes a dysregulation of the immune response in schizophrenia [30, 31]. T-helper 1 cells (TH-1) and certain macrophages/monocytes (M1) produce cytokines (IL-2, IL-12, IL-18, IFN-α, TNF- α) and this immune response is called type-1 immune response. T-helper 2 cells (TH-2) and certain macrophages/monocytes (M2) create cytokines (IL-4, IL-10, IL-13) and this immune response is termed type-2 immune response. Type-1 and type-2 cytokines irritate each other with a down-regulation in schizophrenia [30].
The various roles of microglial cells in neuropsychiatric disorders
Microglial cells play an important role in the pathology of neuropsychiatric disorders such as schizophrenia and major depressive disorder (MDD) [31,32,33,34,35]. Microglial cells are also involved in the cerebral cortex with autism spectrum disorders (ASDs), which are also regarded as neurodevelopmental disorders [36, 37]. Activated microglia and neuroinflammation are also pathological hallmarks of neurodegenerative diseases such as DAT and PD [38]. Original research on postmortem and PET studies demonstrating the presence or absence of microglial activation using different microglial markers in brain regions of interest, predominantly, in patients with schizophrenia, bipolar disorder, and affective disorders, respectively, is summarized in Table 1.
The role of microglial cells in schizophrenia
Glucocorticoid levels are increased during stress, and influence microglial response with pro- and anti-inflammatory activity [9]. Stress-induced release of glutamate from neurons results in the stimulation of microglial cells through the activation of N-methyl-D-aspartate receptors (NMDARs) by glucocorticoids [8, 9].
Microglial cells induce the loss of cortical gray matter in patients with schizophrenia by pruning synapses, phagocytosing stressed neurons, and preventing the release of neurotrophic factors such as BDNF [39].
In a systematic review by Trépanier et al. [40], many of the studies (11 of 22) reported an increase in the expression of microglial markers in the postmortem brains from patients with schizophrenia compared with those of control subjects. Moreover, a meta-analysis by van Kesteren et al. [41] revealed an increase in the number of microglia in specific brain areas, such as the temporal cortex, in postmortem brains of patients with schizophrenia compared with those of control subjects. Calprotectin is expressed in microglial cells and is considered a nonspecific marker of inflammation. Calprotectin expression is significantly increased in patients with schizophrenia compared with healthy control subjects [42]. The level of ionized calcium-binding adaptor molecule 1 (IBA1), a marker showing specific immunostaining of resting and activated microglia, is increased in specific brain areas such as the amygdala, hippocampus, nucleus accumbens, and PFC in patients with schizophrenia [9, 43]. A lateralization of IBA1 immunopositive microglial cells was observed towards the right anterior midcingulate cortex in patients with schizophrenia and bipolar disorder compared with healthy control subjects [44]. A study by Bloomfield et al. [45] using translocator-protein positron emission tomography (TSPO PET) demonstrated that microglial activity is increased in total and frontal and temporal lobe gray matter in patients with schizophrenia and ultrahigh risk individuals compared with healthy control subjects. Nevertheless, an 18 kDa TSPO PET study revealed no increase in microglial activity in the dorsolateral PFC and hippocampus in first-episode psychosis patients compared with healthy control subjects [46]. Moreover, another TSPO PET study by Di Biase et al. [47] did not show any differences among individuals at risk for schizophrenia, patients with schizophrenia and healthy control subjects. A meta-analysis by Marques et al. [48] of TSPO PET studies and microglial activation exposed a moderate effect on gray matter relative to other brain tissue in schizophrenia when using binding potential as an outcome measure but no difference when using volume of distribution as an outcome measure. The TSPO PET study by Conen et al. [49] found no microglial activation in patients with recent onset and established schizophrenia compared with healthy control subjects in the ACC, PFC, parietal cortex, and brainstem, but significant changes in the thalamus and putamen.
Activated microglial cells are linked to the increased expression of peripheral benzodiazepine binding sites (PBBS). (R)-PK1195 is a specific ligand for the PBBS and combines with carbon-11 in PET studies in schizophrenia, which can be used as a novel PET biomarker of activated microglial cells in schizophrenia [50].
The discrepancies in findings related to microglia activation in schizophrenia obtained by PET- and postmortem studies may have been caused by effects of confounding factors (e.g., tissue quality and aging) and comorbid factors (e.g., use of antipsychotic medication, suicidal tendencies, smoking, or drug abuse) [24].
Human leukocyte antigen (HLA-DR) is a class II antigen, and different gene loci code for the alpha (α)- and beta (ß)-chains. Other important genes, namely, TNF-α and tumor necrosis factor beta (TNF-ß) are distributed throughout the HLA-complex [51]. Steiner et al. [52] (2008) found that the density of HLA-DR, which reacts with activated microglia, was increased in the PFCs of suicidal patients. Bayer et al. [53] reported that three late-onset out of 14 patients with schizophrenia, 1 out of 6 patients with affective disorders (AD), and four out of 8 patients with dementia of Alzheimer’s type (DAT) exhibited increased HLA-DR immunostaining in the frontal cortex and hippocampus. Radewicz et al. [54] observed an increase in HLA-DR density in the frontal and temporal cortices in patients with schizophrenia compared with control subjects, and Wierzba-Bobrowicz et al. [55] detected a greater expression of HLA-DR in the anterior cingulate and temporal cortices in patients with schizophrenia compared with control subjects. Additionally, Krause et al. [56] found that the number of HLA-DR immunopositive cells was increased in patients with schizophrenia compared with healthy control subjects. Steiner et al. [57] reported decreased cerebral lateralization of HLA-DR in patients with schizophrenia compared with healthy control subjects. Fillman et al. [58] found an increased HLA-DR in the dorsolateral prefrontal cortex in patients with schizophrenia compared with control subjects. A study by Busse et al. [59] also demonstrated that HLA-DR immunostaining was increased in the posterior hippocampus in patients with paranoid schizophrenia compared with patients with residual schizophrenia and healthy control subjects. Moreover, patients with residual schizophrenia showed a greater concentration of CD3+ and CD20+ lymphocytes in the posterior hippocampus [59]. No significant differences in microglial density using the microglial marker IBA1 in the medial frontal gyrus in patients with bipolar disorder compared with healthy control subjects have been demonstrated [60]. A correlation has also been found between HLA-DR and alcoholism or even alcoholism withdrawal [61,62,63,64].
These discrepancies in the results of postmortem studies could be explained by methodological differences such as the immunohistochemical markers used, the methods used to count microglial cells, the brain regions studied, the cortical layers in which the expression of the markers was measured, and technical issues such as the postmortem interval and brain pH. Other confounding variables including age differences, sex, age at death, cause of death, clinical variables, illness history and duration, medication history, diagnosis method and inclusion and exclusion criteria also exist [14, 65]. Recently, microglial activation rather than microglial density has been investigated [65]. Microglial activation reflects innate immune memory (history of life events) including prenatal and perinatal influences and genetic background [14, 66]. Furthermore, the use of a single microglial marker (such as HLA-DR) for phenotypic identification is problematic, since detailed knowledge of the time course of death is crucial [14]. Multiple markers must be used to identify the functional state [14]. The association between microglial activation and reduced synaptic plasticity, and neuropil alterations and reduced neuroplasticity needs to be studied in a multimodal manner combining imaging studies and postmortem brain analyses of patients with schizophrenia with various immunological markers [14].
Research conducted by Goudriaan et al. [67] has demonstrated that genes that are involved in microglial activation do not contribute genetically to schizophrenia susceptibility. However, there is an association between the HLA-DRB1 gene and schizophrenia in the human population [68,69,70].
Maternal infection during pregnancy is known to be an environmental risk factor for the development of neuropsychiatric disorders such as schizophrenia and ASDs in offspring. The causative role of immunological processes that interfere with brain development in schizophrenia have been discussed [71,72,73]. Sex differences in processes related to microglial cells exist and might contribute to the sex differences in neuropsychiatric disorders such as schizophrenia [74, 75] and in a neonatal rat animal model of early-life infection [76].
Although Smolders et al. [77] demonstrated that a single or double injection of poly(I:C) in pregnant mice does not result in a fetal microglial activation during mid- or late embryonic development, other research groups have found an involvement of microglial cells in maternal immune activation. In an animal study using pregnant mice injected with poly(I:C) on gestational day 9 as an animal model of schizophrenia, the number and shape of microglia in several brain regions were assessed in the offspring on postnatal day (PND) 30 using immunofluorescence with an anti-IBA1 antibody [78]. There were more microglial cells in the hippocampus and striatum in the offspring of poly(I:C)-injected mice than in the offspring of vehicle-treated mice. Additionally, these microglial cells were morphologically characterized by reduced arborization, which is indicative of a greater activation. This is supported by studies showing that microglial cells from control animals show greater microglial arborization, which is indicative for a noninflammatory state of microglia than those from offspring from poly(I:C) treated mice, which were characterized by no or few branched cells, indicating an activated and inflammatory or phagocytic microglial state [79, 80]. Furthermore, in a pilot study comparing the offspring of poly(I:C) treated mice on PND10 and PND100, the mouse pups displayed no microglial alterations in response to prenatal exposure to poly(I:C), whereas adolescent mice showed a significant increase in the number of microglial cells in the frontal association cortex, ventral striatum, dentate gyrus of the hippocampus and secondary visual cortex. In contrast, in adult mice (PND100), immunological activation was only observed in the frontal association cortex [81]. Prenatal poly(I:C) treatment in mice prevents an increase in the number of microglial cells in the cortex [82]. These studies support the hypothesis that maternal infection during embryogenesis contributes to an increase in the number of microglial cells in offspring, which in subsequent periods of life results in decreases in dopamine and the stimulation of high-affinity dopamine receptors (involving DRD3 and DRD5). These alterations in dopamine levels may, therefore, cause a neuroinflammatory response, which contributes to the pathogenesis of schizophrenia [83].
The involvement of microglial cells in depression and suicide
Suicide is a major public health problem [84] with a prevalence of 5.92% and is supported by ongoing neurobiological research [85], suicide behavior is listed as an independent mental disorder in the fifth edition of the Diagnostic Statistical Manual of Mental Disorders-DSM V [86]. The dorsal raphe nucleus (DRN) comprises the caudal and rostral subregions, which are further divided into the dorsal, ventral, interfascicular, ventrolateral, and caudal subregions [87]. The DRN sends serotonergic projections to the striatum, frontal cortex, thalamus, lateral septal nuclei, nucleus accumbens, habenular complex, and hippocampus [88,89,90,91,92,93]. The dorsal raphe nucleus is implicated in the pathology of schizophrenia, major depression, suicidal behavior [94,95,96,97,98,99,100,101,102,103,104] and even alcoholism [105]. Disruption of serotonergic functions is implicated in the pathophysiology of stress, MDD, and suicide [106,107,108,109,110], while serotonin deficiency contributes to suicidal behavior and MDD [111,112,113,114,115,116,117,118,119,120,121,122,123,124,125,126]. Additionally, a link between smoking, suicide, and low serotonin levels has been demonstrated [127]. Nicotine-induced microglial activation in reward seeking brain regions such as nucleus accumbens and basolateral amygdala increases cocaine reinforcement in adolescent rats but not adult rats [128].
Microglial cell activation correlates with transcriptional activity in the DRN neurons in the non suicidal depressed subgroup [104]. In addition, microglial activation induced by interferon-alpha (IFN-α) is associated with depressive-like behavior and the expression of proinflammatory surface markers such as major histocompatibility complex-II (MHC-II) and CD86 in a mouse model of immune-mediated depression, demonstrating that microglia are polarized towards the M1-phenotype [129]. A study by Steiner et al. [52] demonstrated that microglial density is increased in the dorsolateral prefrontal cortex, anterior cingulate cortex, and mediodorsal thalamus in suicidal patients with schizophrenia and suicidal patients with depression compared with healthy control subjects. Schnieder et al. [130] also observed that microglial density is augmented in the white matter of the PFC in suicidal patients with schizophrenia and depression compared with nonsuicidal subjects. Various studies (e.g., postmortem analysis, PET imaging studies, and analysis of the cerebrospinal fluid and serum/plasma of patients) have provided evidence for a role of microglial cells in suicide, which can be used for a new therapeutic target for suicide prevention [131]. Adolescence is a significant period for the development of neuropsychiatric diseases. During this period microglial synaptic pruning occurs in the PFC and contributes to pathological alterations that are evident in schizophrenia [132]. An increase in the number of microglial cells leads to depression through the neuroinflammatory pathway. Activated microglia are also involved in post-stroke depression [133]. Additionally, a decrease in the number of microglial cells causes depression by inducing neuronal degeneration, leading to inhibition of neurogenesis in the hippocampus [134].
Most studies have demonstrated an association between suicidal behavior and alterations in IL-2, IL-6, IL-8, TNF-α and VEGF levels [135]. Elevation of IL-6 levels is one of the most prominent findings in patients exhibiting suicidal behavior. However, future research should focus on the association between cytokines, suicidal behavior, and depression [136, 137]. Moreover, the number of primed microglial cells is increased in the white matter of the dorsal anterior cingulate cortex in depressed suicidal patients compared with healthy control subjects [138].
In an animal model of depression, prenatal stress may be linked to increased activity of microglial cells in the hippocampus and frontal cortex and depression-like disturbances [139, 140]. The hippocampus, especially the CA1 region, is characterized by an elevated number of microglial cells, which are activated by stress [7, 141]. The adult microglial transcription factor MafB is involved in the expression of the adult gene program during inflammatory responses under stress as demonstrated by experiments with knockout mice [142]. Another brain region that is affected by microglial cell activation is the postnatal amygdala, which is involved in emotion, as maternal immune activation causes the activation of microglial cells in the amygdala [143].
The neuroprotective role of microglial cells has been extensively discussed [144,145,146,147]. For example, microglial cells exert neuroprotective effects through anti-inflammatory responses in depression [148, 149] and schizophrenia [150]. Microglia also have neuroprotective effects after ischemia [151]. On one hand, microglial cells are responsible for maintaining neuronal structure and plasticity via clearance of cellular debris, neurogenesis, anti-inflammatory responses, and synaptic pruning [149]. On the other hand, neurons play a vital role in the functions of microglial cells by maintaining inflammatory gene production, the oxidative stress response, and synaptic pruning [149, 152]. Therefore, if the balance between microglial cells and neurons is disturbed, neuropsychiatric disorders may result [149, 153,154,155]. Microglial cells have also been demonstrated to play a neurodegenerative role [156]. For example, a disturbance of the interplay between microglial cells and cytokines may cause neurodegenerative processes in neuropsychiatric diseases such as schizophrenia [34]. Peripheral blood mononuclear cells were combined with neural progenitor cells and pluripotent stem cells to produce microglia-like cells, which express specific markers and function as microglial cells [157]. Microglial cells and monocytes interact with each other to regulate the expression of genes, cytokines, and surface markers through the blood–brain barrier and neuronal networks, and these interactions might be useful for the developing peripheral biomarkers for psychiatric disorders [157].
Prenatal immune challenge by poly(I:C) causes microglial cell alterations in different structures such as microglial clusters, including changes in morphology, the arborization index and the number of dark microglia in the dentate gyrus of the hippocampus, mainly in male mice. These changes are features of cellular stress [158]. Hinwood et al. [159] observed that microglia facilitate the effects of stress on neurons in the medial PFC. Stress exposure correlates with increased microglial activation in the PFC but not antigen presentation [159]. Similarly, microglia are responsible for the impact of stress on neuronal branching in the PFC. Treatment with minocycline eliminates these effects [160]. In patients with depression, repeated stress exposure can lead to microglial inflammation or even suicidal behavior [161]. It has been shown that increased apoptosis causes a reduction in microglial cell number [26].
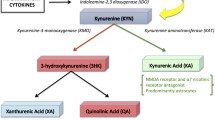
In the CNS, the kynurenine (KYN) pathway is affected by astrocytes, microglial cells, and macrophages. KYN can be converted to kynurenic acid or quinolinic acid [162,163,164]. Abnormal KYN levels are implicated in neurodevelopmental disorders through the proliferation, specification, and differentiation of radial glial cells [165] and the pathophysiology of schizophrenia and affective disorders [166,167,168,169,170,171]. By exploring the inflammatory process and hence the kynurenine pathway, novel therapeutic targets and biomarkers for suicidal depressed patients [172] and patients with schizophrenia [173] might be developed. For example, Steiner et al. [174] reported an increase in the level of quinolinic acid, which is produced by microglial cells, in subregions of the anterior cingulate gyrus in depressed patients. Furthermore, investigations by Busse et al. [59, 175] revealed that quinolinic acid levels are decreased in the hippocampus of suicidal depressed patients compared with healthy control subjects and in patients with paranoid schizophrenia compared with patients with residual schizophrenia [176].
Early postnatal lipopolysaccharide (LPS) administration in a rat animal model of early immune stimulation causes a reduction in hippocampal volume, activation of the KYN pathway of tryptophan metabolism, astrogliosis, and a decrease in tyrosine hydroxylase levels in the substantia nigra demonstrating a link between early immune stimulation and neuropsychiatric disorders [177]. LPS-induced maternal immune activation causes an increase in cytokine levels in the fetal brain, which results in elevation of microglial activation, astrogliosis and cytoarchitectural changes in the postnatal amygdala [143].
The effects of antidepressants, antipsychotics, and electroconvulsive therapy (ECT) on microglial cells
The long-term influences of antidepressants and antipsychotics on microglial cells in various brain regions should not be ignored, as they might offer new options for drug treatment. Therefore, we will discuss the effects of antidepressants, antipsychotics, and electroconvulsive therapy on microglial cells. Typical and atypical antipsychotics suppress microglial activity by inhibiting the secretion of cytokines [178]. Specific interleukins (Il-10, IL-10, and TGF-α) are state markers and increase during acute phases of schizophrenia, but are normalized with antipsychotic medication. Both IL-12, IL-2, interferon- α, and TNF-α are characterized as trait markers and continue to be high during acute phases independent of antipsychotic medication [179]. Typical and atypical antipsychotics influence the intracellular Ca2+ mobilization and signaling in the endoplasmic reticulum (ER) of microglial cells in different ways. Thus, these processes might be targets for antipsychotic therapy [180].
Specifically, inhibitors of microglial activity such as minocycline are regarded as potential antipsychotic drugs [181,182,183]. Riazi et al. [184] showed that minocycline decreased the effects of inflammation and abolished the effects of peripheral inflammation in the hippocampi of rats. For example, minocycline selectively inhibits the expression of M1-polarized microglia in vivo and in vitro in amyotrophic lateral sclerosis (ALS) [185].
Cotel et al. [186] reported that chronic antipsychotic use increases microglial activation and proliferation in the rat brain. Cotel et al. [186] revealed that antipsychotic treatment not only reduced the gray matter volume in the hippocampus, anterior cingulate cortex, corpus striatum, and secondary somatosensory cortex but also increased the density of microglial cells in the hippocampus and somatosensory cortex but surprisingly not in the anterior cingulate cortex. The reason why the microglial density was not increased in the anterior cingulate cortex by chronic antipsychotic use requires further research. In rat experiments, clozapine was shown to increase microglial activation in the striatum and hippocampus, mainly the CA2 and CA3 regions [187]. In contrast, antipsychotics inhibit the release of proinflammatory cytokines and NO by microglial cells [188, 189]. In summary, microglial activity plays a role in the pathophysiology of schizophrenia and anti-inflammatory drugs such as minocycline might be useful for targeting the increased microglial activation in schizophrenia and thus be new treatment options [190, 191]. Regulation of microglial activation site-specifically and function-specifically might be a novel target for the development of new drugs for patients with schizophrenia [192, 193].
For example, studying single-cell screening from blood serum taken from drug-naïve patients with schizophrenia the phenotypic modification of microglial cell signaling in vitro was demonstrated [193]. Overactivated epitopes in blood sera of patients with schizophrenia can be ameliorated by microglial proinflammatory activation inhibitors such as minocycline to improve negative symptoms in schizophrenia [194]. Antagonists of the ATP-gated P2X7 receptor, which is a microglial receptor found in the CNS, might be used as monotherapies or adjunct therapies for the treatment of schizophrenia, bipolar disorder or depression [194]. Activated microglia can be used as biomarkers for predicting the course in bipolar disorder and pharmacological responses [195]. Stertz et al. [196] argued that the inflammatory changes that occur in bipolar disorder patients are associated with disease progression rather than causative. Additionally, the selective serotonin reuptake inhibitor (SSRI) fluoxetine protects neurons against the neurotoxic effects of microglial cells [197]. SSRIs increase TNF-α levels, and when used at low concentrations for a long period of time, SSRIs have slight proinflammatory effect [198]. In contrast, Horikawa et al. [199] reported that SSRIs inhibit the production of NO and TNF-α in microglial cells. Additionally, the SSRI fluoxetine and its metabolite norfluoxetine reduce the viability of microglia and increase the expression of the apoptotic marker cleaved-caspase 3 in microglial cells [200]. Depression is also considered a microglial disease and drugs that either suppress or stimulate microglial cells are regarded as novel drugs in the treatment of depression [147]. Various antidepressants and antipsychotics overturn microglial activation. Antidepressants inhibit the release of proinflammatory cytokines from activated microglial cells [201]. The antidepressant drug venlafaxine can partially protect the viability of microglial cells [202]. Antidepressants cause microglial cell disturbances in the dorsal raphe nucleus [104]. A positive correlation between antidepressants and microglial density in non-suicidal depressed patients in the dorsal raphe nucleus has been reported [104] (Figs. 1 and 2).

Microglial reaction in the DRN (caudal subdivision) in suicidal depressed patient as shown by HLA-DR antigen, scale bar 50 µm

Microglial reaction in the DRN (caudal subdivision) in non-suicidal depressed patient as shown by HLA-DR antigen, scale bar 50 µm
Glutamate has neurotoxic effects on activated microglial cells [203]. For example, an inhibition of glutamine synthetase increases the activity of activated microglial cells [204]. However, a general blockade of microglial cells as a therapeutic intervention might have risky effects, since glutamate and activated microglial cells are involved in dendritic apoptosis. An excessive synaptic pruning during late adolescence and early adulthood might cause severe negative symptoms and a cognitive decline in patients with schizophrenia [205, 206].
Additionally, the use of induced microglial cells (iMG) from the peripheral blood [207] and the use of human-induced pluripotent stem cells for microglial cell culture methods [208] might represent novel directions for psychopharmacological screening or schizophrenia modeling. Cannabis and cannabidiol reduce the oligodendrocyte, astrocyte, and microglial activity in schizophrenia [209, 210]. For example, activated cannabinoid type -2 (CB2) receptors located on microglial cells decrease the effects of activated microglial cells such as neuroinflammation and synaptic pruning [211]. ECT either reduces microglial cell density in the hippocampus [212] or activates microglial cells [213]. It has been demonstrated that microglial cell activation in the hippocampus is inhibited by ECT in a rat model [214].
Evolutionary role of microglial cells in neuropsychiatric disorders
Microglia probably arose during the Cambrian period (500–540 Ma ago) with the propagation of multicellular animals. Microglia have been evolutionarily conserved in both invertebrates and vertebrates [215,216,217]. It has been argued that early microglia contributed to the protection of the CNS in emerging vertebrates, and informed axonal coverage, thus increasing action potential transmission and speed and thereby improving CNS efficiency [218].
It is worth noting that human neuronal density is much lower than that of chimpanzees and other apes and primates [219]. This means a greater volume of glial cells, likely also microglia in proportion to neurons. Such an arrangement allows for greater role of glial cells in processing of information in the human brain. It has been shown that working memory is different in humans than in chimpanzees. In humans, there is evidence that genes of abnormal spindle-like microcephaly-associated protein (APSM), neuronal cell adhesion molecule (NRCAM) and sialic acid-binding Ig like lectin 11 (SIGLEC11) are involved in neural proliferation, promotion of neural connections, and glial expression, respectively [220, 221]. There is a difference in ASPM and SIGLEC-11 in the human lineage expression compared to chimpanzee lineage. SIGLEC-11 is a human-specific microglial gene that is not found in other primates [222]. A study by Wang et al. [223] using human brain tissue demonstrated that human SIGLEC-11 ectopically expressed by a lentiviral vector system in cultured murine microglial cells interacts with polysialic acid (PSA) residues on neurons, reduces LPS-induced gene transcription of proinflammatory mediators, impairs phagocytosis and alleviates microglial neurotoxicity.
The pathogen host defence (PATHOS-D) theory suggests that the immune responses that occur in MDD are proinflammatory and that specific risk alleles for MDD generate a proinflammatory response, which is a driving force in human evolution [224]. Divergence from microglial stability due to inflammatory conditions that trigger microglial activity (e.g. infections, neurodegenerative diseases, stress, brain trauma) are risk factors for depression [147].
It should be noted that during the last 2 million years there have been notable changes in the neurohormonal regulation of the human brain due to self-amplifying feedback processes that contributed not only to the size and capacity of the modern human brain but also to its susceptibility to psychopathologies. H. erectus (1–8 Ma) exhibited a marked increase in physical activity levels (PALs) and total energy expenditure (TEE) compared to australopithecine and the great ape lineages [225, 226]. Selective processes enhanced aerobic mechanisms and modified the thermoregulatory response (i.e., hairlessness in humans facilitated evaporative sweating and increased UV exposure) [227, 228] to optimize the bodies of archaic hominins for persistent hunting in hot environments, thereby increasing the production of growth factors, such as BDNF [229, 230]. Selection for characteristics important for persistent hunting and endurance is biased towards muscle composition changes and the development of more efficient energy systems involving peroxisome proliferator-activated receptor γ coactivator (PGC-1α) and myocyte enhancer factor-2 (MEF2) gene regulation, which stimulate BDNF factor activity [227]. In archaic hominins, BDNF informed brain evolution in cortical and subcortical areas [231, 232]. BDNF also contributes to neurogenesis in the hippocampus, memory and learning. Synaptic plasticity is associated with prenatal brain development [233, 234] and neuroprotection, and ~ 80% of synaptic plasticity is dependent on exercise [235]. Furthermore, BDNF mediates microglial proliferation, promotes microglia and astrocyte activation and has potential to increase neuroinflammation [233]. It can be speculated that changes in thermoregulatory processes that were crucial for increasing the PALs of Homo erectus onwards may have come at an evolutionary cost, altering the expression of BDNF dependent microglia and consequentially that of proinflammatory CNS markers implicated in neuropsychiatric disorders [232]. Thus, there is evidence for a role of microglial cells in the evolution of neuropsychiatric disorders. Therefore, this view suggests that evolutionary psychiatry may become a novel and distinct area of research and doctrine in the field of biological psychiatry [236]. Due to the mismatch between prehistoric and modern environments, humans are becoming susceptible to inflammatory markers due to chronic stress. Evolutionary psychiatry offers an explanatory model for understanding modern psychiatric disorders and their evolutionary antecedents. Psychiatry has reached a critical point where it needs to develop therapeutic methods that integrate evolutionary thinking. This will be especially important in the advent of novel biotechnologies that may alter psychoneuroendocrinological mechanisms in ways that are beyond our current understanding.
Abbreviations
- AD:
-
Affective disorders
- ALS:
-
Amyotrophic lateral sclerosis
- APSM:
-
Abnormal spindle-like microcephaly-associated protein
- AMPA:
-
(S)-2-amino-3-(3-hydroxy-5-methyl-4-isoxazole)-propionic acid
- APOE:
-
Apolipoprotein type E
- ASDs:
-
Autism spectrum disorders
- ATP:
-
Adenosine triphosphate
- BDNF:
-
Brain-derived neurotrophic factor
- CXCL1:
-
Fractalkine
- CD:
-
Cluster of differentiation
- CNS:
-
Central nervous system
- CSF:
-
Cerebrospinal fluid
- DAT:
-
Dementia of Alzheimer’s type
- DRN:
-
Dorsal raphe nucleus
- DRD:
-
Dopamine receptors
- ECT:
-
Electroconvulsive therapy
- ER:
-
Endoplasmic reticulum
- HLA-DR:
-
Human leukocyte antigen
- IBA1:
-
Ionized calcium-binding adaptor molecule 1
- IFN-α:
-
Interferon-alpha
- IL:
-
Interleukin
- KYN:
-
Kynurenine
- LPS:
-
Lipopolysaccharide
- MHC:
-
Major histocompatibility complex
- MDD:
-
Major depressive disorder
- NMDAR:
-
N-methyl-D-aspartate receptors
- NO:
-
Nitric oxide
- NOX2:
-
NADPH oxidase 2
- NRCAM:
-
Neuronal cell adhesion molecule
- PAL:
-
Physical activity level
- PATHOS-D:
-
Pathogen host defence
- PFC:
-
Prefrontal cortex
- PD:
-
Parkinson’s disease
- PSA:
-
Polysialic acid
- PET:
-
Positron emission tomography
- PND:
-
Postnatal day
- PGE2:
-
Prostaglandin E2
- SN:
-
Substantia nigra
- SIGLEC11:
-
Sialic acid-binding Ig like lectin 11
- SIRT 2:
-
Silent information regulator 2
- SSRI:
-
Selective serotonin reuptake inhibitors
- SZ:
-
Schizophrenia
- TEE:
-
Total energy expenditure
- TGF-α:
-
Transforming growth factor alpha
- TH-1:
-
T-helper cells 1
- TH-2:
-
T-helper cells 2
- TNF-α:
-
Tumor necrosis factor alpha
- TSPO PET:
-
Translocator protein positron emission tomography
References
Mosser CA, Baptista S, Arnoux I, Audinat E (2017) Microglia in CNS development: Shaping the brain for the future. Prog Neurobiol 149–150:1–20. https://doi.org/10.1016/j.pneurobio.2017.01.002
Ben Achour SB, Pascual O (2010) Glia: the many ways to modulate synaptic plasticity. Neurochem Int 57:440–445
Walter L, Neumann H (2009) Role of microglia in neuronal degeneration and regeneration. Semin Immunpathol 31:513–525
Napoli I, Neumann H (2009) Microglial clearance function in health and disease. Neuroscience 158:1030–1038
Nakagawa Y, Chiba K (2014) Role of microglial M1/M2 polarisation in relapse and remission of psychiatric disorders and diseases. Pharmaceuticals 7:1028–1048
Na KS, Jung HY, Kim YK (2014) The role of pro-inflammatory cytokines in the neuro-transmission and neurogenesis in schizophrenia. Prog Neuro-Psychopharmacol Biol Psychiatry 48:277–286
Brites D, Fernandes A (2015) Neuroinflammation and depression: microglia activation, extravesicular microvesicles and microRNA dysregulation. Front Cell Neurosci 9:476
Mayhew J, Beart PM, Walker FR (2015) Astrocyte and microglial control of glutamatergic signaling: A primer on understanding the disruptive role of chronic stress. J Neuroendocrin 27:498–506
Howes OD, McCutcheon R (2017) Inflammation and the neural diathesis-stress hypothesis of schizophrenia: a reconceptualization. Transl Psychiatry 7:e1024
Lenz KM, McCarthy MM (2015) A starring role for microglia in brain sex differences. Neuroscientist 21:306–321
Orihuela R, McPherson CA, Harry GJ (2016) Microglial M1/M2 polarization and metabolic states. Br J Pharmacol 173:649–665
Karve IP, Taylor JM, Crack PJ (2016) The contribution of astrocytes and microglia to traumatic brain injury. Br J Pharmacol 173:692–702
Thibaut F (2017) Neuroinflammation: new vistas for neuropsychiatric research. Dialogues Clin Neurosci 19:3–4
De Picker LJ, Morrens M, Chance SA, Boche D (2017) Microglia and brain plasticity in acute psychosis and schizophrenia illness course: a meta-review. Front Psychiatry 8:238
Meyer U, Feldon J, Dammann O (2013) Schizophrenia and autism: Both shared and disorder-specific pathogenesis via perinatal inflammation. Prog Neuro-Psychopharmacol Biol Psychiatry 42:20–34
Lurie DI (2018) An integrative approach to neuroinflammation in psychiatric disorders and neuropathic pain. J Exper Neurosci 12:1–11
Najjar S, Pearlman DM (2015) Neuroinflammation and white matter pathology in schizophrenia: systematic review. Schizophr Res 161:102–122
Chew LJ, Fusar-Poli P, Schmitz T (2013) Oligodendrocyte alterations and the role of microglia in white matter injury: relevance to schizophrenia. Dev Neurosci 35:102–129
Doorduin J, de Vries EFJ, Willemsen ATM, de Groot JC, Dierck RA, Klein HC (2009) Neuroinflammation in schizophrenia-related psychosis: A PET study. J Nucl Med 50:1801–1807
Skaper SD, Facci L, Giusti P (2014) Neuroinflammation, microglia and mast cells in the pathophysiology of neurocognitive disorders: a review. CNS Neurol Disord Drug Targets 13:1654–1666
Filiou MD, Arefin AS, Moscato P, Graeber MB (2014) Neuroinflammation differs categorically from inflammation: transcriptomes of Alzheimer´s disease, Parkinson´s disease, schizophrenia and inflammatory diseases compared. Neurogenetics 15:201–212
Kim YK, Na KS, Myint AM, Leonard BE (2016) The role of pro-inflammatory cytokines in neuroinflammation and the neuroendocrine system in major depression. Prog Neuro-Psychopharmacol Biol Psychiatry 64:277–284
Mallya AP, Deutsch AY (2018) (Micro)Glia as effectors of cortical volume loss in schizophrenia. Schizophr Bull 44:948–957
Szepesi Z, Manouchehrian O, Bachiller S, Deierborg T (2018) Bidirectional microglia-neuron communication in health and disease. Front Cell Neurosci 12:323
Volk DW (2017) Role of microglia disturbances and immune-related marker abnormalities in cortical circuitry dysfunction in schizophrenia. Neurobiol Dis 99:58–65
Blank T, Prinz M (2013) Microglia as modulators of cognition and neuropsychiatric disorders. Glia 61:62–70
Bollinger JL, Wohleb ES (2019) The formative role of microglia in stress-induced synaptic deficits and associated behavioral consequences. Neurosci Lett. 711:134369
Zhao H, Alm A, Chen Q, Eusman MA, Pal A, Eguchi S et al (2017) The role of microglia in the pathobiology of neuropathic pain development: what do we know? Br J Anasth 118:504–516
Uranova NA, Vikhreva OV, Rakhmanova VI, Orlovskaya DD (2020) Dystrophy of oligodendrocytes and adjacent microglia in prefrontal gray matter in schizophrenia. Front Psychiatry 11:204
Müller N, Myint AM, Schwarz MJ (2009) The impact of neuron in the dysregulation on neuroprotection and neurotoxicity in psychiatric disorders-relation to drug treatment. Dialog Clin Neurosci 11:319–332
Réus GZ, Fries GR, Stertz L, Badaway M, Passos IC, Barichello T, Kupczinski F, Quevedo J (2015) The role of inflammation and microglial activation in the pathophysiology of psychiatric disorders. Neuroscience 300:141–154
Bernstein HG, Steiner J, Guest PC, Dobrowolny H, Bogerts B (2015) Glial cells as key players in schizophrenia pathology. Schizophr Res 161:4–18
Leza JC, Garcia-Bueno B, Bioque M, Arango C, Parellada M, Do K, O’Donnell P, Bernardo M (2015) Inflammation in schizophrenia: A question of balance. Neuroscience Biobehav. Rev. 55:612–626
Hong H, Kim BS, Im H-I (2016) Pathophysiological role of neuroinflammation in neurodegnerative diseases and psychiatric disorders. Int Neurolurol J 20(Suppl 1):S2-7
Tay TL, Bléchade C, D’Andrea I, St-Pierre MK, Henry MS, Roumier A, Tremblay ME (2018) Microglia gone rogue: impacts on psychiatric disorders across the lifespan. Front Mol Neurosci 10:421
Morgan JT, Chana G, Pardo CA, Achim C, Semendeferi K, Buckwalter J, Courchsese E, Everall IP (2010) Microglial activation and increased microglial density observed in the dorsolateral prefrontal cortex in autism. Biol Psychiatry 68:368–376
Tetreault NA, Hakeem AY, Jiang S, Williams BA, Allman E, Wold BJ, Allman JM (2012) Microglia in the cerebral cortex in autism. J Autism Dev Disord 42:2569–2584
Bilbo S, Stevens B (2017) Microglia: the brain’s first responders. Cerebrum 2017:cer-14-17
Racki V, Petric D, Kucic N, Grezeta N, Jurdana K, Roncevic-Grzeta I (2016) Cortical gray matter loss in schizophrenia: Could microglia be the culprit? Med. Hypotheses 88:18–21
Trépanier MO, Hopperton KE, Mizrahi R, Mechawar N, Bazinet RP (2016) Postmortem evidence of cerebral inflammation in schizophrenia: a systematic review. Mol Psychiatry 21:1009–1026
van Kesteren CFMG, Gremmels H, de Witte LD, Hol EM, Van Gool AR, Falkai PG, Kahn RS, Sommer IEC (2017) Immune involvement in the pathogenesis of schizophrenia: a meta-analysis on postmortem brain studies. Transl Psychiatry 7:e1075
Foster R, Kandanearatchi A, Beasley C, Williams B, Khan N, Fagerhol MF, Everall IP (2006) Calprotectin in microglia from frontal cortex is up-regulated in schizophrenia. Evidence for an inflammatory process?. Eur J Neurosci 24:3561–3566
Imai Y, Ibata I, Ito D, Ohsawa K, Kohsaka S (1996) A novel gene 1 in the major histocompatibility complex class III region encoding an EF hand protein expressed in a monocytic lineage. Biochem Biophys Res Commun 224:855–862
Petrasch-Parwez E, Schöbel A, Benali A, Moinfar Z, Förster E, Brüne M, Juckel G (2020) Lateralization of increased density of Iba 1-immunopositive microglial cells in the anterior midcingulate cortex of schizophrenia and bipolar density. Eur Arch Psychiatry Clin Neurosci. https://doi.org/10.1007/s00406-020-01107-0
Bloomfield PS, Selvaraj S, Veronese M, Rizzo G, Bertoldo A, Owen DR, Bloomfield MAP, Bonoldi I, Kalk N, Turkheimer F, McGuire P, de Paola V, Howes OD (2016) Microglial activity in people at ultra risk of psychosis and in schizophrenia: An [11C] PBR28 PET brain imaging study. Am J Psychiatry 173:44–52
Hafizi S, Tseng HH, Rao N, Selvanathan T, Kenk M, Bazinet RP, Suridjan I, Wilson AA, Meyer JH, Remington G, Houle S, Rusjan PM, Mizrahi R (2017) Imaging microglial activation in untreated first-episode psychosis: a PET study with [18] FEPPA. Am J Psychiatry 174:118–124
Di Biase MA, Zalesky A, O’Keefe G, Laskaris L, Baune BT, Weickert CS, Olver J, McGorry PD (2017) PET imaging of putative microglial activation in individuals at ultra-high risk for psychosis, recently diagnosed and chronically ill with schizophrenia. Transl. Psychiatry 7:e1225
Marques TR, Ashok AH, Pillinger T, Veronese M, Turkheimer FE, Dazzan P, Sommer IEC, Howes OD (2019) Neuroinflammation in schizophrenia: meta-analysis of in vivo microglial imaging studies. Psychol Medicine 49:2186–2196
Conen S, Gregory CJ, Hinz R, Smallman R, Corsi-Zuelli F, Deakin B, Talbot PS (2020) Neuroinflammation as measured positron emission tomography in patients with recent onset and established schizophrenia: implications for immune pathogenesis. Mol Psychiatry. https://doi.org/10.1038/s41380-020-0829-y
Banati R, Hickie IB (2009) Therapeutic signposts: using biomarkers to guide better treatment of schizophrenia. MJA 190:S26–S32
Burmester GR, Pezutto A (2003) Color atlas of immunology. Thieme Publishers Stuttgart New York. pp. 1–322
Steiner J, Bielau H, Brisch R, Danos P, Ullrich O, Mawrin C, Bernstein HG, Bogerts B (2008) Immunological aspects in the neurobiology of suicide: Elevated microglial density in schizophrenia and depression is associated with suicide. J Psychiatry Res 42:151–157
Bayer TA, Buslei R, Havas L, Falkai P (1999) Evidence for activation of microglia in patients with psychiatric illnesses. Neurosci Lett 271:126–128
Radewicz K, Garey LJ, Gentleman S, Reynolds R (2000) Increase in HLA-DR immunoreactive microglia in frontal and temporal cortex of chronic schizophrenics. J Neuropathol Exp Neurol 59:137–150
Wierzba-Bobrowicz T, Lewandowska E, Lechowicz W, Stepien T, Pasenik E (2005) Qualitative analysis of activated microglia, ramified and damage of processes in the frontal and temporal lobes of chronic schizophrenics. Folia Neuropathol 43:157–165
Krause D, Wagner J, Matz J, Weidinger E, Obermeier M, Riedel M, Gruber R, Schwarz M, Mueller N (2012) Monocytic HLA DR antigens in schizophrenic patients. Neurosci Res 72:87–93
Steiner J, Mawrin C, Ziegeler A, Bielau H, Ullrich O, Bernstein HG, Bogerts B (2006) Distribution of HLA-DR-positive microglia in schizophrenia reflects impaired cerebral lateralization. Acta Neuropathol 112:305–3016
Fillman SG, Cloonan N, Catts VS, Miller LC, Wong J, McCrossin T, Cairns M, Weickert CS (2013) Increased inflammatory markers identified in the dorsolateral prefrontal cortex of individuals with schizophrenia. Mol Psychiatry 18:206–214
Busse S, Busse M, Schiltz K, Bielau H, Gos T, Brisch R, Mawrin C, Schmitt A, Jordan W, Müller UJ, Bernstein HG, Bogerts B (2012) Different distribution patterns of lymphocytes and microglia in the hippocampus of patients with residual schizophrenia versus paranoid schizophrenia: Further evidence for disease course-related immune alterations. Brain Behav Imm 26:1273–1279
Sneeboer MAM, Snijders GJLJ, Berdowski WM, Fernandez-Andreu A, Psychiatric Donor Program of the Netherlands Brain Bank (NBB-Psych), van Mierlo HC, van Berlekom AB, Litjens M, Kahn RS, Hol EM, de Witte LD (2019) Microglia in post-mortem brain tissue of patients with bipolar disorder are not immune activated. Transl Psychiatry 9:153
Shigeta Y, Ishii H, Takagi S, Yoshitake Y, Hirano T, Takata H et al (1980) HLA antigens as immunogenetic markers of alcoholism and alcoholic liver diseases. Pharmacol Biochem Beh 13:89–94
Cook RT, Gurvey MJ, Booth BM, Goeken JA, Stewart B, Noel M (1991) Activated CD-8 cells and HLA-DR expression in alcoholics without overt liver disease. J Clin Immunol 11:246–253
Laso FJ, Madruga JF, San Miguel JF, López M, Alvarez M, Orfao A (1996) Long lasting immunological effects of ethanol after withdrawal. Cytometry 26:275–280
Pan Y, Wang KS, Wang L, Wu LY (2013) Common variants in HLA-DRA gene are associated with alcohol dependence in two Caucasian samples. J Mol Neurosci 49:574–581
Laskaris LE, Biase MA, Everall I, Chana G, Christopoulos A, Skafidas E, Cropley VI, Pantelis C (2016) Microglial activation and progressive brain changes in schizophrenia. Br J Pharmacol 173:666–680
Al-Haddad BJS, Oler E, Armistead B, Elsayed NA, Weinberger DR, Bernier R, Burd I, Kapur R, Jacobsson B, Wang C, Mysorekar I, Rajagopal L, Waldorf KMA (2019) The fetal origins of mental illness. Am J Obstet Gynecol 221:549–562
Goudriaan A, de Leeuw C, Ripke S, Hultman CM, Sklar P, Sullivan PF, Smit AB, Posthuma D, Verheijen MHG (2014) Specific glial functions contribute to schizophrenia susceptibility. Schizophr Bull 40:925–935
Arinami T, Otsuka Y, Hamaguchi H, Itokawa M, Aoki J, Shibuya H, Okubo Y, Jwawaki Y, Ota K, Enguchi H, Tagaya H, Yano S, Shimuzu H, Torio M (1998) Evidence supporting an association between the DRB1 gene and schizophrenia in Japanese. Schizophr Res 32:81–86
Sasaki T, Matsushita M, Nanko S, Fukuda R, Kennedy JL, Tokunga K (1999) Schizophrenia and HLA-DRB1 gene in the Japanese population. Am J Psychiatry 156:771–773
Wright P, Nimgaonkar VL, Donaldson PT, Murray RM (2001) Schizophrenia and HLA: a review. Schizophr Res 47:1–12
Müller N, Ackenheil M (1998) Psychoneuroimmunology and the cytokine action in the CNS: implications for psychiatric disorders. Prog Neuropsychopharmacol Biol Psychiatry 22:1–33
Shi L, Fatemi SH, Sidwell RW, Patterson PH (2003) Maternal influenza infection causes marked behavioral and pharmacological changes in the offspring. J Neurosci 23:297–302
Cowan M, Petri WA Jr (2018) Microglia: Immune regulators of neurodevelopment. Front Immunol 9:2576
Jiang NM, Cowan M, Moonah SN, Petri WA Jr (2018) The impact of systematic inflammation on neurodevelopment. Trends Mol Med 24(9):794–804
Nelson LH, Saulsbery AI, Lenz KM (2019) Small cells with big implications: microglia and sex differences in brain development, plasticity and behavioral health. Prog Neurobiol 176:103–109
Osborne BF, Turano A, Caulfield JI, Schwarz JM (2019) Sex- and region-specific differences in microglia phenotype and characterization of the peripheral immune response following early-life infection in male and female rats. Neurosci Lett 692:1–9
Smolders S, Smolders SMT, Swinnen N, Gärtner A, Rigo J-M, Legendre P, Brone B (2015) Maternal immune activation evoked by polyinosinic:polycytidylic acid does not evoke microglial cell activation in the embryo. Front Cell Neurosci 9:301
Juckel G, Manitz MP, Brüne M, Friebe A, Heneka MT, Wolf RJ (2011) Microglial activation in a neuroinflammational animal model of schizophrenia - a pilot study. Schizophr Res 131:96–100
Shapiro LA, Perez ZD, Foresti ML, Arisi GM, Ribak CE (2009) Morphological andultrastructural features of Iba1-immunolabeled microglial cells in the hippocampal dentate gyrus. Brain Res 1266:29–36
Stence N, Waite M, Dailey ME (2001) Dynamics of microglial activation: a confocal time-lapse analysis in hippocampal slices. Glia 33:256–266
Manitz MP, Esslinger M, Wachholz S, Plümper J, Friebe A, Juckel G, Wolf RJ (2013) The role of microglia during life span in neuropsychiatric disease — an animal study. Schizophr Res 143:221–222
Buschert J, Sakalem ME, Saffari R, Hohoff C, Rothermundt M, Arolt V, Zhang W, Ambrée O (2016) Prenatal immune activation in mice blocks the effects of environmental enrichment on explatory behavior and microglia density. Prog Neuro-Psychopharmacol Biol Psych 67:10–20
Vidal PM, Pacheco R (2020) The cross-talk between the dopaminergic and the immune system involved in schizophrenia. Front Pharmacol 11:394
World Health Organization (2014) Preventing suicide: a global imperative. pp. 1–92
Ernst C, Mechawar N, Turecki G (2009) Suicide neurobiology. Prog Neurobiol 89:315–333
American Psychiatric Association (2013) Diagnostic and statistical manual of mental disorders, 5th edn. American Psychiatric Publishing, Arlington
Baker K, Halliday GM, Törk I (1990) Cytoarchitecture of the human dorsal raphe nucleus. J Comp Neurol 301:147–161
Stockmeier CA, Shapiro LA, Haycock JW, Thompson PA, Lowy MT (1996) Quantitative subregional distribution of serotonin1A receptors and serotonin transporters in the human dorsale raphe. Brain Res 727:1–12
Valentino RJ, Commons KG (2005) Peptides that fine-tune the serotonin system. Neuropeptides 39:1–8
Lowry CA, Hale MW, Evans AK, Heerkens J, Staub DR, Gasser PJ, Shekhaw A (2008) Serotonergic systems, anxiety, and affective disorder: Focus on the dorsal raphe nucleus. Ann N Y Acad Sci 1148:86–94
Ranft K, Dobrowolny H, Krell D, Bielau H, Bogerts B, Bernstein HG (2010) Evidence for structural abnormalities of the human habenular complex in affective disorders but not in schizophrenia. Psychol Med 40:557–567
Zhao H, Zhang BL, Yang SJ, Rusak B (2015) The role of lateral habenula-dorsal raphe nucleus circuits in higher brain functions and psychiatric illness. Behav Brain Res 277:89–98
Kasper JM, McCue DL, Milton AJ, Szwed A, Sampson CM, Huang M, Carlton S, Meltzer HY, Cunningham KA, Hornell JD (2016) Gamma-aminobutyric acidergic projections from the dorsal raphe to the nucleus accumbens are regulated by neuromedin U. Biol Psychiatry 80:878–887. https://doi.org/10.1016/j.biopsycho.2016.02.031
Bolderini M, Underwood MD, Mann JJ, Arango V (2005) More tryptophan hydroxylase in the brainstem dorsal raphe nucleus in depressed suicides. Brain Res 1041:19–28
Bonkale WL, Murdock S, Janosky JE, Austin MC (2004) Normal levels of tryptophan hydroxylase immunoreactivity in the dorsal raphe of depressed suicide victims. J Neurochem 88:958–964
Bielau H, Mawrin C, Krell D, Agelink MW, Trübner K, Davis R, Gos T, Bogerts B, Bernstein HG, Baumann B (2005) Differences in activation of the dorsal raphe nucleus depending on performance of suicide. Brain Res 1039:43–52
Craven RM, Priddle TH, Coopert SJ, Crow TJ, Esiri MM (2005) The dorsal raphe nucleus in schizophrenia: a post-mortem study of 5-hydroxytryptamine neurons. Neuropathol Appl Neurobiol 31:258–269
Bonkale WL, Turecki G, Austin MC (2006) Increased tryptophan hydroxylase immunoreactivity in the dorsal raphe nucleus of alcohol-dependent, depressed suicide subjects is restricted to the dorsal subnucleus. Synapse 60:81–85
Bach-Mizrachi H, Underwood MD, Kassir SA, Bakalian MJ, Sibille E, Tamir H, Arango V, Mann J (2006) Neuronal tryptohan hydroxylase mRNA expression in the human dorsal and median raphe nuclei. Neuropsychopharmacol 31:814–824
Bach-Mizrachi H, Underwood MD, Tin A, Ellis SP, Mann JJ, Arango V (2008) Elevated expression of tryptophan hydroxylase-2 mRNA at the neuronal level. Mol Psychiatry 13:507–513
Gos T, Krell D, Brisch R, Bielau H, Trübner K, Steiner J, Bernstein HG, Baumann B (2008) Demonstration of decreased activity of dorsal raphe nucleus neurons in depressed suicidal patients by the AgNOR staining method. J Affect Disord 111:251–260
Krzyzanowska M, Steiner J, Karnecki K, Kaliszan M, Brisch R, Wiekowski M, Braun K, Jankowski Z, Gos T (2016) Decreased ribosomal DNA transcription in dorsal raphe differentiates between suicidal and non-suicidal death. Eur Arch Psychiatry Clin Neurosci 266:217–224
Krzyzanowska M, Steiner J, Brisch R, Mawrin C, Busse S, Karnecki K, Jankowski Z, Gos T (2016) Decreased ribosomal DNA transcription in dorsal raphe nucleus is specific for suicide regardless of psychiatric diagnosis. Psychiatry Res 241:43–46
Brisch R, Steiner J, Mawrin C, Krzyzanowska M, Jankowski Z, Gos T (2017) Microglia in the dorsal raphe nucleus plays a potential role in both suicide facilitation and prevention in affective disorders. Eur Arch Psychiatry Clin Neurosci 267:403–415
Bach H, Arango V, Kassir SA, Tsaava TT, Dwork AJ, Mann J, Underwood MD (1997) Alcoholics have more tryptophan hydroxylase 2 mRNA and protein in the dorsal and median raphe nuclei. Alcohol Clin Exp Res 2014(38):1894–1901
Stockmeier CA (1997) Neurobiology of serotonin in depression and suicide. Ann N Y Acad Sci 723:37–45
Jasinska AJ, Lowry CA, Burmester M (2012) Serotonin transporter gene, stress and raphe-raphe interactions: a molecular mechanism of depression. Trends Neurosci 35:395–402
Albert PR, Benkelfast C (2013) The neurobiology of depression-revisiting the serotonin hypothesis. II Genetic, epigenetic, and clinical studies. Phil Trans R Soc B 368:20120535
Mann JJ (2013) The serotonergic system in mood disorders and suicidal behavior. Phil Trans R Soc B 368:20120537
Hahn A, Haeusler D, Kraus C, Höflich AS, Kranz GS, Baldinger P et al (2014) Attenuated serotonin transporter association between dorsal raphe and ventral striatum in major depression. Hum Brain Mapping 35:3857–3866
Bobillier P, Seguin S, Petitjean F, Salvert D, Touret M, Jouvet M (1976) The raphe nuclei of the rat brain stem: a topographical atlas of their efferent projections as revealed by autoradiography. Brain Res 113(3):449–486. https://doi.org/10.1016/0006-8993(76)90050-0
Soares JC, Mann J (1997) The functional neuroanatomy of mood disorders. J Psychiatr Res 31:393–432
Underwood MD, Khaibulina AA, Ellis SP, Moran A, Rice PM, Mann J, Arango V (1999) Morphometry of the dorsal raphe nucleus serotonergic neurons in suicide victims. Biol Psychiatry 46:473–483
Arango V, Underwood MD, Boldrini M, Tamir H, Kassir SA, Hsiung SC, Chen JJ, Mann JJ (2001) Seotonin 1A receptors, serotonin transporter binding and serotonin transporter mRNA expression in the brainstem of depressed suicide victims. Neuropsychopharmacol 25:892–903
Nestler EJ, Barrot M, DiLeone RJ, Eisch AJ, Gold SJ (2002) Neurobiology of depression. Neuron 34:13–25
Hornung JP (2003) The human raphe nuclei and the serotonergic system. J Chem Neuroanatomy 26:331–343
Mann JJ (2003) Neurobiology of suicidal behavior. Nature Rev Neurosci 4:819–823
Nemeroff CB, Vale WW (2005) The neurobiology of depression: in roads to treatment and new drug discovery. J Clin Psychiatry 66(Suppl 7):5–13
Bolderini M, Underwood MD, Mann JJ, Arango V (2008) Serotonin-1A autoreceptor binding in the dorsal raphe nucleus of depressed suicides. J Psychiatry Res 42:433–442
Matthews PR, Harrison PJ (2012) A morphometric, immunohistochemical, and in situ hybridization study of the dorsal raphe nucleus in major depression, bipolar disorder, schizophrenia, and suicide. J Affect Disord 137:125–134
Quesseveur G, Reperant C, David DJ, Gardier AM, Sanchez C, Guiard BP (2013) 5-HT2A receptor inactivation potentiates the acute antidepressant-like activity of escitalopram: involvement of the noradrenergic system. Exp Brain Res 226:285–295
Challis C, Berton O (2015) Top-down control of serotonin systems by the prefrontal cortex: a path toward restored socioemotional in depression. ALS Chem Neurosci 6:1040–1054
Rahn KA, Cao YJ, Hendrix CW, Kaplin AI (2015) The role of 5-HT1A receptors in mediating acute negative effects of antidepressants: implications in pediatric depression. Transl Psychiatry 5:e563
Sullivan GM, Oquendo MA, Milak JM, Miller JM, Burke A, Ogden RT, Parsey RV, Mann JJ (2015) Positron emission tomography quantification of serotonin1A receptor binding in suicide attempters with major depressive disorder. JAMA Psychiat 72:169–178
Dankoski EC, Carroll S, Wightman RM (2016) Acute selective serotonin reuptake inhibitors regulate the dorsal raphe nucleus causing amplification of terminal serotonin release. J Neurochem 136:1131–1141
Wang L, Zhou C, Zhu D, Wang X, Fang L, Zhong J, Mao Q, Sun L, Gong X, Xia J, Lian B, Xie P (2016) Serotonin-1A receptor alterations in depression: a meta-analysis of molecular imaging studies. BMC Psychiatry 16:319
Malone KM, Waternaux C, Haas GL, Cooper TB, Li S, Mann J (2003) Cigarette smoking, suicidal behavior, and serotonin function in major psychiatric disorders. Am J Psychiatry 160:773–779
Linker KE, Elabd MG, Tawadrous P, Cano M, Green KN, Wood MA, Leslie FM (2020) Microglial activation increases cocaine self-administration following adolescent nicotine exposure. Nat Commun 11:306
Wachholz S, Eßlinger M, Plümper J, Manitz MP, Juckel G, Friebe A (2016) Microglia activation is associated with IFN-α induced depressive-like behavior. Brain Behav Imm 55:105–113
Schnieder TP, Trencevska I, Rosoklija G, Stankov A, Mann J, Smiley J, Dwork AJ (2014) Microglia of prefrontal white matter in suicide. J Neuropathol Exp Neurol 73:880–890
Suzuki H, Ohgidani M, Kuwano N, Chrétien F, Lorin de la Grandmaison G, Onaya M, Tominaga I, Setoyama D, Kang D, Mimura M, Kanba S, Kato TH (2019) Suicide and microglia: recent findings and future perspectives based on human studies. Front Cell Neurosci 13:31
Mallya AP, Wang HD, Lee HNR, Deutsch AY (2019) Microglial pruning of synapses in the prefrontal cortex during adolescence. Cereb Cortex 29:1631–1643. https://doi.org/10.1093/cercor/bhy061
Jawaid A, Krajewska J, Pawliczak F, Kandra V, Schulz PE (2016) A macro role for microglia in poststroke depression. JAGS 64:459–461
Singhal G, Baune T (2017) Microglia: an interface between the loss of neuroplasticity and depression. Front Cell Neurosci 11:270
Serafini G, Pompili M, Seretti ME, Stefani H, Palermo M, Coryell W, Girardi P (2013) The role of inflammatory cytokines in suicidal behavior: A systematic review. Eur Neuropsychopharmacol 23:1672–1686
Mina VAL, Lacerda-Pinheiro SF, Maia LC, Pinheiro RFF Jr, Meireles CB, de Souza SIR, Reis AOA, Bianco B, Rolim MLN (2015) The influence of inflammatory cytokines in physiopathology of suicidal behavior. J Affect Disord 172:219–230
Gananca L, Oquendo MA, Tyrka AR, Cisneros-Trujilo S, Mann JJ, Sublette ME (2016) The role of cytokines in the pathophysiology of suicidal behavior. Psychoneuroendocrinology 63:296–310
Torres-Platas SG, Cruceanu C, Chen GG, Turecki G (2014) Evidence for increased microglial priming and macrophage recruitment in the dorsal anterior cingulate white matter of depressed suicides. Brain Behav Immun 42:50–59
Slusarczyk J, Trojan E, Glombik K, Budziszewska B, Kubera M, Lason W, Papiolek-Barczyl K, Mika J, Wedzong K, Basta-Kaim A (2015) Prenatal stress is a vulnerability factor for altered morphology and biological activity of microglial cells. Front Cell Neurosci 9:82
Stein DJ, Vasconcelos MF, Albrecht-Souza L, Ceresér KMM, de Almeida RMM (2017) Microglial over-activation contributes to anxiety- and depressive-like behaviors. Front Behav Neurosci 11:207
Turano A, Lawrence JH, Schwarz JM (2017) Activation of neonatal microglia can be influenced by other neural cells. Neurosci Lett 657:32–37
Matcovitch-Natan O, Winter DR, Giladi A, Aguilar SV, Spinrad A, Sarrazin S, Ben-Yehuda H, David E, González FZ, Perrin P, Keren-Shaul H, Gury M, Lara-Astaiso D, Thaiss CA, Cohen M, Halpern KB, Baruch K, Deczkowska A, Lorenzo-Vivas E, Itzkovitz S, Elinav E, Sieweke MH, Schwartz M, Amit I (2016) Microglia development follows a stepwise program to regulate brain homeostasis. Science. https://doi.org/10.1126/science.aad8670
O’Loughin E, Pakan JM, Yilmazer-Hanke D, McDermott KW (2017) Acute in utero exposure to lipopolysaccharide induces inflammation in the pre- and postnatal brain and alters the glial cytoarchitecture in the developing amygdala. J. Neuroinflamm. 14:212
Polazzi E, Monti B (2010) Microglia and neuroprotection: From in vitro studies to therapeutic applications. Prog Neurobiol 92:293–315
Hetman M, Pietrzak M (2012) Emerging roles of the neuronal nucleolus. Trends Neurosci 35:305–314
Parlatto R, Kreiner G (2013) Nucleolar activity in neurodegenerative diseases: a missing piece of the puzzle? J Mol Med 91:541–547
Yirmiya R, Rimmerman N, Reshef R (2015) Depression as a microglial disease. Trends Neurosci 38:637–658
Bray JC, Reyes KC, Roberts AJ, Ransohoff RM, Gruol DL (2013) Synaptic plasticity in the hippocampus shows resistance to acute ethanol exposure in transgenic mice with astrocyte-targeted enhanced CCL2 expression. Neuropharmacol 67:115–125
Tian L, Hui CW, Bisht K, Tan Y, Sharma K, Chen S, Zhang X, Tremblay M-E (2017) Microglia under psychosocial stressors along the aging trajectory: consequences on neuronal circuicts, behavior, and brain diseases. Prog Neuro-Psychopharmacol Biol Psychiatry 79:27–39. https://doi.org/10.1016/j.pnpbp.2017.01.007
Prata J, Santos SG, Almeida MI, Coelho R, Barbosa MA (2017) Bridging autism spectrum disorders and schizophrenia through inflammation and biomarkers – pre-clinical and clinical investigations. J Neuroinflammation 14:179
Neumann J, Gunzer M, Gutzeit HO, Ullrich O, Reymann KG, Dinkel K (2006) Microglia provide neuroprotection after ischemia. FASEB 20:714–716. https://doi.org/10.1096/fj.05-4882fje
Neumann H, Kotter MR, Franklin RJM (2009) Debris clearance by microglia: an essential link between degeneration and regeneration. Brain 132:288–295
Biber K, Neumann H, Inoue K, Boddecke HW (2007) Neuronal on and off signals control microglia. Trends Neurosci 30:596–602
Kettenmann H, Hanisch UK, Noda M, Verkhratsky A (2011) Physiology of microglia. Physiol Rev 91:461–553
DiSabato DJ, Quan N, Godbout JP (2016) Neuroinflammation: the devil is in the detail. J Neurochem 139(Suppl. 2):136–153
Sellgren CM, Sheridan SD, Gracias J, Xuan D, Fu T, Perlis RH (2017) Patient-specific models of microglia-mediated engulfment of synapses and neural progenitors. Mol Psychiatry 22:170–177
Takahashi Y, Yu Z, Sakai M, Tomita H (2016) Linking activation of microglia and peripheral monocytic cells to the pathophysiology of psychiatric disorders. Front Cell Neurosci 10:144
Hui CW, St.-Pierre A, El Hajj H, Remy Y, Hebert S, (2018) Prenatal immune challenge in mice leads to partly sex-dependent behavioral, microglial, and molecular abnormalities associated with schizophrenia. Front Mol Neurosci 11:13
Hinwood M, Morandini J, Day TA, Walker FR (2012) Evidence that microglia mediate the neurobiological effects of chronic psychological stress on the medial prefrontal cortex. Cereb Cortex 22:1442–1454
Hinwood M, Tynan RJ, Charnley JL, Beynon SB, Day TA, Walker FR (2013) Chronic stress induced remodeling of the prefrontal cortex: structural re-organization of microglia and the inhibitory effect of minocycline. Cereb Cortex 13:1784–1797
Wohleb ES, Delpech JC (2016) Dynamic cross-talk between microglia and peripheral monocytes underlies stress-induced neuroinflammation and behavioral consequences. Prog-Neuropsychopharmacol Biol Psychiatry 79(1):40–48
Müller N, Myint A-M, Schwarz MJ (2009) The impact of neuroimmunedysregulation on neuroprotection and neurotoxicity in psychiatric disorders-reaction to drug treatment. Dial Clin Neurosci 11:319–332
Müller N, Myint A-M, Schwarz MJ (2012) Inflammation in schizophrenia. Adv Protein Chem Struct Biol 88:49–68
Jo WK, Zhang Y, Emrich HM, Dietrich DE (2015) Glia in the cytokine-mediated onset of depression: fine tuning the immune response. Front Cell Neurosci 9:268
Bagasrawala I, Zecevic N, Radonjic NV (2016) N-methyl D-aspartate receptor antagonist kynurenic acid affects human cortical development. Front Neurosci 10:435
Kegel ME, Bhat M, Skogh E, Samuelsson M, Lundberg K, Dahl ML, Sellgren C, Schwieler L, Engberg G, Schuppe-Kiostinen I, Erhardt S (2014) Imbalanced kynurenine pathway in schizophrenia. Int J Tryptophan Res 7:15–22
Larsson MK, Schwieler L, Goiny M, Erhardt S, Engberg G (2015) Chronic antipsychotic treatment in the rat-effects on brain interleukin-8 and kynurenic acid. Int J Tryptophan Res 8:49–52
Erhardt S, Schwieler L, Imbeault S, Engberg G (2017) The kynurenine pathway in schizophrenia and bipolar disorder. Neuropharmacology 112:297–306
Notarangelo FM, Pocivavsek A (2017) Elevated kynurenine pathway metabolism during neurodevelopment: implications for brain and behavior. Neuropsychopharmacology 112:275–285
Pitman E, Iwata Y, Caravaggio F, Nakajima S, Chung JK, Gerretsen P, Kim J, Takeuchi H, Chakravarty MM, Remington G, Graff-Guerro A (2017) Kynurenic acid in schizophrenia: a systematic review and meta-analysis. Schizophr Bull 43:764–767
Wurfel BE, Drevets BE, Bliss SA, McMillin JR, Suzuki H, Ford BN, Morris HM, Teague TK, Dantzer R, Savitz JB (2017) Serum kynurenic acid is reduced in affective psychosis. Transl. Psychiatry 7:e1115
Brundin L, Erhardt S, Bryleva EY, Achtyes ED, Postolache TT (2011) The role of inflammation in suicidal behavior. Acta Psychiatr Scand 132:192–203
Müller N, Myint AM, Krause D, Weidinger E, Schwarz MJ (2013) Anti-inflammatory treatment in schizophrenia. Prog Neuro-Psychopharmacol Biol Psychiatry 42:146–153
Steiner J, Walter M, Gos T, Guillemin GJ, Bernstein HG, Sarnyai Z, Mawrin C, Brisch R, Bielau H, Meyer zu Schwabendissen L, Bogerts B, Myint AM (2011) Severe depression is associated with increased quinolinic acid in subregions of the anterior cingulate gyrus. Evidence for an immune-modulated glutamatergic neurotransmission? J Neuroinflamm. 8:94
Busse M, Busse S, Myint AM, Gos T, Dobrowolny H, Müller UJ, Bogerts B, Bernstein HG, Steiner J (2015) Decreased quinolinic acid in the hippocampus of depressive patients: evidence for local anti-inflammatory and neuroprotective responses? Eur Arch Psychiatry Clin Neurosci 265:321–329
Steiner J, Bogerts B, Sarnyai Z, Walter M, Gos T, Bernstein HG, Myint AM (2012) Bridging the gap between the immune and glutamate hypotheses of schizophrenia and major depression: Potential role of glial NMDA receptor modulators and impaired blood-brain barrier integrity. World J Biol Psychiatry 13:482–492
Kubesova A, Tejkalova H, Syslova K, Kacer P, Vondrousova J, Tyls F, Fujakova M, Palenicek T, Horacek J (2015) Biochemical, histopathological and morphological profiling of a rat model of early immune stimulation: relation to psychopathology. PLoS ONE 10:e0115439
Kato TA, Monji A, Mizoguchi Y, Hashioka S, Horikawa H, Seki Y (2011) Anti-inflammatory properties of antipsychotics via microglia modulations. Are antipsychotics a fire extinguisher in the brain of schizophrenia? Mini Rev Med Chem 11:565–574
Miller BJ, Buckley P, Seabolt W, Mellor A, Kirkpatrick B (2011) Meta-analysis of cytokine alterations in schizophrenia: Clinical status and antipsychotic effects. Biol Psychiatry 70:663–671
Mizoguchi Y, Kato T, Horikawa H, Monji A (2014) Microglial intracellular Ca2+ signaling as a target of antipsychotic actions for the treatment of schizophrenia. Front Cell Neurosci 8:370
Hashimoto K (2008) Microglial activation in schizophrenia and minocycline treatment. Prog Neuro-Psychopharmacol Biol Psychiatry 32:1758–1759
Hashioka S, McGeer PL, Miyaoka T, Wake R, Horiguchi J (2015) Can inhibition of microglial activation cure schizophrenia. Schizophr Res 168:583–584
Giovanoli S, Engler H, Engler A, Richetto J, Feldon J, Riva MA, Schedlowski M, Meyer U (2016) Preventive effects of minocycline in a neurodevelopmental two-hit model with relevance to schizophrenia. Transl. Psychiatry 6:e772
Riazi K, Galic MA, Kentner AC, Reid AY, Sharkey KA, Pittman QJ (2015) Microglia-dependent alteration of glutamatergic synaptic transmission and plasticity in the hippocampus during peripheral inflammation. J Neurosci 35:4942–4952
Kobayashi K, Imagama S, Ohgomori T, Hirano K, Uchimuru K, Sakamoto K, Herakawa A, Takeuchi H, Suzumura A, Ishiguro N, Kordomatsu K (2013) Minocycline selectively inhibits M1 polarization of microglia. Cell Dise 4:e25
Cotel MC, Lenartowicsz EM, Nateasan S, Modo MM, Cooper JD, Williams SCR, Kapur S, Vernon AC (2015) Microglial activation in the rat brain following chronic antipschotic treatment at clinically relevant doses. Eur Neuropsychopharmacol 25:2098–2107
Ribeiro BMM, Santos do Carno MR, Souze Freire R, Flavio N, Rocha, M, Borella VCM, (2013) Evidences for a progressive microglial activation and increase in iNOS expression in rats submitted to a neurodevelopmental model of schizophrenia: Reversal by clozapine. Schizophr Res 151:12–19
Kenk M, Selvanathan T, Rao N, Suridan I, Rusjan P, Remington G, Meyer JH, Wilson AA, Houle S, Mizrahi R (2015) Imaging neuroinflammation in gray and white matter in schizophrenia: An in-vivo PET study with [18F]-FEPPA. Schizophr Bull 41:85–93
Monji A, Kato T, Kanba S (2009) Cytokines and schizophrenia: microglia hypothesis of schizophrenia. Psychiatry Clin Neurosci 63:257–265
Frick LR, Williams K, Pittenger C (2013) Microglial dysregulation in psychiatric disease. Clin Dev Immunol 2013:10
Inta D, Lang UE, Borgwardt S, Meyer-Lindenberg A, Gass P (2017) Microglia activation and schizophrenia: Lessons from the effects of minocycline on postnatal neurogenesis, neuronal survival and synaptic pruning. Schizophr Bull 43:493–496
Monji A, Kato TA, Mizoguchi Y, Horikawa H, Seki Y, Kasai M, Yamauchi Y, Yamada S, Kanba S (2013) Neuroinflammation in schizophrenia especially focused on the role of microglia. Prog Neuro-Psychopharmacol Biol Psychiatry 42:115–121
van Rees GF, Lago SG, Cox DA, Tomasi KJ, Rustogi N, Weigelt K, Ozcan S, Cooper J, Drexhage H, Leweke FM, Bahn S (2018) Evidence of microglial activation following exposure to serum from first-onset drug-naïve schizophrenic patients. Brain Behav Immun 67:364–373
Bhattacharya A (2018) Recent advances in CNS P2X7 physiology and pharmacology: focus on neuropsychiatric disorders. Front Phamacol 9:30
Dong XH, Zhen XC (2015) Glial Pathology in bipolar disorder. CNS Neurosci Ther 21:393–397
Stertz L, Magalhaes PVS, Kapczinski F (2013) Is bipolar disorder an inflammatory condition: the relevance of microglial activation. Curr Opn Psychiatry 26:19–26
Zhang F, Zhou H, Wilson BC, Shi JS, Hong JS, Gao HM (2012) Fluoxetine protects against microglial activation-mediated neurotoxicity. Parkinsonism Relat Disord 18S1:S213-S217
Tynan RJ, Weidenhofer J, Hinwood M, Cairns MJ, Day TA, Walker FR (2012) A comparative examination of the anti-inflammatory effects of SSRI and SNRI antidepressants on LPS stimulated microglia. Brain Beh Imm 26:469–479
Horikawa H, Kato TA, Mizoguchi Y, Monji A, Seki Y, Ohkuri T, Gotoh L, Yonaha M, Ueda T, Hashioka S, Kanba S (2010) Inhibitory effects of SSRI on IFN-y induced microglial activation through the regulation of intracellular calcium. Prog Neuro-Psychopharmacol Biol Psychiatry 34:1306–1316
Dhami KS, Churchward MA, Baker GB, Todd KG (2019) Fluoxetine and its metabolite norfluoxetine induce microglial apoptosis. J Neurochem 148:761–778
Leonard BE (2014) Impact of inflammation on neurotransmitter changes in major depression. An insight into the action of antidepressants. Prog Neuro-Psychopharmacol Biol Psychiatry 48:261–267
Dubovicky M, Csazar E, Melichercikova K, Kuniakova M, Rackova L (2014) Modulation of microglial function by the antidepressant drug venlafaxine. Interdiscip Toxicol 7:201–207
Takeuchi H (2010) Neurotoxicity by microglia: Mechanisms and potential therapeutic strategy. Clin Exper Neuroimmunol 1:12–21
Palmieri EM, Menga A, Lebrun A, Hooper DC, Butterfield DA, Mazzone M, Castegna A (2017) Blockade of glutamine synthetase enhances inflammatory response in microglial cells. Antioxid Redox Signal 26:351–363
Parellada E, Gasso P (2021) Glutamate and microglia activation as a driver of dendritic apoptosis: a core pathophysiological mechanism to understand schizophrenia. Trans Psychiatry https://doi.org/10.1038/s41398-021-01385-9
Ohgidani M, Kato TA, Kanba S (2015) Introducing directly induced microglia-like (iMG) cells from fresh human monocytes: a novel translational research tool for psychiatric disorders. Front Cell Neurosci 9:184
Prytkova I, Brennand KJ (2017) Prospects for modeling abnormal neuronal function in schizophrenia using human induced pluripotent stem cells. Front Cell Neurosci 11:360
Gomes FV, Llorente R, Delbel EA, Viveros MP, López-Gallardo M, Guimarães FS (2015) Decreased glial reactivity could be involved in the antipsychotic-like effect of cannabiol. Schizophr Res 164:155–183
Lisboa SF, Gomes FV, Guimaraes FS, Campos AC (2016) Microglial cells as a link between cannabinoids and the immune hypothesis of psychiatric disorders. Front Neurol 7:5
de Almeida V, Martins-de-Souza D (2018) Cannabinoids and glial cells: possible mechanism to understand schizophrenia. Eur Arch Psychiatry Clin Neurosci 268:727–737
Cortez IL, Rodrigues da Silva NR, Guimaraes FS, Gomes FV (2020) Are CB2 receptors a new target for schizophrenia treatment. Front. Psychiatry 11:587154
Jinno S, Kosaka T (2008) Reduction of Iba1-expressing microglial process density in the hippocampus following electroconvulsive shock. Expr Neurol 212:440–447
Jansson L, Wennström M, Johanson A, Tingström A (2009) Glial cell activation in response to electroconvulsive seizures. Prog Neuropsychopharmacol Biol Psychiatry 133:119–128
Limoa E, Hashioka S, Miyaoka T, Tsuchie K, Arauchi R, Azis IA, Wake R, Hayashida M, Araki T, Furuya M, Liaury K, Tanra AJ (2016) Electroconvulsive shock attenuated microgliosis and astrogliosis in the hippocampus and ameliorated schizophrenia-like behavior of Gunn rat. J Neuroinflamm 13:320
Hartenstein V, Giangrande A (2018) Connecting the nervous and the immune systems in evolution. Comm Biol 1:64. https://doi.org/10.1038/s42003-018-0070-2
Schlegelmilch T, Henke K, Peri F (2011) Microglia in the developing brain: From immunity to behaviour. Curr Opin Neurobiol 21:5–10
Swinnen N, Smolders S, Avila A, Notelaers K, Paesen R, Ameloot M et al (2013) Complex invasion pattern of the cerebral cortex by microglial cells during development of the mouse embryo. Glia 61:150–163
Verkhratsky A, Ho MS, Parpura V (2019) Evolution of neuroglia. In: Verkhratsky A, Ho MS, Zorec R, Parpura V (eds) Neuroglia in Neurodegenerative Diseases. Springer, USA, pp 15–44
Haug H (1987) Brain sizes, surfaces and neuronal sizes of the cortex cerebri: A stereological investigation of man and his variability and a comparison with some mammals (primates, whales, marsupials, insectivores and one elephant). Am J Anat 180:126–142
Evans PD, Anderson JR, Vallender EJ, Gilbert SL, Malcom CM, Dorus S et al (2004) Adaptive evolution of ASPM, a major determinant of cerebral cortical size in humans. Hum Mol Genet 13:489–494
Martín-Loeches M (2010) Uses and abuses of the enhanced-working-memory hypothesis in explaining modern thinking. Curr Anthropol 51:S67–S75
Hayakawa T, Angata T, Lewis AL, Mikkelsen TS, Varki NM, Varki A (2005) A human-specific gene in microglia. Science 309:1693–1693
Wang Y, Neumann H (2010) Alleviation of neurotoxicity by microglial Human Siglec-11. J Neurosci 30:3482–3488. https://doi.org/10.1523/JNEUROSCI.3940-09.2010
Raison CL, Miller AH (2013) The evolutionary significance of depression in Pathogen Host Defence (PATHOS-D). Mol Psychiatry 18:15–37
Panter-Brick C (2002) Sexual division of labor: energetic and evolutionary scenarios. Am J Hum Biol 14:627–640
Brisch R, Saniotis A, Wolf R, Bielau H, Bernstein HG, Steiner J, Bogerts B, Braun K, Jankowski Z, Kumaratilake J, Henneberg M, Gos T (2014) The role of dopamine in schizophrenia from a neurobiological and evolutionary perspective: old fashioned, but still in vogue. Front Psychiatry 5:47
Tyler H, Polk JD (2019) BDNF, endurance activity, and mechanisms underlying the evolution of hominin brains. Am J Phys Anthropol 168(Suppl 67):47–62. https://doi.org/10.1002/ajpa.23762
Ruxton GD, Wilkinson DM (2011) Avoidance of overheating and selection for both hair loss and bipedality in hominins. Proc Nat Acad Sci 108:20965–20969
Mattson MP, Wan R (2008) Neurotrophic factors in autonomic nervous system plasticity and dysfunction. NeuroMol Med 10:157–168
Noakes T, Spedding M (2012) Run for your life. Nature 487:295–296
Raichlen DA, Polk JD (2013) Linking brains and brawn: Exercise and the evolution of human neurobiology. Proc R Soc B Biol Sci 280:1750
Saniotis A, Henneberg M (2013) Evolutionary medicine and future humanity: will evolution have the final word? Humanities 2:278–291. https://doi.org/10.3390/h2020278
Ding H, Chen J, Su M et al (2020) BDNF promotes activation of astrocytes and microglia contributing to neuroinflammation and mechanical allodynia in cyclophosphamide-induced cystitis. J Neuroinflamm 17:19. https://doi.org/10.1186/s12974-020-1704-0
Lipsky RH, Marini AM (2007) Brain-derived neurotrophic factor in neuronal survival and behavior-related plasticity. Ann N Y Acad Sci 1122:130–143
Rasmussen P, Brassard P, Adser H, Pedersen MV, Leick L, Hart E, Secher NH, Pedersen BK, Pilegaard H (2009) Evidence for a release of brain-derived neurotrophic factor from the brain during exercise. Exper Physiol 94:1062–1069
Saniotis A (2019) Evolutionary psychiatry enhancing our current knowledge of psychopathologies. Arch Psychiatry Psychotherapy 3:13–14
Acknowledgements
This work was supported by the Medical University of Gdańsk and the Stanley Medical Foundation.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
Wrote the paper: RB SW AS RW MH. Searched the references: RB JK AS. Provided the photos: TG. Edited the manuscript: RB AS MH RW JS JK.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that the research was conducted in absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Brisch, R., Wojtylak, S., Saniotis, A. et al. The role of microglia in neuropsychiatric disorders and suicide. Eur Arch Psychiatry Clin Neurosci 272, 929–945 (2022). https://doi.org/10.1007/s00406-021-01334-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00406-021-01334-z