
Abstract
In recent years, the classification of adult-type diffuse gliomas has undergone a revolution, wherein specific molecular features now represent defining diagnostic criteria of IDH-wild-type glioblastomas, IDH-mutant astrocytomas, and IDH-mutant 1p/19q-codeleted oligodendrogliomas. With the introduction of the 2021 WHO CNS classification, additional molecular alterations are now integrated into the grading of these tumors, given equal weight to traditional histologic features. However, there remains a great deal of heterogeneity in patient outcome even within these established tumor subclassifications that is unexplained by currently codified molecular alterations, particularly in the IDH-mutant astrocytoma category. There is also significant intercellular genetic and epigenetic heterogeneity and plasticity with resulting phenotypic heterogeneity, making these tumors remarkably adaptable and robust, and presenting a significant barrier to the design of effective therapeutics. Herein, we review the mechanisms and consequences of genetic and epigenetic instability, including chromosomal instability (CIN), microsatellite instability (MSI)/mismatch repair (MMR) deficits, and epigenetic instability, in the underlying biology, tumorigenesis, and progression of IDH-mutant astrocytomas. We also discuss the contribution of recent high-resolution transcriptomics studies toward defining tumor heterogeneity with single-cell resolution. While intratumoral heterogeneity is a well-known feature of diffuse gliomas, the contribution of these various processes has only recently been considered as a potential driver of tumor aggressiveness. CIN has an independent, adverse effect on patient survival, similar to the effect of histologic grade and homozygous CDKN2A deletion, while MMR mutation is only associated with poor overall survival in univariate analysis but is highly correlated with higher histologic/molecular grade and other aggressive features. These forms of genomic instability, which may significantly affect the natural progression of these tumors, response to therapy, and ultimately clinical outcome for patients, are potentially measurable features which could aid in diagnosis, grading, prognosis, and development of personalized therapeutics.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
As a group, diffusely infiltrating gliomas occur at a rate of 4.5 cases per 100,000 people in the United States annually (a mean of 16,800 cases) and comprise approximately 19% of all central nervous system (CNS) tumors [162, 163]. Originally described in 1865 by Rudolph Virchow, the diagnosis and classification of gliomas have undergone a number of revisions and updates in the intervening decades [56, 198]; however, until 2016, these diagnoses and tumor grades were based solely on histologic features [126]. In the 5th edition of the WHO Classification of Central Nervous System Tumors, released in 2021, adult-type diffuse gliomas were classified with a combination of histologic and molecular features as oligodendrogliomas (~ 7% of diffuse gliomas) in the presence of mutation in either IDH1 or IDH2 (hereafter grouped simply as “IDH mutation”) and co-deletion of chromosomal arms 1p and 19q, IDH-mutant astrocytoma (~ 11%) in the presence of an IDH mutation and retained 1p/19q, and IDH-wild-type glioblastoma (~ 82%) in the absence of either of these molecular alterations [127, 163]. IDH-mutant gliomas generally arise in the cerebral cortex with only rare exceptions in posterior fossa structures or spinal cord, in contrast to tumors defined by other drivers, including H3 K27M-mutant diffuse midline gliomas, and different progenitor cells may be more susceptible to malignant transformation by mutually exclusive oncogenic drivers [111, 199].
First identified as a molecular feature of diffusely infiltrating gliomas in 2008, IDH mutation confers a significantly better prognosis compared to their histologic grade-matched IDH-wild-type counterparts [127, 166, 231]. The majority of IDH mutations are IDH1 R132H mutations, which can be identified with immunohistochemical stains [16, 22, 35, 36]. IDH mutation has been shown to be an early event in gliomagenesis [49, 168, 210], and results in the production of the oncometabolite 2-hydroxyglutarate (2-HG) [43, 52, 68, 210, 222, 230], which can be identified with magnetic resonance spectroscopy [9, 43]. The production of 2-HG inhibits α-ketoglutarate-dependent dioxygenases and induces widespread DNA hypermethylation, resulting in the glioma CpG island methylator phenotype (G-CIMP), causing significant transcriptional alterations, metabolic abnormalities, cellular dysregulation, and a poorly differentiated state [52, 69, 128, 139, 148, 160, 188, 189, 204, 210]. This methylation state can be used to distinguish IDH-mutant astrocytoma from other forms of diffusely infiltrating gliomas and other CNS neoplasms, and predict clinical outcomes [33, 34, 74, 108, 183, 227]. This global DNA methylation profiling also separates IDH-mutant astrocytomas into low- and high-grade clusters [33], as well as G-CIMP-low and G-CIMP-high clusters [38]. This G-CIMP-low subgroup has a lower level of methylation at some CpG sites and comprises a minority of IDH-mutant astrocytoma cases with significantly worse overall survival, relative to G-CIMP-high astrocytomas, although there is a general trend toward conversion from G-CIMP-high to -low with recurrence and progression in grade [38, 55, 121, 134].
In addition to diagnosis, additional molecular features have become increasingly important in tumor grading. The presence of EGFR amplification, TERT promoter mutation, and/or simultaneous gain of chromosome 7 and loss of chromosome 10 (+ 7/− 10) are currently considered molecular grade 4 features in IDH-wild-type glioblastoma, and homozygous loss of CDKN2A/B is considered a molecular grade 4 feature in IDH-mutant astrocytoma and a grade 3 feature in oligodendroglioma [127], although rare exceptions have been reported [11, 211]. A number of other genetic alterations have also been considered for diagnostic and grading schemes, including hemizygous CDKN2A/B loss and CDKN2A/B mutations [91, 110, 229, 234], as well as a number of other molecular features [10, 46, 72, 149, 177, 190, 224]. Unlike oligodendrogliomas and glioblastomas, which both frequently feature mutations in the promoter region of TERT to maintain telomere length, IDH-mutant astrocytomas frequently have mutations in ATRX, promoting an alternative lengthening of telomere (ALT) phenotype [152, 235]. Another key molecular feature associated with the clinical course of diffuse gliomas is O6-methylguanine-DNA methyltransferase (MGMT), which encodes a DNA repair protein that removes methyl groups from guanine residues caused by the administration of temozolomide (TMZ), the most common chemotherapeutic agent used in the treatment of malignant gliomas [79, 90, 156]. When the promoter region of MGMT is hypermethylated, however, the cellular level of MGMT protein is decreased, leaving the effects of TMZ unchallenged, leading to tumor cell death via intact mismatch repair (MMR) mechanisms [64, 71, 213].
Another factor, imperative to understanding the pathogenesis of these tumors, is intratumoral heterogeneity, which has been definitively shown in recent years with single-cell sequencing techniques and geographically targeted biopsies/autopsy specimens, and recapitulated in organoid models [2, 4, 80, 95, 129, 133, 147, 167, 207, 214, 237]. As a general rule, diffuse gliomas are composed of numerous subclones comprised of cells with different epigenetically regulated transcriptional states, different mutational or copy number states, and different microenvironmental states and interactions [156, 201]. This principle becomes more complex with tumor recurrence and treatment, and even genetic alterations thought to be foundational to tumorigenesis, such as IDH mutation, may become subclonal and relegated to “passenger mutation” status with tumor evolution, while other newly acquired mutations may emerge in dominant clones and drive tumor biology [17, 65, 100, 132, 138, 142, 215]. This heterogeneity makes diffusely infiltrating gliomas incredibly diverse and adaptable and is believed to contribute significantly to resistance to immune regulation and therapeutics.
Herein, we review processes that contribute to the rapid development of intercellular heterogeneity, including forms of epigenetic instability, chromosomal instability, and mismatch repair (MMR) defects/microsatellite instability (MSI). These processes are well-known to drive cancer formation and progression in a wide variety of cancer types through production of a population of tumor cells with increased genetic and phenotypic diversity, followed by Darwinian selection of the tumor clones most suited to survival, invasion, progression, metastasis, and resistance to therapy (Fig. 1), but their contribution to gliomagenesis and malignant progression of gliomas has only recently been uncovered. Genetic instability in its various forms has been identified in all subtypes of adult-type diffusely infiltrating gliomas, including IDH-wild-type glioblastoma, IDH-mutant astrocytoma, and oligodendroglioma. In the IDH-mutant astrocytoma category, in particular, there is evidence that these processes significantly impact biologic behavior [17, 175, 179, 209], suggesting that forms of genetic and epigenetic instability may be key to understanding the natural history of IDH-mutant astrocytoma, and may have significant prognostic implications as well as the potential to serve as therapeutic targets.

Overview of the mechanisms and consequences of chromosomal instability (CIN) and microsatellite instability (MSI)/mismatch repair (MMR) mutations and the resulting intratumoral heterogeneity. Figure created by Melissa Logies
Defining tumor heterogeneity using single-cell transcriptomics
In the past decade, single-cell sequencing technology has revolutionized the way we look at cancer, providing extraordinary granularity of intratumoral and intertumoral heterogeneity as it relates to cellular diversity. A notable contribution of this technology has been toward the deconvolution of cellular states linked to developmental lineages or molecular processes and the discovery of rare cell types. Single-cell transcriptomics (scRNA-seq) and open chromatin accessibility (scATAC-seq) have been the two most widely used -omics modalities thus far, enabling unprecedented dissection of tumor composition and functional state, in both IDH-wild-type and IDH-mutant diffuse gliomas. In contrast, single-cell DNA-seq has been more technically challenging and thus less informative of single-cell level resolution mutational heterogeneity and clonal evolution in gliomas [207]. The earliest scRNA-seq glioma studies elegantly demonstrated the power of this technology to resolve tumor heterogeneity in GBM, as well as to infer specific copy number variations (CNVs) using gene co-expression data [167]. Subsequent transcriptomic studies have focused on consolidating GBM heterogeneity into distinct but shared cellular states [77, 155, 218, 219, 236], including developmentally coopted hierarchies that recapitulate gliogenesis [50, 170].
Profiling of IDH-mutant gliomas using single-cell sequencing has demonstrated a more conserved tripartite tumor hierarchy with less overall heterogeneity, compared to IDH-wild-type GBM. In grade 2 IDH-mutant oligodendrogliomas, scRNA-seq defined a developmental hierarchy in tumor cells, with a small population of stem-like proliferative glioma cells resembling neural stem/progenitor biology at one apex along with two differentiation trajectories, one towards mature oligodendrocyte-like and the other towards mature astrocyte-like apices [208]. A similar hierarchy was reconstructed in IDH-mutant astrocytomas in a follow-up study, suggesting shared developmental lineages between these otherwise genetically distinct IDH-mutant gliomas [214]. Interestingly, the proportion of stem-like malignant cells expanded with higher grade, in both IDH-mutant astrocytoma and oligodendroglioma [214]. As well, the “oligodendrocyte-like” differentiation state was shown to be “stalled” with an aberrantly blocked myelination transcriptional program [223]. Such analyses have been enhanced by longitudinal studies in primary/recurrent IDH-mutant gliomas specimens, tracking changes and stability in persistent and recurrent tumor subpopulations [21]. Additionally, multi-omic analyses, including scRNA-seq plus scATAC-seq [3, 12, 223] or scRNA-seq plus single-cell DNA methylome [39, 103], have enhanced transcriptional cell state diversity analysis and helped to reconstruct more accurate transcription factor regulatory networks. Single-cell transcriptomics has informed heterogeneity not only in tumor cells but also in the surrounding infiltrated non-neoplastic parenchyma, which includes microglia/macrophages, oligodendroglial-lineage cells, astrocytes, and neurons, collectively called the tumor microenvironment (TME) [154]. Important for GBM, it was shown that macrophage–tumor cell interaction not only drives MES transition in tumor cells but in macrophages themselves [41, 89]. The most notable differences observed so far between IDH-mutant and IDH-wild-type high-grade gliomas relate to their myeloid TME composition and functional properties [1, 70, 169]. With the exciting emergence of IDH inhibitors showing therapeutic efficacy in early clinical trials [144,145,146], this technology can be used to probe important mechanistic and translational questions related to treatment response and resistance, with the first of such studies providing an early glimpse into the biology of response and resistance in human IDH-mutant oligodendrogliomas after treatment with vorasidenib [194].
While the use of single-cell DNA sequencing to characterize mutations has been technically challenging, scRNA-seq and scATAC-seq enable the inference of genetic CNVs using several different computational pipelines [76, 153, 157, 167]. Inference of CNVs allows for the accurate annotation of tumor cell identity in scRNA-seq data. Furthermore, it has enabled us to define CNV heterogeneity within different tumor niches in IDH-mutant and IDH-wild-type high-grade gliomas, including between necrotic tumor core and infiltrative tumor margin (Fig. 2a). It represents an adaptable, orthogonal method to explore still unanswered questions in the field, related to clonal tumor evolution and glioma cell-of-origin. With the emergence of user-friendly bioinformatic toolsets, reduced cost, and the ability to perform single-cell sequencing and spatial transcriptomics on FFPE samples, this powerful technology will likely become a diagnostic companion to every neuropathologist in the near future.

Measurement of chromosomal instability in IDH-mutant astrocytomas. Chromosomal instability can be identified via a single-cell sequencing, b methylation profiling, c identification of incongruous “chromosomal complexity” or high levels of copy number alterations spread throughout the genome, and d mRNA profiling. Panel c is adapted from Lyon et al., 2021 [133], and is used with permission under Creative Commons Attribution 4.0 International Licenses (http://creativecommons.org/licenses/by/4.0/)
Chromosomal instability (CIN)
Chromosomal instability (CIN) is an ongoing process of relatively rapid chromosomal alterations resulting in numerical abnormalities (aneuploidy) and/or structural abnormalities, and may involve catastrophic, large-scale fragmentation and reassembly of one or more chromosomes in a single event (a process known as chromothripsis), as well as unequal segregation on extrachromosomal DNA (ecDNA) [48, 57, 130, 159, 191, 193, 196, 216]. CIN is a continuing process that can lead to the rapid loss or alteration of tumor suppressor genes, a gain of oncogenes, disruption of genetic and epigenetic architecture, and/or formation of gene fusions, followed by clonal evolution (Fig. 1) in the face of cellular stressors, which may include clonal competition due to resource availability, tumor microenvironment, immune system interactions, and therapeutic challenges [17, 102, 156, 217]. Recent work has shown that individual cells isolated from cancers with proven chromosomal instability may fully recapitulate the intercellular heterogeneity found in the parental population within approximately 22 generations [86]. It is important to stress that although CIN frequently results in aneuploidy, these two terms are not synonymous, and the presence of aneuploidy is not necessarily evidence of CIN [8]. For example, oligodendrogliomas are defined by co-deletion of chromosomal arms 1p and 19q (in addition to IDH mutation), but this represents a form of stable aneuploidy, mediated by an unbalanced translocation between chromosomes 1 and 19, with subsequent loss of the derivative chromosomal fragment der(1;19) (q10;p10) [82, 98, 171].
CIN has been well-described in several cancers, perhaps most famously in colorectal cancer, where mutations in the APC gene result in dysregulation of WNT signaling as well as interference with microtubule function during mitosis, leading to a CIN phenotype [29, 81, 158]. Numerous cancers and syndromes have CIN as a key component, including multiple carcinoma types, hematologic malignancies, and Fanconi’s anemia [13, 42, 44, 60, 117, 185, 203], and recent work has suggested that approximately 80% of samples across a wide variety of human cancers have some detectable degree of CIN as a component of their disease process [60, 212]. Mutations in a number of genes with functions involving DNA/chromosome repair, mitotic control and fidelity (including centrosome function, spindle assembly, and mitotic fidelity/checkpoints), and apoptosis, among others, have been positively implicated in the development of chromosomal instability, although given the number of proteins involved in mitosis and cell replication, it has been hypothesized that loss of function in any one of as many as 2,000 genes could potentially result in some form of chromosomal instability [14, 30, 177, 197, 205, 206, 216].
Given that CIN is an ongoing process and pathologic specimens generally represent only a single point in time, detection of CIN may be difficult, and a number of potential direct and indirect methods of measurement have been proposed [118]. The most direct and conclusive method for identifying CIN in a tumor sample is cell culture where changes in karyotype and/or single-cell copy number alteration (CNA) in successive generations of cells can be directly observed; however, due to the time and costs associated with this method, this is generally not practical for routine clinical evaluation [86, 116, 117]. Indirect methods to infer CIN include evaluating histologic features such as anaphase segregation errors (including chromatin bridges and lagging chromosomes), double minute/circular extrachromosomal DNA (ecDNA), and micronucleus formation [19, 20, 57, 159, 181, 195, 232]. Other techniques involve the assessment of intercellular variation in aneuploidy with comparative genomic hybridization (CGH) [118], fluorescent in situ hybridization (FISH) [44, 185, 203], and single-cell sequencing (Fig. 2a) with inferred CNA levels between cells [147, 167, 237]. Additional methods in IDH-mutant astrocytomas and other neoplasm types have involved global DNA methylation signatures (Fig. 2b) [114, 125], CNA profiling to identify “chromosomal complexity”/“copy number heterogeneity” (Fig. 2c) [150, 173, 174, 176, 228], mRNA expression signature patterns (Fig. 2d) [37, 175], and other computational molecular signatures of CIN [60].
In diffuse gliomas, the impact of CIN is less clear than in other systemic cancers. Multiple recent studies have shown that IDH-mutant astrocytomas accumulate increasing CNA with recurrence [133, 177], as they progress from a mean CNA level of 9.3 ± 0.5% (~ 290 Megabase pairs [Mbp]) in CNS WHO grade 2 to 20.9 ± 1.8% (~ 650 Mbp) in CNS WHO grade 4 [48, 173,174,175,176,177, 190], and/or with the development of defined high-grade molecular features, such as homozygous CDKN2A loss [150]. This accumulation of chromosomal complexity occurs in a much more pronounced manner compared to glioblastoma [175] or oligodendroglioma [178], and unlike oligodendroglioma or glioblastoma, this total/overall CNA level (a snapshot of the level of genomic disruption at the time of surgery) tends to be scattered across the genome with relatively few consistent recurrent sites of alteration [73, 150, 174, 176, 212, 228]. While not a definite signal of CIN, this progressive rise in CNA in parallel with tumor progression is indicative of an ongoing process of genome alteration with accumulating gains and losses.
Previous studies in large cohorts of IDH-mutant astrocytomas and IDH-wild-type glioblastomas have generated several lines of evidence suggesting that elevated CNA levels and CIN may be present early in the development of a select subset of tumors (prior to developing high-grade histologic or molecular hallmarks), and that this finding has predictive power, identifying glioma subsets with more aggressive behavior and worse clinical outcomes in terms of the patient’s recurrence-/progression-free survival and overall survival. While overall CNA levels tend to be significantly elevated in cases with either grade 4 histology or equivalent molecular features [150, 175], there is a subset of grade 2–3 IDH-mutant astrocytomas (approximately 15% of cases) in which incongruously elevated overall CNA levels are associated with dismal survival (progression-free survival (PFS) ≤ 12 months and overall survival ≤ 24 months), and this is true when selecting specifically for elevated overall CNA or poor clinical outcomes and excluding cases with other known high-grade molecular features [174,175,176]. We have shown that overall CNA level at the time of initial surgery is an independent prognostic factor in IDH-mutant astrocytoma, in which cases with CNA comprising ≥ 15% of the total genome (approximately 465 Mbp) have significantly worse Karnofsky performance status, progression-free survival, and overall survival compared to their counterparts with relatively low CNA [125, 150, 151, 174,175,176], a finding which was validated in other glioma cohorts and other cancers, although the particular CNA threshold varied somewhat [10, 190, 212]. IDH-wild-type glioblastomas followed a similar trend; the vast majority of cases had high overall copy number variation, but the few cases with < 10% total copy number variation had a significantly better clinical outcome in terms of both progression-free and overall survival [172, 175, 212].
Other data have shown that patients with IDH-mutant astrocytomas harboring mutations in genes known to be involved with maintenance of chromosomal stability in other cancer types (APC, BLM, BRCA1/2, the Fanconi anemia family genes, among numerous others) have higher overall CNA at the time of initial surgery and correspondingly poor clinical outcomes regardless of histologic grade [150, 174, 175, 177]. Cases with mRNA expression patterns and global DNA methylation patterns associated with known CIN had higher overall CNA levels and worse clinical outcomes compared to cases with mRNA and methylation patterns consistent with chromosomal stability, in terms of progression-free survival (median PFS of 29–40 months vs. 70–88 months, respectively) and overall survival (median OS of 41–63 months vs. 87–140 months, respectively) [37, 125, 175, 177]. In addition, genetic analysis of multiple biopsies, resections, and autopsy specimens within individual patients with diffuse glioma has demonstrated significant regional heterogeneity of chromosomal alterations and heterogeneity of specific epigenetic alterations (including MGMT promoter methylation), as well as mounting overall CNA levels in the recurrent tumor samples, an effect that was more pronounced and was more predictive of clinical behavior in IDH-mutant tumors than other glioma types, although this may be driven in part by treatment, including radiotherapy [133]. There have been several studies utilizing single-cell sequencing technology to investigate genomic heterogeneity in multiple types of diffusely infiltrating gliomas; however, these studies have generally had a relatively small number of included tumors and have not been focused on uncovering CIN, although inferred CNA analysis in these studies demonstrates copy number profile heterogeneity in some cases [147, 167, 207, 237].
Mismatch repair (MMR) protein defects and microsatellite instability (MSI)
The mismatch repair (MMR) system is an evolutionarily conserved system comprised in humans of the paired proteins, MSH2–MSH6, which recognize base-pair mutations and mismatches, and then recruit the MLH1–PMS2 complex, as well as DNA ligase I, DNA polymerase (POLD1/POLE), PCNA, and EXO1 (among other proteins) to excise and repair incorrect base pairing, ensuring fidelity of DNA replication [99, 120, 137]. Inactivation of the genes involved in this process (primarily MSH2, MSH6, MLH1, and PMS2) predictably results in an increased rate of somatic mutations, high tumor mutational burden (TMB; termed “hypermutation” when there are ≥ 10 mutations/Mb), and microsatellite instability [187]. Microsatellite instability is a consequence of MMR defects and occurs with an increased rate of mutation in repeated sequences of DNA (microsatellites) across the genome [192], as well as an increased rate of mutations affecting proto-oncogenes and tumor suppressors with subsequent clonal evolution, similar to that seen in CIN (Fig. 1) [17, 102, 156, 209, 217]. Mutation, deletion, or promoter methylation of these MMR genes is present in approximately 12% of ovarian cancers, 15% of colorectal cancer, 22% of gastric cancers, and up to 30% of endometrial cancers, as well as heritable cancer syndromes, such as Lynch syndrome (monoallelic germline mutation of an MMR gene) and constitutional mismatch repair deficiency (CMMRD; biallelic germline mutation of an MMR gene), which have an increased risk of numerous cancers [40, 84, 105, 106]. In diffusely infiltrating gliomas, mutations in MMR genes may occur as part of a germline syndrome [28, 59, 75, 84, 105, 106, 161, 200], as sporadic mutations [27, 32, 179, 209], or as the result of temozolomide therapy, either due to the selection of temozolomide-resistant MMR-deficient clones or by temozolomide-induced mutation in one of the MMR genes [6, 17, 31, 45, 53, 94, 104, 179, 209, 213, 233].
A recent case series by Suwala et al. described a distinct subtype of IDH-mutant astrocytoma with MMR mutations occurring in the setting of Lynch syndrome or CMMRD, termed primary mismatch repair-deficient IDH-mutant astrocytoma (PMMRDIA) [200]. These tumors occurred primarily in children and young adults (although this cohort included patients up to 54 years), had a hypermutant phenotype and microsatellite instability, high-grade morphology, low rates of MGMT promoter methylation, and dismal prognosis with a median survival of only 15 months. Interestingly, these tumors were epigenetically distinct from IDH-mutant astrocytomas, with reduced global methylation levels, and were distinguishable with methylation profiling from secondary MMR-deficient tumors, which tended to match better with their MMR-retained IDH-mutant astrocytoma counterparts [59, 200]. This may also be true of IDH-wild-type glioblastomas [87], suggesting that there may be fundamental differences in the biology of tumors with acquired and germline MMR mutations.
While sporadic MMR mutations are relatively rare in diffuse gliomas, those occurring in recurrent gliomas in the setting of TMZ administration are much more frequent [17, 27, 32, 38, 179, 209], and appear to be more common for IDH-mutant astrocytomas (47%) than IDH-wild-type glioblastomas (16%) or oligodendrogliomas (25%) [17, 94]. MMR mutations occurring after TMZ administration may occur either through the selection of MMR-deficient subclones already present in the heterogeneous tumor cell population or by TMZ directly inducing mutation in an MMR gene, particularly in tumor cells with methylated MGMT [17, 31, 45, 53, 94, 102, 107, 179, 209, 213, 217, 233]. These MMR mutations precede TMB increase [17, 179, 209], and subsequent clonal expansion of the surviving MMR-deficient subclone may then contribute to further TMZ resistance [51, 209]. However, this TMZ resistance may potentially be countered with poly(ADP-ribose) polymerase inhibitors (PARPi) [92], inhibitors of the RecQ DNA helicase WRN [18, 40, 122], and newer chemotherapeutic agents specifically designed to induce DNA damage and death of MGMT-deficient tumor cells in an MMR-independent manner [123].
MMR mutations can be detected through routine NGS panels or immunohistochemistry (IHC) with loss of staining for one or more MMR proteins (Fig. 3) [7, 28, 140, 141, 179, 200]. IHC may be particularly useful as non-neoplastic brain cells (native glial cells, neurons, and endothelial cells) retain nuclear reactivity in cases of acquired MMR mutation, while these cells should also be negative in cases with germline mutations [141, 179, 200], although notably in many cases mutations resulting in MMR-deficiency are only present at a subclonal level, which may be evident with IHC staining [140, 209]. No matter the mechanism of loss of MMR function, these tumors often have distinct and striking morphology with diverse cellular appearances including areas with primitive neuronal component, ependymoma-like areas with perivascular pseudorosettes, large bizarre nuclei, multinucleate giant cells, and frequent atypical mitotic figures including Creutzfeldt-like cells and tripolar mitoses [84, 105, 140, 179, 200].

Mismatch repair protein deficits. Photomicrographs demonstrate loss of a MSH2 and b MSH6 expression with retention of both proteins in non-neoplastic background cells and retention of c PMS2 and d MLH1 in tumor cells. All images are taken at ×200 total magnification, scale bar = 100 µm
MMR-mutation and the resulting hypermutant phenotype have been associated with worse clinical outcomes in terms of more frequent and rapid recurrences, tumor progression, and patient death in IDH-mutant astrocytomas compared to grade-matched counterparts in many, but not all studies [17, 124, 179, 200, 209]. Some authors have suggested that germline MMR mutations may be sufficient to warrant WHO grade 4 status, regardless of other histologic or molecular features [200]. Whether they are inherited or acquired, these mutations are frequently associated with high-grade histologic and molecular features and are often associated with progression in grade when acquired between sampling, but they may confer poor prognosis even when identified in IDH-mutant astrocytomas with no otherwise worrisome features, suggesting they may serve as a useful biomarker in certain circumstances [179, 200, 209]. MMR mutations also increase the number of neoantigens (mutant proteins potentially viewed as novel by the immune system) [15, 109, 112, 135], with some data suggesting that as many as 42% of nonsynonymous mutations in exon regions may result in neoantigen formation [17]. This property makes immune checkpoint inhibitor therapy an attractive adjuvant treatment option for these tumors [24, 47, 53, 83, 87, 93, 101, 112, 131, 180, 182, 209], although not all hypermutant cancers respond [112, 143, 180, 209]. Other studies have suggested that immune therapy may only remove MMR-mutant subclones [140, 180, 220], so this therapy may be most effective in patients with germline mutations as the MMR mutations are more uniform in these cases [24, 200].
Epigenetic plasticity, epigenetic instability, the G-CIMP phenotype, and MGMT promoter methylation
Conceptually, epigenetics involves the control of and changes to gene function and expression that does not involve changes to the actual DNA sequence, including modifications such as acetylation and methylation of DNA and histones, which result in the activation, silencing, and other modulation of gene expression [62]. In part, epigenetic alterations control the “fate” of the cell under normal conditions; all cells in an organism theoretically contain the same DNA content, however, the epigenetically driven gene expression pattern is thought to result in cellular differentiation from stem cells [62]. Many tumor types, including gliomas, are thought to contain cancer stem cells, which retain a more dedifferentiated state as well as multiple clones of differentiated cells with phenotypic variation and plasticity, which may include features favorable to tumor cell survival, including ability to evade the immune system, resistance to therapy and hypoxia, better ability to infiltrate the surrounding neuropil, and advantageous metabolic states [66, 67, 156]. As described above, IDH mutation and the resulting production of the oncometabolite 2-HG results in a global hypermethylated state in IDH-mutant astrocytomas, which can be detected with DNA methylation profiling and can be used to discriminate these tumors from other histologically similar neoplasms, including oligodendroglioma and glioblastoma [33]. The G-CIMP methylation phenotype can further be distinguished into relatively highly methylated (G-CIMP-high) and lowly methylated (G-CIMP-low) groups [38, 55, 134]. The vast majority of IDH-mutant astrocytomas are classified as G-CIMP-high, and have a superior prognosis to the G-CIMP-low subgroup, which accounts for only 6–17% of primary and recurrent tumors, and is characterized by more frequent CNA and chromothripsis, a more stem-cell-like molecular signature, and epigenetic features similar to IDH-wild-type glioblastoma [38, 56, 63, 150, 175].
Primary IDH-mutant astrocytomas are predominantly lower grade (2021 CNS WHO grades 2–3) and are predominantly classified as G-CIMP-high, an epigenetic phenotype retained in ~ 70% of recurrent cases. There is also an intermediate G-CIMP subgroup in recurrent cases, representing those which are slowly evolving from low to high grade, and trending from G-CIMP-high to G-CIMP-low ("epigenetic plasticity”), which is associated with malignant progression [55], similar in concept to rising CNA levels with increasing grades of IDH-mutant astrocytoma [175]. There was also a small “epigenetically unstable” subgroup that converted rapidly from G-CIMP-high to G-CIMP-low status with recurrence that exhibited significantly worse clinical outcomes (with a predictive panel of 7 CpG sites) [55], similar to incongruously high CNA in low-grade IDH-mutant astrocytomas with rapid recurrence and patient death [10, 151, 172, 174, 176, 190]. Notably, a portion of IDH-mutant astrocytomas, including tumors which were designated WHO grade 4 at initial presentation, were relatively stable; ~ 35% maintained a G-CIMP-high status and ~ 15% moved to an intermediate epigenetic phenotype, suggesting that this progression is related to but not completely dependent upon grade [55].
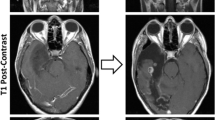
Another important epigenetic marker associated with treatment and outcome in IDH-mutant astrocytoma is MGMT promoter methylation [79, 90]. Although many neuropathology practices do not routinely re-test MGMT promoter methylation at recurrence or in multiple regions during surgery, recent reports have indicated that among other genetic and epigenetic changes, there may be spatial and/or temporal differences in MGMT promoter methylation status (Fig. 4a), particularly after treatment with TMZ [17, 23, 58, 61, 78, 133, 156, 165, 225]. There may be significant epigenetic heterogeneity at this particular locus, and progression from hypermethylated to hypomethylated MGMT promoter status in recurrent tumors may reflect the selection of existing hypomethylated MGMT cells and serve as a mechanism for further resistance to TMZ [25, 156]. Analysis of pooled cohorts of diffusely infiltrating gliomas with paired MGMT promoter status evaluation in both paired primary and recurrent tumors [17, 38, 104, 133] demonstrates that ~ 15% of cases have discrepancies in MGMT promoter methylation status and may either develop or lose promoter methylation at the time of recurrence (Fig. 4b), although the available sample size is too small to make definitive conclusions on the prognostic or treatment effects of this change.

Spatial and temporal MGMT promoter methylation status change. a Case study demonstrating MGMT promoter methylation at the site of initial biopsy in the right temporal lobe and immediately adjacent tumor at autopsy with loss of MGMT promoter methylation in more distant brain regions in the right medial frontal lobe and corpus callosum [133], and b relative frequency of change in MGMT promoter methylation status in initial and recurrent diffuse glioma samples [17, 133, 173, 175, 176]
Other recent work has suggested a possible mechanism for epigenetic instability. During DNA replication, epigenetic modifications, including histone methylation, must be preserved in the daughter DNA strands [184]; this process is complex but broadly is accomplished by the distribution of recycled parental histones split between two daughter copies with the synthesis of new histones in an approximately 50:50 ratio on each chromosome and subsequent duplication of epigenetic modifications onto the newly synthesized histones [88, 136, 184]. This process is complicated by a number of factors, including the availability of nucleotides, DNA damage, and formation of secondary DNA structures (such as guanine-rich quadruplexes, which may be crucial to genetic instability with ATRX inactivation) [54, 115, 119, 164, 184, 202, 221], which pause replication and histone recycling/incorporation [96, 97]. When histone recycling is then disconnected from DNA replication these regions are populated primarily by newly synthesized histones which lack the epigenetic modification of the parental DNA and, as a result, will tend toward loss of methylation in these particular regions [184]. The failure of this process and the resulting loss of fidelity in histone methylation becomes more likely with significant DNA damage, including that associated with CIN and MSI. A number of genes are known to be specifically associated with this process, and mutations in BLM, FANCJ, PRIMPOL, REV1, and WRN, among others, have been associated with this form of epigenetic instability [119, 184, 186]. Interestingly, mutations in the first four of these genes were identified in cases with loss of MGMT promoter methylation in recurrences (Fig. 4b), suggesting that this process may be related to changing MGMT promoter methylation in at least some cases.
Prognostic and therapeutic implications of instability and heterogeneity
Numerous studies on the various forms of genetic and epigenetic instability in IDH-mutant astrocytomas (and other diffusely infiltrating gliomas and CNS neoplasms) have yielded somewhat mixed results. In general, however, these molecular mechanisms tend toward increasing intercellular heterogeneity, which appears to drive progression and aggressiveness in these tumors after Darwinian selection of clones with increased survival potential, including resistance to therapy and immune regulation, similar to the effect of these mechanisms in other systemic cancer types [205, 216]. Univariate analysis, performed on three combined cohorts of publicly available IDH-mutant astrocytoma cases (n = 518) [17, 38, 104], demonstrated a significantly detrimental effect of increasing histologic grade (p < 0.0001), homozygous CDKN2A deletion (p < 0.0001), CDK4 amplification (p < 0.0001), CCND2 amplification (p < 0.0001), chromosomal instability (p < 0.0001), MMR mutation (p = 0.0003), and subclonality of IDH1 mutation (p = 0.0457), while MGMT promoter methylation was not significantly associated with patient survival (Fig. 5a). In multivariate analysis, histologic grade (p = 0.0019), homozygous CDKN2A deletion (p = 0.0004), and CIN (p = 0.0320) were significantly associated with impaired survival in IDH-mutant astrocytoma patients, suggesting that these features are independently associated with poor outcomes in this diffuse glioma subset, while factors such as MGMT methylation status, MMR mutation, and IDH1 clonality were not (Fig. 5b). Although the presence of MMR mutation was not independently associated with poor outcome in multivariate analysis, these mutations were significantly correlated with higher initial histologic and molecular grade as well as progression to WHO grade 4 [179], and some authors have suggested that some instances of MMR mutation, particularly cases with germline mutation, may warrant an automatic WHO grade 4 designation [200]. Similarly, CIN was considered for inclusion as a molecular prognostic factor by The Consortium to Inform Molecular and Practical Approaches to CNS Tumor Taxonomy–Not Official WHO (cIMPACT-NOW) in 2020; however, it was decided at the time that more evidence was needed as well as consensus on a threshold of CNA [26]. As technology that can readily detect these processes becomes more widespread, future WHO grading recommendations may be revised to include a more granular assessment of these genetic and epigenetic processes to further refine astrocytoma prognosis. Leveraging single-cell transcriptomics technologies to infer copy number alterations with single-cell resolution represents one such promising endeavor to better define the CIN landscape with high granularity and spatial resolution (Fig. 2a).

Forest plots of Cox proportional hazard regression analysis. a Univariate and b multivariate analysis of hazard ratios (HR) and 95% confidence intervals (CI) of features in IDH-mutant astrocytomas performed on three distinct publicly available cohorts (n = 518) [17, 38, 104]. In the multivariate model, histologic grade, homozygous CDKN2A deletion, and chromosomal instability are independently associated with significantly worse clinical outcomes. Individual molecular features were not available for all cases; analysis was performed using MedCalc Statistical Software version 22.007 (MedCalc Software Ltd, Ostend, Belgium)
In addition to prognostic and predictive importance, the identification of these instability patterns may provide a significant therapeutic opportunity [235]. Given that the hypermutator phenotype that frequently accompanies MMR mutation produces numerous neoantigens [109, 112, 135], some studies have found a benefit in treatment with immune checkpoint inhibitors, although the evidence is mixed, and these therapies may only affect hypermutant subclones with MMR-mutations [17, 24, 93, 101, 140, 182, 209, 220]. While CIN has been associated with resistance to traditional therapeutic strategies [113], dozens of drugs are currently in clinical trials or have been previously approved by the Food and Drug Administration (FDA) for other cancer types aimed at disrupting the process of CIN or further inducing CIN to lethal levels [177, 193, 205]. Compounds aimed at reducing CIN generally promote mitotic checkpoints and inhibit cell division, while therapies which enhance CIN further destabilize the cell replication process (microtubule and centrosome dynamics, mitotic kinases, mitotic checkpoints, chromatin modification, etc.) to push DNA damage past a viability threshold in cells that are already prone to developing CIN, although concerns remain for the potential of this strategy to promote more malignant and aggressive tumor cell clones [193, 205].
Conclusions
Tumorigenesis and molecular evolution in adult-type diffusely infiltrating gliomas, including IDH-mutant astrocytomas, is a complex process that involves widespread genetic and epigenetic changes that occur in highly complex and evolving tumor microenvironments. While gliomas are generally defined and classified according to a small number of discrete molecular alterations, the genetic landscape is far more complicated, with numerous detrimental alterations affecting a wide range of fundamental cellular processes, and there may be significant cell-to-cell heterogeneity. As these tumors are widely considered to be surgically incurable and standard chemotherapy and radiation therapy ultimately have not proven successful at curing the majority of cases, future rational treatment of these tumors must resolve these issues and address heterogeneity both in neoplastic cells as well as in the non-neoplastic tumor microenvironments in which these tumors emerge and grow if it has any hope of success [5, 85, 104, 156, 188, 226, 235]. In addition, the processes of genetic and epigenetic instability outlined here should be studied further in large cohorts to determine more precise prognostic implications and how this ongoing process of DNA and histone alterations will affect response to therapy, particularly as numerous studies have shown an independent effect on clinical outcome and potential mechanisms of escape from therapy associated with these processes, and there is a need for retrospective analysis and prospective studies designed with these processes in mind.
Data availability
Not applicable.
References
Abdelfattah N, Kumar P, Wang C, Leu JS, Flynn WF, Gao R et al (2022) Single-cell analysis of human glioma and immune cells identifies S100A4 as an immunotherapy target. Nat Commun 13:767. https://doi.org/10.1038/s41467-022-28372-y
Abdullah KG, Bird CE, Buehler JD, Gattie LC, Savani MR, Sternisha AC et al (2022) Establishment of patient-derived organoid models of lower-grade glioma. Neuro Oncol 24:612–623. https://doi.org/10.1093/neuonc/noab273
Al-Ali R, Bauer K, Park JW, Al Abdulla R, Fermi V, von Deimling A et al (2019) Single-nucleus chromatin accessibility reveals intratumoral epigenetic heterogeneity in IDH1 mutant gliomas. Acta Neuropathol Commun 7:201. https://doi.org/10.1186/s40478-019-0851-y
Al-Dalahmah O, Argenziano MG, Kannan A, Mahajan A, Furnari J, Paryani F et al (2023) Re-convolving the compositional landscape of primary and recurrent glioblastoma reveals prognostic and targetable tissue states. Nat Commun 14:2586. https://doi.org/10.1038/s41467-023-38186-1
Aldape K, Brindle KM, Chesler L, Chopra R, Gajjar A, Gilbert MR et al (2019) Challenges to curing primary brain tumours. Nat Rev Clin Oncol 16:509–520. https://doi.org/10.1038/s41571-019-0177-5
Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV et al (2013) Signatures of mutational processes in human cancer. Nature 500:415–421. https://doi.org/10.1038/nature12477
Alphones S, Chatterjee U, Singh A, Das A, Zameer L, Achari R et al (2021) Immunohistochemical screening for mismatch repair protein deficiency in paediatric high-grade gliomas—institutional experience and review of literature. Childs Nerv Syst 37:2521–2530. https://doi.org/10.1007/s00381-021-05229-1
Andrade JR, Gallagher AD, Maharaj J, McClelland SE (2023) Disentangling the roles of aneuploidy, chromosomal instability and tumour heterogeneity in developing resistance to cancer therapies. Chromosome Res 31:28. https://doi.org/10.1007/s10577-023-09737-5
Andronesi OC, Rapalino O, Gerstner E, Chi A, Batchelor TT, Cahill DP et al (2013) Detection of oncogenic IDH1 mutations using magnetic resonance spectroscopy of 2-hydroxyglutarate. J Clin Invest 123:3659–3663. https://doi.org/10.1172/JCI67229
Aoki K, Nakamura H, Suzuki H, Matsuo K, Kataoka K, Shimamura T et al (2018) Prognostic relevance of genetic alterations in diffuse lower-grade gliomas. Neuro Oncol 20:66–77. https://doi.org/10.1093/neuonc/nox132
Arita H, Matsushita Y, Machida R, Yamasaki K, Hata N, Ohno M et al (2020) TERT promoter mutation confers favorable prognosis regardless of 1p/19q status in adult diffuse gliomas with IDH1/2 mutations. Acta Neuropathol Commun 8:201. https://doi.org/10.1186/s40478-020-01078-2
Babikir H, Wang L, Shamardani K, Catalan F, Sudhir S, Aghi MK et al (2021) ATRX regulates glial identity and the tumor microenvironment in IDH-mutant glioma. Genome Biol 22:311. https://doi.org/10.1186/s13059-021-02535-4
Bakhoum SF, Danilova OV, Kaur P, Levy NB, Compton DA (2011) Chromosomal instability substantiates poor prognosis in patients with diffuse large B-cell lymphoma. Clin Cancer Res 17:7704–7711. https://doi.org/10.1158/1078-0432.CCR-11-2049
Bakhoum SF, Thompson SL, Manning AL, Compton DA (2009) Genome stability is ensured by temporal control of kinetochore-microtubule dynamics. Nat Cell Biol 11:27–35. https://doi.org/10.1038/ncb1809
Ballhausen A, Przybilla MJ, Jendrusch M, Haupt S, Pfaffendorf E, Seidler F et al (2020) The shared frameshift mutation landscape of microsatellite-unstable cancers suggests immunoediting during tumor evolution. Nat Commun 11:4740. https://doi.org/10.1038/s41467-020-18514-5
Balss J, Meyer J, Mueller W, Korshunov A, Hartmann C, von Deimling A (2008) Analysis of the IDH1 codon 132 mutation in brain tumors. Acta Neuropathol 116:597–602. https://doi.org/10.1007/s00401-008-0455-2
Barthel FP, Johnson KC, Varn FS, Moskalik AD, Tanner G, Kocakavuk E et al (2019) Longitudinal molecular trajectories of diffuse glioma in adults. Nature 576:112–120. https://doi.org/10.1038/s41586-019-1775-1
Behan FM, Iorio F, Picco G, Goncalves E, Beaver CM, Migliardi G et al (2019) Prioritization of cancer therapeutic targets using CRISPR-Cas9 screens. Nature 568:511–516. https://doi.org/10.1038/s41586-019-1103-9
Bhatia A, Kumar Y (2013) Cancer cell micronucleus: an update on clinical and diagnostic applications. APMIS 121:569–581. https://doi.org/10.1111/apm.12033
Bhatia A, Kumar Y (2014) Relevance of microscopic indicators of chromosomal instability in routine reporting of malignancies. Diagn Cytopathol 42:181–188. https://doi.org/10.1002/dc.23012
Blanco-Carmona E, Narayanan A, Hernandez I, Nieto JC, Elosua-Bayes M, Sun X et al (2023) Tumor heterogeneity and tumor-microglia interactions in primary and recurrent IDH1-mutant gliomas. Cell Rep Med 4:101249. https://doi.org/10.1016/j.xcrm.2023.101249
Bleeker FE, Lamba S, Leenstra S, Troost D, Hulsebos T, Vandertop WP et al (2009) IDH1 mutations at residue p. R132 (IDH1(R132)) occur frequently in high-grade gliomas but not in other solid tumors. Hum Mutat 30:7–11. https://doi.org/10.1002/humu.20937
Bomsztyk K, Mar D, Denisenko O, Powell S, Vishnoi M, Delegard J et al (2024) Analysis of gliomas DNA methylation: assessment of pre-analytical variables. bioRxiv. https://doi.org/10.1101/2024.03.26.586350
Bouffet E, Larouche V, Campbell BB, Merico D, de Borja R, Aronson M et al (2016) Immune checkpoint inhibition for hypermutant glioblastoma multiforme resulting from germline biallelic mismatch repair deficiency. J Clin Oncol 34:2206–2211. https://doi.org/10.1200/JCO.2016.66.6552
Brandes AA, Franceschi E, Paccapelo A, Tallini G, De Biase D, Ghimenton C et al (2017) Role of MGMT methylation status at time of diagnosis and recurrence for patients with glioblastoma: clinical implications. Oncologist 22:432–437. https://doi.org/10.1634/theoncologist.2016-0254
Brat DJ, Aldape K, Colman H, Figrarella-Branger D, Fuller GN, Giannini C et al (2020) cIMPACT-NOW update 5: recommended grading criteria and terminologies for IDH-mutant astrocytomas. Acta Neuropathol 139:603–608. https://doi.org/10.1007/s00401-020-02127-9
Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR et al (2013) The somatic genomic landscape of glioblastoma. Cell 155:462–477. https://doi.org/10.1016/j.cell.2013.09.034
Buecher B, Cacheux W, Rouleau E, Dieumegard B, Mitry E, Lievre A (2013) Role of microsatellite instability in the management of colorectal cancers. Dig Liver Dis 45:441–449. https://doi.org/10.1016/j.dld.2012.10.006
Bugter JM, Fenderico N, Maurice MM (2021) Mutations and mechanisms of WNT pathway tumour suppressors in cancer. Nat Rev Cancer 21:5–21. https://doi.org/10.1038/s41568-020-00307-z
Cahill DP, Lengauer C, Yu J, Riggins GJ, Willson JK, Markowitz SD et al (1998) Mutations of mitotic checkpoint genes in human cancers. Nature 392:300–303. https://doi.org/10.1038/32688
Cahill DP, Levine KK, Betensky RA, Codd PJ, Romany CA, Reavie LB et al (2007) Loss of the mismatch repair protein MSH6 in human glioblastomas is associated with tumor progression during temozolomide treatment. Clin Cancer Res 13:2038–2045. https://doi.org/10.1158/1078-0432.CCR-06-2149
Cancer Genome Atlas Research N, Brat DJ, Verhaak RG, Aldape KD, Yung WK, Salama SR et al (2015) Comprehensive, integrative genomic analysis of diffuse lower-grade gliomas. N Engl J Med 372:2481–2498. https://doi.org/10.1056/NEJMoa1402121
Capper D, Jones DTW, Sill M, Hovestadt V, Schrimpf D, Sturm D et al (2018) DNA methylation-based classification of central nervous system tumours. Nature 555:469–474. https://doi.org/10.1038/nature26000
Capper D, Stichel D, Sahm F, Jones DTW, Schrimpf D, Sill M et al (2018) Practical implementation of DNA methylation and copy-number-based CNS tumor diagnostics: the Heidelberg experience. Acta Neuropathol 136:181–210. https://doi.org/10.1007/s00401-018-1879-y
Capper D, Weissert S, Balss J, Habel A, Meyer J, Jager D et al (2010) Characterization of R132H mutation-specific IDH1 antibody binding in brain tumors. Brain Pathol 20:245–254. https://doi.org/10.1111/j.1750-3639.2009.00352.x
Capper D, Zentgraf H, Balss J, Hartmann C, von Deimling A (2009) Monoclonal antibody specific for IDH1 R132H mutation. Acta Neuropathol 118:599–601. https://doi.org/10.1007/s00401-009-0595-z
Carter SL, Eklund AC, Kohane IS, Harris LN, Szallasi Z (2006) A signature of chromosomal instability inferred from gene expression profiles predicts clinical outcome in multiple human cancers. Nat Genet 38:1043–1048. https://doi.org/10.1038/ng1861
Ceccarelli M, Barthel FP, Malta TM, Sabedot TS, Salama SR, Murray BA et al (2016) Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell 164:550–563. https://doi.org/10.1016/j.cell.2015.12.028
Chaligne R, Gaiti F, Silverbush D, Schiffman JS, Weisman HR, Kluegel L et al (2021) Epigenetic encoding, heritability and plasticity of glioma transcriptional cell states. Nat Genet 53:1469–1479. https://doi.org/10.1038/s41588-021-00927-7
Chan EM, Shibue T, McFarland JM, Gaeta B, Ghandi M, Dumont N et al (2019) WRN helicase is a synthetic lethal target in microsatellite unstable cancers. Nature 568:551–556. https://doi.org/10.1038/s41586-019-1102-x
Chen Z, Hambardzumyan D (2021) Macrophage-tumor cell intertwine drives the transition into a mesenchymal-like cellular state of glioblastoma. Cancer Cell 39:743–745. https://doi.org/10.1016/j.ccell.2021.05.003
Cheung RS, Taniguchi T (2017) Recent insights into the molecular basis of Fanconi anemia: genes, modifiers, and drivers. Int J Hematol 106:335–344. https://doi.org/10.1007/s12185-017-2283-4
Choi C, Ganji SK, DeBerardinis RJ, Hatanpaa KJ, Rakheja D, Kovacs Z et al (2012) 2-hydroxyglutarate detection by magnetic resonance spectroscopy in IDH-mutated patients with gliomas. Nat Med 18:624–629. https://doi.org/10.1038/nm.2682
Choi CM, Seo KW, Jang SJ, Oh YM, Shim TS, Kim WS et al (2009) Chromosomal instability is a risk factor for poor prognosis of adenocarcinoma of the lung: fluorescence in situ hybridization analysis of paraffin-embedded tissue from Korean patients. Lung Cancer 64:66–70. https://doi.org/10.1016/j.lungcan.2008.07.016
Choi S, Yu Y, Grimmer MR, Wahl M, Chang SM, Costello JF (2018) Temozolomide-associated hypermutation in gliomas. Neuro Oncol 20:1300–1309. https://doi.org/10.1093/neuonc/noy016
Cimino PJ, Zager M, McFerrin L, Wirsching HG, Bolouri H, Hentschel B et al (2017) Multidimensional scaling of diffuse gliomas: application to the 2016 World Health Organization classification system with prognostically relevant molecular subtype discovery. Acta Neuropathol Commun 5:39. https://doi.org/10.1186/s40478-017-0443-7
Cloughesy TF, Mochizuki AY, Orpilla JR, Hugo W, Lee AH, Davidson TB et al (2019) Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat Med 25:477–486. https://doi.org/10.1038/s41591-018-0337-7
Cohen A, Sato M, Aldape K, Mason CC, Alfaro-Munoz K, Heathcock L et al (2015) DNA copy number analysis of Grade II-III and Grade IV gliomas reveals differences in molecular ontogeny including chromothripsis associated with IDH mutation status. Acta Neuropathol Commun 3:34. https://doi.org/10.1186/s40478-015-0213-3
Cohen AL, Holmen SL, Colman H (2013) IDH1 and IDH2 mutations in gliomas. Curr Neurol Neurosci Rep 13:345. https://doi.org/10.1007/s11910-013-0345-4
Couturier CP, Ayyadhury S, Le PU, Nadaf J, Monlong J, Riva G et al (2020) Single-cell RNA-seq reveals that glioblastoma recapitulates a normal neurodevelopmental hierarchy. Nat Commun 11:3406. https://doi.org/10.1038/s41467-020-17186-5
Crunkhorn S (2022) Targeting drug-resistant glioblastoma. Nat Rev Drug Discov 21:711. https://doi.org/10.1038/d41573-022-00146-7
Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM et al (2009) Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462:739–744. https://doi.org/10.1038/nature08617
Daniel P, Sabri S, Chaddad A, Meehan B, Jean-Claude B, Rak J et al (2019) Temozolomide induced hypermutation in glioma: evolutionary mechanisms and therapeutic opportunities. Front Oncol 9:41. https://doi.org/10.3389/fonc.2019.00041
De S, Michor F (2011) DNA secondary structures and epigenetic determinants of cancer genome evolution. Nat Struct Mol Biol 18:950–955. https://doi.org/10.1038/nsmb.2089
de Souza CF, Sabedot TS, Malta TM, Stetson L, Morozova O, Sokolov A et al (2018) A distinct DNA methylation shift in a subset of glioma CpG island methylator phenotypes during tumor recurrence. Cell Rep 23:637–651. https://doi.org/10.1016/j.celrep.2018.03.107
DeAngelis LM, Mellinghoff IK (2011) Virchow 2011 or how to ID(H) human glioblastoma. J Clin Oncol 29:4473–4474. https://doi.org/10.1200/JCO.2011.37.5873
deCarvalho AC, Kim H, Poisson LM, Winn ME, Mueller C, Cherba D et al (2018) Discordant inheritance of chromosomal and extrachromosomal DNA elements contributes to dynamic disease evolution in glioblastoma. Nat Genet 50:708–717. https://doi.org/10.1038/s41588-018-0105-0
Della Puppa A, Persano L, Masi G, Rampazzo E, Sinigaglia A, Pistollato F et al (2012) MGMT expression and promoter methylation status may depend on the site of surgical sample collection within glioblastoma: a possible pitfall in stratification of patients? J Neurooncol 106:33–41. https://doi.org/10.1007/s11060-011-0639-9
Dodgshun AJ, Fukuoka K, Edwards M, Bianchi VJ, Das A, Sexton-Oates A et al (2020) Germline-driven replication repair-deficient high-grade gliomas exhibit unique hypomethylation patterns. Acta Neuropathol 140:765–776. https://doi.org/10.1007/s00401-020-02209-8
Drews RM, Hernando B, Tarabichi M, Haase K, Lesluyes T, Smith PS et al (2022) A pan-cancer compendium of chromosomal instability. Nature 606:976–983. https://doi.org/10.1038/s41586-022-04789-9
Dunn J, Baborie A, Alam F, Joyce K, Moxham M, Sibson R et al (2009) Extent of MGMT promoter methylation correlates with outcome in glioblastomas given temozolomide and radiotherapy. Br J Cancer 101:124–131. https://doi.org/10.1038/sj.bjc.6605127
Dupont C, Armant DR, Brenner CA (2009) Epigenetics: definition, mechanisms and clinical perspective. Semin Reprod Med 27:351–357. https://doi.org/10.1055/s-0029-1237423
Erson-Omay EZ, Henegariu O, Omay SB, Harmanci AS, Youngblood MW, Mishra-Gorur K et al (2017) Longitudinal analysis of treatment-induced genomic alterations in gliomas. Genome Med 9:12. https://doi.org/10.1186/s13073-017-0401-9
Esteller M, Hamilton SR, Burger PC, Baylin SB, Herman JG (1999) Inactivation of the DNA repair gene O6-methylguanine-DNA methyltransferase by promoter hypermethylation is a common event in primary human neoplasia. Cancer Res 59:793–797
Favero F, McGranahan N, Salm M, Birkbak NJ, Sanborn JZ, Benz SC et al (2015) Glioblastoma adaptation traced through decline of an IDH1 clonal driver and macro-evolution of a double-minute chromosome. Ann Oncol 26:880–887. https://doi.org/10.1093/annonc/mdv127
Feinberg AP, Koldobskiy MA, Gondor A (2016) Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat Rev Genet 17:284–299. https://doi.org/10.1038/nrg.2016.13
Feinberg AP, Levchenko A (2023) Epigenetics as a mediator of plasticity in cancer. Science 379:eaaw3835. https://doi.org/10.1126/science.aaw3835
Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A et al (2010) Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 18:553–567. https://doi.org/10.1016/j.ccr.2010.11.015
Flavahan WA, Drier Y, Liau BB, Gillespie SM, Venteicher AS, Stemmer-Rachamimov AO et al (2016) Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature 529:110–114. https://doi.org/10.1038/nature16490
Friedrich M, Sankowski R, Bunse L, Kilian M, Green E, Ramallo Guevara C et al (2021) Tryptophan metabolism drives dynamic immunosuppressive myeloid states in IDH-mutant gliomas. Nat Cancer 2:723–740. https://doi.org/10.1038/s43018-021-00201-z
Fu D, Calvo JA, Samson LD (2012) Balancing repair and tolerance of DNA damage caused by alkylating agents. Nat Rev Cancer 12:104–120. https://doi.org/10.1038/nrc3185
Galbraith K, Garcia M, Wei S, Chen A, Schroff C, Serrano J et al (2024) Prognostic value of DNA methylation subclassification, aneuploidy, and CDKN2A/B homozygous deletion in predicting clinical outcome of IDH mutant astrocytomas. Neuro Oncol. https://doi.org/10.1093/neuonc/noae009
Galbraith K, Kumar A, Abdullah KG, Walker JM, Adams SH, Prior T et al (2020) Molecular correlates of long survival in IDH-wildtype glioblastoma cohorts. J Neuropathol Exp Neurol 79:843–854. https://doi.org/10.1093/jnen/nlaa059
Galbraith K, Vasudevaraja V, Serrano J, Shen G, Tran I, Abdallat N et al (2023) Clinical utility of whole-genome DNA methylation profiling as a primary molecular diagnostic assay for central nervous system tumors—a prospective study and guidelines for clinical testing. Neurooncol Adv 5:vdad076. https://doi.org/10.1093/noajnl/vdad076
Galuppini F, Opocher E, Tabori U, Mammi I, Edwards M, Campbell B et al (2018) Concomitant IDH wild-type glioblastoma and IDH1-mutant anaplastic astrocytoma in a patient with constitutional mismatch repair deficiency syndrome. Neuropathol Appl Neurobiol 44:233–239. https://doi.org/10.1111/nan.12450
Gao R, Bai S, Henderson YC, Lin Y, Schalck A, Yan Y et al (2021) Delineating copy number and clonal substructure in human tumors from single-cell transcriptomes. Nat Biotechnol 39:599–608. https://doi.org/10.1038/s41587-020-00795-2
Garofano L, Migliozzi S, Oh YT, D’Angelo F, Najac RD, Ko A et al (2021) Pathway-based classification of glioblastoma uncovers a mitochondrial subtype with therapeutic vulnerabilities. Nat Cancer 2:141–156. https://doi.org/10.1038/s43018-020-00159-4
Gempt J, Withake F, Aftahy AK, Meyer HS, Barz M, Delbridge C et al (2022) Methylation subgroup and molecular heterogeneity is a hallmark of glioblastoma: implications for biopsy targeting, classification and therapy. ESMO Open 7:100566. https://doi.org/10.1016/j.esmoop.2022.100566
Gerson SL (2004) MGMT: its role in cancer aetiology and cancer therapeutics. Nat Rev Cancer 4:296–307. https://doi.org/10.1038/nrc1319
Gill BJ, Pisapia DJ, Malone HR, Goldstein H, Lei L, Sonabend A et al (2014) MRI-localized biopsies reveal subtype-specific differences in molecular and cellular composition at the margins of glioblastoma. Proc Natl Acad Sci USA 111:12550–12555. https://doi.org/10.1073/pnas.1405839111
Green RA, Kaplan KB (2003) Chromosome instability in colorectal tumor cells is associated with defects in microtubule plus-end attachments caused by a dominant mutation in APC. J Cell Biol 163:949–961. https://doi.org/10.1083/jcb.200307070
Griffin CA, Burger P, Morsberger L, Yonescu R, Swierczynski S, Weingart JD et al (2006) Identification of der(1;19)(q10;p10) in five oligodendrogliomas suggests mechanism of concurrent 1p and 19q loss. J Neuropathol Exp Neurol 65:988–994. https://doi.org/10.1097/01.jnen.0000235122.98052.8f
Gubin MM, Schreiber RD (2015) CANCER. The odds of immunotherapy success. Science 350:158–159. https://doi.org/10.1126/science.aad4140
Guerrini-Rousseau L, Varlet P, Colas C, Andreiuolo F, Bourdeaut F, Dahan K et al (2019) Constitutional mismatch repair deficiency-associated brain tumors: report from the European C4CMMRD consortium. Neurooncol Adv 1:vdz033. https://doi.org/10.1093/noajnl/vdz033
Guo J, Fathi Kazerooni A, Toorens E, Akbari H, Yu F, Sako C et al (2024) Integrating imaging and genomic data for the discovery of distinct glioblastoma subtypes: a joint learning approach. Sci Rep 14:4922. https://doi.org/10.1038/s41598-024-55072-y
Guscott MA, Gómez-Peregrina D, Andersen AM, Soliman TN, Horrach CV, Bakker B et al (2023) Tracking genome evolution in single cell clones reveals the rates and features of copy number alterations generated by ongoing chromosomal instability in cancer. bioRxiv. https://doi.org/10.1101/2023.09.27.559836
Hadad S, Gupta R, Oberheim Bush NA, Taylor JW, Villanueva-Meyer JE, Young JS et al (2023) “De novo replication repair deficient glioblastoma, IDH-wildtype” is a distinct glioblastoma subtype in adults that may benefit from immune checkpoint blockade. Acta Neuropathol 147:3. https://doi.org/10.1007/s00401-023-02654-1
Hansen KH, Bracken AP, Pasini D, Dietrich N, Gehani SS, Monrad A et al (2008) A model for transmission of the H3K27me3 epigenetic mark. Nat Cell Biol 10:1291–1300. https://doi.org/10.1038/ncb1787
Hara T, Chanoch-Myers R, Mathewson ND, Myskiw C, Atta L, Bussema L et al (2021) Interactions between cancer cells and immune cells drive transitions to mesenchymal-like states in glioblastoma. Cancer Cell 39(779–792):e711. https://doi.org/10.1016/j.ccell.2021.05.002
Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M et al (2005) MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med 352:997–1003. https://doi.org/10.1056/NEJMoa043331
Hickman RA, Gedvilaite E, Ptashkin R, Reiner AS, Cimera R, Nandakumar S et al (2023) CDKN2A/B mutations and allele-specific alterations stratify survival outcomes in IDH-mutant astrocytomas. Acta Neuropathol 146:845–847. https://doi.org/10.1007/s00401-023-02639-0
Higuchi F, Nagashima H, Ning J, Koerner MVA, Wakimoto H, Cahill DP (2020) Restoration of temozolomide sensitivity by PARP inhibitors in mismatch repair deficient glioblastoma is independent of base excision repair. Clin Cancer Res 26:1690–1699. https://doi.org/10.1158/1078-0432.CCR-19-2000
Hodges TR, Ott M, Xiu J, Gatalica Z, Swensen J, Zhou S et al (2017) Mutational burden, immune checkpoint expression, and mismatch repair in glioma: implications for immune checkpoint immunotherapy. Neuro Oncol 19:1047–1057. https://doi.org/10.1093/neuonc/nox026
Hunter C, Smith R, Cahill DP, Stephens P, Stevens C, Teague J et al (2006) A hypermutation phenotype and somatic MSH6 mutations in recurrent human malignant gliomas after alkylator chemotherapy. Cancer Res 66:3987–3991. https://doi.org/10.1158/0008-5472.CAN-06-0127
Jacob F, Salinas RD, Zhang DY, Nguyen PTT, Schnoll JG, Wong SZH et al (2020) A patient-derived glioblastoma organoid model and biobank recapitulates inter- and intra-tumoral heterogeneity. Cell 180(188–204):e122. https://doi.org/10.1016/j.cell.2019.11.036
Jasencakova Z, Groth A (2010) Replication stress, a source of epigenetic aberrations in cancer? BioEssays 32:847–855. https://doi.org/10.1002/bies.201000055
Jasencakova Z, Scharf AN, Ask K, Corpet A, Imhof A, Almouzni G et al (2010) Replication stress interferes with histone recycling and predeposition marking of new histones. Mol Cell 37:736–743. https://doi.org/10.1016/j.molcel.2010.01.033
Jenkins RB, Blair H, Ballman KV, Giannini C, Arusell RM, Law M et al (2006) A t(1;19)(q10;p10) mediates the combined deletions of 1p and 19q and predicts a better prognosis of patients with oligodendroglioma. Cancer Res 66:9852–9861. https://doi.org/10.1158/0008-5472.CAN-06-1796
Jiricny J (2006) The multifaceted mismatch-repair system. Nat Rev Mol Cell Biol 7:335–346. https://doi.org/10.1038/nrm1907
Johannessen TA, Mukherjee J, Viswanath P, Ohba S, Ronen SM, Bjerkvig R et al (2016) Rapid conversion of mutant IDH1 from driver to passenger in a model of human gliomagenesis. Mol Cancer Res 14:976–983. https://doi.org/10.1158/1541-7786.MCR-16-0141
Johanns TM, Miller CA, Dorward IG, Tsien C, Chang E, Perry A et al (2016) Immunogenomics of hypermutated glioblastoma: a patient with germline POLE deficiency treated with checkpoint blockade immunotherapy. Cancer Discov 6:1230–1236. https://doi.org/10.1158/2159-8290.CD-16-0575
Johnson BE, Mazor T, Hong C, Barnes M, Aihara K, McLean CY et al (2014) Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science 343:189–193. https://doi.org/10.1126/science.1239947
Johnson KC, Anderson KJ, Courtois ET, Gujar AD, Barthel FP, Varn FS et al (2021) Single-cell multimodal glioma analyses identify epigenetic regulators of cellular plasticity and environmental stress response. Nat Genet 53:1456–1468. https://doi.org/10.1038/s41588-021-00926-8
Jonsson P, Lin AL, Young RJ, DiStefano NM, Hyman DM, Li BT et al (2019) Genomic correlates of disease progression and treatment response in prospectively characterized gliomas. Clin Cancer Res 25:5537–5547. https://doi.org/10.1158/1078-0432.CCR-19-0032
Kim B, Tabori U, Hawkins C (2020) An update on the CNS manifestations of brain tumor polyposis syndromes. Acta Neuropathol 139:703–715. https://doi.org/10.1007/s00401-020-02124-y
Kim H, Lim KY, Park JW, Kang J, Won JK, Lee K et al (2022) Sporadic and Lynch syndrome-associated mismatch repair-deficient brain tumors. Lab Invest 102:160–171. https://doi.org/10.1038/s41374-021-00694-3
Kim H, Zheng S, Amini SS, Virk SM, Mikkelsen T, Brat DJ et al (2015) Whole-genome and multisector exome sequencing of primary and post-treatment glioblastoma reveals patterns of tumor evolution. Genome Res 25:316–327. https://doi.org/10.1101/gr.180612.114
Kling T, Ferreyra Vega S, Suman M, Denes A, Lipatnikova A, Lagerstrom S et al (2024) Refinement of prognostication for IDH-mutant astrocytomas using DNA methylation-based classification. Brain Pathol. https://doi.org/10.1111/bpa.13233
Kloor M, von Knebel DM (2016) The immune biology of microsatellite-unstable cancer. Trends Cancer 2:121–133. https://doi.org/10.1016/j.trecan.2016.02.004
Kocakavuk E, Johnson KC, Sabedot TS, Reinhardt HC, Noushmehr H, Verhaak RGW (2023) Hemizygous CDKN2A deletion confers worse survival outcomes in IDHmut-noncodel gliomas. Neuro Oncol 25:1721–1723. https://doi.org/10.1093/neuonc/noad095
Korshunov A, Schrimpf D, Ryzhova M, Sturm D, Chavez L, Hovestadt V et al (2017) H3-/IDH-wild type pediatric glioblastoma is comprised of molecularly and prognostically distinct subtypes with associated oncogenic drivers. Acta Neuropathol 134:507–516. https://doi.org/10.1007/s00401-017-1710-1
Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD et al (2015) PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med 372:2509–2520. https://doi.org/10.1056/NEJMoa1500596
Lee AJ, Endesfelder D, Rowan AJ, Walther A, Birkbak NJ, Futreal PA et al (2011) Chromosomal instability confers intrinsic multidrug resistance. Cancer Res 71:1858–1870. https://doi.org/10.1158/0008-5472.CAN-10-3604
Lee MD, Jain R, Galbraith K, Chen A, Lieberman E, Patel SH et al (2024) T2-FLAIR mismatch sign predicts DNA methylation subclass and CDKN2A/B status in IDH-mutant astrocytomas. Clin Cancer Res. https://doi.org/10.1158/1078-0432.CCR-24-0311
Lemmens B, van Schendel R, Tijsterman M (2015) Mutagenic consequences of a single G-quadruplex demonstrate mitotic inheritance of DNA replication fork barriers. Nat Commun 6:8909. https://doi.org/10.1038/ncomms9909
Lengauer C, Kinzler KW, Vogelstein B (1998) Genetic instabilities in human cancers. Nature 396:643–649. https://doi.org/10.1038/25292
Lengauer C, Kinzler KW, Vogelstein B (1997) Genetic instability in colorectal cancers. Nature 386:623–627. https://doi.org/10.1038/386623a0
Lepage CC, Morden CR, Palmer MCL, Nachtigal MW, McManus KJ (2019) Detecting chromosome instability in cancer: approaches to resolve cell-to-cell heterogeneity. Cancers (Basel). https://doi.org/10.3390/cancers11020226
Lerner LK, Sale JE (2019) Replication of G quadruplex DNA. Genes (Basel). https://doi.org/10.3390/genes10020095
Li GM (2008) Mechanisms and functions of DNA mismatch repair. Cell Res 18:85–98. https://doi.org/10.1038/cr.2007.115
Li KK, Shi ZF, Malta TM, Chan AK, Cheng S, Kwan JSH et al (2019) Identification of subsets of IDH-mutant glioblastomas with distinct epigenetic and copy number alterations and stratified clinical risks. Neurooncol Adv 1:vdz015. https://doi.org/10.1093/noajnl/vdz015
Lieb S, Blaha-Ostermann S, Kamper E, Rippka J, Schwarz C, Ehrenhofer-Wolfer K et al (2019) Werner syndrome helicase is a selective vulnerability of microsatellite instability-high tumor cells. Elife. https://doi.org/10.7554/eLife.43333
Lin K, Gueble SE, Sundaram RK, Huseman ED, Bindra RS, Herzon SB (2022) Mechanism-based design of agents that selectively target drug-resistant glioma. Science 377:502–511. https://doi.org/10.1126/science.abn7570
Liu G, Bu C, Guo G, Zhang Z, Sheng Z, Deng K et al (2023) Molecular and clonal evolution in vivo reveal a common pathway of distant relapse gliomas. iScience 26:107528. https://doi.org/10.1016/j.isci.2023.107528
Liu Y, Sathe AA, Abdullah KG, McBrayer SK, Adams SH, Brenner AJ et al (2022) Global DNA methylation profiling reveals chromosomal instability in IDH-mutant astrocytomas. Acta Neuropathol Commun 10:32. https://doi.org/10.1186/s40478-022-01339-2
Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK et al (2016) The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol 131:803–820. https://doi.org/10.1007/s00401-016-1545-1
Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D et al (2021) The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro Oncol 23:1231–1251. https://doi.org/10.1093/neuonc/noab106
Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O et al (2012) IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 483:474–478. https://doi.org/10.1038/nature10860
Lucas A, Eberwine JH, Bagley SJ, Fan Y, Nasrallah MP, Brem S (2021) Commentary: “Zooming in” on glioblastoma: understanding tumor heterogeneity and its clinical implications in the era of single-cell ribonucleic acid sequencing. Neurosurgery 89:E262–E263. https://doi.org/10.1093/neuros/nyab288
Luijten MNH, Lee JXT, Crasta KC (2018) Mutational game changer: chromothripsis and its emerging relevance to cancer. Mutat Res Rev Mutat Res 777:29–51. https://doi.org/10.1016/j.mrrev.2018.06.004
Lukas RV, Rodon J, Becker K, Wong ET, Shih K, Touat M et al (2018) Clinical activity and safety of atezolizumab in patients with recurrent glioblastoma. J Neurooncol 140:317–328. https://doi.org/10.1007/s11060-018-2955-9
Luo S, Zhu S, Liao J, Zhang Y, Hou X, Luo T et al (2021) IDH clonal heterogeneity segregates a subgroup of non-1p/19q codeleted gliomas with unfavourable clinical outcome. Neuropathol Appl Neurobiol 47:394–405. https://doi.org/10.1111/nan.12671
Lyon JF, Vasudevaraja V, Mirchia K, Walker JM, Corona RJ, Chin LS et al (2021) Spatial progression and molecular heterogeneity of IDH-mutant glioblastoma determined by DNA methylation-based mapping. Acta Neuropathol Commun 9:120. https://doi.org/10.1186/s40478-021-01221-7
Malta TM, de Souza CF, Sabedot TS, Silva TC, Mosella MS, Kalkanis SN et al (2018) Glioma CpG island methylator phenotype (G-CIMP): biological and clinical implications. Neuro Oncol 20:608–620. https://doi.org/10.1093/neuonc/nox183
Mandal R, Samstein RM, Lee KW, Havel JJ, Wang H, Krishna C et al (2019) Genetic diversity of tumors with mismatch repair deficiency influences anti-PD-1 immunotherapy response. Science 364:485–491. https://doi.org/10.1126/science.aau0447
Margueron R, Reinberg D (2010) Chromatin structure and the inheritance of epigenetic information. Nat Rev Genet 11:285–296. https://doi.org/10.1038/nrg2752
Martin-Lopez JV, Fishel R (2013) The mechanism of mismatch repair and the functional analysis of mismatch repair defects in Lynch syndrome. Fam Cancer 12:159–168. https://doi.org/10.1007/s10689-013-9635-x
Mazor T, Chesnelong C, Pankov A, Jalbert LE, Hong C, Hayes J et al (2017) Clonal expansion and epigenetic reprogramming following deletion or amplification of mutant IDH1. Proc Natl Acad Sci USA 114:10743–10748. https://doi.org/10.1073/pnas.1708914114
McBrayer SK, Mayers JR, DiNatale GJ, Shi DD, Khanal J, Chakraborty AA et al (2018) Transaminase inhibition by 2-hydroxyglutarate impairs glutamate biosynthesis and redox homeostasis in glioma. Cell 175(101–116):e125. https://doi.org/10.1016/j.cell.2018.08.038
McCord M, Lukas RV, Amidei C, Demars N, Gelb A, Buck J et al (2021) Disappearance of MMR-deficient subclones after controlled IL-12 and PD-1 inhibition in a glioma patient. Neurooncol Adv 3:vdab045. https://doi.org/10.1093/noajnl/vdab045
McCord M, Steffens A, Javier R, Kam KL, McCortney K, Horbinski C (2020) The efficacy of DNA mismatch repair enzyme immunohistochemistry as a screening test for hypermutated gliomas. Acta Neuropathol Commun 8:15. https://doi.org/10.1186/s40478-020-0892-2
McGranahan N, Favero F, de Bruin EC, Birkbak NJ, Szallasi Z, Swanton C (2015) Clonal status of actionable driver events and the timing of mutational processes in cancer evolution. Sci Transl Med 7:283ra254. https://doi.org/10.1126/scitranslmed.aaa1408
McGranahan N, Furness AJ, Rosenthal R, Ramskov S, Lyngaa R, Saini SK et al (2016) Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 351:1463–1469. https://doi.org/10.1126/science.aaf1490
Mellinghoff IK, Lu M, Wen PY, Taylor JW, Maher EA, Arrillaga-Romany I et al (2023) Vorasidenib and ivosidenib in IDH1-mutant low-grade glioma: a randomized, perioperative phase 1 trial. Nat Med 29:615–622. https://doi.org/10.1038/s41591-022-02141-2
Mellinghoff IK, Penas-Prado M, Peters KB, Burris HA 3rd, Maher EA, Janku F et al (2021) Vorasidenib, a dual inhibitor of mutant IDH1/2, in recurrent or progressive glioma; results of a first-in-human phase i trial. Clin Cancer Res 27:4491–4499. https://doi.org/10.1158/1078-0432.CCR-21-0611
Mellinghoff IK, van den Bent MJ, Blumenthal DT, Touat M, Peters KB, Clarke J et al (2023) Vorasidenib in IDH1- or IDH2-mutant low-grade glioma. N Engl J Med 389:589–601. https://doi.org/10.1056/NEJMoa2304194
Meyer M, Reimand J, Lan X, Head R, Zhu X, Kushida M et al (2015) Single cell-derived clonal analysis of human glioblastoma links functional and genomic heterogeneity. Proc Natl Acad Sci USA 112:851–856. https://doi.org/10.1073/pnas.1320611111
Miller JJ, Cahill DP, Arrillaga-Romany I (2022) Enhancing demethylation-induced differentiation in IDH-mutant glioma. Neuro Oncol 24:724–725. https://doi.org/10.1093/neuonc/noac056
Mirchia K, Richardson TE (2020) Beyond IDH-mutation: emerging molecular diagnostic and prognostic features in adult diffuse gliomas. Cancers (Basel). https://doi.org/10.3390/cancers12071817
Mirchia K, Sathe AA, Walker JM, Fudym Y, Galbraith K, Viapiano MS et al (2019) Total copy number variation as a prognostic factor in adult astrocytoma subtypes. Acta Neuropathol Commun 7:92. https://doi.org/10.1186/s40478-019-0746-y
Mirchia K, Snuderl M, Galbraith K, Hatanpaa KJ, Walker JM, Richardson TE (2019) Establishing a prognostic threshold for total copy number variation within adult IDH-mutant grade II/III astrocytomas. Acta Neuropathol Commun 7:121. https://doi.org/10.1186/s40478-019-0778-3
Mukherjee J, Johannessen TC, Ohba S, Chow TT, Jones L, Pandita A et al (2018) Mutant IDH1 cooperates with ATRX loss to drive the alternative lengthening of telomere phenotype in glioma. Cancer Res 78:2966–2977. https://doi.org/10.1158/0008-5472.CAN-17-2269
Muller S, Cho A, Liu SJ, Lim DA, Diaz A (2018) CONICS integrates scRNA-seq with DNA sequencing to map gene expression to tumor sub-clones. Bioinformatics 34:3217–3219. https://doi.org/10.1093/bioinformatics/bty316
Muller S, Kohanbash G, Liu SJ, Alvarado B, Carrera D, Bhaduri A et al (2017) Single-cell profiling of human gliomas reveals macrophage ontogeny as a basis for regional differences in macrophage activation in the tumor microenvironment. Genome Biol 18:234. https://doi.org/10.1186/s13059-017-1362-4
Neftel C, Laffy J, Filbin MG, Hara T, Shore ME, Rahme GJ et al (2019) An integrative model of cellular states, plasticity, and genetics for glioblastoma. Cell 178(835–849):e821. https://doi.org/10.1016/j.cell.2019.06.024
Nicholson JG, Fine HA (2021) Diffuse glioma heterogeneity and its therapeutic implications. Cancer Discov 11:575–590. https://doi.org/10.1158/2159-8290.CD-20-1474
Nikolic A, Singhal D, Ellestad K, Johnston M, Shen Y, Gillmor A et al (2021) Copy-scAT: deconvoluting single-cell chromatin accessibility of genetic subclones in cancer. Sci Adv 7:eabg6045. https://doi.org/10.1126/sciadv.abg6045
Nishisho I, Nakamura Y, Miyoshi Y, Miki Y, Ando H, Horii A et al (1991) Mutations of chromosome 5q21 genes in FAP and colorectal cancer patients. Science 253:665–669. https://doi.org/10.1126/science.1651563
Noorani I, Mischel PS, Swanton C (2022) Leveraging extrachromosomal DNA to fine-tune trials of targeted therapy for glioblastoma: opportunities and challenges. Nat Rev Clin Oncol 19:733–743. https://doi.org/10.1038/s41571-022-00679-1
Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP et al (2010) Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 17:510–522. https://doi.org/10.1016/j.ccr.2010.03.017
Onishi S, Yamasaki F, Kuraoka K, Taguchi A, Takayasu T, Akagi K et al (2023) Diagnostic and therapeutic challenges of glioblastoma as an initial malignancy of constitutional mismatch repair deficiency (CMMRD): two case reports and a literature review. BMC Med Genomics 16:6. https://doi.org/10.1186/s12920-022-01403-9
Ostrom QT, Price M, Neff C, Cioffi G, Waite KA, Kruchko C et al (2022) CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2015–2019. Neuro Oncol 24:v1–v95. https://doi.org/10.1093/neuonc/noac202
Ostrom QT, Price M, Neff C, Cioffi G, Waite KA, Kruchko C et al (2023) CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2016–2020. Neuro Oncol 25:iv1–iv99. https://doi.org/10.1093/neuonc/noad149
Papadopoulou C, Guilbaud G, Schiavone D, Sale JE (2015) Nucleotide pool depletion induces G-quadruplex-dependent perturbation of gene expression. Cell Rep 13:2491–2503. https://doi.org/10.1016/j.celrep.2015.11.039
Parkinson JF, Wheeler HR, Clarkson A, McKenzie CA, Biggs MT, Little NS et al (2008) Variation of O(6)-methylguanine-DNA methyltransferase (MGMT) promoter methylation in serial samples in glioblastoma. J Neurooncol 87:71–78. https://doi.org/10.1007/s11060-007-9486-0
Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P et al (2008) An integrated genomic analysis of human glioblastoma multiforme. Science 321:1807–1812. https://doi.org/10.1126/science.1164382
Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H et al (2014) Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344:1396–1401. https://doi.org/10.1126/science.1254257
Philip B, Yu DX, Silvis MR, Shin CH, Robinson JP, Robinson GL et al (2018) Mutant IDH1 promotes glioma formation in vivo. Cell Rep 23:1553–1564. https://doi.org/10.1016/j.celrep.2018.03.133
Poon CC, Gordon PMK, Liu K, Yang R, Sarkar S, Mirzaei R et al (2019) Differential microglia and macrophage profiles in human IDH-mutant and -wild type glioblastoma. Oncotarget 10:3129–3143. https://doi.org/10.18632/oncotarget.26863
Ramos SI, Mussa ZM, Falk EN, Pai B, Giotti B, Allette K et al (2022) An atlas of late prenatal human neurodevelopment resolved by single-nucleus transcriptomics. Nat Commun 13:7671. https://doi.org/10.1038/s41467-022-34975-2
Reifenberger J, Reifenberger G, Liu L, James CD, Wechsler W, Collins VP (1994) Molecular genetic analysis of oligodendroglial tumors shows preferential allelic deletions on 19q and 1p. Am J Pathol 145:1175–1190
Richardson TE, Hatanpaa KJ, Walker JM (2021) Molecular characterization of “true” low-grade IDH-wildtype astrocytomas. J Neuropathol Exp Neurol 80:431–435. https://doi.org/10.1093/jnen/nlab023
Richardson TE, Patel S, Serrano J, Sathe AA, Daoud EV, Oliver D et al (2019) Genome-wide analysis of glioblastoma patients with unexpectedly long survival. J Neuropathol Exp Neurol 78:501–507. https://doi.org/10.1093/jnen/nlz025
Richardson TE, Sathe AA, Kanchwala M, Jia G, Habib AA, Xiao G et al (2018) Genetic and epigenetic features of rapidly progressing IDH-mutant astrocytomas. J Neuropathol Exp Neurol 77:542–548. https://doi.org/10.1093/jnen/nly026
Richardson TE, Sathe AA, Xing C, Mirchia K, Viapiano MS, Snuderl M et al (2021) Molecular signatures of chromosomal instability correlate with copy number variation patterns and patient outcome in IDH-mutant and IDH-wildtype astrocytomas. J Neuropathol Exp Neurol. https://doi.org/10.1093/jnen/nlab008
Richardson TE, Snuderl M, Serrano J, Karajannis MA, Heguy A, Oliver D et al (2017) Rapid progression to glioblastoma in a subset of IDH-mutated astrocytomas: a genome-wide analysis. J Neurooncol 133:183–192. https://doi.org/10.1007/s11060-017-2431-y
Richardson TE, Walker JM, Abdullah KG, McBrayer SK, Viapiano MS, Mussa ZM et al (2022) Chromosomal instability in adult-type diffuse gliomas. Acta Neuropathol Commun 10:115. https://doi.org/10.1186/s40478-022-01420-w
Richardson TE, Williams M, Galbraith K, Mirchia K, Kumar A, Xing C et al (2020) Total copy number variation, somatic mutation burden, and histologic grade correlate with clinical outcome in oligodendroglioma. Clin Neuropathol 39:238–242. https://doi.org/10.5414/NP301260
Richardson TE, Yokoda RT, Rashidipour O, Vij M, Snuderl M, Brem S et al (2023) Mismatch repair protein mutations in isocitrate dehydrogenase (IDH)-mutant astrocytoma and IDH-wild-type glioblastoma. Neurooncol Adv 5:vdad085. https://doi.org/10.1093/noajnl/vdad085
Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ et al (2015) Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 348:124–128. https://doi.org/10.1126/science.aaa1348
Rosswog C, Bartenhagen C, Welte A, Kahlert Y, Hemstedt N, Lorenz W et al (2021) Chromothripsis followed by circular recombination drives oncogene amplification in human cancer. Nat Genet 53:1673–1685. https://doi.org/10.1038/s41588-021-00951-7
Samstein RM, Lee CH, Shoushtari AN, Hellmann MD, Shen R, Janjigian YY et al (2019) Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet 51:202–206. https://doi.org/10.1038/s41588-018-0312-8
Santana-Santos L, Kam KL, Dittmann D, De Vito S, McCord M, Jamshidi P et al (2022) Validation of whole genome methylation profiling classifier for central nervous system tumors. J Mol Diagn 24:924–934. https://doi.org/10.1016/j.jmoldx.2022.04.009
Sarkies P, Reams C, Simpson LJ, Sale JE (2010) Epigenetic instability due to defective replication of structured DNA. Mol Cell 40:703–713. https://doi.org/10.1016/j.molcel.2010.11.009
Sato H, Uzawa N, Takahashi K, Myo K, Ohyama Y, Amagasa T (2010) Prognostic utility of chromosomal instability detected by fluorescence in situ hybridization in fine-needle aspirates from oral squamous cell carcinomas. BMC Cancer 10:182. https://doi.org/10.1186/1471-2407-10-182
Schiavone D, Jozwiakowski SK, Romanello M, Guilbaud G, Guilliam TA, Bailey LJ et al (2016) PrimPol is required for replicative tolerance of G quadruplexes in vertebrate cells. Mol Cell 61:161–169. https://doi.org/10.1016/j.molcel.2015.10.038
Sha D, Jin Z, Budczies J, Kluck K, Stenzinger A, Sinicrope FA (2020) Tumor mutational burden as a predictive biomarker in solid tumors. Cancer Discov 10:1808–1825. https://doi.org/10.1158/2159-8290.CD-20-0522
Shi DD, Anand S, Abdullah KG, McBrayer SK (2022) DNA damage in IDH-mutant gliomas: mechanisms and clinical implications. J Neurooncol. https://doi.org/10.1007/s11060-022-04172-8
Shi DD, Savani MR, Levitt MM, Wang AC, Endress JE, Bird CE et al (2022) De novo pyrimidine synthesis is a targetable vulnerability in IDH mutant glioma. Cancer Cell 40(939–956):e916. https://doi.org/10.1016/j.ccell.2022.07.011
Shirahata M, Ono T, Stichel D, Schrimpf D, Reuss DE, Sahm F et al (2018) Novel, improved grading system(s) for IDH-mutant astrocytic gliomas. Acta Neuropathol 136:153–166. https://doi.org/10.1007/s00401-018-1849-4
Shoshani O, Brunner SF, Yaeger R, Ly P, Nechemia-Arbely Y, Kim DH et al (2021) Chromothripsis drives the evolution of gene amplification in cancer. Nature 591:137–141. https://doi.org/10.1038/s41586-020-03064-z
Sinicrope FA, Sargent DJ (2012) Molecular pathways: microsatellite instability in colorectal cancer: prognostic, predictive, and therapeutic implications. Clin Cancer Res 18:1506–1512. https://doi.org/10.1158/1078-0432.CCR-11-1469
Siri SO, Martino J, Gottifredi V (2021) Structural chromosome instability: types, origins, consequences, and therapeutic opportunities. Cancers (Basel). https://doi.org/10.3390/cancers13123056
Spitzer A, Gritsch S, Nomura M, Jucht A, Fortin J, Raviram R et al (2024) Mutant IDH inhibitors induce lineage differentiation in IDH-mutant oligodendroglioma. Cancer Cell. https://doi.org/10.1016/j.ccell.2024.03.008
Spoor JKH, den Braber M, Dirven CMF, Pennycuick A, Bartkova J, Bartek J et al (2023) Investigating chromosomal instability in long-term survivors with glioblastoma and grade 4 astrocytoma. Front Oncol 13:1218297. https://doi.org/10.3389/fonc.2023.1218297
Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ et al (2011) Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 144:27–40. https://doi.org/10.1016/j.cell.2010.11.055
Stirling PC, Bloom MS, Solanki-Patil T, Smith S, Sipahimalani P, Li Z et al (2011) The complete spectrum of yeast chromosome instability genes identifies candidate CIN cancer genes and functional roles for ASTRA complex components. PLoS Genet 7:e1002057. https://doi.org/10.1371/journal.pgen.1002057
Stoyanov GS, Dzhenkov DL (2018) On the concepts and history of glioblastoma multiforme—morphology, genetics and epigenetics. Folia Med (Plovdiv) 60:48–66. https://doi.org/10.1515/folmed-2017-0069
Sturm D, Witt H, Hovestadt V, Khuong-Quang DA, Jones DT, Konermann C et al (2012) Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 22:425–437. https://doi.org/10.1016/j.ccr.2012.08.024
Suwala AK, Stichel D, Schrimpf D, Kloor M, Wefers AK, Reinhardt A et al (2021) Primary mismatch repair deficient IDH-mutant astrocytoma (PMMRDIA) is a distinct type with a poor prognosis. Acta Neuropathol 141:85–100. https://doi.org/10.1007/s00401-020-02243-6
Suzuki H, Aoki K, Chiba K, Sato Y, Shiozawa Y, Shiraishi Y et al (2015) Mutational landscape and clonal architecture in grade II and III gliomas. Nat Genet 47:458–468. https://doi.org/10.1038/ng.3273
Svikovic S, Sale JE (2017) The effects of replication stress on S phase histone management and epigenetic memory. J Mol Biol 429:2011–2029. https://doi.org/10.1016/j.jmb.2016.11.011
Takami S, Kawasome C, Kinoshita M, Koyama H, Noguchi S (2001) Chromosomal instability detected by fluorescence in situ hybridization in Japanese breast cancer patients. Clin Chim Acta 308:127–131. https://doi.org/10.1016/s0009-8981(01)00473-9
Tateishi K, Wakimoto H, Iafrate AJ, Tanaka S, Loebel F, Lelic N et al (2015) Extreme vulnerability of IDH1 mutant cancers to NAD+ depletion. Cancer Cell 28:773–784. https://doi.org/10.1016/j.ccell.2015.11.006
Thompson LL, Jeusset LM, Lepage CC, McManus KJ (2017) Evolving therapeutic strategies to exploit chromosome instability in cancer. Cancers (Basel). https://doi.org/10.3390/cancers9110151
Thompson SL, Bakhoum SF, Compton DA (2010) Mechanisms of chromosomal instability. Curr Biol 20:R285-295. https://doi.org/10.1016/j.cub.2010.01.034
Tirosh I, Suva ML (2018) Dissecting human gliomas by single-cell RNA sequencing. Neuro Oncol 20:37–43. https://doi.org/10.1093/neuonc/nox126
Tirosh I, Venteicher AS, Hebert C, Escalante LE, Patel AP, Yizhak K et al (2016) Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature 539:309–313. https://doi.org/10.1038/nature20123
Touat M, Li YY, Boynton AN, Spurr LF, Iorgulescu JB, Bohrson CL et al (2020) Mechanisms and therapeutic implications of hypermutation in gliomas. Nature 580:517–523. https://doi.org/10.1038/s41586-020-2209-9
Turcan S, Rohle D, Goenka A, Walsh LA, Fang F, Yilmaz E et al (2012) IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 483:479–483. https://doi.org/10.1038/nature10866
Umphlett M, Bilal KH, Martini ML, Suwala AK, Ahuja S, Rashidipour O et al (2022) IDH-mutant astrocytoma with EGFR amplification-genomic profiling in four cases and review of literature. Neurooncol Adv 4:vdac067. https://doi.org/10.1093/noajnl/vdac067
van Dijk E, van den Bosch T, Lenos KJ, El Makrini K, Nijman LE, van Essen HFB et al (2021) Chromosomal copy number heterogeneity predicts survival rates across cancers. Nat Commun 12:3188. https://doi.org/10.1038/s41467-021-23384-6
van Thuijl HF, Mazor T, Johnson BE, Fouse SD, Aihara K, Hong C et al (2015) Evolution of DNA repair defects during malignant progression of low-grade gliomas after temozolomide treatment. Acta Neuropathol 129:597–607. https://doi.org/10.1007/s00401-015-1403-6
Venteicher AS, Tirosh I, Hebert C, Yizhak K, Neftel C, Filbin MG et al (2017) Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science. https://doi.org/10.1126/science.aai8478
Vij M, Yokoda RT, Rashidipour O, Tran I, Vasudevaraja V, Snuderl M et al (2023) The prognostic impact of subclonal IDH1 mutation in grade 2–4 astrocytomas. Neurooncol Adv 5:vdad069. https://doi.org/10.1093/noajnl/vdad069
Vishwakarma R, McManus KJ (2020) Chromosome instability; implications in cancer development, progression, and clinical outcomes. Cancers (Basel). https://doi.org/10.3390/cancers12040824
Wang J, Cazzato E, Ladewig E, Frattini V, Rosenbloom DI, Zairis S et al (2016) Clonal evolution of glioblastoma under therapy. Nat Genet 48:768–776. https://doi.org/10.1038/ng.3590
Wang L, Babikir H, Muller S, Yagnik G, Shamardani K, Catalan F et al (2019) The phenotypes of proliferating glioblastoma cells reside on a single axis of variation. Cancer Discov 9:1708–1719. https://doi.org/10.1158/2159-8290.CD-19-0329
Wang L, Jung J, Babikir H, Shamardani K, Jain S, Feng X et al (2022) A single-cell atlas of glioblastoma evolution under therapy reveals cell-intrinsic and cell-extrinsic therapeutic targets. Nat Cancer 3:1534–1552. https://doi.org/10.1038/s43018-022-00475-x
Wang Q, Hu B, Hu X, Kim H, Squatrito M, Scarpace L et al (2017) Tumor evolution of glioma-intrinsic gene expression subtypes associates with immunological changes in the microenvironment. Cancer Cell 32(42–56):e46. https://doi.org/10.1016/j.ccell.2017.06.003
Wang Y, Yang J, Wild AT, Wu WH, Shah R, Danussi C et al (2019) G-quadruplex DNA drives genomic instability and represents a targetable molecular abnormality in ATRX-deficient malignant glioma. Nat Commun 10:943. https://doi.org/10.1038/s41467-019-08905-8
Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA et al (2010) The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 17:225–234. https://doi.org/10.1016/j.ccr.2010.01.020
Wei Y, Li G, Feng J, Wu F, Zhao Z, Bao Z et al (2023) Stalled oligodendrocyte differentiation in IDH-mutant gliomas. Genome Med 15:24. https://doi.org/10.1186/s13073-023-01175-6
Weller M, Felsberg J, Hentschel B, Gramatzki D, Kubon N, Wolter M et al (2024) Improved prognostic stratification of patients with isocitrate dehydrogenase-mutant astrocytoma. Acta Neuropathol 147:11. https://doi.org/10.1007/s00401-023-02662-1
Wenger A, Ferreyra Vega S, Kling T, Bontell TO, Jakola AS, Caren H (2019) Intratumor DNA methylation heterogeneity in glioblastoma: implications for DNA methylation-based classification. Neuro Oncol 21:616–627. https://doi.org/10.1093/neuonc/noz011
Whitfield BT, Huse JT (2022) Classification of adult-type diffuse gliomas: Impact of the World Health Organization 2021 update. Brain Pathol 32:e13062. https://doi.org/10.1111/bpa.13062
Wiestler B, Capper D, Sill M, Jones DT, Hovestadt V, Sturm D et al (2014) Integrated DNA methylation and copy-number profiling identify three clinically and biologically relevant groups of anaplastic glioma. Acta Neuropathol 128:561–571. https://doi.org/10.1007/s00401-014-1315-x
Wu CC, Jain R, Neto L, Patel S, Poisson LM, Serrano J et al (2019) MR imaging phenotype correlates with extent of genome-wide copy number abundance in IDH mutant gliomas. Neuroradiology 61:1023–1031. https://doi.org/10.1007/s00234-019-02219-8
Xi S, Huang Q, Zeng J (2024) A novel grading system combining histological grade and CDKN2A homozygous and hemizygous deletion to predict prognosis in IDH-mutant astrocytoma. J Neuropathol Exp Neurol. https://doi.org/10.1093/jnen/nlad112
Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH et al (2011) Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell 19:17–30. https://doi.org/10.1016/j.ccr.2010.12.014
Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W et al (2009) IDH1 and IDH2 mutations in gliomas. N Engl J Med 360:765–773. https://doi.org/10.1056/NEJMoa0808710
Ye CJ, Sharpe Z, Alemara S, Mackenzie S, Liu G, Abdallah B et al (2019) Micronuclei and genome chaos: changing the system inheritance. Genes (Basel). https://doi.org/10.3390/genes10050366
Yip S, Miao J, Cahill DP, Iafrate AJ, Aldape K, Nutt CL et al (2009) MSH6 mutations arise in glioblastomas during temozolomide therapy and mediate temozolomide resistance. Clin Cancer Res 15:4622–4629. https://doi.org/10.1158/1078-0432.CCR-08-3012
Yokoda RT, Cobb WS, Yong RL, Crary JF, Viapiano MS, Walker JM et al (2023) CDKN2A mutations have equivalent prognostic significance to homozygous deletion in IDH-mutant astrocytoma. J Neuropathol Exp Neurol 82:845–852. https://doi.org/10.1093/jnen/nlad063
Yong RL, Tsankova NM (2015) Emerging interplay of genetics and epigenetics in gliomas: a new hope for targeted therapy. Semin Pediatr Neurol 22:14–22. https://doi.org/10.1016/j.spen.2014.12.004
Yu K, Hu Y, Wu F, Guo Q, Qian Z, Hu W et al (2020) Surveying brain tumor heterogeneity by single-cell RNA-sequencing of multi-sector biopsies. Natl Sci Rev 7:1306–1318. https://doi.org/10.1093/nsr/nwaa099
Zhao Y, Carter R, Natarajan S, Varn FS, Compton DA, Gawad C et al (2019) Single-cell RNA sequencing reveals the impact of chromosomal instability on glioblastoma cancer stem cells. BMC Med Genomics 12:79. https://doi.org/10.1186/s12920-019-0532-5
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
T.E.R., J.M.W., and N.M.T. wrote the first draft of the manuscript. All authors contributed to the conceptualization, literature review, and editing/revising of this manuscript. All authors have read and approved of the final submitted manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflict of interest to report.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Richardson, T.E., Walker, J.M., Hambardzumyan, D. et al. Genetic and epigenetic instability as an underlying driver of progression and aggressive behavior in IDH-mutant astrocytoma. Acta Neuropathol 148, 5 (2024). https://doi.org/10.1007/s00401-024-02761-7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00401-024-02761-7