
Abstract
Glioneuronal tumors are a heterogenous group of CNS neoplasms that can be challenging to accurately diagnose. Molecular methods are highly useful in classifying these tumors—distinguishing precise classes from their histological mimics and identifying previously unrecognized types of tumors. Using an unsupervised visualization approach of DNA methylation data, we identified a novel group of tumors (n = 20) that formed a cluster separate from all established CNS tumor types. Molecular analyses revealed ATRX alterations (in 16/16 cases by DNA sequencing and/or immunohistochemistry) as well as potentially targetable gene fusions involving receptor tyrosine-kinases (RTK; mostly NTRK1-3) in all of these tumors (16/16; 100%). In addition, copy number profiling showed homozygous deletions of CDKN2A/B in 55% of cases. Histological and immunohistochemical investigations revealed glioneuronal tumors with isomorphic, round and often condensed nuclei, perinuclear clearing, high mitotic activity and microvascular proliferation. Tumors were mainly located supratentorially (84%) and occurred in patients with a median age of 19 years. Survival data were limited (n = 18) but point towards a more aggressive biology as compared to other glioneuronal tumors (median progression-free survival 12.5 months). Given their molecular characteristics in addition to anaplastic features, we suggest the term glioneuronal tumor with ATRX alteration, kinase fusion and anaplastic features (GTAKA) to describe these tumors. In summary, our findings highlight a novel type of glioneuronal tumor driven by different RTK fusions accompanied by recurrent alterations in ATRX and homozygous deletions of CDKN2A/B. Targeted approaches such as NTRK inhibition might represent a therapeutic option for patients suffering from these tumors.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Glioneuronal tumors represent a histologically and molecularly heterogeneous group of rare neoplasms of the central nervous system (CNS) [4, 15]. Accurate diagnosis of these tumors is often challenging due to overlapping morphological and immunohistochemical features. The recently published fifth edition of the World Health Organization (WHO) classification of CNS tumors comprises 15 different types of glioneuronal and neuronal tumors, which correspond most frequently to WHO grade 1 or 2 [15].
In recent years, CNS tumor classification has been greatly influenced by novel technologies such as DNA methylation analysis and led to the identification of numerous previously unrecognized, relatively rare types of brain tumors, some of them with a wide range of histopathological appearances [1, 5, 6, 19, 20, 24]. Furthermore, comprehensive molecular profiling of glioneuronal tumors revealed several ‘druggable’ targets mainly affecting the mitogen-activated protein kinase (MAPK) pathway, including BRAF as well as FGFR and NTRK gene family alterations as found e.g. in ganglioglioma [18], dysembryoplastic neuroepithelial tumor [17], rosette-forming glioneuronal tumor [9, 16, 23], diffuse leptomeningeal glioneuronal tumor [8], extraventricular neurocytoma [16, 26], polymorphous low-grade neuroepithelial tumors of the young (PLNTY) [11], or the recently described group of glioneuronal tumors driven by different kinase-fusions [25, 28].
Here, we describe a molecularly distinct type of glioneuronal tumors using a DNA methylation-based approach in combination with targeted next-generation DNA and RNA sequencing. In addition to molecular characterization, tumors were investigated through an extensive histomorphological and immunohistochemical workup and through retrospective analysis of the available clinical data.
Materials and methods
Collection of tissue samples and clinical data
Case selection of this series was based on unsupervised visualization of genome-wide DNA methylation data (more than 100,000 CNS tumor samples) that revealed an epigenetically distinct cluster of tumors (n = 20). Tumor samples and retrospective clinical data were obtained from multiple international institutes, with a subset of cases initially uploaded to the www.molecularneuropathology.org platform. Collection and analysis of tissue samples and clinical data was performed in accordance with local ethics regulations (ethical vote S-318/2022).
Histology and immunohistochemistry
Histological and immunohistochemical review was performed on a subset of tumor samples with available material for extensive histopathological workup (n = 18). Hematoxylin–eosin (H&E) and reticulin staining was performed according to standard protocols. Immunohistochemistry was carried out on a Ventana BenchMark ULTRA Immunostainer (Ventana Medical Systems, Tucson, AZ, USA). Following Antibodies were used: ATRX (mouse monoclonal, clone BSB-108, dilution 1:2000, Bio SB, Santa Barbara, CA, USA), CD34 (mouse monoclonal, clone QBEnd/10, undiluted, Roche Ventana, Basel, Switzerland), Class III ß-tubulin (mouse monoclonal, clone TU-20, dilution 1:100, Abcam, Cambridge, UK), GFAP (mouse monoclonal, clone GA5, dilution 1:2000, Cell Signaling, Danvers, MA, USA), Ki-67 (mouse monoclonal, clone MIB-1, dilution 1:100, Dako Agilent, Santa Clara, CA, USA), MAP2 (mouse monoclonal, clone HM-2, dilution 1:15,000, Sigma-Aldrich, St. Louis, MO, USA), Neu-N (mouse monoclonal, clone A60, dilution 1:100, Merck, Darmstadt, Germany), NSE (mouse monoclonal, clone MIG-N3, dilution 1:4, Linaris, Dossenheim, Germany), Olig2 (rabbit monoclonal, clone EPR2673, dilution 1:50, Abcam), Synaptophysin (rabbit monoclonal,clone MRQ-40, dilution 1:50, Merck). Class III ß-tubulin, Neu-N, NSE, Olig2, MAP2, Synaptophysin and GFAP immunohistochemistry was performed with the Ventana UltraView DAB IHC Detection Kit. For ATRX, CD34 and Ki-67 the Ventana OptiView DAB IHC Detection Kit (Ventana Medical Systems) were used. Slides were scanned with an Aperio AT2 scanner and visualized with Aperio Image Scope v12.4.3.7001 (Aperio, Leica Biosystems, Deer Park, IL, USA). For one case available material only allowed H&E staining while immunohistochemical information was submitted via the cooperating center. For six cases histological and immunohistochemical review was performed in collaborating institutes and information or scans were digitally evaluated.
DNA methylation profiling and copy number analysis
DNA methylation analysis was performed using formalin-fixed paraffin-embedded (FFPE) or fresh-frozen tissue samples. Raw data was generated at the Department of Neuropathology Heidelberg, the German Cancer Research Center (DKFZ) or at respective collaborating institutes using the Infinium MethylationEPIC (850k) or Infinium HumanMethylation450 (450k) BeadChip array (Illumina, San Diego, CA, USA) according to the manufacturer’s instructions and as previously described [5]. All computational analyses were performed using R version 4.6.1 (R Development Core Team 2020, https://www.R-project.org). O6-methylguanine-DNA methyltransferase (MGMT) -promoter methylation status was evaluated using the method described by Bady et al. [3]. For an unsupervised hierarchical cluster analysis of tumors and reference samples, the 10,000 most variably methylated CpG sites across the dataset according to median absolute deviation were selected. Clustering was done using the Euclidean distance and Ward linkage after adjustment for FFPE versus frozen material. To perform unsupervised non-linear dimension reduction, the remaining probes after standard filtering were used to calculate the 1-variance weighted Pearson correlation between samples. The resulting distance matrix was used as input for t-SNE analysis (t-distributed stochastic neighbor embedding). The following non-default parameters were applied: is distance = T, theta = 0, pca = F, max_iter = 10,000 perplexity = 30.
RNA sequencing and fusion calling
RNA sequencing for the purpose of gene fusion calling was performed in 16/20 (80%) of the cases. For 14 cases RNA sequencing was performed on a NextSeq 500 or NovaSeq 6000 instrument (Illumina) at the Department of Neuropathology Heidelberg as previously described [27]. In addition, one case (#14) was analyzed at the Department of Pathology Copenhagen (Rigshospitalet, Copenhagen University Hospital, Denmark) using an Archer FusionPlex Solid tumor panel (Invitae, Boulder, CO, USA) on an Ion Torrent S5 Prime platform (Thermo Fisher, Waltham, MA, USA). One case (#2) was analyzed at the Department of Pathology at Boston Children’s Hospital and Harvard Medical School (Boston, MA, USA) as previously described [29].
Targeted next-generation DNA sequencing and mutational analysis
DNA sequencing were performed in all cases with available material for DNA extraction (15/20 cases, 75%). The majority of cases (12/14, 86%) were sequenced at the Department of Neuropathology Heidelberg using a capture-based next-generation DNA sequencing approach on a NextSeq 500 or NovaSeq 6000 instrument (Illumina) applying a custom brain tumor panel as previously described [21]. For one case (#14) mutational analysis was performed at the Department of Pathology Copenhagen (Rigshospitalet, Copenhagen University Hospital, Denmark) using an AmpliSeq NGS neuropanel on an Ion Torrent S5 Prime platform (Thermo Fisher). Additionally, one case (#2) was sequenced at the Department of Pathology, Boston Children’s Hospital and Harvard Medical School (Boston, MA, USA) as previously described [29].
Statistical analysis
Survival analysis was performed using GraphPad Prism 9 (GraphPad Software, La Jolla, CA, USA). Data on survival could be retrospectively retrieved for 18 of 20 (90%) patients. Overall survival (OS) and progression-free survival (PFS) probabilities were displayed using Kaplan–Meier method.
Results
Epigenetic profiling reveals a molecularly distinct group of neuroepithelial tumors
By unsupervised visualization of genome-wide DNA methylation data (t-SNE analysis) from more than 100,000 CNS tumor samples, we identified a distinct group of tumors (n = 20) that formed a cluster separate from all established DNA methylation classes. All of these cases occurred in the CNS and originally received various different histological diagnoses as further described below. A focused t-distributed stochastic neighbor embedding (t-SNE) analysis using a reference cohort of 718 neuroepithelial tumors including different glial and glioneuronal tumors confirmed the distinct clustering (Fig. 1). No similarity was seen with the recently described tumor type ‘glioneuronal tumor kinase-fused A’ (Supplementary Fig. 1) [25].

Unsupervised, non-linear t-distributed stochastic neighbor embedding (t-SNE) projection of DNA methylation array profiles from 738 tumor samples. DNA methylation profiling reveals a molecular distinct group of glioneuronal tumors (GTAKA). Reference DNA methylation classes: dysembryoplastic neuroepithelial tumor (DNET), rosette-forming glioneuronal tumor (RGNT), diffuse leptomeningeal glioneuronal tumor subtype 1 (DLGNT_1), diffuse leptomeningeal glioneuronal tumor subtype 2 (DLGNT_2), extraventricular neurocytoma (EVNCYT), papillary glioneuronal tumor (PGNT), ganglioglioma (GG), polymorphous low-grade neuroepithelial tumor of the young (PLNTY), myxoid glioneuronal tumor, PDGFRA-mutant (MYXGNT), diffuse glioneuronal tumor with oligodendroglioma-like features and nuclear clusters (DGONC), glioneuronal tumor kinase-fused (GNT_KinF_A), central neurocytoma (CN), cerebellar liponeurocytoma (LIPN), desmoplastic infantile ganglioglioma / desmoplastic infantile astrocytoma (DIG/DIA), angiocentric glioma MYB/MYBL1-altered (LGG_MYB), pilocytic astrocytoma hemispheric (PA_CORT), pilocytic astrocytoma infratentorial (PA_INF), pilocytic astrocytoma midline (PA_MID), pleomorphic xanthoastrocytoma (PXA), infant-type hemispheric glioma (IHG), high-grade astrocytoma with piloid features (HGAP), subependymal giant cell astrocytoma (SEGA), diffuse midline glioma, H3 K27-altered (DMG_K27), diffuse hemispheric glioma, H3 G34-mutant (DHG_G34), glioblastoma, IDH-wildtype, RTK1 subtype (HGG_RTK1), glioblastoma, IDH-wildtype, RTK2 subtype (HGG_RTK2), diffuse pediatric-type high-grade glioma, RTK1 subtype (HGG_pedRTK1), diffuse pediatric-type high-grade glioma, RTK2 subtype (HGG_pedRTK2) and diffuse pediatric-type high-grade glioma, MYCN subtype (HGG_pedMYCN), supratentorial ependymoma, ZFTA-fused (EPN_ZFTA), supratentorial ependymoma, YAP1-fused (EPN_YAP), supratentorial ependymoma, NOS (EPN_ST), posterior fossa group A ependymoma (EPN_PFA), posterior fossa group B ependymoma (EPN_PFB), myxopapillary enpendymoma (EPN_MPE)
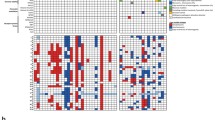
Analysis of copy number variations derived from the DNA methylation data showed a homozygous deletion at chr9p21 including CDKN2A/B in 11 of 20 (55%) cases (Table 1 and Fig. 2a). In addition, a heterozygous deletion of CDKN2A/B was observed in one of the cases (Table 1 and Supplementary Table 1, online resource). Further recurrent copy number alterations included: segmental or whole gains of chr9q in 12/20 (60%) and chr17q in 12/20 (60%) of the cases, as well as a segmental or whole loss of chr19q in 11/20 (55%) cases. In addition, several tumors (7 out of 20) showed indication for a gene fusion based on their copy number profile (Fig. 2a and Supplementary Table 1, online resource). This included three NTRK fusions with focal gain or loss located at the gene locus of the respective fusion partner (Fig. 2b) as well as one case with a KIAA1549::BRAF fusion, two cases with a FGFR1::TACC1 fusion and one case with CLIP2::EGFR fusion (Supplementary Table. 1, online resource). All recurrent copy number variations detected are given in a summary chart in the supplement (Supplementary Table 1 and Supplementary Figs. 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, online resource).

Copy-number profile derived from DNA methylation array data showing a homozygous deletion of CDKN2A/B as well as structural alterations affecting chromosome 9q and 10p around the NTRK2 and KIF5B loci (a). Visualization of the KIF5B::NTRK2 gene fusion confirmed by RNA sequencing (b). Circos plot of the different gene fusions detected in the series (lines link fusion gene partners according to chromosomal location; c)
RNA sequencing identifies oncogenic gene fusions involving different receptor tyrosine-kinases as a frequent event in this novel group of neuroepithelial tumors
RNA sequencing for the purpose of gene fusion detection could be performed in 16 of 20 cases and revealed rearrangements involving different receptor tyrosine-kinases (RTKs) in all samples (Table 1 and Fig. 2c). Eleven of the cases harbored rearrangements involving the NTRK gene family, with the most common fusion partner being NTRK2 (n = 9). In addition, one NTRK1 and one NTRK3 fusion was detected in this series (Supplementary Table 1, online resource). In all fusions NTRK1-3 served as the 3′ partner and the tyrosine kinase domain was conserved as a part of the fusion product (Fig. 2b). NTRK fusion partners were highly variable and included: SPECC1L, BEND5, NACC2, SOX6, KIF5B, CLIP2, BCR, GTF2I and HNRNPU (Fig. 2c). Further oncogenic rearrangements detected by RNA sequencing included an FGFR1::TACC1, CLIP2::EGFR, KIAA1549::BRAF, MYO5A::FER and CTTNBP2::MET fusion (Fig. 2c). Detailed information about fusion partners, breakpoints and indication of the gene fusions based on the copy number variations are given in the supplement (Supplementary Table 1 and Supplementary Figs. 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, online resource).
Mutational analysis identifies recurrent alterations in ATRX accompanied by an immunohistochemical loss
Targeted next-generation DNA sequencing was performed in 15 of 20 cases (75%) and revealed recurrent alterations in ATRX in 12 of 15 cases (80%). The mutational spectrum includes different frameshift, missense and nonsense mutations (Fig. 3a). In three of the cases an ATRX alteration was not detected by sequencing, however, an immunohistochemical loss of nuclear ATRX expression was present in tumor cells (Fig. 3b, c). In two cases nuclear ATRX expression was retained, but inactivating ATRX mutations were detected. All detected ATRX alterations as well as the immunohistochemical status of the single cases are also provided in Supplementary Table 1 (online resource). No additional recurrent alteration was detected by DNA sequencing.

Visualization of the ATRX mutation profile in the investigated cohort was created using the online tool ProteinPaint available at https://proteinpaint.stjude.org/ (a). Immunohistochemical loss of ATRX expression in the tumor cells of case #01 and #18 (b, c). Scale bars 200 µm
Histological and immunohistochemical review reveals glioneuronal tumors with clear cell morphology, condensed nuclei and anaplastic features
Histologically, tumor cells typically showed perinuclear clearing (14/16, 87%), at first sight mimicking oligodendroglioma or neurocytoma (Fig. 4a). Nuclei were mostly isomorphous and round, often with remarkably condensed chromatin (Fig. 4a, b). Multinucleated cells were observed frequently (11/15, 73%; Fig. 4c). Tumor cells were embedded in a fibrillary background matrix. Scattered, possibly entrapped ganglion cells were found in a subset of cases (56%). Anaplastic features like microvascular proliferation (95%), a high mitotic activity with an average mitotic count of up to 3.9 mitoses/mm2 (median 4 mitoses/mm2, range 0.4–6.7 mitoses/mm2, n = 12) and necrosis (32%) were common (Fig. 4b, d, e and Supplementary Table 2, online resource). Tumors were highly vascularized (Fig. 4f). Angiocentric arrangement mimicking ependymoma was seen in single cases (Fig. 4g). Tumor cell density was variable, in some cases showing a biphasic pattern with low-grade areas of moderate cell density (Fig. 4h), low mitotic activity and hyalinized vessels as well as areas of high cell density with numerous mitoses, microvascular proliferation and necrosis. Collectively, cell dense anaplastic areas were more frequently observed. Reticulin staining did not show an appreciable network in the tumor tissue apart from the vessels (Fig. 4i). Eosinophilic granular bodies and/or Rosenthal fibers were absent. Microcalcifications were seen in only one of the tumors. Immunohistochemistry revealed a diffuse positivity for synaptophysin with a weak or moderate cytoplasmic reactivity, while the tumor cell matrix showed a consistently strong staining (Fig. 5a). NeuN staining revealed scattered ganglion cells and only weak or negative staining of most tumor cells (Fig. 5b). Class III ß-tubulin and NSE were consistently positive in the tumors, although sometimes restricted to only a proportion of neoplastic cells (Fig. 5c and d). GFAP staining showed strong positivity of the tumor cell matrix with variable cytoplasmic reactivity between the cases and/or different areas within the same tumor (Fig. 5e). Olig2 was usually strongly expressed (Fig. 5f). MAP2 was positive (Fig. 5g), except in areas suggestive of hypoxia. CD34 expression was restricted to the vessels (Fig. 5h). Ki-67 labeling index was variable between different cases as well as within different areas of the same tumor. Lower grade areas showed only few positive nuclei while anaplastic areas showed a Ki-67 of up to 26% on average (median up to 20%, range 5–65%; Fig. 5i and Supplementary Table 2, online resource). Immunohistochemical ATRX expression was lost in 11 of 13 cases (85%) as described above. Detailed description of histological and immunohistochemical findings can be viewed in the supplement (Supplementary Table 2 and Supplementary Fig. 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, online resource).

Typical histological features of tumors within this series showing tumor cells with inconspicuous nuclei and perinuclear clearing (a, b). Some cases show multinucleated cells (c, indicated by arrows). Cell density is variable (b, h). Tumors are densely vascularized (note the typical pattern, as f and i show two different cases). Microvascular proliferation is frequently observed (b, e). Necrosis is present in a subset of cases (d). An angiocentric arrangement was seen in single cases (g). Scale bars 200 µm

Immunohistochemistry reveals diffuse positivity for synaptophysin with a weak or moderate cytoplasmic reactivity, while the tumor cell matrix shows a consistently strong staining (a). The majority of tumor cells shows negative or weak NeuN staining, although scattered positive cells are frequently present (b). Class III ß-tubulin (c) and NSE (d) are positive in the tumors, although sometimes restricted to a proportion of neoplastic cells. GFAP staining shows strong positivity of the tumor cell matrix with variable cytoplasmic reactivity between the cases and/or different areas within the same tumor (e). Olig2 staining is usually strongly positive (f). MAP2 staining is positive in all tumors (g). CD34 expression is restricted to the vessels (h). Ki-67 labeling index varies between the different cases, here showing a moderate proliferative activity (f). Scale bars 200 µm
Clinical data indicate aggressive tumors that typically occur in children or young adults
Analysis of available clinical information revealed a mean age of the patients at the time of diagnosis of 28.8 years (median age: 19 years, range: 4–76 years, n = 20). Notably, 75% of patients were younger than 34 years at the time of diagnosis (Fig. 6a). The cohort consisted of 12 female patients and 8 male patients, resulting in a sex ratio of 2:3 (male:female; Fig. 6b; not significant). Data on tumor location was available in 19 of 20 of the cases (Fig. 6c). While 18 of 19 tumors occurred in the brain (95%), one showed a spinal location. Most tumors (84%) were found in the supratentorial compartment with only two cases occurring infratentorially (11%). Initial diagnosis of the tumors was very variable with some cases having a descriptive diagnosis (Supplementary Table 1, online resource). Most frequent tumor category was high-grade glioma (n = 8) including glioblastoma (n = 4) and anaplastic oligodendroglioma (n = 2). A proportion of tumors were described as ependymoma or clear cell ependymoma (n = 4). Only a subset of cases was diagnosed as glioneuronal tumor (n = 3).

Clinical features of the investigated cohort. Age at diagnosis of the patients with a median age of 19 years (a), patient sex distribution (b) and distribution of tumor location (c). Progression-free (d) and overall survival (e) of the 18 patients from the investigated cohort for whom follow-up data were available. MRI (axial, T2) performed 12 months after surgery showing a small enhancing nodule along the cavity of resection (f). No sign of progression after 37 months of Larotrectinib (axial, T2; g)
Survival data was available for 18 of 20 patients. 15 patients experienced tumor recurrence after an average time of 22.9 months (median: 12.5, range: 2–53). 11 of 18 patients (61%) were alive at last follow-up with a mean follow-up time of 57.4 months (median: 33, range: 11–228). Seven patients (39%) have deceased with a mean survival time of 59.3 months (median: 30.5, range: 17–228; Fig. 6d, e).
From the available clinical data only one patient is known who received targeted NTRK inhibition (larotrectinib) after first recurrence, with no sign of progression on MRI after 37 months of ongoing therapy (Fig. 6f, g). Clinical data are summarized in the supplement (Supplementary Table 1, online resource).
Discussion
Here, we describe a novel rare type of tumor that is characterized by a distinct epigenetic pattern and recurrent gene fusions mainly involving the NTRK gene family. ATRX alterations as well as typical morphological characteristics of isomorphic tumor cells with condensed nuclei and anaplastic features are additional unifying pattern of tumors within this group.
A first key finding of this study relates to the high frequency of oncogenic fusions detected in this series. While all tumors analyzed harbored a rearrangement involving different receptor tyrosine-kinases, most cases were found to show structural variants targeting NTRK1-3. In particular NTRK2 was found with numerous different fusion partners. Contrary to previous reports [29], rearrangements involving NTRK2 were not seen predominantly in the pediatric setting. In line with previous findings, all NTRK gene family fusions involved the 5′ end of the fusion partner and the 3′ end of NTRK preserving the tyrosine kinase domain resulting in constitutive activation of TRK signaling which ultimately leads to tumor proliferation and resistance to apoptosis [7]. In addition to NTRK1-3, all remaining cases also showed potentially targetable gene fusions mainly affecting the MAPK pathway. This offers therapeutic opportunities for targeted inhibition. In particular NTRK inhibition has proven to be mostly well tolerated and effective, although data on primary CNS tumors is still limited in comparison to other tumor types [2, 13]. Based on these findings, larotrectinib and entrectinib have recently received entity-agnostic FDA approval for NTRK fusion-positive tumors. In our series only one patient received targeted NTRK inhibition (larotrectinib) at first recurrence, with no signs of progression by MRI after 37 months of ongoing therapy.
Another important finding of this study was the high number of ATRX alterations detected in this series, which not only expands the spectrum of tumor types showing a recurrent loss of ATRX expression but also highlights a valuable diagnostic marker in particular in comparison to glioneuronal tumors such as extraventricular neurocytoma, diffuse leptomeningeal glioneuronal tumor or the recently described group of glioneuronal tumors driven by different kinase-fusions [25, 28]. ATRX is recurrently mutated in IDH-mutant astrocytoma [14], H3 K27-altered diffuse midline glioma [22], H3 G34-mutant diffuse hemispheric glioma [12] and high-grade astrocytoma with piloid features (HGAP) [20]. In particular HGAP would be one of the most probable differential diagnoses of this new group of tumors in terms of histopathology and molecular profile. These tumors, in addition to MAPK alterations, are usually characterized by anaplastic features and frequently harbor homozygous deletions of CDKN2A/B as well as loss of ATRX expression [20]. However, piloid features such as eosinophilic granular bodies and/or Rosenthal fibers typically found in HGAP were absent in the present series. In addition, the high frequency of gene fusions in particular rearrangements involving the NTRK gene family as observed in this study is not very typical for HGAPs. Further differences apply to the tumor location. While HGAP is mainly located infratentorially (74%), tumors within our cohort were found most frequently in the supratentorial compartment [20]. Further differential diagnoses include infant-type hemispheric glioma and glioneuronal tumors driven by different kinase-fusions that are characterized by the presence of RTK fusions as well including the NTRK family gene [6, 10, 25]. However, these tumors typically arise in early childhood / in a pediatric setting and do not show ATRX alterations or homozygous deletions of CDKN2A/B [6, 10, 25, 28].
An important issue raised in the context of the histopathological and molecular data generated here is where these tumors fit best into the current WHO CNS tumor taxonomy. Given the heterogeneous morphology of the tumors with a wide range of original diagnoses it seems difficult to classify them at first sight. However, since co-expression of different glial and neuronal markers have been characteristic for tumors within this series, it seems reasonable to classify them as ‘glioneuronal’, which also fits to the morphological appearance of the tumors.
While most glioneuronal tumors show a relatively benign clinical behavior and can often be cured by surgery when amenable to complete resection [4, 15], tumors within this novel group are characterized by a more aggressive biology. This is underlined by the clinical course (although follow-up data are limited), the anaplastic features as well as a high frequency of homozygous deletions of CDKN2A/B in these tumors. Future studies are needed to give more reliable prognostic information.
In summary, the identification of this novel type of glioneuronal tumor emphasizes once again the great benefit of molecular characterization in CNS tumor diagnostics and treatment. The high frequency of targetable fusion events found in theses tumors provides further significant opportunities for patient care. Given their molecular characteristics in addition to anaplastic features, we suggest the term glioneuronal tumor with ATRX alteration, kinase fusion and anaplastic features (GTAKA) to describe these tumors.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Alhalabi KT, Stichel D, Sievers P, Peterziel H, Sommerkamp AC, Sturm D et al (2021) PATZ1 fusions define a novel molecularly distinct neuroepithelial tumor entity with a broad histological spectrum. Acta Neuropathol 142:841–857. https://doi.org/10.1007/s00401-021-02354-8
Alvarez-Breckenridge C, Miller JJ, Nayyar N, Gill CM, Kaneb A, D’Andrea M et al (2017) Clinical and radiographic response following targeting of BCAN-NTRK1 fusion in glioneuronal tumor. NPJ Precis Oncol 1:5. https://doi.org/10.1038/s41698-017-0009-y
Bady P, Sciuscio D, Diserens AC, Bloch J, van den Bent MJ, Marosi C et al (2012) MGMT methylation analysis of glioblastoma on the Infinium methylation BeadChip identifies two distinct CpG regions associated with gene silencing and outcome, yielding a prediction model for comparisons across datasets, tumor grades, and CIMP-status. Acta Neuropathol 124:547–560. https://doi.org/10.1007/s00401-012-1016-2
Bale TA, Rosenblum MK (2022) The 2021 WHO classification of tumors of the central nervous system: an update on pediatric low-grade gliomas and glioneuronal tumors. Brain Pathol 32:e13060. https://doi.org/10.1111/bpa.13060
Capper D, Jones DTW, Sill M, Hovestadt V, Schrimpf D, Sturm D et al (2018) DNA methylation-based classification of central nervous system tumours. Nature 555:469–474. https://doi.org/10.1038/nature26000
Clarke M, Mackay A, Ismer B, Pickles JC, Tatevossian RG, Newman S et al (2020) Infant high-grade gliomas comprise multiple subgroups characterized by novel targetable gene fusions and favorable outcomes. Cancer Discov 10:942–963. https://doi.org/10.1158/2159-8290.CD-19-1030
Cocco E, Scaltriti M, Drilon A (2018) NTRK fusion-positive cancers and TRK inhibitor therapy. Nat Rev Clin Oncol 15:731–747. https://doi.org/10.1038/s41571-018-0113-0
Deng MY, Sill M, Chiang J, Schittenhelm J, Ebinger M, Schuhmann MU et al (2018) Molecularly defined diffuse leptomeningeal glioneuronal tumor (DLGNT) comprises two subgroups with distinct clinical and genetic features. Acta Neuropathol 136:239–253. https://doi.org/10.1007/s00401-018-1865-4
Gessi M, Moneim YA, Hammes J, Goschzik T, Scholz M, Denkhaus D et al (2014) FGFR1 mutations in Rosette-forming glioneuronal tumors of the fourth ventricle. J Neuropathol Exp Neurol 73:580–584. https://doi.org/10.1097/NEN.0000000000000080
Guerreiro Stucklin AS, Ryall S, Fukuoka K, Zapotocky M, Lassaletta A, Li C et al (2019) Alterations in ALK/ROS1/NTRK/MET drive a group of infantile hemispheric gliomas. Nat Commun 10:4343. https://doi.org/10.1038/s41467-019-12187-5
Huse JT, Snuderl M, Jones DT, Brathwaite CD, Altman N, Lavi E et al (2017) Polymorphous low-grade neuroepithelial tumor of the young (PLNTY): an epileptogenic neoplasm with oligodendroglioma-like components, aberrant CD34 expression, and genetic alterations involving the MAP kinase pathway. Acta Neuropathol 133:417–429. https://doi.org/10.1007/s00401-016-1639-9
Korshunov A, Capper D, Reuss D, Schrimpf D, Ryzhova M, Hovestadt V et al (2016) Histologically distinct neuroepithelial tumors with histone 3 G34 mutation are molecularly similar and comprise a single nosologic entity. Acta Neuropathol 131:137–146. https://doi.org/10.1007/s00401-015-1493-1
Laetsch TW, DuBois SG, Mascarenhas L, Turpin B, Federman N, Albert CM et al (2018) Larotrectinib for paediatric solid tumours harbouring NTRK gene fusions: phase 1 results from a multicentre, open-label, phase 1/2 study. Lancet Oncol 19:705–714. https://doi.org/10.1016/S1470-2045(18)30119-0
Liu XY, Gerges N, Korshunov A, Sabha N, Khuong-Quang DA, Fontebasso AM et al (2012) Frequent ATRX mutations and loss of expression in adult diffuse astrocytic tumors carrying IDH1/IDH2 and TP53 mutations. Acta Neuropathol 124:615–625. https://doi.org/10.1007/s00401-012-1031-3
Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D et al (2021) The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro Oncol 23:1231–1251. https://doi.org/10.1093/neuonc/noab106
Lucas CG, Gupta R, Doo P, Lee JC, Cadwell CR, Ramani B et al (2020) Comprehensive analysis of diverse low-grade neuroepithelial tumors with FGFR1 alterations reveals a distinct molecular signature of rosette-forming glioneuronal tumor. Acta Neuropathol Commun 8:151. https://doi.org/10.1186/s40478-020-01027-z
Pages M, Debily MA, Fina F, Jones DTW, Saffroy R, Castel D et al (2022) The genomic landscape of dysembryoplastic neuroepithelial tumours and a comprehensive analysis of recurrent cases. Neuropathol Appl Neurobiol 48:e12834. https://doi.org/10.1111/nan.12834
Pekmezci M, Villanueva-Meyer JE, Goode B, Van Ziffle J, Onodera C, Grenert JP et al (2018) The genetic landscape of ganglioglioma. Acta Neuropathol Commun 6:47. https://doi.org/10.1186/s40478-018-0551-z
Pratt D, Abdullaev Z, Papanicolau-Sengos A, Ketchum C, Panneer Selvam P, Chung HJ et al (2022) High-grade glioma with pleomorphic and pseudopapillary features (HPAP): a proposed type of circumscribed glioma in adults harboring frequent TP53 mutations and recurrent monosomy 13. Acta Neuropathol 143:403–414. https://doi.org/10.1007/s00401-022-02404-9
Reinhardt A, Stichel D, Schrimpf D, Sahm F, Korshunov A, Reuss DE et al (2018) Anaplastic astrocytoma with piloid features, a novel molecular class of IDH wildtype glioma with recurrent MAPK pathway, CDKN2A/B and ATRX alterations. Acta Neuropathol 136:273–291. https://doi.org/10.1007/s00401-018-1837-8
Sahm F, Schrimpf D, Jones DT, Meyer J, Kratz A, Reuss D et al (2016) Next-generation sequencing in routine brain tumor diagnostics enables an integrated diagnosis and identifies actionable targets. Acta Neuropathol 131:903–910. https://doi.org/10.1007/s00401-015-1519-8
Schwartzentruber J, Korshunov A, Liu XY, Jones DT, Pfaff E, Jacob K et al (2012) Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 482:226–231. https://doi.org/10.1038/nature10833
Sievers P, Appay R, Schrimpf D, Stichel D, Reuss DE, Wefers AK et al (2019) Rosette-forming glioneuronal tumors share a distinct DNA methylation profile and mutations in FGFR1, with recurrent co-mutation of PIK3CA and NF1. Acta Neuropathol 138:497–504. https://doi.org/10.1007/s00401-019-02038-4
Sievers P, Henneken SC, Blume C, Sill M, Schrimpf D, Stichel D et al (2021) Recurrent fusions in PLAGL1 define a distinct subset of pediatric-type supratentorial neuroepithelial tumors. Acta Neuropathol 142:827–839. https://doi.org/10.1007/s00401-021-02356-6
Sievers P, Sill M, Schrimpf D, Friedel D, Sturm D, Gardberg M et al (2022) Epigenetic profiling reveals a subset of pediatric-type glioneuronal tumors characterized by oncogenic gene fusions involving several targetable kinases. Acta Neuropathol. https://doi.org/10.1007/s00401-022-02492-7
Sievers P, Stichel D, Schrimpf D, Sahm F, Koelsche C, Reuss DE et al (2018) FGFR1:TACC1 fusion is a frequent event in molecularly defined extraventricular neurocytoma. Acta Neuropathol 136:293–302. https://doi.org/10.1007/s00401-018-1882-3
Stichel D, Schrimpf D, Casalini B, Meyer J, Wefers AK, Sievers P et al (2019) Routine RNA sequencing of formalin-fixed paraffin-embedded specimens in neuropathology diagnostics identifies diagnostically and therapeutically relevant gene fusions. Acta Neuropathol 138:827–835. https://doi.org/10.1007/s00401-019-02039-3
Tauziede-Espariat A, Volodia Dangouloff R, Figarella-Branger D, Uro-Coste E, Nicaise Y, Andre N et al (2022) Clinicopathological and molecular characterization of three cases classified by DNA-methylation profiling as “Glioneuronal Tumors, NOS, Subtype A.” Acta Neuropathol 144:1179–1183. https://doi.org/10.1007/s00401-022-02490-9
Torre M, Vasudevaraja V, Serrano J, DeLorenzo M, Malinowski S, Blandin AF et al (2020) Molecular and clinicopathologic features of gliomas harboring NTRK fusions. Acta Neuropathol Commun 8:107. https://doi.org/10.1186/s40478-020-00980-z
Acknowledgements
We thank U. Vogel, S. Sprengart, L. Dörner, A. Habel and J. Meyer for skillful technical assistance and the microarray unit of the DKFZ Genomics and Proteomics Core Facility for providing Illumina DNA methylation array-related services. This study was supported by the Hertie Network of Excellence in Clinical Neuroscience. P. Sievers is supported by the Else Kröner Fresenius Stiftung and a fellow of the Hertie Academy of Excellence in Clinical Neuroscience. D.T.W. Jones, F. Sahm and S. Pfister gratefully acknowledge support for the Everest Centre for Low-Grade Paediatric Brain Tumour Research (The Brain Tumour Charity (UK), GN-000707). Australian and New Zealand participation was supported by project funding from the Australian Government through Cancer Australia, the Robert Connor Dawes Foundation, Carrie’s Beanies 4 Brain Cancer Foundation and the Victorian Government’s Operational Infrastructure Support Program.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bogumil, H., Sill, M., Schrimpf, D. et al. Glioneuronal tumor with ATRX alteration, kinase fusion and anaplastic features (GTAKA): a molecularly distinct brain tumor type with recurrent NTRK gene fusions. Acta Neuropathol 145, 667–680 (2023). https://doi.org/10.1007/s00401-023-02558-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-023-02558-0