
Abstract
Pediatric spinal low-grade glioma (LGG) and glioneuronal tumours are rare, accounting for less 2.8–5.2% of pediatric LGG. New tumour types frequently found in spinal location such as diffuse leptomeningeal glioneuronal tumours (DLGNT) have been added to the World Health Organization (WHO) classification of tumours of the central nervous system since 2016, but their distinction from others gliomas and particularly from pilocytic astrocytoma (PA) are poorly defined. Most large studies on this subject were published before the era of the molecular diagnosis and did not address the differential diagnosis between PAs and DLGNTs in this peculiar location. Our study retrospectively examined a cohort of 28 children with LGGs and glioneuronal intramedullary tumours using detailed radiological, clinico-pathological and molecular analysis. 25% of spinal PAs were reclassified as DLGNTs. PA and DLGNT are nearly indistinguishable in histopathology or neuroradiology. 83% of spinal DLGNTs presented first without leptomeningeal contrast enhancement. Unsupervised t-distributed stochastic neighbor embedding (t-SNE) analysis of DNA methylation profiles showed that spinal PAs formed a unique methylation cluster distinct from reference midline and posterior fossa PAs, whereas spinal DLGNTs clustered with reference DLGNT cohort. FGFR1 alterations were found in 36% of spinal tumours and were restricted to PAs. Spinal PAs affected significantly younger patients (median age 2 years old) than DLGNTs (median age 8.2 years old). Progression-free survival was similar among the two groups. In this location, histopathology and radiology are of limited interest, but molecular data (methyloma, 1p and FGFR1 status) represent important tools differentiating these two mitogen-activated protein kinase (MAPK) altered tumour types, PA and DLGNT. Thus, these molecular alterations should systematically be explored in this type of tumour in a spinal location.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Primary spinal cord tumours represent 2.8–5.2% of the primary central nervous system (CNS) tumours in children and young adults and are most often diagnosed as pilocytic astrocytomas (PA) [29, 30, 36]. It is very likely from the literature that a subset of previously described PA in spinal location might correspond to diffuse leptomeningeal glioneuronal tumours (DLGNT), as up to 62% of published cases of DLGNTs are located in the spine and are often initially misdiagnosed as PA [10, 12, 17, 20, 21, 26, 27, 32, 35, 38, 39, 44, 46]. Distinguishing a PA from a DLGNT represents a diagnostic challenge, particularly in this spinal location, since these two tumours (respectively, circumscribed astrocytic and disseminated glioneuronal) [48], share a similar clinical presentation (a spinal tumuor associated or not with leptomeningeal dissemination at diagnosis) [1, 5, 20, 28, 32, 35], histopathological (oligodendroglial-like morphology with Olig2 and synaptophysin expressions), and molecular features (Mitogen-Associated Protein Kinase (MAPK) pathway alteration) [48]. 1p deletion was previously reported in 59–100% of DLGNTs among this reported series [12, 34]. From a series of 32 DLGNT with DNA-methylation profiling, the 1p deletion was observed in 100% of cases and thus was considered an essential diagnostic criteria in the 2021 WHO classification [48]. However, very little cytogenetic data (only 3 cases reported with 1p19q codeletion) concerning chromosome 1 are available in existing literature (108 reported spinal PAs) (Supplementary Table 1, online resource) [4, 6, 8, 23, 24, 36, 40]. Moreover, there is no series in the literature comparing radiological features and clinical behaviors of these two histomolecular diagnoses in this spinal location. Thus, we performed an integrated radiological, clinico-pathological and molecular (including DNA methylation profiling) characterization of a series of 28 intramedullary LGGs and glioneuronal tumours from patients under the age of 21 years in order to define relevant radiological, pathological and molecular features that would differentiate between PAs from DLGNTs.
Materials and methods
Subject selection
We retrieved from the GHU-Paris-Neuro Sainte-Anne database 28 paediatric or adolescents and young adult (AYA) patients operated for a primary spinal tumour with a pathological diagnostic of LGG or glioneuronal tumuor (GNT), non-ependymal, IDH and H3 wildtype, between 2008 and 2020. Written informed consent to participate in this study was provided by the participants’ legal guardian. Data collection was approved under the public health declaration number as folows: DC-2020-3840. Clinical data were retrospectively collected for each case. They included sex, age at diagnosis, tumour location, age at first surgery, extent of surgical resection (gross total resection vs. biopsy or partial resection), follow up (including date of first progression radiologically confirmed, treatments, date of last follow up and survival status).
Radiology data
Initially, post-operative and follow-up magnetic resonance imaging (MRI) were retrieved from Necker Enfants-Malades Hospital database for every patient. Imaging protocols were very heterogeneous as they were realized at different times and in different radiology centers. Spinal studies usually included sagittal SE (Spin Echo) T1-weighted, SE T2-weigthed and SE T1-weighted after gadolinium injection images. Cerebral explorations included T2-weighted, T2-FLAIR (fluid-attenuated inversion recovery), diffusion and T1-weighted with gadolinium injection images. All MRIs were centrally reviewed in consensus by one experienced paediatric neuroradiologist (VDR) and by one radiology resident (WY). Tumour location, extension (height according to the number of adjacent vertebrae), presence of nodular leptomeningeal metastases and leptomeningeal thin enhancement were assessed. Radiological progression was recorded as local recurrence, growth of existing metastases or the appearance of new metastases.
FISH analyses
Fluorescence in situ hybridization (FISH) studies were performed on interphase nuclei according to the standard procedures and the manufacturer’s instructions and previously published methods [18]. The ploidy for the chromosomes 1 and 19q as well as copy number of the BRAF gene was assessed using the following centromeric and locus-specific probes: Vysis LS1 1p36/1q25 and LS1 19p13/19q13 FISH Probe Kit (Abbott Molecular, USA), ZytoLight® SPEC BRAF Dual Color Break Apart (Zytovision, Germany). NTRK2 rearrangement was assessed using the following centromeric and locus-specific probe: ZytoLight® SPEC NTRK2 Dual color Break Apart (Zytovision, Germany). Signals were scored in at least 100 non-overlapping intact interphase nuclei per case. Gene copy number per nucleus was recorded as follows: one copy, two copies, copy number gain (3–7). Copy gain and deletion were considered if they were detected in more than 10% and 30% of nuclei, respectively. Results were recorded using a DM600 imaging fluorescence microscope (Leica Biosystems, Richmond, IL) fitted with appropriate filters, a CCD camera, and digital imaging software from Leica (Cytovision, v7.4). Normal cells (endothelial cells or normal glial or neuronal cells from the adjacent parenchyma) were used as positive internal controls for locus-specific probes 1p36/1q25 and 19p13/19q13. For BRAF and NTRK2 break apart probes, a positive case with confirmed fusion was used.
Targeted array SNP genotyping and RNA sequencing data
Molecular data from DNA analysis or RNA sequencing were retrieved from pathological reports. Most experiments were performed according to previously described methods [31, 42]. Briefly, total DNA was extracted with the use of the QIAampDNA mini-kit® (Qiagen Inc., Courtaboeuf, France) according to manufacturer’s protocols. Briefly, tissues were disrupted in lysis buffer. After removing paraffin, the DNA was purified via sequential centrifugation through membrane spin columns. The purity and quantity of DNA were assessed by measuring the absorbance ratio at 260/280 nm with a NanoDrop® Spectrophotometer (LabTech, Palaiseau, France). A brain tumour gene mutation panel was developed using the MassARRAY iPlex technology and MassARRAY online design tools (Agena Bioscience), including the following mutations: IDH1 R132HLSGC; IDH2 R172KTMGWS; BRAF V600EGAKRD; EGFR A289TSVD, D770ins; FGFR1 K656EQ, N546K; H3F3A K27M, G34VRW; HIST1H3B K27MT; HISTH3C K27M, TERT promoter c228at, c250t. The MassARRAY iPlex procedure involves a three-step process consisting of the initial polymerase chain reaction (PCR), inactivation of unincorporated nucleotides by shrimp alkaline phosphatase and a single-base primer extension. Then, the products are nano-dispensed onto a matrix-loaded silicon chip (SpectroChipII, Agena Bioscience, San Diego, California, USA). Finally, the mutations are detected by MALDI–TOF (matrix-assisted laser desorption-ionization–time of flight) mass spectrometry. Data analysis was performed using MassARRAY Typer Analyzer software 4.0.4.20 (Agena Bioscience, San Diego, California, USA), which facilitates visualization of data patterns as well as the raw spectra.
RNA was extracted from two 8-μm-thick formol-fixed paraffin-embedded (FFPE) material sections using the high Pure FFPE RNA Isolation Kit (Roche Diagnostics, Boulogne-Billancourt, France) according to the manufacturer’s instruction. RNA concentrations were measured on a Qubit 4 Fluorometer (Thermo Fisher Scientific, USA) with the Invitrogen Qubit RNA BR Kit (Thermo Fisher Scientific). The percentage of RNA fragments > 200 nt (fragment distribution value; DV200) was evaluated by capillary electrophoresis (Agilent 2100 Bioanalyzer). A DV200 > 30% was required to process the next steps in the analysis. Next-generation sequencing (NGS)-based RNA sequencing was performed using the Illumina TruSight RNA Fusion Panel on a NextSeq550 instrument according to the manufacturer’s instructions (Illumina, San Diego, CA, USA). This targeted RNA sequencing panel covers 507 fusion-associated genes, to assess the most known cancer-related fusions. The TruSight RNA Fusion Panel gene list is available at https://www.illumina.com/content/dam/illumina-marketing/documents/products/gene_lists/gene_list_trusight_rna_fusion_panel.xlsx. A total of 7690 exonic regions are targeted with 21,283 probes. Libraries were prepared according to the Illumina instructions for the TruSight RNA Fusion Panel kit. STAR_v2.6.1a or Bowtie software was used to produce aligned reads in relation to the Homo sapiens reference genome (UCSC hg19) [13]. Manta v1.4.0, TopHat2 and Arriba tools were used for fusion calling [9].
Droplet digital PCR
FGFR1 tyrosine kinase domain (TKD) duplication and hotspot mutations (N546K/K656E) were assessed by previously described droplet digital polymerase chain reaction (ddPCR) [2, 15, 16]. Extracted DNA was quantified using the IDQUANTq kit (ID-Solutions, Grabels, France) with the magnetic induction cycler (Mic) PCR Machine Cycler from Bio Molecular Systems (Göttingen, Germany). After quantification, DNA concentration was adjusted. Eight microliters of DNA comprising 1–5 ng and 14 µl of PCR mix (ready to use) were used for each ddPCR assay. A similar amplification program (50 °C 2 min; 95 °C 10 min; 40 × 95 °C 30 ss–60 °C 1 min; 98 °C 10 min) was used for all targets. The QX200 Droplet Digital PCR System (Bio-Rad, Hercules, California, USA) was used with the AutoDG droplet generator (Bio-Rad). Quantasoft Analysis Pro Software v1.0.596 (Bio-Rad) was used for the qualitative and quantitative analyses. Fractional abundance and copy number variations (CNV) were calculated with the cut-off values and detection thresholds defined by Appay et al. [2]. The cut-off value of positive results for mutant detection were two positive droplets. Detection thresholds were set when the number of positives droplets was strictly above the limit of blank at 95% confidence interval defined for each assay depending on the number of replicates.
DNA methylation profiling data
Nineteen tissue samples (FFPE or freshly frozen if available), for which 500 ng of DNA was extracted were analyzed. DNA was extracted using the QIAamp® DNA Tissue kit or QIAamp® DNA FFPE Tissue Kit (Qiagen, Hilden, Germany) for FFPE samples. DNA from FFPE samples was restored using the Infinium HD FFPE Restore Kit (Illumina, San Diego, California, USA). Bisulfite conversion was performed using the Zymo EZ DNA methylation Kit (Zymo Research, Irvine, California, USA). Standard quality controls confirmed DNA quality/quantity and bisulfite conversion. DNA was then processed using the either Illumina Infinium Methylation EPIC or HumanMethylation450 BeadChip (Illumina, San Diego, California, USA) arrays according to the manufacturer’s instructions. The iScan control software was used to generate raw data files in.idat format, analyzed using GenomeStudio software version v2011 and checked for quality measures according to the manufacturer’s instructions.
Affiliation predictions to CNS tumour classes were obtained from a DNA methylation-based classification of CNS tumours from DKFZ (Deutsches Krebsforschungszentrum—German Cancer Research Center) based on a random forest algorithm available on the web platform www.molecularneuropathology.org. Version v12.5 of the algorithm was used for the present study. The output of this classifier is a score (calibrated score, CS) indicating the resemblance to the reference CNS tumour class in the algorithm. We choose a dimension reduction technique for data visualization: the t-SNE algorithm (t-distributed stochastic neighbor embedding). This non-linear method allows the visualization of data in the form of scatter plots and is well suited for the analysis of raw methylation data. Distinct samples from the same tumour type will usually lead to compact clusters. However, there is no distance threshold that can serve to determine if one sample of interest belongs to one particular cluster; we thus consider that a sample belongs to one class of reference if it overlaps the corresponding cluster or fell in the close vicinity. This method is frequently used in cancer research and to study DNA methylation profiling data in CNS tumours. It was used in the original paper on the classification of central nervous system tumours based on DNA methylation profiles by Capper et al. [7]. Parameters used in this study are the same as those from the DKFZ.Data from EPIC and 450k methylation array were analysed with R language (v4.0.4). The minfi package was used to load idat file and preprocessed with function preprocess.illumina with dye bias correction and background correction. We removed probes located on sex chromosomes, not uniquely mapped to the human reference genome (hg19), probes containing single nucleotide polymorphisms and probes that are not present in both EPIC and 450k methylation array. A batch effect correction was done with removebatchEffect function from limma package, to remove difference between FFPE and frozen samples. The probes were sorted by standard deviation with 10,000 most variable probes were kept for the clustering analysis. These probes were used to calculate the 1-variance weighted Pearson correlation between samples. The distance matrix was used as input for t-distributed stochastic neighbor embedding (t-SNE) from Rtsne package, with the following non-default parameters: theta = 0, pca = F, max_iter = 2500 and perplexity = 20. Visualization was done using ggplot2 packages.
CNV analysis of KIAA1549:BRAF fusion and 1p deletion
KIAA1549:BRAF and 1p deletion were searched by visual inspection of CNV profiles generated by the molecularneuropathology.org platform as described in Stichel et al. [43]. Visual inspection indicated a deletion of 1p if a complete loss of chromosome 1p arm was present. A gain of 7q34 region was indicative of the BRAF duplication and KIAA1549:BRAF fusion.
Histopathological analysis, immunohistochemistry and integrated diagnostic
Samples were stained with hematoxylin-phloxin-saffron (HPS) according to standard protocols. Original slides from all tissue samples were centrally reviewed (ATE, AM) on a Nikon Eclipse E600 (Nikon, Japan) light microscope with Nikon Plan Fluor objectives. Due to often limited examinable surface, mitotic activity was monitored on five high-power fields (HPF, 40 ×/0.75) corresponding to 1.6 mm2. The following primary antibodies were used: Glial Fibrillary Acidic Protein (GFAP) (1:200, clone 6F2, Dako, Glostrup, Denmark), Olig2 (1:3000, clone C-17, Santa Cruz Biotechnology, Dallas, USA), CD34 (1:40, clone QBEnd10, Dako, Glostrup, Denmark), Chromogranin A (1:200, clone LK2 H10, Diagnostic Biosystem, Pleasanton, USA), Neurofilament Protein (1:100, clone 2F 11, Dako, Glostrup, Denmark), Synaptophysin (1:150, clone DAK-SYNAP, Dako, Glostrup, Denmark), Ki-67 (1:200, clone MIB-1, Dako, Glostrup, Denmark). Integrated diagnoses were performed according to the current WHO classification [48].
Statistics
Quantitative variables are expressed by median and compared using Mann–Whitney tests. Qualitative variables are expressed by proportions and percentages and compared using Fischer’s exact test. Those analyses were performed in GraphPad Prism version 7.0a. The Reverse Kaplan–Meier method was used to determine the median follow up [37]. Time to treatment initiation (TTI) was calculated as the duration of time between diagnosis and the initiation of first treatment. Progression-free survival, termed PFS-R, was defined as the time between first treatment start and first radiologically confirmed progression on magnetic resonance imaging (MRI). Overall survival (OS) was calculated as the time from the date of diagnosis to the date of death from any cause or the date of last follow-up. Patient were considered disease-free (DF) if gross total resection was achieved and without any signs of disease recurrence at last follow-up. The appearance of new leptomeningeal contrast enhancement and/or of distant metastases, and/or the growth of the main tumour site, as assessed by MRI, were considered as disease progression (DP). Censored variables were analyzed using the Kaplan–Meier method and comparison were assessed using the log-rank test performed in R version 4.1.2 [33]. All statistical significance was considered at a 5% alpha level. Values between brackets [] are 95% confidence intervals unless stated otherwise.
Results
25% of spinal PAs were reclassified as DLGNTs
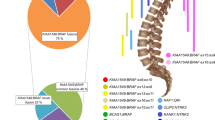
Integrated diagnoses took into account WHO criteria and DNA methylation profiling analysis which includes t-SNE analysis [7, 48]. PA diagnosis was made for cases presenting as either a low-grade piloid astrocytic neoplasm with solitary MAPK alteration and without 1p deletion (13/15 cases), or with a DNA methylation profile clustering with PA on t-SNE analysis despite a low calibrated score (as defined as < 0.9 in Capper et al. [7]) for PA methylation class (MC) (2 infantile cases with FGFR1 without 1p deletion). DLGNT diagnosis was made for oligodendroglioma-like tumours with Olig2 and neuronal immunoreactivity and a MAPK alteration associated with chromosome arm 1p deletion. Spinal tumours were reclassified as follows: (1) DLGNTs accounted for 36% (10/28) of the cohort (initially diagnosed by original pathologists as 7 PA, one ganglioglioma, one oligodendroglioma not otherwise specified (NOS), and one glioneuronal tumour NOS). PAs accounted for 54% (15/28) (initially diagnosed by original pathologists as 13 PAs, one pilomyxoid astrocytoma, one oligodendroglioma). Three cases remained unclassified and, therefore, were diagnosed as spinal pediatric LGG NOS because molecular analysis could not be fully performed or failed technically due to low DNA quality and/or quality (Fig. 1 and Supplementary Table 2, online resource).

Characteristics of primary pediatric primary spinal low grade glioma and glioneuronal tumours. PA diagnosis was made for cases presenting as, either a low-grade piloid astrocytic neoplasm with solitary MAPK alteration and without 1p deletion (13/15 cases), or with a DNA methylation profile clustering with PA on t-SNE analysis despite a low calibrated score (< 0.9) for PA methylation class” and DLGNT diagnosis was made for oligodendroglioma-like tumours with Olig2 and neuronal immunoreactivity and a MAPK alteration associated with chromosome arm 1p deletion. A diagnosis of LGG NOS was made when molecular analysis (particularly 1p status and DNA methylation profiling) could not be fully performed or failed technically due to low DNA quality and/or quality. Copy number alterations were assessed by FISH assays and corroborated by methylation data. Molecular alterations were detected by FISH assays for BRAF rearrangement, FGFR1 alterations were assessed by ddPCR or NGS, RAF1 and NTRK2 fusions were assessed by RNA sequencing. DLGNT subtype 1 refers to a methylation class according to Deng et al. assigned by the version 12.5 of DKFZ classifier. High Grade glioma pediatric type RTK is also a methylation class assigned by the classifier that is further split up into 3 subtypes A, B and C that are currently not fully understood. The methylation class infratentorial pilocytic astrocytoma represents pilocytic astrocytoma that are located in the posterior fossa and that carry MAPK pathway alterations. CE contrast enhancement, DLGNT diffuse leptomeningeal glioneuronal tumour, LGG low-grade glioma, MAPK mitogen-activated protein kinase, MC v12.5 methylation class according to the version 12.5 of the molecularneuropathology.org CNS tumour classifier, N neuropile islands, NOS not otherwise specified, PA pilocytic astrocytoma
MAPK alterations in spinal DLGNTs do not include FGFR1 mutations or internal tandem duplication
Whereas PAs and DLGNTs share a frequent KIAA1549:BRAF fusion (64% (n = 9/14) in PAs and 70% (n = 7/10) in DLGNTs), FGFR1 alterations (3 TKD mutations and 2 internal tandem duplication, ITD) were exclusively observed in PA [33% (n = 5/15)] (Fig. 1, case #30 illustrated in Fig. 2a–e). Among DLGNT, 30% (n = 3/10) had rare fusion transcripts: one QKI:RAF1 fusion and two NTRK2 fusion (NTRK2:AGAP1 and TNS3:NTRK2) (Fig. 1, case #26 illustrated in Fig. 2f–j). 1q gain was observed in 3 cases (Fig. 1 and Supplementary Table 2, online resource). There was a complete correspondence between the visual analysis of the CNV plots obtained from the DKFZ (Deutsches Krebsforschungszentrum—German Cancer Research Center) classifier and the FISH results, both for the chromosome 1 CNVs and BRAF rearrangement (examples are provided in Supplementary Fig. 1, online resource).

Radiological and histopathological aspects of pilocytic astrocytoma and diffuse leptomeningeal glioneuronal tumour. a–e Illustration of case #30, a PA FGFR1 altered with leptomeningeal contrast enhancement at initial diagnosis and neuropil islands: sagittal T2-weighted (a) and T1-weighted after contrast injection (b, c) images showing a heterogeneous cystic and nodular tumour with partial contrast enhancement, and leptomeningeal enhancement along the cauda equina nerve roots, synaptophysin positive neuropil islands (arrow head, d, e) in neurofilament negative areas (zoom box e). f–j Illustration of case #26, a DLGNT with NTRK2:AGAP1 fusion and without leptomeningeal contrast enhancement: sagittal T2-weighted (f) and T1-weighted after contrast injection (g, h) images showing a heterogeneous cystic and nodular tumour with partial contrast enhancement, without leptomeningeal involvement, synaptophysin positive neuropil islands (arrow head i, j). d, e Scale bar = 50 µm; i, j: scale bar = 100 µm. DLGNT diffuse leptomeningeal glioneuronal tumour, PA pilocytic astrocytoma
Spinal PAs are epigenetically distinct from others PAs and DLGNTs
A t-SNE dimensionality reduction analysis was performed on the DNA methylation profiles of 13 spinal PAs and 6 spinal DLGNTs, in comparison with cases from the DFKZ reference cohort (Fig. 3a, b). The 13 spinal PAs cases analyzed by DNA methylation profiling formed a unique cluster distinct from hemispheric pilocytic astrocytomas and gangliogliomas (LGG_PA_GG_ST), posterior fossa pilocytic astrocytomas (LGG_PA_PF) and midline pilocytic astrocytomas (LGG_PA_MID) as well as from DLGNTs. The 6 DLGNT cases clustered together with the DKFZ DLGNT reference cohort, whatever their location and their chromosome arm 1q status. Methylation classes and calibrated scores are detailed in Fig. 1 and Supplementary Table 2, online resource. Of note, one case (#18) with KIAA1549:BRAF and a 1p deletion had a MC of PA_INF with a calibrated score of 0.38 but the t-SNE nonetheless reclassified it as a DLGNT (Figs. 1, 2). Another case (#06), with a FGFR1 alteration, without 1p deletion, and a MC of diffuse pediatric high-grade glioma, RTK1 type, subtype A with a calibrated score of 0.25 was reclassified as a spinal PA by t-SNE analysis (Figs. 1, 3 and Supplementary Fig. 3, online resource).

t-SNE clustering analysis of pediatric primary spinal low grade glioma and glioneuronal tumours against the DKFZ reference dataset. The 13 spinal PAs cases analyzed by DNA methylation profiling formed a unique cluster distinct from hemispheric pilocytic astrocytomas and gangliogliomas (LGG_PA_GG_ST), posterior fossa pilocytic astrocytomas (LGG_PA_PF) and midline pilocytic astrocytomas (LGG_PA_MID) as well as from DLGNTs (a, b). Cases: n = 19. ANA_PA: methylation class anaplastic pilocytic astrocytoma (n = 21); CONTR_ADENOPIT: methylation class control tissue, pituitary gland anterior lobe (n = 9); CONTR_HYPTHAL: methylation class control tissue, hypothalamus (n = 9); CONTR_PONS: methylation class control tissue, pons (n = 12); CONTR_REACT: methylation class control tissue, reactive tumour microenvironment (n = 23); CONTR_WM: methylation class control tissue, white matter (n = 9); DLGNT: methylation class diffuse leptomeningeal glioneuronal tumour (n = 8; 5/8 reference cohort samples had a spinal location, they were centrally reviewed in Heidelberg and initial diagnoses were diffuse leptomeningeal glioneuronal tumour, age at diagnosis ranged from 5 to 39 years old, including 2 adults of 26 and 39 years old); DMG_K27: methylation class diffuse midline glioma H3 K27M mutant (n = 78); EPN_SPINE: methylation class ependymoma, spinal (n = 27); GBM_MID: methylation class glioblastoma, IDH wildtype, subclass midline (n = 14); GBM_MYCN: methylation class glioblastoma, IDH wildtype, subclass MYCN (n = 16); LGG_GG: methylation class low grade glioma, ganglioglioma (n = 21); LGG_PA_GG_ST: methylation class low grade glioma, subclass hemispheric pilocytic astrocytoma and ganglioglioma (n = 24); LGG_PA_MID: methylation class low grade glioma, subclass midline pilocytic astrocytoma (n = 38); LGG_PA_PF: methylation class low grade glioma, subclass posterior fossa pilocytic astrocytoma (n = 114); LGG_RGNT: methylation class low grade glioma, rosette forming glioneuronal tumour (n = 9); PXA: methylation class (anaplastic) pleomorphic xanthoastrocytoma (n = 44); SUBEPN_SPINE: methylation class methylation class subependymoma, spinal (n = 9).
Histopathology alone do not distinguish PAs and DLGNTs in spinal locations
Microcystic changes and piloid component were significantly more frequently observed in PAs (respectively, p = 0.003 and p = 0.005) (Fig. 1, Table 1 and Supplementary Fig. 3, online resource). Neuropil islands were observed in 40% (n = 4/10) of DLGNT (Fig. 1, Table 1, Fig. 2i, j) and only one in PA (7%, p = 0.120) (Table 1, Fig. 2d, e). Rosenthal fibers were present in 73% of PA and 40% of DLGNT; however, no statistical difference was observed (p = 0.122) (Table 1 and Supplementary Fig. 3, online resource). No case displayed eosinophilic granular bodies. The expression of neuronal markers (synaptophysin and chromogranin A) was significantly more often observed in DLGNTs than PAs (respectively, p = 0.009 and p = 0.011) (Fig. 1, Table 1). There was no noticeable difference in terms of vasculature between the two tumour types (Table 1). Glial markers, Olig2 and GFAP, were expressed in both tumour types (Table1). No statistical difference of mitotic count or Ki67 index was demonstrated between PAs and DLGNTs (Table 1). Median mitotic count was 1 [0; 9] mitoses per 1.6 mm2 for PAs and 2.5 [0; 7] mitoses per 1.6 mm2 for DLGNTs. The median Ki67 index of proliferation was 4% [3; 15] in PAs and it was 6% [1; 10] in DLGNTs (Table 1).
Radiological characteristics of spinal PAs and DLGNTs
MRI did not allow distinction between PA and DLGNT, regarding primary tumour characteristics as well as leptomeningeal dissemination (Table 1). Both tumour types were intramedullary cystic and solid with high T2-weighted signal intensity and heterogeneous contrast enhancement (CE). At initial diagnosis, the intramedullary tumour extension was similar in both tumour types and was most often located in the cervico-thoracic region (median number vertebrae involved was 7 in PAs and 6 in DLGNTs, p = 0.693, Fig. 1, Table 1, and Supplementary Table 2, online resource). Metastatic dissemination at the time of initial diagnosis was observed in 26% (n = 4/15) PAs and 44% (n = 4/9) DLGNTs (Fig. 1). At the initial diagnosis, a thin leptomeningeal CE was observed in 1/13 PAs (8%) (case #30, Fig. 3a–c), and in 4/9 DLGNTs (44%). Five DLGNTs did not have a leptomeningeal CE at initial diagnosis (56%) (case #26, Fig. 3f–h). PA and DLGNT demonstrated no statistical difference in terms of metastasis and leptomeningeal CE. Radiological progression was a frequent event in both groups, and was most commonly accounted for a local progression of the disease (Table 1, Supplementary Table 2, online resource). Patients who were initially diagnosed as non-metastatic did not show leptomeningeal dissemination during follow-up (median follow-up was 61 [2.4; 134.2]). Progression of existing metastasis or occurrence of new metastases occurred respectively in 20% (n = 2/10) and 10% (n = 1/10) of DLGNTs (Table 1, Supplementary Table 2, online resource).
Demography and survival analyses
Sex ratio in the entire cohort of 28 cases was 1.5 (male: female), with no significant differences between DLGNTs and PAs. Overall median age at diagnosis was 4 years [1; 18]. 46% (n = 13/28) of the cases were infants (≤ 3 years), 46% (n = 13/28) were children (4–17 years) and 8% (n = 2/28) were AYA (18–25 years). PAs occurred in significantly younger children than DLGNTs: median age at diagnosis was 2 [range 0.7; 13] years for PA and 8.5 [range 1.9; 18] years for DLGNTs (p = 0.016) (Table 1 and Supplementary Table 2, online resource). Overall median follow-up was 61 [range 2–134] months. Two patients died, one from disease progression 14 months after diagnosis (a NOS case) and the second from Sars-Cov2 infection 3 years and 4 months after diagnosis (a DLGNT case). First-line treatment modalities comprised surgery, chemotherapy or both, and were partitioned similarly between the DLGNT and the PA groups (Supplementary Table 3, online resource). The mean TTI was equal for both groups (Fig. 4a). Since the therapeutic approach was similar in both groups, the survival rates were easily compared. The overall median PFS-R was 19 months [9; NA], without significant differences between the PA and DLGNT groups (respectively, 21[4; NA] and 16[9; NA] months) (Fig. 4b). Within the PA group, the 5 FGFR1 altered cases and the 9 KIAA1449:BRAF rearranged cases showed no differences in terms of survival (Fig. 4c).

Survival analyses. No statistical differences could be evidenced between PA and DLGNT for the time to treatment (TTI, a) and the Progression-Free Survival (PFS-R, b) nor between the KIAA1549:BRAF altered PA versus FGFR1 altered PA (PA PFS-R, c)
Discussion
DNA methylation profiling classifier from DKFZ subdivides PAs into three subclasses according to their anatomical location: infratentorial, supratentorial and midline, which suggests a regional specificity of DNA methylation profiles in PA [7]. Taken together, our data build on previous works suggesting that PAs from different CNS locations may arise from region-specific progenitors (Fig. 3) [22, 25, 41, 45]. Although MC midline PAs do not include spinal locations, our study is the first to demonstrate that pediatric primary spinal PAs form a unique cluster, distinct from the DKFZ reference cohort, as previously suggested by Lebrun et al. (Figs. 3, 5) [23].
With extensive characterization of spinal tumours including molecular and radiological analysis, our study shows for the first time that a significant proportion of spinal PAs should be reclassified as DLGNTs. We confirmed that 1p deletion is specific to DLGNTs and never occurred in spinal PAs. This was reinforced by one case in this present study with 1p deletion classified as a PA_INF (calibrated score of 0.38), whereas t-SNE reclassified this case as a DLGNT (Figs. 1 and 2, Supplementary Table 2, online resource). When samples are limited, FISH analysis may be useful in assessing 1p status. All DLGNT cases in this series were classified by the v12.5 of the DKFZ classifier as DLGNT subtype 1 which is enriched in pediatric patients and spinal location [12] (Fig. 1 and Supplementary Table 2, online resource). In the present study, KIAA1549:BRAF is present in spinal PAs and DLGNTs. RAF1 and NTRK2 fusions were seen in spinal DLGNTs, as previously described (Fig. 5) [12]. Interestingly, in other studies, these fusions were also documented in PA of other locations than the spine [47, 49]. FGFR1 TKD alterations were found only in pediatric spinal PAs and not in DLGNTs, suggesting it is an important differential diagnostic criterion [4, 11, 19]. In the literature, FGFR1 mutation has been reported in only one tumour diagnosed as DLGNT, but this diagnosis was doubtful because of the presence of a H3K27M mutation, the lack of 1p status information, and the absence of DNA methylation profiling [14]. All these results confirm that molecular data (particularly 1p and FGFR1 status) are crucial in the diagnosis of spinal tumours, whereas histopathology and radiology are of little help (Fig. 5). This study found that the only significant histological variables were microcystic changes, piloid features, and the expression of neuronal markers, which is in line with previous data [10, 48]. Interestingly, neuropil islands were observed in one PA (Fig. 2d, e).

Summary of PA and DLGNT main characteristics. Del deletion, L leptomeningeal contrast enhancement, pLGG pediatric low grade glioma, yo years old
From a clinical perspective, we showed that spinal PAs affect significantly younger patients (median age 2 years old) than DLGNT (median age 8.5 years old) (Table 1 and Fig. 5). In previous published works [4, 6, 8, 23, 36, 40] where the two groups were not differentiated, the median age for spinal LGG ranged from 6 to 14 years. With the exception of age at the time of diagnosis, spinal PAs and DLGNTs show no clear demographic or clinical differences.
Similar to past publications [1, 5], we confirmed in our study that spinal PAs have frequent leptomeningeal involvement (thin leptomeningeal enhancement or nodular metastases) at initial diagnosis (7% of PAs vs 44% of DLGNTs) (Figs. 2, 5). According to the literature, up to 37% of spinal DLGNTs present with a leptomeningeal involvement [12, 17, 21, 26, 27, 35, 38, 39, 44], whereas in the present study, 83% (n = 5/6) of epigenetically defined DLGNTs presented initially without leptomeningeal contrast enhancement. This was in line with previous studies suggesting that leptomeningeal dissemination should not be considered as an essential diagnostic criterion for DLGNTs [3, 10, 12]. During the evolution of the disease, progression in PAs and DLGNTs was mostly local, and new metastasis appeared in only one DLGNT case (case #23). The therapeutic strategies in this cohort for spinal LGG were identical to the ones applied to encephalic LGG. None of the first-line strategies took into account the molecular biology. In this retrospective study including mainly tumours initially diagnosed as PA, we found no difference in the type of treatments used for spinal PAs versus DLGNTs. Overall survival could not be used to decipher whether one tumour type of spinal LGG better responds to the treatment, because only one patient died of the disease (a DLGNT). We used the progression-free survival as severity marker, which was not influenced by the tumour type or molecular alterations. Comparing these results with tumours in other locations would, however, be hazardous since the impact and the type of surgery can be strikingly different according to the location. The limitations of the study are the small number of patient and its retrospective nature, which is due to the rarity of these tumour types and the rarity of spinal tumoural locations, resulting in insufficient power of this series. Also, the study includes only pediatric cases and does not allow comparison with adult cases (even rarer).
In summary, pediatric spinal PAs represent a distinct methylation class from PAs in other locations of PA, suggesting that they arise from distinct region-specific progenitors. Because histopathological and radiological criteria remain too similar and not sufficiently discriminant, lack of 1p deletion is essential before proposing a diagnosis of PA in spinal location. The presence of a FGFR1 TKD alteration also favors the diagnostic of PA. Finally, the terms “diffuse” and “leptomeningeal” of DLGNT seem to be a less and less adapted terminology for this tumour type that could be better characterized by the co-occurrence of MAPK and 1p deletion.
References
Abel TJ, Chowdhary A, Thapa M, Rutledge JC, Geyer JR, Ojemann J et al (2006) Spinal cord pilocytic astrocytoma with leptomeningeal dissemination to the brain. Case report and review of the literature. J Neurosurg 105:508–514. https://doi.org/10.3171/ped.2006.105.6.508
Appay R, Fina F, Barets D, Gallardo C, Nanni-Metellus I, Scavarda D et al (2020) Multiplexed droplet digital PCR assays for the simultaneous screening of major genetic alterations in tumors of the central nervous system. Front Oncol 10:579762. https://doi.org/10.3389/fonc.2020.579762
Appay R, Pages M, Colin C, Jones DTW, Varlet P, Figarella-Branger D (2020) Diffuse leptomeningeal glioneuronal tumor: a double misnomer? A report of two cases. Acta Neuropathol Commun 8:95. https://doi.org/10.1186/s40478-020-00978-7
Becker AP, Scapulatempo-Neto C, Carloni AC, Paulino A, Sheren J, Aisner DL et al (2015) KIAA1549: BRAF gene fusion and FGFR1 hotspot mutations are prognostic factors in pilocytic astrocytomas. J Neuropathol Exp Neurol 74:743–754. https://doi.org/10.1097/NEN.0000000000000213
Bell E, Kanodia AK, Gunaratne B, Edgar A (2018) Leptomeningeal dissemination of spinal pilocytic astrocytoma: a rare entity. BMJ Case Rep 2018:bcr-2018-226955. https://doi.org/10.1136/bcr-2018-226955
Biczok A, Strübing FL, Eder JM, Egensperger R, Schnell O, Zausinger S et al (2021) Molecular diagnostics helps to identify distinct subgroups of spinal astrocytomas. Acta Neuropathol Commun 9:119. https://doi.org/10.1186/s40478-021-01222-6
Capper D, Jones DTW, Sill M, Hovestadt V, Schrimpf D, Sturm D et al (2018) DNA methylation-based classification of central nervous system tumours. Nature 555:469–474. https://doi.org/10.1038/nature26000
Carey SS, Sadighi Z, Wu S, Chiang J, Robinson GW, Ghazwani Y et al (2019) Evaluating pediatric spinal low-grade gliomas: a 30-year retrospective analysis. J Neurooncol 145:519–529. https://doi.org/10.1007/s11060-019-03319-4
Chen X, Schulz-Trieglaff O, Shaw R, Barnes B, Schlesinger F, Källberg M et al (2016) Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 32:1220–1222. https://doi.org/10.1093/bioinformatics/btv710
Chiang JCH, Harreld JH, Orr BA, Sharma S, Ismail A, Segura AD et al (2017) Low-grade spinal glioneuronal tumors with BRAF gene fusion and 1p deletion but without leptomeningeal dissemination. Acta Neuropathol (Berl) 134:159–162. https://doi.org/10.1007/s00401-017-1728-4
Collins VP, Jones DTW, Giannini C (2015) Pilocytic astrocytoma: pathology, molecular mechanisms and markers. Acta Neuropathol (Berl) 129:775–788. https://doi.org/10.1007/s00401-015-1410-7
Deng MY, Sill M, Chiang J, Schittenhelm J, Ebinger M, Schuhmann MU et al (2018) Molecularly defined diffuse leptomeningeal glioneuronal tumor (DLGNT) comprises two subgroups with distinct clinical and genetic features. Acta Neuropathol (Berl) 136:239–253. https://doi.org/10.1007/s00401-018-1865-4
Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S et al (2013) STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29:15–21. https://doi.org/10.1093/bioinformatics/bts635
Dyson K, Rivera-Zengotita M, Kresak J, Weaver K, Stover B, Fort J et al (2016) FGFR1 N546K and H3F3A K27M mutations in a diffuse leptomeningeal tumour with glial and neuronal markers. Histopathology 69:704. https://doi.org/10.1111/his.12983
Fina F, Barets D, Colin C, Bouvier C, Padovani L, Nanni-Metellus I et al (2017) Droplet digital PCR is a powerful technique to demonstrate frequent FGFR1 duplication in dysembryoplastic neuroepithelial tumors. Oncotarget 8:2104–2113. https://doi.org/10.18632/oncotarget.12881
Fina F, Henaff D, Bresson A, Juline V, Romain A, Carole C et al (2018) 377PD—multiplex digital PCR for the diagnostic of pilocytic astrocytoma and glioneuronal tumors. Ann Oncol. https://doi.org/10.1093/annonc/mdy273.365
Gardiman MP, Fassan M, Orvieto E, D’Avella D, Denaro L, Calderone M et al (2010) Diffuse leptomeningeal glioneuronal tumors: a new entity? Brain Pathol Zurich Switz 20:361–366. https://doi.org/10.1111/j.1750-3639.2009.00285.x
Gareton A, Tauziède-Espariat A, Dangouloff-Ros V, Roux A, Saffroy R, Castel D et al (2020) The histomolecular criteria established for adult anaplastic pilocytic astrocytoma are not applicable to the pediatric population. Acta Neuropathol (Berl) 139:287. https://doi.org/10.1007/s00401-019-02088-8
Jones DTW, Hutter B, Jäger N, Korshunov A, Kool M, Warnatz H-J et al (2013) Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic astrocytoma. Nat Genet 45:927–932. https://doi.org/10.1038/ng.2682
Kang JH, Buckley AF, Nagpal S, Fischbein N, Peters KB (2018) A diffuse leptomeningeal glioneuronal tumor without diffuse leptomeningeal involvement: detailed molecular and clinical characterization. J Neuropathol Exp Neurol 77:751–756. https://doi.org/10.1093/jnen/nly053
Karlowee V, Kolakshyapati M, Amatya VJ, Takayasu T, Nosaka R, Sugiyama K et al (2017) Diffuse leptomeningeal glioneuronal tumor (DLGNT) mimicking Whipple’s disease: a case report and literature review. Childs Nerv Syst ChNS 33:1411–1414. https://doi.org/10.1007/s00381-017-3405-2
Lambert SR, Witt H, Hovestadt V, Zucknick M, Kool M, Pearson DM et al (2013) Differential expression and methylation of brain developmental genes define location-specific subsets of pilocytic astrocytoma. Acta Neuropathol (Berl) 126:291–301. https://doi.org/10.1007/s00401-013-1124-7
Lebrun L, Bizet M, Melendez B, Alexiou B, Absil L, Van Campenhout C et al (2021) Analyses of DNA methylation profiling in the diagnosis of intramedullary astrocytomas. J Neuropathol Exp Neurol 80:663–673. https://doi.org/10.1093/jnen/nlab052
Lebrun L, Meléndez B, Blanchard O, Nève ND, Campenhout CV, Lelotte J et al (2020) Clinical, radiological and molecular characterization of intramedullary astrocytomas. Acta Neuropathol Commun 8:1–14
Lee DY, Gianino SM, Gutmann DH (2012) Innate neural stem cell heterogeneity determines the patterning of glioma formation in children. Cancer Cell 22:131–138. https://doi.org/10.1016/j.ccr.2012.05.036
Manoharan N, Ajuyah P, Senapati A, Wong M, Mullins A, Rodriguez M et al (2021) Diffuse leptomeningeal glioneuronal tumour (DLGNT) in children: the emerging role of genomic analysis. Acta Neuropathol Commun 9:147. https://doi.org/10.1186/s40478-021-01248-w
Nambirajan A, Suri V, Kedia S, Goyal K, Malgulwar PB, Khanna G et al (2018) Paediatric diffuse leptomeningeal tumor with glial and neuronal differentiation harbouring chromosome 1p/19q co-deletion and H3.3 K27M mutation: unusual molecular profile and its therapeutic implications. Brain Tumor Pathol 35:186–191. https://doi.org/10.1007/s10014-018-0325-0
Neutel D, Teodoro T, Coelho M, Pimentel J, Albuquerque L (2014) Spinal cord astrocytoma mimicking multifocal myelitis. J Spinal Cord Med 37:429–431. https://doi.org/10.1179/2045772313Y.0000000187
Ostrom QT, Cioffi G, Waite K, Kruchko C, Barnholtz-Sloan JS (2021) CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2014–2018. Neuro Oncol 23:iii1–iii105. https://doi.org/10.1093/neuonc/noab200
Ostrom QT, Patil N, Cioffi G, Waite K, Kruchko C, Barnholtz-Sloan JS (2020) CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2013–2017. Neuro Oncol 22:iv1–iv96. https://doi.org/10.1093/neuonc/noaa200
Pages M, Beccaria K, Boddaert N, Saffroy R, Besnard A, Castel D et al (2018) Co-occurrence of histone H3 K27M and BRAF V600E mutations in paediatric midline grade I ganglioglioma. Brain Pathol. https://doi.org/10.1111/bpa.12473
Perez-Vega C, Akinduro OO, Cheek BJ, Beier AD (2021) Spinal cord diffuse leptomeningeal glioneuronal tumor presenting without leptomeningeal dissemination. Pediatr Neurosurg 56:563–568. https://doi.org/10.1159/000518802
R Core Team (2016) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. http://www.R-project.org/
Rodriguez FJ, Schniederjan MJ, Nicolaides T, Tihan T, Burger PC, Perry A (2015) High rate of concurrent BRAF-KIAA1549 gene fusion and 1p deletion in disseminated oligodendroglioma-like leptomeningeal neoplasms (DOLN). Acta Neuropathol (Berl) 129:609–610. https://doi.org/10.1007/s00401-015-1400-9
Rossi S, Rodriguez F, Dei Tos A, Di Paola F, Bendini M, Jenkins R et al (2009) Re: Diffuse leptomeningeal glioneuronal tumors: a new entity? Brain Pathol Zurich Switz 19:745. https://doi.org/10.1111/j.1750-3639.2009.00330.x (author reply 746)
Ryall S, Zapotocky M, Fukuoka K, Nobre L, Guerreiro Stucklin A, Bennett J et al (2020) Integrated molecular and clinical analysis of 1,000 pediatric low-grade gliomas. Cancer Cell 37:569-583.e5. https://doi.org/10.1016/j.ccell.2020.03.011
Schemper M, Smith TL (1996) A note on quantifying follow-up in studies of failure time. Control Clin Trials 17:343–346. https://doi.org/10.1016/0197-2456(96)00075-x
Schniederjan MJ, Alghamdi S, Castellano-Sanchez A, Mazewski C, Brahma B, Brat DJ et al (2013) Diffuse leptomeningeal neuroepithelial tumor: 9 pediatric cases with chromosome 1p/19q deletion status and IDH1 (R132H) immunohistochemistry. Am J Surg Pathol 37:763–771. https://doi.org/10.1097/PAS.0b013e31827bf4cc
Schwetye KE, Kansagra AP, McEachern J, Schmidt RE, Gauvain K, Dahiya S (2017) Unusual high-grade features in pediatric diffuse leptomeningeal glioneuronal tumor: comparison with a typical low-grade example. Hum Pathol 70:105–112. https://doi.org/10.1016/j.humpath.2017.06.004
Shankar GM, Lelic N, Gill CM, Thorner AR, Van Hummelen P, Wisoff JH et al (2016) BRAF alteration status and the histone H3F3A gene K27M mutation segregate spinal cord astrocytoma histology. Acta Neuropathol (Berl) 131:147–150. https://doi.org/10.1007/s00401-015-1492-2
Sharma MK, Mansur DB, Reifenberger G, Perry A, Leonard JR, Aldape KD et al (2007) Distinct genetic signatures among pilocytic astrocytomas relate to their brain region origin. Cancer Res 67:890–900. https://doi.org/10.1158/0008-5472.CAN-06-0973
Siegfried A, Rousseau A, Maurage C-A, Pericart S, Nicaise Y, Escudie F et al (2019) EWSR1-PATZ1 gene fusion may define a new glioneuronal tumor entity. Brain Pathol. https://doi.org/10.1111/bpa.12619
Stichel D, Schrimpf D, Sievers P, Reinhardt A, Suwala AK, Sill M et al (2021) Accurate calling of KIAA1549-BRAF fusions from DNA of human brain tumours using methylation array-based copy number and gene panel sequencing data. Neuropathol Appl Neurobiol 47:406–414. https://doi.org/10.1111/nan.12683
Tan GIL, Merchant K, Tan EEK, Low DCY, Ng LP, Seow WT et al (2019) A germline variant of TP53 in paediatric diffuse leptomeningeal glioneuronal tumour. Childs Nerv Syst ChNS 35:1021–1027. https://doi.org/10.1007/s00381-019-04128-w
Tchoghandjian A, Fernandez C, Colin C, El Ayachi I, Voutsinos-Porche B, Fina F et al (2009) Pilocytic astrocytoma of the optic pathway: a tumour deriving from radial glia cells with a specific gene signature. Brain J Neurol 132:1523–1535. https://doi.org/10.1093/brain/awp048
Tiwari S, Yadav T, Pamnani J, Mathew JM, Elhence P, Praneeth K et al (2020) Diffuse leptomeningeal glioneuronal tumor: a unique leptomeningeal tumor entity. World Neurosurg 135:297–300. https://doi.org/10.1016/j.wneu.2019.12.119
Torre M, Vasudevaraja V, Serrano J, DeLorenzo M, Malinowski S, Blandin A-F et al (2020) Molecular and clinicopathologic features of gliomas harboring NTRK fusions. Acta Neuropathol Commun 8:107. https://doi.org/10.1186/s40478-020-00980-z
WHO Classification of Tumours Editorial Board (2021) Central nervous system tumours (WHO classification of tumours series), 5th edn, vol 6. International Agency for Research on Cancer, Lyon (France). https://publications.iarc.fr/601. Accessed 11 Oct 2022
Zhang J, Wu G, Miller CP, Tatevossian RG, Dalton JD, Tang B et al (2013) Whole-genome sequencing identifies genetic alterations in pediatric low-grade gliomas. Nat Genet 45:602–612. https://doi.org/10.1038/ng.2611
Acknowledgements
We thank The RENOCLIP-LOC network (Réseau national de neuro-oncologie clinicoPathologique pour les cancers rares du système nerveux central) funded by the French Institut National du Cancer (INCa) grant (Decision no. 2019-29), the SFCE (Société Française de Lutte contre les Cancers et Leucémies de l’Enfant et de l'Adolescent), The Charity Etoile de Martin and Enfants Cancers Santé (ECS) and Liv et Lumière, for their financial support.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
401_2022_2512_MOESM2_ESM.xlsx
Supplementary file2 Supplementary table 2: Data ADP: alive, with disease progression; AND: alive, no evidence of disease; ASD: alive, stable disease; CT: chemotherapy; DLGNT: diffuse leptomeningeal glioneuronal tumour; DLGNT_1: DLGNT subclass 1; DOC: dead of other cause; DOD; dead of the disease; F: female; GG: ganglioglioma; GNT NOS: glioneuronal tumour not otherwise specified; HGG: high-grade glioma; M: male; MC v12.5: methylation class assigned from DNA methylation-based central nervous system tumour classifier version 12.5; NA: not available; NOS: not otherwise specified; oligo NOS: oligodendroglioma not otherwise specified; oligo. comp. PA: oligodendroglial component from a pilocytic astrocytoma; PA: pilocytic astrocytoma; PA_INF: infratentorial pilocytic astrocytoma; PMA: pilomyxoid astrocytoma; SU: surgery; TKD ITD: tyrosine kinase domain internal tandem duplication; TKD mut: tyrosine kinase domain hotspot mutation; vus: variant of unknown signification (XLSX 19 KB)
401_2022_2512_MOESM3_ESM.docx
Supplementary file3 Supplementary table 3: proportion of cases first treated by chemotherapy (CT), surgery (SU) or both (SU+CT) DLGNT: diffuse leptomeningeal glioneuronal tumour; PA: pilocytic astrocytoma (DOCX 13 KB)
401_2022_2512_MOESM4_ESM.tif
Supplementary file4 Supplementary Figure 1: a: CNV plots from DNA methylation profiling data of a spinal pilocytic astrocytoma showing BRAF rearrangement indicated by a narrow gain of the 7q34 region and no 1p deletion. b: CNV plots from DNA methylation profiling data of a diffuse leptomeningeal glioneuronal tumour showing 1p deletion and by a narrow gain of the 7q34 region indicative of BRAF rearrangement (TIF 528 KB)
401_2022_2512_MOESM5_ESM.tif
Supplementary file5 Supplementary Figure 2: a: Rosenthal fibers observed in a pilocytic astrocytoma (case #16). b: microcystic changes in a pilocytic astrocytoma (case #10). c: oligodendroglial-like component in a diffuse leptomeningeal glioneuronal tumour (case #24). d: neuropil island in a diffuse leptomeningeal glioneuronal tumour (case #26). a, b, c: scale bar = 50µm; d: scale bar = 100 µm (TIF 6280 KB)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Métais, A., Bouchoucha, Y., Kergrohen, T. et al. Pediatric spinal pilocytic astrocytomas form a distinct epigenetic subclass from pilocytic astrocytomas of other locations and diffuse leptomeningeal glioneuronal tumours. Acta Neuropathol 145, 83–95 (2023). https://doi.org/10.1007/s00401-022-02512-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-022-02512-6