
Abstract
In Alzheimer’s disease (AD), where amyloid-β (Aβ) and tau deposits in the brain, hyperexcitation of neuronal networks is an underlying disease mechanism, but its cause remains unclear. Here, we used the Collaborative Cross (CC) forward genetics mouse platform to identify modifier genes of neuronal hyperexcitation. We found LAMP5 as a novel regulator of hyperexcitation in mice, critical for the survival of distinct interneuron populations. Interestingly, synaptic LAMP5 was lost in AD brains and LAMP5 interneurons degenerated in different AD mouse models. Genetic reduction of LAMP5 augmented functional deficits and neuronal network hypersynchronicity in both Aβ- and tau-driven AD mouse models. To this end, our work defines the first specific function of LAMP5 interneurons in neuronal network hyperexcitation in AD and dementia with tau pathology.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Alzheimer’s disease (AD) is the most common neurological disease and an increasing global health problem [7]. Cognitive decline in AD is linked to synaptic and neuronal loss as well as the deposition of Αβ in extracellular plaques and the microtubule-associated protein tau in intraneuronal fibrillar tangles [16, 27, 34]. While Aβ is derived from the transmembrane Aβ-precursor protein (APP) by proteolytic cleavage and accumulates in AD brains due to increased formation and/or reduced clearance [24], tau undergoes aberrant phosphorylation that interferes with its ability to maintain its physiological functions and makes it prone to aggregation forming toxic species [16]. With current therapeutic options being of limited efficacy, therapies targeting neuronal network dysfunction has gained traction [39], though molecular/cellular targets remain poorly defined.
Others and we have previously reported tau-dependent neuronal hyperexcitation as a pathomechanism contributing to functional deficits in Aβ-induced AD mouse models [16, 18, 29, 32]. Furthermore, neuronal hyperexcitation contributes to deficits in tau-dependent dementia models [9, 25, 30]. This includes neuronal network dysfunction with spontaneous non-convulsive seizures, that have been reported in both AD patients and mouse models [3, 17, 46]. However, the cellular events that mediate neuronal network dysfunction remain unclear.
Here, we used forward genetics in mice to identify modifier genes of neuronal hyperexcitation and reveal novel pathways that contribute to neuronal network dysfunction in AD. The combination of both Aβ and tau-dependent transgenic mouse models of AD with validation in human donor brain tissue led us to identify the loss of distinct interneuronal sub-populations likely contributing to neuronal network failure in AD pathogenesis.
Materials and methods
Mice
Animal experiments were approved by the Animal Ethics Committee of Macquarie University. APP23 [36], TAU58 [44], APP/PS1 [2] and Mapt-null (tau–/–) mice [42] were previously described. The Collaborative Cross (CC) platform has been reported in detail before [6]. CC mice were obtained from the Australian Resources Centre (Perth, Australia) and directly used for experiments. Lamp5Δ/Δ mice were generated using CRISPR/Cas9 genome editing [49]. Briefly, cytoplasmic microinjection of Cas9 mRNA (50 ng/µl) and Lamp5 gene-specific guide RNAs (sgRNA1 5′-GTGGCCGGTAGAGTGAGCTA-3′ and sgRNA2 5′-GTTCTTCCGTGAGTAGCGTC-3′; 12.5 ng/µl each) into fertilized C57BL/6 zygotes induced a target deletion of 250 bp comprising the transcriptional start in exon 1 of the Lamp5 locus (Fig. 1c). First-generation (F1) heterozygotes offspring were obtained from a founder with confirmed homozygous deletions around the ATG initiation codon by Sanger sequencing. F1 Lamp5Δ/+ mice were sibling-mated to generate all three genotypes: homozygous knockouts Lamp5Δ/Δ, heterozygous knockouts Lamp5Δ/+ and non-mutant Lamp5+/+ mice (Supplementary Fig. 1, online resource). All lines were maintained on a C57Bl/6 background and identified by polymerase chain reaction (PCR). Oligonucleotide primers used for genotyping are described in Supplementary Table 1 (online resource). Mice had ad libitum access to water and food and were housed on a 12 h day-night cycle.

LAMP5 limits neuronal hyperexcitation. a Latency to develop more severe seizures (left) and ranking of mean seizure severity score (right) reached within 10 min after intraperitoneal administration of 50 mg/kg pentylenetetrazole (PTZ) in 59 collaborative cross (CC; black) and 4 inbred strains (red). Symbol size indicate number of mice that reached the indicated seizure severity score. b QTL mapping using mean seizure severity identified a seizure susceptibility locus on the murine chromosome 2 (top/middle) including contribution of the 8 CC background strains to this locus. c Gene targeting approach to generate Lamp5Δ/Δ mice by deleting 250 bp of the exon1/intron1 junction including the translational start codon (ATG) of the murine Lamp5 locus, as confirmed by genomic sequencing. d Western blotting confirms absence of LAMP5 in Lamp5Δ/Δ mice. e Mean seizure severity (left) and latency to more severe seizures (right) in response to PTZ in Lamp5+/+, Lamp5Δ/+, Lamp5Δ/Δ mice (n = 10–12; *p < 0.05; **p < 0.01; ***p < 0.001; one-way ANOVA). Latency symbol sizes indicate proportion of mice reaching each seizure score
Seizures
Mice were intraperitoneally injected with 50 mg/kg pentylenetetrazole (PTZ, Sigma-Aldrich) in phosphate-buffered saline (PBS) to induce seizures [18]. Animals were video monitored for ten minutes and times to reach individual seizure stage was recorded. Abbreviated Seizure Severity Stage: 0, no seizures; 1, immobility; 2, forelimb clonus; 3, generalized clonus; 4, tonic clonic seizures or worse. Then mice were terminated.
Quantitative trail locus mapping
All data entry was performed on Excel and later processed in R (version 3.2.3) on RStudio (version 0.99.491) platform. The following packages were used for data processing and visualisation: reshape2, ggplot2, sangerseqR, Biostrings, BiocGenerics, parallel, S4Vectors, stats4, IRanges, XVector. Quantitative trait locus (QTL) mapping was performed using a previously published [31] and pre-established web application platform called GeneMiner (http://130.95.9.22/Geniad2/). Different permutations of phenotype data were imputed to screen for meaningful inputs.
Human samples
Human brain tissue sections were obtained from the New South Wales Brain Bank and the Toronto UHN Neurodegenerative Disease brain collection. Clinical and neuropathological details are described in Supplementary Table 2 (online resource). All cases were recruited with informed consent through regional brain donor programs. The brain donor programs hold ethics approval through the University Health Network (CAPCR-ID: 20-5258 and University of Toronto (Nr. 39459), Research Ethics Board and Human Ethics Committees of the South Eastern and Illawarra Area Health Service, the University of New South Wales and the University of Sydney. All procedures complied with the statement on human experimentation issued by the National Health and Medical Research Council of Australia. All cases included in this study received a routine pathological diagnosis according to neuropathological consensus recommendations. Alzheimer’s disease cases had Braak NFT stage ≥ 5, Thal phase ≥ 4 or CERAD scores ≥ 3 and control cases had Braak NFT stage ≤ 1, Thal phase ≤ 1 or CREAD scores ≤ 1. Co-existing phosphorylated TDP-43 or a-synuclein immunoreactivity was only in a subset of AD cases.
Histology
Mice were anaesthetized and transcardially perfused with cold PBS at specified ages. Brains were removed and post-fixed in 4% (w/v) paraformaldehyde (PFA) in PBS overnight. Tissue was processed in an Excelsior tissue processor (Thermo Fisher) followed by paraffin embedding. Brains were sectioned at 4 μm. Brain sections from animals and human patients were stained with primary antibody against LAMP5 (Thermo Fisher), eGFP (Abcam), Aβ (6E10, BioLegend), tau phosphorylated at Ser 214 (pS214, Abcam) and at Ser 422 (pS422, Abcam). For fluorescence staining, sections were incubated with Alexa Fluor conjugated secondary antibodies (Thermo) and 4′,6-diamidine-2′-phenylindole dihydrochloride (DAPI) for 1 h at room temperature. For chromogenic staining, biotinylated secondary antibodies and avidin–biotin complexed to horseradish peroxidase (HRP) from a detection kit (Vector) were applied after primary antibodies. Slides were then incubated with 3,3′-diaminobenzidine (DAB, Vector), followed by a haematoxylin counterstain, and mounted with DPX mounting medium. Aβ plaque load in mouse brains was determined using Thioflavin S staining following a standard protocol. Briefly, deparaffinated and rehydrated brain sections were incubated in pre-filtered 1% (w/v) aqueous Thioflavin-S for 8 min, followed by washing in 80% ethanol and water, before being mounted with aqueous mounting media (Fluoromount-G, Southern Biotech). All staining were either imaged using a BX51 bright field/epifluorescence microscope equipped with a DP70 colour camera (Olympus) or Axio Imager Z1 (Zeiss) or scanned on an Axio Scan.Z1 slide scanner (Zeiss) or Huron TissueScope LE120 slide scanner. Using HuronViewer, five randomly selected areas of interest (AOI, 1000 × 1000 pixel size) from the human putamen and globus pallidus from each case were captured at 21× magnification and exported to GIMP (https://www.gimp.org). The total number of LAMP5 pixels in each AOI was analysed following colour deconvolution and threshold adjustment to remove the background. The total number of pixels in each AOI was averaged across the five AOIs and expressed as % pixel area for each case. For human hippocampus, LAMP5 positive projections were counted manually on five randomly captured images at 20× magnification. For human cortex, LAMP5 positive cells were counted automatically using the Fiji ImageJ image processing software (NIH).
Aβ levels and pathology
Aβ42 levels were examined by ELISA as previously described [18].
Lamp5 promoter reporter
The pLamp5-GFP reporter construct was designed by cloning 1753 bp upstream of the transcriptional start of the murine Lamp5 gene locus that contained the annotated promoter 5′ of an enhanced green fluorescence protein (eGFP) open reading frame in an AAV expression vector. Cloning was done by VectorBuilder (USA). Packaging of AAV vector was performed as described [12]. Titers were determined by quantitative polymerase chain reaction (qPCR). Twenty microliters (1 × 1011 vector genomes) of AAV-pLamp5-GFP was bilaterally intravenously injected into the brains of cryo-anaesthetised neonatal mice as described [23].
Behaviour and memory test
Open Field (OF) testing was done as previously described [20]. Briefly, mice were placed individually in an open arena (40 cm × 40 cm) in dimly lit sound-insulated enclosures and recorded for 10 min. Movements were tracked and analysed using the AnyMaze software (Stölting). Spatial learning and memory formation was assessed in the Morris water maze (MWM) as described before [30, 45]. Briefly, the test equipment consisted of a camera, a Perspex platform (10 cm diameter) and a white circular pool (140 cm diameter and 50 cm height) filled with water containing diluted non-irritant white acrylic-based paint dye. The pool was virtually divided into four equal size quadrants, four different visual cues were placed equidistant of the four quadrants. The platform was hidden in the target quadrant (Q1) and submerged 1 cm below the water. The room was low-lit. Testing was done on eight consecutive days, consist of three tests. Day 1–6 was spatial acquisition with each mouse performing four trials started from a different position in the maze. Individual mice were put into the water facing the wall and allowed 60 s to find the hidden platform where they remained for an additional 60 s. The escape latency was recorded as the time to find the platform. If a mouse failed to reach the platform within 60 s, it was guided to the platform. Probe trials were done on day 7 when each mouse was subjected to two rounds of swim trial (30 s each) without the hidden platform in the maze. The platform entries and time spent in each quadrant were recorded. Day 8 concluded the test with a visual cued test for which the platform was placed back inside, with a visible flag on top, and four trials were performed. The latency to reach the platform was recorded and mice with an average time greater than 20 s were excluded. Movements were tracked and analysed using the AnyMaze software (Stölting).
Electroencephalography
Hippocampal EEG recording was performed in freely moving mice as previously described by us [17]. Briefly, wire EEG electrodes connected to remote telemetric transmitters (DSI) were implanted in the hippocampus (x 2.0, y − 2.0, z − 2.0 from bregma) and a reference electrode was placed above the cerebellum (x 0, y − 6.0, z 0 from bregma) in anaesthetized mice. Electrodes were fixed in place using polyacrylate followed by wound closure. Ten days after surgery, EEGs and activity of animals was recorded using a DSI wireless receiver setup (DSI) with amplifier matrices using the Dataquest A.R.T. recording software at 500 Hz sampling rate [48]. EEG recording was stopped after 48 h. Animals were perfused with ice-cold PBS and brains extracted for biochemical and histological analysis as stated above. Correct implantation of electrodes was confirmed by haematoxylin–eosin staining of serial sections of paraffin-embedded brains. Only recordings from mice with correct placement of electrodes were used for further analysis.
EEG recordings were analysed using the NeuroScore software v3.2.1 (DSI) with an integrated spike detection module. The number of spikes, spike train duration and number of spikes per train were obtained. Movement artifacts during recordings were detected automatically and validated further by manual inspection of recordings to ensure that only artifact-free episodes were used for analysis. Spectral analysis (i.e. analysis of signal power at individual frequencies expressed as square of the fast Fourier transform (FFT) magnitude) of interictal sequences was performed using the integrated FFT spectral analysis function of NeuroScore. Gamma (30–100 Hz) and theta (4–12 Hz) waveform spectral contributions were quantified by area under the curve (AUC) analysis in 4 one-hour-length sequences (pre-filtered to remove artifact and hypersynchronous spike sequences) per recording. Cross-frequency coupling of theta phase and gamma amplitude was conducted using MATLAB (Mathworks) as previously described [14, 40]. Phase-amplitude distributions and modulation indexes were determined from 8 one-minute-length sequences (artifact- and hypersynchronous spike-free).
Western blotting
Western blotting was performed as previously described [19]. Primary antibodies were to LAMP5 (Thermo Fisher) and GAPDH (Sigma). Primary antibodies were detected with species specific HRP-coupled secondary antibodies and visualised with the HRP substrate (Bio-Rad) on a ChemiDoc imager (BioRad).
Statistical analysis
Statistical analysis was done using Graphpad Prism Version 9.2.0. Student’s t-tests were performed for pairwise comparison, one-way analysis of variance (ANOVA) tests was used to compare more than two datasets and two-way ANOVA tests were applied to compare groups across time. Linear regression and correlation analysis were conducted by sum-of-squares minimisation. Survival data were analysed by log-rank Mantel–Cox testing. P values < 0.05 were considered significant. All values are presented as mean ± standard error of the mean (SEM). Details on individual test parameters and n numbers are provided in figure legends.
Results
LAMP5 limits neuronal hyperexcitation in vivo
We have previously shown that induced neuronal hyperexcitation is a reliable proxy for identifying molecular processes involved in tau-dependent network aberrations in AD models [1, 15, 18]. Here, we combined our model of induced brain hyperexcitation [15, 18] with the Collaborative Cross (CC) forward genetics mouse platform [6]. The CC is a large collection of ~ 500 fully genotyped inbred strains originating from intercrossing of 8 founder strains A/J (AJ), C57BL/6J (B6), 129S1/SvImJ (129S1), NOD/ShiLtJ (NOD), NZO/HlLtJ (NZO), CAST/EiK (CAST), PWK/PhJ (PWK) and WSB/EiJ (WSB) (Supplementary Fig. 2, online resource). Single nucleotide polymorphism (SNP) annotation established the relative contribution of each founder to the genome of individual CC strains, allowing rapid identification of phenotype-modifying genes through comparison of effect size and genome makeup of multiple CC lines [26]. Accordingly, we induced excitotoxic seizures with pentylenetetrazol (PTZ; 50 mg/kg i.p.) in at least 10 mice of each of 59 CC strains and the 4 available founder strains, resulting in different severity and latency of progression of seizures between strains (Fig. 1a). Using mean seizure severity for quantitative trait locus (QTL), mapping identified a 135 Mbp susceptibility locus on chromosome 2 with major WSB allele and minor AJ and CAST contributions, while the B6 allele conferred protection (Fig. 1b; Supplementary Table 3; Supplementary Fig. 3, online resource). After examination of coding variants from all 8 founder strains and exclusion of splice sites, only 2 genes remained in the locus of interest; (i) Lamp5 with a 9-nucleotide insertion (dbSNP: rs224715897) resulting in early truncation at aa 22, a likely a loss of function, and (ii) Plcb1 (dbSNP: rs258044618) resulting in a missense mutation that was well-tolerated according to the SIFT algorithm [28]. Neuronal Lamp5 was largely uncharacterised but more recently emerged as a marker of inhibitory interneurons [13]. Using CRISPR/Cas9 genome editing [49], we deleted the transcriptional start in exon 1 of the Lamp5 locus in C57Bl/6 mice (Fig. 1c; Supplementary Fig. 1, online resource), resulting in loss of LAMP5 protein expression in homozygous Lamp5Δ/Δ brains (Fig. 1d). Induction of excitotoxic seizure confirmed a gene-dosage dependent increase in severity and reduced seizure latency in Lamp5Δ/+ and Lamp5Δ/Δ mice, respectively, as compared to Lamp5+/+ wild-type littermate controls (Fig. 1e), consistent with a novel seizure-limiting role of LAMP5.
Loss of LAMP5 interneurons in AD brains and models
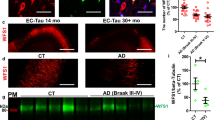
Given our previous experience with translatability of molecular pathways that modulated induced seizures to pathomechanism in AD models [15, 18], we hypothesized that LAMP5 may similarly modulate neuronal hyperexcitation in AD pathogenesis. Recent single-cell sorting studies of mouse brains have identified a non-overlapping subpopulation of inhibitory interneurons expressing LAMP5 but its function(s) is yet-to-be-defined [11, 13, 37]. Cortical LAMP5-positive (LAMP5+) interneurons vary in abundance and distribution between species with mice having less and primates/humans having more [22]. Therefore, we stained frontal cortex and hippocampus sections of human AD, tau-only Frontotemporal lobar degeneration (FTLD-tau), as well as neurologically healthy controls (CTR) lacking neuropathological signs of neurodegenerative diseases and AD, with LAMP5-specific antibodies (Fig. 2; Supplementary Table 2; Supplementary Fig. 4 and 5, online resource). Specificity of antibody staining of neurons, their projections and synaptic terminals was confirmed using Lamp5Δ/Δ brain sections (Fig. 2c). All AD and FTLD-tau cases presented with a marked reduction of LAMP5+ neurons and their projections/synaptic boutons in the frontal cortex as compared to CTR (Fig. 2a). Consistently, LAMP5 containing neuronal projections were significantly reduced in the hippocampus of AD and FTLD-tau cases compared to CTR (Fig. 2b). Similarly, globus pallidus, where LAMP5 is highly expressed in neuronal projections, showed significant LAMP5 reduction in AD (Supplementary Fig. 6, online resource). To understand whether this is a result of the underlying Aβ and/or tau pathology, we determined LAMP5 expression in brains of APP23 mice with transgenic neuronal expression of human mutant APP [36] and APP/PS1 mice co-expressing human mutant APP and presenilin 1 (PS) [2], both forming Aβ, as well as TAU58 mice that express neuronal human mutant tau and form neurofibrillary pathology [44]. Similar to human AD and FTLD-tau, we found an age-dependent significant loss of LAMP5+ neurons in brain sections of APP23, APP/PS1 and TAU58 mice as compared to non-transgenic (non-tg) littermates (Fig. 3a). Interestingly, aged tau-deficient APP23/tau−/− brains showed comparable loss of LAMP5+ neurons as do APP23 mice, suggesting the Aβ induced loss of LAMP5 expression in AD is not tau-dependent. Notably, we previously reported only partially improved survival and recovery of behavioural deficits in APP23/tau−/− mice compared to APP23 alone [15, 18]. LAMP5 loss may explain this incomplete rescue of APP23/tau−/−. It was critical to establish whether reduced LAMP5 signals were due to reduced protein expression or actual loss of LAMP5+ interneuron populations. Therefore, we injected Lamp5Δ/Δ mice and Lamp5+/+ littermates with adeno-associated viruses (AAVs) encoding a Lamp5 promoter-driven green fluorescence protein (pLamp5-GFP) reporter. This labelled a distinct neuronal population in Lamp5+/+ controls consistent with spatial and temporal distribution of cortical and hippocampal LAMP5+ interneurons (Fig. 3b, Supplementary Fig. 7, online resource). At 1 months of age, reporter signals were comparable in Lamp5+/+ and Lamp5Δ/Δ cortex and hippocampus, suggesting normal development of LAMP5+ interneurons in these mice. In contrast, 3 months-old Lamp5Δ/Δ mice presented with marked reduction of pLamp5-GFP reporter-expressing neurons and dystrophic neurites consistent with loss of LAMP5+ interneurons due to LAMP5 reduction. This suggests that a reduction and loss of LAMP5 causes degeneration of LAMP5+ interneurons in cortex and dentate gyrus and hence, loss of their inhibitory functions.

Loss of LAMP5 interneurons in human AD and FTLD. a Representative staining of LAMP5 (brown; arrowheads in CTR) in the frontal cortex from human AD, FTLD-tau and control (CTR) brains. Overview (top row) and higher magnification (broken boxes) of layer I/II (middle row) and layer IV (bottom row) are shown. Insets show individually stained neurons. Quantification of numbers of LAMP5+ neurons (*p < 0.05; ***p < 0.001; Student t test). Scale bar, top row 500 μm, middle and bottom row 50 μm. b Representative staining of LAMP5 (brown; arrowheads in CTR) in the hippocampus (CA4) from human AD, FTLD-tau and control (CTR) brains. Quantification of LAMP5+ projections (**p < 0.01; Student t test). Scale bar, 50 μm. c Sagittal sections of wild-type (Lamp5+/+) and LAMP5 deficient (Lamp5Δ/Δ) mouse brains stained for LAMP5 (brown) confirms antibody specificity. Higher magnification show frontal cortex (broken box). Scale bar, 500 μm

Loss of LAMP5 interneurons in Aβ and tau-expressing mouse models of AD and FTLD-tau. a Representative staining of LAMP5 in 12-month-old of control (non-tg), APP23, APP/PS1, TAU58 and APP23/tau–/– brains. Quantification of numbers of LAMP5+ interneurons in the cortex of non-tg, APP23, APP/PS1, TAU58 and APP23/tau–/– mice (*p < 0.05, Student t test). Scale bar, 100 μm. b Representative imaging of AAV-mediated eGFP reporter activity controlled by the murine Lamp5 promoter in wild-type (Lamp5+/+) and Lamp5Δ/Δ mice at 1 and 3 months of age. Insets show higher magnification of eGFP reporter-positive neurons, including lack of dendritic arborization in 3-month-old Lamp5Δ/Δ brains. Quantification of cells with Lamp5 promoter-driven eGFP reporter expression in the cortex (CTX) and dentate gyrus (DG) of Lamp5+/+ and Lamp5Δ/Δ mice (*p < 0.05; Student t test). Scale bar, 100 μm
LAMP5 reduction accelerates deficits of APP23 mice
To directly address whether LAMP5 reduction is an effect of Aβ neuropathology or contribute to neuronal network aberrations in AD, we crossed APP23 mice onto a Lamp5Δ/Δ background to deplete LAMP5 expression (Fig. 4a). APP23, like other APP transgenic lines, are characterized by a significant premature mortality that has been attributed to early onset neuronal hyperexcitation [15, 18, 32]. Accordingly, male and more so, female APP23/Lamp5+/+ mice showed a significantly reduced survival as compared to non-tg littermates (Fig. 4b, Supplementary Fig. 8, online resource). Survival of APP23/Lamp5Δ/+ mice was comparable to APP23/Lamp5+/+ littermates until 150 days and thereafter showed a trend towards increased mortality. In contrast, APP23/Lamp5Δ/Δ mice presented with earlier and more rapid mortality and poor overall survival, suggesting a gene dosage effect. Survival of Lamp5Δ/Δ and Lamp5Δ/+ mice was not compromised. Aβ formation and plaque load in aged APP23/Lamp5Δ/+ mice were comparable to APP23/Lamp5+/+ littermates (Supplementary Fig. 9, online resource). Learning and memory formation was impaired in APP23/Lamp5Δ/+ compared to APP23/Lamp5+/+ mice at 12 months of age (Fig. 4c, Supplementary Fig. 10, online resource). Memory testing of APP23/Lamp5Δ/Δ mice was prohibited by their high premature mortality rate prior to deficits become overt in APP23 mice [43]. Before memory deficits manifest, 2-month old APP23 mice presented with hyperactivity and slower habituation in the open field task as compared to non-tg littermates (Supplementary Fig. 10, online resource). This was augmented in APP23/Lamp5Δ/Δ mice with significantly increased hyperactivity over the entire test period. Lamp5Δ/Δ and Lamp5Δ/+ did not affect overall activity but delayed habituation as compared to non-tg mice. Consistent with our previous findings [14], hippocampal electroencephalography (EEG) recording showed significantly increased spontaneous discharges and spike trains in APP23/Lamp5+/+ mice as compared to non-tg controls (Fig. 4d). Spectral power analysis showed previously reported increased high frequency power (β < γ) in APP23/Lamp5+/+ mice compared to non-tg controls, which was similarly increased in Lamp5Δ/Δ mice (Supplementary Fig. 11, online resource). Interestingly, LAMP5 depletion alone was sufficient to produce frequent spontaneous discharge events and increased high frequency power in the absence of Aβ (Fig. 4d, Supplementary Fig. 11, online resource). Both spontaneous discharges and spike trains were further increased in APP23/Lamp5Δ/Δ mice. In addition, spectral power at gamma (30–100 Hz) frequency was markedly increased in APP23/Lamp5Δ/+ and more so in APP23/Lamp5Δ/Δ mice as compared to APP23/Lamp5+/+ littermates, revealing profound neuronal network hypersynchronicity upon LAMP5 reduction in APP23 mice. Accordingly, θ/γ cross frequency coupling (CFC), an EEG measure linked to memory in humans [4, 5, 10, 41] which is compromised in APP23 [14] mice, was further disrupted in APP23/Lamp5Δ/+ and APP23/Lamp5Δ/Δ mice compared to APP23/Lamp5+/+ littermates (Fig. 4e). Taken together, LAMP5 reduction accelerated and worsened mortality and functional deficits associated with Aβ in AD mice.

LAMP5 reduction augments deficits in AD mice. a Crossbreeding scheme of APP23 and Lamp5Δ/Δ strains to obtain experimental colony. Color-coding of indicated genotypes apply to all figure panels. b Survival of Lamp5+/+ (n = 39), Lamp5Δ/+ (n = 158), Lamp5Δ/Δ (n = 90), APP23/Lamp5+/+ (n = 40), APP23/Lamp5Δ/+ (n = 118), APP23/Lamp5Δ/Δ (n = 59) mice (**p < 0.01; ****p < 0.0001; ns, not significant; Mantel–Cox test). c Memory testing at 12 months of age in the Morris water maze: example shown of day 6 swim traces per genotype (left), mean latency to find escape platform on individual days of the acquisition trials (middle) and platform zone entries during probe trials without escape platform on day 7 (right) (*p < 0.05; ***p < 0.001; ns, not significant; two-way ANOVA (Tukey post hoc) for acquisition trials; one-way ANOVA for probe trials). d Hippocampal electroencephalography (EEG) recording in freely moving mice at 4.5 months of age: example of EEG traces showing episodes of spontaneous hyperexcitatory discharges (spikes: grey bars; spike trains: green bars) in Lamp5Δ/Δ and APP23 crossings (left), quantification of spontaneous hyperexcitatory discharges (= spikes) per hour (middle) and occurrence of artifact-corrected spike trains during 24 h EEG recordings (right) (*p < 0.05; **p < 0.01; ns, not significant; one-way ANOVA (Tukey post hoc)). e Interictal cross-frequency coupling (CFC) of theta phase and gamma amplitude was disrupted in Lamp5Δ/Δ and APP23 crossings (left). Phase amplitude blots of interictal EEG recording with reduced CFC in APP23/Lamp5Δ/+ and more so in APP23/Lamp5Δ/Δ mice (middle). Modulation index of CFC (right) (*p < 0.05; ***p < 0.001; ns, not significant; one-way ANOVA (Tukey post hoc))
Lamp5 reduction augments impairments in TAU58 mice
Having identified the loss of LAMP5+ interneurons in human FTLD-tau brains and tau transgenic mice (Figs. 2a, 3a), we further assessed if tau pathology develops in LAMP5+ interneurons per se. Consistent with a recent study showing accumulation of phosphorylated tau in GABAergic interneurons in human AD and AD mice brains [50], phosphorylated tau was detected in LAMP5+ interneurons in the cortex of 3-months old TAU58 mice (Fig. 5a). Considering other neuronal populations harbouring tau pathology are not lost in TAU58 mice, LAMP5+ interneurons may be more vulnerable to toxic tau species. To determine the functional consequences of LAMP5 depletion on tau-induced deficits and pathology in the absence of Aβ pathology, we crossed the Lamp5Δ/Δ line with TAU58 mice which express P301S human tau in neurons (Fig. 5b) [44]. TAU58 mice develop tau pathology with progressive functional deficits, including impaired learning and neuronal network dysfunction [30]. LAMP5 depletion did not change tau pathology in TAU58/Lamp5Δ/Δ, TAU58/Lamp5Δ/+ and TAU58/Lamp5+/+ mice (Supplementary Fig. 12, online resource), nor was their overall survival compromised (Supplementary Fig. 13a, online resource). However, learning of TAU58/Lamp5Δ/Δ mice was significantly impaired compared to TAU58/Lamp5Δ/+ and TAU58/Lamp5+/+ mice and non-tau transgenic controls (Fig. 5c, Supplementary Fig. 13, online resource). Consistent with learning deficits, EEG recording of TAU58/Lamp5Δ/+ and more so TAU58/Lamp5Δ/Δ mice revealed increased numbers of spontaneous discharges (Fig. 5d). Furthermore, high frequency spectral power (β/γ) was significantly elevated, while low frequency spectral power (θ) was reduced in TAU58/Lamp5Δ/Δ compared to TAU58/Lamp5+/+ littermates (Supplementary Fig. 14, online resource). Accordingly, CFC was severely compromised in TAU58/Lamp5 Δ/Δ mice (Fig. 5e). Hence, loss of LAMP5 augmented the impact of tau pathology on neuronal network function in FTLD-mutant tau transgenic mice, consistent with the loss of inhibitory interneuron function.

LAMP5 aggravates impairments of P301S tau transgenic mice. a Representative staining of pS214 (green) and LAMP5 (red) in 3-month-old TAU58 brains. Arrows indicate cells with pS214 and LAMP5 colocalization. Scale bar, 50 μm. b Crossbreeding scheme of TAU58 and Lamp5Δ/Δ strains to obtain experimental colony. Color-coding of indicated genotypes apply to all figure panels. c Memory testing at 6 months of age in the Morris water maze: example shown of day 6 swim traces per genotype (left), mean latency to find escape platform on individual days of the acquisition trials (middle) and platform zone entries during probe trials without escape platform on day 7 (right) (*p < 0.05; **p < 0.01; two-way ANOVA (Tukey post hoc) for acquisition trials; one-way ANOVA for probe trials). d Hippocampal electroencephalography (EEG) recording in freely moving mice at 6.5 months of age: example shown of EEG traces showing episodes of spontaneous hyperexcitatory discharges (spikes: grey bars; spike trains: green bars) in Lamp5Δ/Δ and TAU58 crossings, and increase amplitudes in TAU58/Lamp5Δ/+ and TAU58/Lamp5Δ/Δ mice (left), quantification of spontaneous hyperexcitatory discharges (= spikes) per hour (middle) and occurrence of artifact-corrected spike trains during 24 h EEG recordings (right) (*p < 0.05; **p < 0.01; ns, not significant; one-way ANOVA (Tukey post hoc)). (e) Interictal cross-frequency coupling (CFC) of theta phase and gamma amplitude was disrupted in Lamp5Δ/Δ, TAU58/Lamp5Δ/+ and more so in TAU58/Lamp5Δ/Δ mice (left). Phase amplitude blots of interictal EEG recording with reduced CFC in TAU58/Lamp5Δ/+ and more so in TAU58/Lamp5Δ/Δ mice (middle). Modulation index of CFC (right) (***p < 0.001; ns, not significant; one-way ANOVA (Tukey post hoc))
Discussion
In summary, we identified Lamp5 as a novel modifier gene that limits neuronal hyperexcitation and contributes to physiological brain network functions. This is in line with LAMP5 expression in a small subpopulation of hippocampal and cortical inhibitory interneurons [13, 22]. Aβ/Tau pathology-dependent loss of LAMP5+ interneurons in AD mouse models and human AD/FTLD-tau may therefore directly contribute to the neuronal network aberrations and symptoms reported in the disease models and affected humans. While previous LAMP5 studies mainly focus on its role in cancer [8, 35, 47], insights into its function in the brain remain limited, despite its brain-specific expression pattern [21]. Interestingly, Lamp5 knockout mice displayed increased amplitudes of auditory brainstem responses [21], indicative of enhanced neuronal activity in the cochlear system. Lamp5 knockout also has decreased striatal GABAergic neurotransmitter release probability [38]. Although the C. elegans LAMP5 homolog UNC-46 localized to presynaptic terminals of inhibitory GABAergic interneurons [33] similar to LAMP5 in mice [21, 38] and humans (this study), its role of pre-synaptic VGAT delivery was not conserved from C. elegans to mice [21, 38]. No neuronal loss was reported in basal ganglia and brainstem of Lamp5 knockout mice [21, 38], but these studies focused on very young mice in the absence of additional stress, like Aβ and tau in the present study, or investigated the less abundant cortical and hippocampal LAMP5+ neurons. The present study is the first report of a functional role of specific LAMP5+ inhibitory interneurons in disease. Although rare in numbers, the inhibitory function of hippocampal and/or cortical LAMP5+ interneurons appear to be critical for limiting hyperexcitation of executive neuron populations susceptible to Aβ/tau-induced impairment in the pathogenesis of AD and FTLD-tau. This explains, at least in parts, the neuronal network aberration observed in human AD and AD mice. Mechanistically, we found that both Aβ and tau induced loss of LAMP5+ interneurons, engaging either converging or parallel pathways. Whether these specific interneurons or LAMP5 function(s) represent potential therapeutic targets for AD and FTLD-tau remains to be shown.
Data and materials availability
Plasmids and sequences of the pLamp5-GFP reporter are available upon request and will be deposited to Addgene. All data that support the findings in this study are available from the corresponding author upon request.
References
Bi M, Gladbach A, Van Eersel J, Ittner A, Przybyla M, Van Hummel A et al (2017) Tau exacerbates excitotoxic brain damage in an animal model of stroke. Nat Commun 8:473. https://doi.org/10.1038/s41467-017-00618-0
Borchelt DR, Ratovitski T, van Lare J, Lee MK, Gonzales V, Jenkins NA et al (1997) Accelerated amyloid deposition in the brains of transgenic mice coexpressing mutant presenilin 1 and amyloid precursor proteins. Neuron 19:939–945. https://doi.org/10.1016/s0896-6273(00)80974-5
Busche MA, Eichhoff G, Adelsberger H, Abramowski D, Wiederhold KH, Haass C et al (2008) Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer’s disease. Science 321:1686–1689. https://doi.org/10.1126/science.1162844
Buzsaki G, Moser EI (2013) Memory, navigation and theta rhythm in the hippocampal-entorhinal system. Nat Neurosci 16:130–138. https://doi.org/10.1038/nn.3304
Canolty RT, Edwards E, Dalal SS, Soltani M, Nagarajan SS, Kirsch HE et al (2006) High gamma power is phase-locked to theta oscillations in human neocortex. Science 313:1626–1628. https://doi.org/10.1126/science.1128115
Churchill GA, Airey DC, Allayee H, Angel JM, Attie AD, Beatty J et al (2004) The collaborative cross, a community resource for the genetic analysis of complex traits. Nat Genet 36:1133–1137. https://doi.org/10.1038/ng1104-1133
Collaborators GBDD (2019) Global, regional, and national burden of Alzheimer’s disease and other dementias, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol 18:88–106. https://doi.org/10.1016/S1474-4422(18)30403-4
Defays A, David A, de Gassart A, De Angelis RF, Wenger T, Camossetto V et al (2011) BAD-LAMP is a novel biomarker of nonactivated human plasmacytoid dendritic cells. Blood 118:609–617. https://doi.org/10.1182/blood-2010-11-319699
Garcia-Cabrero AM, Guerrero-Lopez R, Giraldez BG, Llorens-Martin M, Avila J, Serratosa JM et al (2013) Hyperexcitability and epileptic seizures in a model of frontotemporal dementia. Neurobiol Dis 58:200–208. https://doi.org/10.1016/j.nbd.2013.06.005
Goutagny R, Jackson J, Williams S (2009) Self-generated theta oscillations in the hippocampus. Nat Neurosci 12:1491–1493. https://doi.org/10.1038/nn.2440
Gouwens NW, Sorensen SA, Baftizadeh F, Budzillo A, Lee BR, Jarsky T et al (2020) Integrated morphoelectric and transcriptomic classification of cortical GABAergic cells. Cell 183(935–953):e919. https://doi.org/10.1016/j.cell.2020.09.057
Harasta AE, Power JM, von Jonquieres G, Karl T, Drucker DJ, Housley GD et al (2015) Septal glucagon-like peptide 1 receptor expression determines suppression of cocaine-induced behavior. Neuropsychopharmacology 40:1969–1978. https://doi.org/10.1038/npp.2015.47
Huang ZJ, Paul A (2019) The diversity of GABAergic neurons and neural communication elements. Nat Rev Neurosci 20:563–572. https://doi.org/10.1038/s41583-019-0195-4
Ittner A, Asih PR, Tan ARP, Prikas E, Bertz J, Stefanoska K et al (2020) Reduction of advanced tau-mediated memory deficits by the MAP kinase p38gamma. Acta Neuropathol 140:279–294. https://doi.org/10.1007/s00401-020-02191-1
Ittner A, Chua SW, Bertz J, Volkerling A, Van Der Hoven J, Gladbach A et al (2016) Site-specific phosphorylation of tau inhibits amyloid-β toxicity in Alzheimer’s mice. Science 354:904–908. https://doi.org/10.1126/science.aah6205
Ittner A, Ittner LM (2018) Dendritic tau in Alzheimer’s disease. Neuron 99:13–27. https://doi.org/10.1016/j.neuron.2018.06.003
Ittner AA, Gladbach A, Bertz J, Suh LS, Ittner LM (2014) p38 MAP kinase-mediated NMDA receptor-dependent suppression of hippocampal hypersynchronicity in a mouse model of Alzheimer inverted question marks disease. Acta Neuropathol Commun 2:149. https://doi.org/10.1186/s40478-014-0149-z
Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J et al (2010) Dendritic function of tau mediates amyloid-β toxicity in Alzheimer’s disease mouse models. Cell 142:387–397. https://doi.org/10.1016/j.cell.2010.06.036
Ittner LM, Koller D, Muff R, Fischer JA, Born W (2005) The N-terminal extracellular domain 23–60 of the calcitonin receptor-like receptor in chimeras with the parathyroid hormone receptor mediates association with receptor activity-modifying protein 1. Biochemistry 44:5749–5754. https://doi.org/10.1021/bi048111o
Ke YD, van Hummel A, Stevens CH, Gladbach A, Ippati S, Bi M et al (2015) Short-term suppression of A315T mutant human TDP-43 expression improves functional deficits in a novel inducible transgenic mouse model of FTLD-TDP and ALS. Acta Neuropathol 130:661–678. https://doi.org/10.1007/s00401-015-1486-0
Koebis M, Urata S, Shinoda Y, Okabe S, Yamasoba T, Nakao K et al (2019) LAMP5 in presynaptic inhibitory terminals in the hindbrain and spinal cord: a role in startle response and auditory processing. Mol Brain 12:20. https://doi.org/10.1186/s13041-019-0437-4
Krienen FM, Goldman M, Zhang Q, del Rosario RCH, Florio M, Machold R et al (2020) Innovations present in the primate interneuron repertoire. Nature 586:262–269. https://doi.org/10.1038/s41586-020-2781-z
Lampe SEG, Kaspar BK, Foust KD (2014) Intravenous injections in neonatal mice. JoVE J Visual Exp. https://doi.org/10.3791/52037
Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC et al (2010) Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science 330:1774. https://doi.org/10.1126/science.1197623
Menkes-Caspi N, Yamin HG, Kellner V, Spires-Jones TL, Cohen D, Stern EA (2015) Pathological tau disrupts ongoing network activity. Neuron 85:959–966. https://doi.org/10.1016/j.neuron.2015.01.025
Morahan G, Balmer L, Monley D (2008) Establishment of “The Gene Mine”: a resource for rapid identification of complex trait genes. Mamm Genome 19:390–393. https://doi.org/10.1007/s00335-008-9134-9
Mucke L, Selkoe DJ (2012) Neurotoxicity of amyloid beta-protein: synaptic and network dysfunction. Cold Spring Harb Perspect Med 2:a006338. https://doi.org/10.1101/cshperspect.a006338
Ng PC, Henikoff S (2003) SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res 31:3812–3814. https://doi.org/10.1093/nar/gkg509
Palop JJ, Chin J, Roberson ED, Wang J, Thwin MT, Bien-Ly N et al (2007) Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer’s disease. Neuron 55:697–711. https://doi.org/10.1016/j.neuron.2007.07.025
Przybyla M, van Eersel J, van Hummel A, van der Hoven J, Sabale M, Harasta A et al (2020) Onset of hippocampal network aberration and memory deficits in P301S tau mice are associated with an early gene signature. Brain 143:1889–1904. https://doi.org/10.1093/brain/awaa133
Ram R, Mehta M, Balmer L, Gatti DM, Morahan G (2014) Rapid identification of major-effect genes using the collaborative cross. Genetics 198:75–86. https://doi.org/10.1534/genetics.114.163014
Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T et al (2007) Reducing endogenous tau ameliorates amyloid ß-induced deficits in an Alzheimer’s disease mouse model. Science 316:750–754. https://doi.org/10.1126/science.1141736
Schuske K, Palfreyman MT, Watanabe S, Jorgensen EM (2007) UNC-46 is required for trafficking of the vesicular GABA transporter. Nat Neurosci 10:846–853. https://doi.org/10.1038/nn1920
Selkoe DJ (2002) Alzheimer’s disease is a synaptic failure. Science 298:789–791. https://doi.org/10.1126/science.1074069
Shi J, Jiang D, Yang S, Sun Y, Wang J, Zhang X et al (2020) Molecular profile reveals immune-associated markers of lymphatic invasion in human colon adenocarcinoma. Int Immunopharmacol 83:106402. https://doi.org/10.1016/j.intimp.2020.106402
Sturchler-Pierrat C, Abramowski D, Duke M, Wiederhold KH, Mistl C, Rothacher S et al (1997) Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc Natl Acad Sci USA 94:13287–13292. https://doi.org/10.1073/pnas.94.24.13287
Tasic B, Yao Z, Graybuck LT, Smith KA, Nguyen TN, Bertagnolli D et al (2018) Shared and distinct transcriptomic cell types across neocortical areas. Nature 563:72–78. https://doi.org/10.1038/s41586-018-0654-5
Tiveron MC, Beurrier C, Ceni C, Andriambao N, Combes A, Koehl M et al (2016) LAMP5 fine-tunes gabaergic synaptic transmission in defined circuits of the mouse brain. PLoS One 11:e0157052. https://doi.org/10.1371/journal.pone.0157052
Toniolo S, Sen A, Husain M (2020) Modulation of brain hyperexcitability: potential new therapeutic approaches in Alzheimer’s disease. Int J Mol Sci 21:9318. https://doi.org/10.3390/ijms21239318
Tort AB, Komorowski R, Eichenbaum H, Kopell N (2010) Measuring phase-amplitude coupling between neuronal oscillations of different frequencies. J Neurophysiol 104:1195–1210. https://doi.org/10.1152/jn.00106.2010
Tort AB, Komorowski RW, Manns JR, Kopell NJ, Eichenbaum H (2009) Theta-gamma coupling increases during the learning of item-context associations. Proc Natl Acad Sci USA 106:20942–20947. https://doi.org/10.1073/pnas.0911331106
Tucker KL, Meyer M, Barde YA (2001) Neurotrophins are required for nerve growth during development. Nat Neurosci 4:29–37. https://doi.org/10.1038/82868
Van Dam D, D’Hooge R, Staufenbiel M, Van Ginneken C, Van Meir F, De Deyn PP (2003) Age-dependent cognitive decline in the APP23 model precedes amyloid deposition. Eur J Neurosci 17:388–396. https://doi.org/10.1046/j.1460-9568.2003.02444.x
Van Eersel J, Stevens CH, Przybyla M, Gladbach A, Stefanoska K, Chan CK et al (2015) Early-onset axonal pathology in a novel P301S-Tau transgenic mouse model of frontotemporal lobar degeneration. Neuropathol Appl Neurobiol 41:906–925. https://doi.org/10.1111/nan.12233
Vorhees CV, Williams MT (2006) Morris water maze: procedures for assessing spatial and related forms of learning and memory. Nat Protoc 1:848–858. https://doi.org/10.1038/nprot.2006.116
Vossel KA, Beagle AJ, Rabinovici GD, Shu H, Lee SE, Naasan G et al (2013) Seizures and epileptiform activity in the early stages of Alzheimer disease. JAMA Neurol 70:1158–1166. https://doi.org/10.1001/jamaneurol.2013.136
Wang WT, Han C, Sun YM, Chen ZH, Fang K, Huang W et al (2019) Activation of the lysosome-associated membrane protein LAMP5 by DOT1L serves as a bodyguard for MLL fusion oncoproteins to evade degradation in leukemia. Clin Cancer Res 25:2795–2808. https://doi.org/10.1158/1078-0432.CCR-18-1474
Weiergraber M, Henry M, Hescheler J, Smyth N, Schneider T (2005) Electrocorticographic and deep intracerebral EEG recording in mice using a telemetry system. Brain Res Brain Res Protoc 14:154–164. https://doi.org/10.1016/j.brainresprot.2004.12.006
Yang H, Wang H, Jaenisch R (2014) Generating genetically modified mice using CRISPR/Cas-mediated genome engineering. Nat Protoc 9:1956–1968. https://doi.org/10.1038/nprot.2014.134
Zheng J, Li H-L, Tian N, Liu F, Wang L, Yin Y et al (2020) Interneuron accumulation of phosphorylated tau impairs adult hippocampal neurogenesis by suppressing GABAergic transmission. Cell Stem Cell 26:331–345. https://doi.org/10.1016/j.stem.2019.12.015
Acknowledgements
The authors would like to thank Ms Yijun Lin and Dr Emilija Robinson for AAV production, Ms Andrea Chan for help with histology, and Ms Jess Spathos and Mr Troy butler for animal husbandry.
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions. This work was supported by funding from the National Health & Medical Research Council (#1095215, #1123564, #1132524, #1136241, #1143848, #1141789, #2001572), the Australian Research Council (#DP170100781, #DP210101957), the Ainsworth Medical Research Innovation Fund and Macquarie University.
Author information
Authors and Affiliations
Contributions
MB, GM and LMI conceived the study. YD, MB, YDK and LMI designed experiments. YD, MB, FD, SLF, GC, JvdH, AvH, AFF, SL and IM did experiments. YD, MB, SLF, GGK, GM, YDK and LMI analysed data. TK, GGK and GM provided essential reagents. YDK and LMI obtained funding for the study. YD, MB, YDK and LMI wrote the first manuscript draft. All authors edited the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no relevant financial or non-financial interests to disclose.
Ethics declaration
Approval was obtained from the ethics committee of Macquarie University. The procedures used in this study adhere to the tenets of the Declaration of Helsinki.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Deng, Y., Bi, M., Delerue, F. et al. Loss of LAMP5 interneurons drives neuronal network dysfunction in Alzheimer’s disease. Acta Neuropathol 144, 637–650 (2022). https://doi.org/10.1007/s00401-022-02457-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-022-02457-w