
Abstract
Pelizaeus–Merzbacher disease (PMD) is an untreatable and fatal leukodystrophy. In a model of PMD with perturbed blood–brain barrier integrity, cholesterol supplementation promotes myelin membrane growth. Here, we show that in contrast to the mouse model, dietary cholesterol in two PMD patients did not lead to a major advancement of hypomyelination, potentially because the intact blood–brain barrier precludes its entry into the CNS. We therefore turned to a PMD mouse model with preserved blood–brain barrier integrity and show that a high-fat/low-carbohydrate ketogenic diet restored oligodendrocyte integrity and increased CNS myelination. This dietary intervention also ameliorated axonal degeneration and normalized motor functions. Moreover, in a paradigm of adult remyelination, ketogenic diet facilitated repair and attenuated axon damage. We suggest that a therapy with lipids such as ketone bodies, that readily enter the brain, can circumvent the requirement of a disrupted blood–brain barrier in the treatment of myelin disease.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Primary defects in lipid metabolism are associated with myelin disease [13] and vice versa, a growing number of neurodegenerative diseases [1, 2, 14, 68], including the hereditary leukodystrophy Pelizaeus–Merzbacher disease (PMD), is associated with a perturbed brain lipid metabolism [29, 57, 63, 73]. In the nervous system, the majority of lipids is found in myelin sheaths, a multilayered stack of membranes synthesized by oligodendrocytes. Myelinating oligodendrocytes contribute to axon integrity by providing trophic support and electrical insulation for impulse propagation [23, 33]. Myelination failure is hence associated with axon damage and dysfunction leading to deficits in cognition and motor abilities.
PMD is most frequently caused by a duplication of the X-linked myelin gene PLP1 (proteolipid protein 1) [11, 24, 40, 59, 74]. Pathological hypomyelination and symptoms such as nystagmus, hypotonia, spasticity, ataxia and retarded cognitive development are characteristic of PMD. At present, PMD lacks any therapeutic option and the symptomatic and supportive medications are directed at seizure or spasticity control. In PMD oligodendrocytes, overexpressed PLP accumulates together with cholesterol which impairs the intracellular transport of proteins and lipids to the growing myelin sheath [31, 57, 62]. In combination with the progressive loss of mutant oligodendrocytes, this leads to dysmyelination and demyelination in PMD [11, 53, 59]. The MRI pattern of diffuse hypomyelination in PMD caused by PLP1 duplication is hence considered consequence of arrested brain maturation and lacks focal or inflammatory demyelination [9, 66, 71]. Despite variable disease severity of PMD duplication patients, hypomyelination improves only to a minor extent over time. At later stages, axon degeneration is also a feature of the PMD pathology, leading to cortical atrophy that likely contributes to neurological impairment [9, 35]. It has been shown in vitro and in vivo that lipid supplementation can enhance myelination in hypomyelinating pathologies and thereby promote repair [6, 21, 55,56,57]. However, under physiological conditions plasma cholesterol cannot cross the blood–brain barrier (BBB) [7]. In a previous study, we found that the BBB is perturbed in a line of Plp1 transgenic mice (Plp1tg-72 [51]), a model of PMD, allowing the entry of cholesterol from the circulation into the CNS. In this model, dietary supplementation with cholesterol strongly improves myelination [57].
Here, we hypothesized that (1) the accessibility to the CNS determines the efficacy of lipid treatment, and (2) in the CNS, the different lipid classes target distinct aspects of the hypomyelinating pathology, i.e. the primary defect in myelin-forming oligodendrocytes and the subsequent damage of hypo/demyelinated axons. This study combines two important therapeutic targets, cholesterol for the support of remyelinating oligodendrocytes and ketone bodies for the metabolic support of the axonal compartment.
Results and discussion
Dietary cholesterol does not rescue hypomyelination in PMD patients
Active demyelination with inflammatory responses in patients with PMD [24] predicts at least minor BBB disturbances. Therefore, in an individual, compassionate use trial, we first asked whether we could translate the dietary treatment approach in two patients with PMD caused by PLP1 duplication. Daily cholesterol supplementation of up to 590 mg/kg was well tolerated and blood lipid and glucose values remained in the normal range (Online Resource Supplemental Fig. 1). No adverse reactions were observed in the PMD patients, in agreement with cholesterol supplementation studies in patients with Smith-Lemli-Opitz disease [18]. However, a therapeutic benefit was not as obvious as in PMD mice. By MRI measurements, hypomyelination remained stable over 2 years of treatment (Online Resource Supplemental Fig. 2) which is consistent with the normal course of PMD disease [59, 66]. Taken together, dietary supplementation with high dose cholesterol was safe in our two PMD patients and long term disease monitoring will decide about the potential therapeutic benefit of this treatment strategy.
In Plp1tg-72 mice, the compromised BBB allowed access of cholesterol from the circulation into the brain [57]. In contrast, in PMD the comparative analysis of CSF and serum did not reveal gross disturbance of BBB integrity (Online Resource Supplemental Fig. 1a), in agreement with the lack of enhancing gadolinium signals in MRI in a PMD patient [43]. In a substrain of Plp1tg-72 mice with preserved BBB integrity (Online Resource Supplemental Fig. 3a-b), the dietary cholesterol supplementation only moderately ameliorated myelin disease (Online Resource Supplemental Fig. 3c-d), most likely reflecting the limited therapeutic success in PMD patients. Of note, PMD patients and both Plp1tg mouse strains show a comparable initial increase in serum cholesterol (Online Resource Supplemental Fig. 1b, Online Resource Supplemental Fig. 3e) [57] as expected from healthy adult individuals [30], suggesting that serum cholesterol alone does not suffice to predict treatment efficacy. Since the substrain of Plp1tg-72 mice more adequately models human PMD, we used this line, termed Plp1tgB below, in subsequent experiments.
Ketogenic diet improves pathology in a PMD model with preserved BBB integrity
The consumption of a high-fat/low-carbohydrate ketogenic diet causes the liver to generate ketone bodies. In the brain, ketone bodies such as beta-hydroxybutyrate facilitate sterol synthesis [32] which is essential for myelin membrane growth [55]. Thus, we asked whether a ketogenic diet that promotes CNS lipid metabolism under conditions of preserved BBB function [22, 32, 50] is also effective in hypomyelinating disease. Plp1tgB and control mice were given a ketogenic diet (KD) or standard a chow diet (SD) between 2 and 12 weeks of age and physiological parameters were monitored weekly (Fig. 1a). Already after 7 days of dietary intervention with KD, blood levels of ketone bodies strongly increased (about fivefold) and serum glucose dropped to about 70% of the level in SD fed mice (Fig. 1b, Online Resource Supplemental Fig. 4a, b). Ketone bodies are imported into the brain by monocarboxylate transporters, mainly MCT1 expressed by endothelial cells [37]. In accordance with ongoing ketosis, mRNA and protein levels of monocarboxylate transporters as well as enzymes of ketone body utilization significantly increased. This included the rate-limiting enzyme of the pathway, 3-oxoacid CoA-transferase 1 (OXCT1, also termed Succinyl-CoA:3-ketoacid coenzyme A transferase 1, SCOT, EC:2.8.3.5) whose abundance was threefold elevated in spinal cord of KD fed Plp1tgB mice, even more obvious by immunolabeling (Fig. 1d, e, Online Resource Supplemental Fig. 4c, d).

Increased ketone body uptake and ketolysis is induced in KD fed animals. a Treatment paradigm, Plp1tgB mice were fed ketogenic diet (KD) or standard chow (SD) between 2 and 12 weeks of age. b Mean blood levels of the ketone body beta-hydroxybutyrate (βHB) ± SEM. Significance was evaluated by 2way ANOVA with Tukey’s post test [N = 4 (wild type, WT), N = 8 (SD fed Plp1tgB), N = 9 (KD fed Plp1tgB)]. c Representative immunofluorescence detecting MCT1 on spinal cord sections of SD or KD fed Plp1tgB mice. Isolectin served to visualize blood vessels. The dashed line marks the tissue outline and the boxed area is enlarged on the right. d Western Blot with quantification of OXCT1 in wild type mice (N = 3), Plp1tgB mice fed SD (N = 3) or KD (N = 4). Equal protein loading was confirmed by staining of actin. Significance was tested using 1way ANOVA with Tukey’s multiple comparison test. e Immunolabeling of OXCT1 and neurofilament heavy chain (NF200) in the corticospinal tract of the spinal cord in Plp1tgB mice fed SD or KD. Dashed line marks the border between gray matter (GM) and white matter (WM) in dorsal spinal cord. The boxed area is enlarged on the right. Indicated are only significant differences between SD and KD fed Plp1tgB mice (*P < 0.05; ***P < 0.001). Scale bars 10 µm
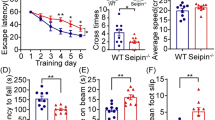
We next determined the degree of oligodendroglial defects, the hallmark of PMD pathology, in the corticospinal tract of the spinal cord from 12 weeks old Plp1tgB mice. As expected, the number of Olig2- and CAII-positive oligodendroglia was reduced in SD fed Plp1tgB mice, but feeding the KD diet rescued this defect in mutants (Fig. 2a). Moreover, untreated Plp1tgB mutant mice showed a robust endoplasmic reticulum stress response with about fourfold elevated ATF6 mRNA and protein levels. Both were significantly ameliorated in KD fed mutant mice (Fig. 2b, Online Resource Supplemental Fig. 4e). In agreement with the reduced oligodendroglial pathology, also astrogliosis and microgliosis were ameliorated in KD fed Plp1tgB compared to SD fed mutants (Online Resource Supplemental Fig. 5). Plp1 mRNA overexpression is the primary defect in the classical form of PMD and in our mouse model [11, 51]. Feeding Plp1tgB mutants with KD did not lower Plp1 expression but rather increased Plp1 mRNA levels slightly (Fig. 2c), presumably reflecting enhanced oligodendrocyte survival. Indeed, the severe hypomyelination of untreated Plp1tgB mice was ameliorated by feeding a KD, as evidenced by fewer unmyelinated axons (g-ratio = 1) and more normally myelinated axons (g-ratio < 0.8), resulting in an overall reduction in mean g-ratio, as determined for the corticospinal tract (Fig. 2d, e). We next investigated whether feeding KD also ameliorated the clinical phenotype of PMD mice. When we assessed motor performance by two different behavioral tests, elevated beam test and rotarod test, untreated Plp1tgB mice showed progressive worsening of motor functions. By contrast, KD fed Plp1tgB animals retained their motor fitness at wild type levels (Fig. 2f, g). For comparison, feeding Plp1tgB mice a medium-chain triglyceride diet, which contains less fat than classical KD, elicited only a mild ketosis in our mice. This dietary regimen did not ameliorate motor performance, suggesting a critical role of ketone bodies rather than a direct effect of triglycerides/lipids (Online Resource Supplemental Fig. 6).

KD ameliorates PMD pathology in Plp1tgB animals. a Olig2 and CAII (arrowheads) immunolabeling of wild type and Plp1tgB mice fed SD and KD with quantification of cell numbers in dorsal white matter of the spinal cord on the right (N = 4–5 (WT), N = 7–8 (Plp1tgB fed SD), N = 8–9 (Plp1tgB fed KD), 1way ANOVA with Tukey’s post test). b Western Blot with quantification of ATF6 in lumbar spinal cord of wild type mice (N = 4), Plp1tgB mice fed SD (N = 3) or KD (N = 4). Equal protein loading was confirmed by reprobing for actin (1way ANOVA with Tukey’s post test). c Quantitative RT-PCR determining Plp1 in spinal cord of Plp1tgB mice fed SD or KD (N = 8, 1way ANOVA with Sidak’s post test) normalized to wild type controls (N = 5, set to 1). d Quantification of myelination in the corticospinal tract from wild type mice, and Plp1tgB mice fed SD or KD (N = 7), showing g-ratio analysis as scatter plot (left panel) and the mean g ratio (right panel, 1way ANOVA with Tukey’s post test). e Relative frequency of sufficiently myelinated fibers (g ratio < 0.8), hypomyelinated fibers (g ratio > 0.8) or unmyelinated fibers (g-ratio = 1) in the CST of Plp1tgB fed SD or KD (N = 7, two-sided Student’s t test of each group). f Rotarod analysis and g elevated beam test performance at 6 to 12 weeks of age (N = 7–8; 2way ANOVA with Sidak’s post test). Indicated are only significant differences between SD and KD fed Plp1tgB mice (*P < 0.05; **P < 0.01; ***P < 0.001). Scale bars 20 µm
Ketogenic diet ameliorates mitochondrial abnormalities in axons of PMD mice
It is unlikely that the moderately improved myelination explains the dramatic improvement in motor functions in KD fed Plp1tgB animals. In PMD patients, motor development is strongly retarded, and progressive axonal loss likely causes the gradual decline of motor functions already achieved [58]. In accordance, the frequent axonal swellings (spheroids) in Plp1tgB mice were strongly reduced when feeding a KD (Fig. 3a). In addition, we observed in Plp1tgB mice that many axons contained enlarged mitochondrial profiles (Fig. 3b), as observed before in other models of PMD [28, 45, 53]. Such morphological alterations can reflect increased activity and/or functional deficits in mitochondria which could both occur in PMD (see below).

KD rescues mitochondria enlargement and ameliorates impulse conduction in Plp1tgB mice. a APP staining of axonal swellings/spheroids (arrowheads) in wild type and Plp1tgB mice fed SD and KD with quantification of cells per 30,000 µm2 corticospinal tract of the spinal cord (N = 6–8; 1way ANOVA with Tukey’s post test). Scale bar 20 µm. b Electron micrographs showing an example of enlarged axonal mitochondria (colored in yellow) in Plp1tgB mice fed SD and a normally sized mitochondrium in KD fed Plp1tgB mice. Scale bar 0.5 μm. c Mean area of axonal mitochondrial in cross-sectioned corticospinal tract of wild type mice, and Plp1tgB animals fed SD or KD (at least 60 mitochondria per animal were counted, N = 6–7 animals; 1way-ANOVA with Tukey’s post test). d Stratification of mitochondrial sizes shown in c with respect to myelinated axons (g-ratio < 1) and unmyelinated axons (g-ratio = 1) (two-sided Student’s t test of each group). e 3D modeling of ACO2 immunolabeled mitochondria in deconvolved confocal images from Plp1tgB mice fed SD or KD with quantification of mitochondrial volumes (SD, 3178 or KD, 2817 mitochondria; 4 animals per condition). Volume data are presented as median with interquartile range (Kolmogorov–Smirnov test). f Density of myelinated axons in the optic nerve of SD and KD fed Plp1tgB animals (mean per 100 square µm ± SEM of N = 4–5 animals counting 13 images per animal; two-sided Student’s t test). g Evaluation of mitochondrial area in axons of the optic nerve analogous to the analysis shown in c (N = 5 animals, 1way-ANOVA with Tukey’s post test). h–n Electrophysiological evaluation of (N = 9–11) optic nerves from SD or KD fed Plp1tgB mice, showing h mean CAP traces ± SEM, I-V curves of i the CAP amplitude and j the CAP area (2way ANOVA with Sidak’s post test), and k nerve conduction velocity (NCV, two-sided Student’s t test). l Mean CAP area ± SEM after challenging optic nerves with 5 Hz stimulation in the presence of 3 mM lactate for 75 min, followed by recovery in the presence of 3 mM lactate and 10 mM glucose (N = 7–8 optic nerves from Plp1tgB animals and 2 WT nerves). m Mean traces extracted from l (position marked by arrowheads) at baseline, at the end of the stimulation challenge, and after recovery. n Quantification of recovery showing mean CAP amplitude and CAP area with individual data points in relation to baseline (two-sided Student’s t test for each analysis) (*P < 0.05; **P < 0.01; ***P < 0.001)
By systematic quantification of electron microscopic images, we detected a significant about 20% area increase of mitochondrial profiles in axons from Plp1tgB animals compared to wild type controls. This enlargement was most evident in unmyelinated axons (Fig. 3c, d), and even detectable in young mutants (Online Resource Supplemental Fig. 7). Importantly, KD completely normalized this phenomenon, which was most evident in axons that remained unmyelinated, and correlated well with the reduced volume of mitochondria, modelled from deconvolved confocal image stacks (Fig. 3c–e, Online Resource Supplemental Fig. 8). We also determined mRNA and protein abundance of several mitochondrial markers in spinal cord lysates to correlate mitochondrial morphology to alterations in fusion or fission of mitochondria but neither of the tested markers showed significant differences between the two treatment groups of Plp1tgB animals, likely because this analysis reflect the sum of neuronal and glial mitochondria (Online Resource Supplemental Fig. 9). Notably, feeding a cholesterol supplemented chow to Plp1tg animals (Plp1tgB or Plp1tg-72) did not reverse the increased mitochondrial sizes, even where myelination was improved (Online Resource Supplemental Fig. 3f). This suggests that high-cholesterol and ketogenic diets supported distinct therapeutic pathways.
The latter prompted us to evaluate the impact of KD on axonal integrity in the absence of myelin and analyzed optic nerves of Plp1tgB animals. When untreated, this fiber tract is almost devoid of myelin at the age of 12 weeks but unmyelinated axons appear morphologically healthy and retinal ganglion cells are not degenerated [20, 57]. Here, the KD diet showed only a modest effect on hypomyelination (Fig. 3f), but ameliorated the enlarged axonal mitochondria, similar to our observations in the corticospinal tract (Fig. 3g).
Next, we assessed the functional significance of mitochondrial abnormalities in hypomyelinated axons by sensitive electrophysiological analyses. Specifically, in acute ex-vivo preparations of the optic nerve, we quantified nerve conduction and functional axon integrity, by comparing compound axon potentials (CAP) as described [54, 69]. As expected for hypomyelinated fibers [4, 54, 64], optic nerves of untreated Plp1tgB mice exhibited an abnormally attenuated evoked response with a strong after-hyperpolarization phase caused by elevated activity of potassium channels (Online Resource Supplemental Fig. 10). In KD fed Plp1tgB mutants, in contrast, CAP amplitude and CAP area was significantly increased compared to their SD fed littermates, implying that more axons contributed to evoked impulse propagation (Fig. 3h–j). Nerve conduction velocity of Plp1tgB mice was slightly increased in KD fed animals, likely reflecting the increased density of myelinated axons (Fig. 3f, k).
Glycolytic oligodendrocytes support the axonal energy metabolism by exporting lactate [23, 36]. To bypass this glial support and rather directly assess mitochondrial function in axons, we challenged the axonal energy metabolism during repeated firing. Specifically, we monitored CAPs in the optic nerve in the absence of glucose and in the presence of a suboptimal lactate concentration (3 mM) during 5 Hz stimulation for 75 min [10], followed by a recovery period in the presence of both lactate and glucose. In wild type optic nerves, this protocol led to a gradual reduction of CAPs to about 25% of baseline values (Fig. 3l), in agreement with recent findings in the corpus callosum [41]. In optic nerves from Plp1tgB mice, however, the resulting CAP decay was even stronger and more rapid than in wild type controls, demonstrating that the PMD pathology affects the ability of axons to utilize lactate to maintain conduction. Feeding a KD to Plp1tgB mice attenuated the CAP decay (of 5 Hz stimulated optic nerves ex vivo), suggesting that optic nerves from KD fed mice are more resilient to this metabolic stress (Fig. 3l, m). Moreover, in this protocol we noted a strong (80%) recovery of the original CAP amplitude in nerves from KD fed mice comparable to wild types, contrasting to only 40% recovery in SD fed mice (Fig. 3l–n). This finding demonstrates that PMD, which primarily perturbs myelination, also indirectly affects axonal energy metabolism. These data also suggest that axonal integrity can be uncoupled from the myelination status by the KD. That is, despite the marginally increased myelination of the optic nerve of Plp1tgB mice when fed the KD, dysmyelinated axons functionally improved, as evidenced by physiological recordings, and this effect was accompanied by morphological normalization of axonal mitochondria.
Ketogenic diet promotes repair in a model of adult remyelination
Because of the therapeutic benefit of KD in the PMD mouse, a model of developmental hypomyelination, we compared in a 4-arm study the therapeutic potential of KD in a model of adult demyelination and remyelination. At the age of 8–10 weeks, wild type mice were demyelinated by feeding cuprizone, contained in a SD, for 4 weeks (acute paradigm) or for 12 weeks (chronic demyelination), followed by cuprizone withdrawal and remyelination with feeding SD or KD (Fig. 4a). Increased blood levels of the ketone body beta-hydroxybutyrate (βHB) and reduced blood glucose levels confirmed ketosis already at 7 days after the diet switch (Online Resource Supplemental Fig. 11a-d).

KD supports repair after cuprizone mediated demyelination. a Treatment paradigms. In an acute paradigm cuprizone (CUP) in SD was fed for 4 weeks, followed by feeding SD or KD for 1 week. In the chronic paradigm cuprizone was fed for 12 weeks, followed by KD or SD for up to 6 weeks. Histochemical quantification of b oligodendroglial cells (Olig2) and c mature oligodendrocytes (CAII) in the corpus callosum (two-sided Student‘s t test of each treatment cohort with N = 4–5 mice). d Relative gene expression of Olig2, Car2 and Plp1 in dissected corpus callosum of mice after 12 + 2 weeks paradigm, normalized to untreated controls (set to 1, N = 4). Significance was evaluated by 1way ANOVA with Tukey’s post test, indicated are only significant differences between cuprizone groups. e Myelin abundance (Gallyas silver impregnation, N = 4–5) and f number of myelinated axons per square 100 µm in the corpus callosum (N = 5–6). Data are expressed as mean with individual values (two-sided Student‘s t test of each treatment cohort). g Representative electron micrographs of the corpus callosum with boxed detail below (mitochondria colored in yellow, axonal outline colored in blue). Scale bar 2 µm. h Mean number of APP spheroids with individual values as a readout of axonal damage (two-sided Student‘s t test of each treatment cohort). i Mean size of axonal mitochondria in the corpus callosum (N = 5 animals, 1way ANOVA with Tukey’s post test). j Beam test to measure motor performance (N = 4–5), showing mean slips with individual data points (4 + 1, two-sided Student‘s t test) or mean ± SEM (12w-12 + 2 weeks, 2way ANOVA with Sidak’s post test) (*P < 0.05; **P < 0.01; ***P < 0.001)
In agreement with our findings in Plp1tgB mice, mRNA expression levels of monocarboxylate transporters and enzymes of ketolysis were robustly increased in the corpus callosum of KD fed mice in comparison to SD fed controls and untreated wild type mice (Online Resource Supplemental Fig. 11e, f), likely reflecting ongoing ketone body utilization. Cuprizone leads to the death of most mature oligodendrocytes in the corpus callosum [39, 49], and after the withdrawal of cuprizone, myelin repair is reflected in an increasing number of oligodendrocyte lineage cells and gradual remyelination. While the number of Olig2 positive cells was not altered in KD fed mice compared to SD fed controls, the rate of oligodendrocyte precursor cell differentiation to CAII positive oligodendrocytes was strongly increased in KD fed mice (Fig. 4b–d, Online Resource Supplemental Fig. 11-12). In accordance, by feeding KD remyelination was accelerated about twofold at only 7 days after acute demyelination, and at 2 weeks after chronic demyelination (Fig. 4e–g).
In contrast, the degree of astrogliosis and microgliosis and expression of enzymes involved in detoxification of reactive oxygen species appeared unaffected by KD (Online Resource Supplemental Fig. 11 g-k, Online Resource Supplemental Fig. 12). Pro-inflammatory eicosanoids could contribute to pathogenesis in multiple sclerosis and expression of respective biosynthetic enzymes such as ALOX5 (arachidonate 5-lipoxygenase) was also increased in the chronic cuprizone model (Online Resource Supplemental Fig. 11 l) in accordance with previous reports [47, 75]. However, in contrast to a dietary intervention study with KD in MS patients [8], their expression was not altered by KD in our study. Axonal defects, as assessed by the density of APP positive axonal spheroids, was reduced by about 50% in KD fed mice compared to SD fed controls (Fig. 4h, Online Resource Supplemental Fig. 12). In addition, the cross-sectional size of axonal mitochondria, which were increased in SD fed mice during remyelination, even normalized (Fig. 4g, i). This suggests that the loss of oligodendroglial integrity rather than gliosis correlates with axonal perturbations. Finally, motor abilities, as quantified by an elevated beam test, also improved in KD fed mice (Fig. 4j). Thus, similar to our findings in developmental dysmyelination, KD also promoted functional repair in acute and chronic adult remyelination paradigms.
In this study, we have used two models of white matter disease with distinct etiologies and pathomechanisms, and found that feeding a KD is remarkably efficient in preserving axonal and oligodendroglial integrity. Plp1tgB mutants model the hereditary leukodystrophy PMD, in which overexpression of the myelin protein PLP induces cellular stress in differentiating oligodendroglia followed by dysmyelination and demyelination [67]. In contrast, the pharmacological cuprizone model induces transient demyelination and also assesses remyelination in adult mice [39, 49]. In both disease models, axon damage is likely secondary to the primary oligodendroglial defect [15, 34, 59], but a detailed understanding of the axonal injury is lacking. A combinatory effect of reduced metabolic support by injured oligodendrocytes and the exposure to pro-inflammatory factors, including reactive oxygen and nitrogen species, is most likely [15, 34]. In the PMD mouse model, we found mitochondrial enlargement not only in dysmyelinated axons but also in myelinated axons before overt demyelination, suggesting metabolic problems in axons at an early preclinical stage.
The KD dramatically changes the global metabolism and induces a plethora of alterations in the body including the CNS [50]. While discussed in many neurodegenerative diseases, KD is currently used in the management of epilepsy [3, 16, 70]. The mechanisms of seizure control are likely diverse, involve the gut microbiota [46] and may act in parallel, including effects on neurotransmission, epigenetic gene regulation, energy metabolism, and anti-inflammatory activity [16, 22, 61]. Our preclinical data reveal that KD provides metabolic support to axons in two distinct myelin pathologies that model key aspects of human myelin disease and mechanisms of recovery. We specifically note that also (chronically) unmyelinated axons appear to profit from KD.
Ketone bodies share some of the molecular targets with certain fatty acids that are contained in or processed from high-fat ketogenic diet regimens [3, 61]. However, in our study, a medium-chain triglyceride diet that only slightly raised blood ketone body levels was therapeutically ineffective, suggesting a critical and direct role of ketone bodies in the therapy of PMD.
We envision the following scenario of causal relations: (1) In PMD, PLP overexpression induces endoplasmic reticulum stress which affects oligodendrocytes and likely disturbs the oligodendroglial trophic support to axons. Chronic damage ultimately leads to loss of Plp1-overexpressing oligodendrocytes and demyelination. In the cuprizone model, pharmacologically induced oligodendrogliopathy causes demyelination. (2) To maintain impulse conduction unmyelinated axons require about five-fold more energy compared to the same tissue volume of myelinated axons [44]. Hence, aberrantly unmyelinated axons suffer from energy deficits because of the insufficient support by mutant oligodendrocytes and increased energy demands. (3) Energy deprived cells typically increase the activity of mitochondria. In addition, demyelination triggers inflammatory responses involving production of reactive oxygen and nitrogen species. Increased mitochondrial activity and oxidative stress are both associated with morphological changes and volume enlargement of mitochondria [38, 48] as observed in axons of both models of myelin disease. Chronic oxidative stress might damage mtDNA and mitochondrial enzymes [53] that could further trigger compensatory volume increases of mitochondria and aggravate degenerative processes. (4) In contrast to glucose, ketone bodies are directly metabolized by mitochondria fueling into the tricarboxylic cycle and oxidative phosphorylation. Since feeding a KD supports the conduction of optic nerve axons even in the absence of substantial myelination, we propose there is a direct supportive effect of KD on mitochondrial integrity in dysmyelinated axons. Bypassing the need for oligodendroglial trophic support could restore axonal mitochondria function (and morphology) and resolve energy deficits of axons. Relieving oligodendrocytes from the task to provide energetic support to axons might also contribute to the increased survival of mutant oligodendrocytes and to the ameliorated PMD pathology.
In contrast to the developmental PMD model, adult remyelination in the cuprizone model strongly benefitted from the KD, in line with previous findings using a fasting mimicking diet [12]. Here, we suggest that a compromised BBB in cuprizone fed animals allows for the entry of circulating cholesterol into the CNS (further enhanced by the KD that contains 0.1% cholesterol) [5, 6]. While the BBB may remain largely intact in classical human PMD, it is perturbed in other myelin diseases with a stronger inflammatory component. We conclude that there is a clear rationale to consider KD or a derivative as a future therapy for myelin diseases as it combines two important therapeutic targets, cholesterol for the support of remyelinating oligodendrocytes and ketone bodies for the metabolic support of the axonal compartment, and future clinical trials will reveal its feasibility in the management of demyelinating episodes.
Materials and methods
Patients
Two patients diagnosed Pelizaeus–Merzbacher disease (PMD) with hemizygote duplication of the complete PLP1 gene were treated with high-dose oral cholesterol, conducted in two different clinical centers. Patient PMD-A was 29 months at start of dietary cholesterol supplementation. The final dose of 200 mg/kg/day was reached within 2 weeks. Cardiologic assessments including ECG, echocardiography and Doppler sonography were performed on annual basis and revealed normal results. Patient PMD-B was 30 months at start of dietary cholesterol supplementation. Over a period of 17 months the initial dose of 125 mg/kg/day was increased to 590 mg/kg/day and maintained for now further 13 months. Carotid artery intima-media thickness was determined to detect possible vascular side-effects of the cholesterol treatment which did not occur. Serum lipid levels of both patients were monitored regularly and remained in normal range [17]. Repeated measurements of blood and CSF glucose levels yielded normal results. CRP values stayed within the normal range. Evaluation of the developmental and clinical disease status [72] included general physical parameters, neurologic and developmental scores (ET6-6; WPPSI, Wechsler Preschool and Primary Scale of Intelligence), motor function (GMFCS, gross motor function classification system), as well as neurophysiologic and ophthalmologic assessments. Reiber analysis of albumin concentrations Qalb ([albumin]CSF/[albumin]serum) served to detect gross blood–brain barrier abnormalities [52] which did not occur. The control subject was diagnosed with neurological asymptomatic adrenoleukodystrohy who undergo regular cranial MRIs as surveillance for onset of inflammatory demyelination.
Magnetic resonance imaging (MRI)
Cranial MRI studies conducted at a 1.5T (Ingenia, Philips) (Hamburg) or a 3T clinical scanner (Tim TRIO, Siemens Healthcare) (Göttingen) comprised conventional T1- and T2-weighted images. In addition, semiquantitative magnetization transfer (MT) imaging was performed using a 3D FLASH (fast low angle shot) sequence with 1.25 mm isotropic resolution and 240 mm field-of-view. MT contrast was imposed upon a proton density weighted reference (TR/TE/α = 25/4.9 ms/5°, 3.5 min measurement using partial acquisition techniques) by applying a 12.8 ms Gaussian MT-pulse of 540° nominal flip angle 2.2 kHz off resonance prior to excitation. By means of a second T1-weighted reference (TR/α = 11 ms/15°, 1.5 min), maps of the percentage MT saturation (MTsat maps) were calculated as described [19, 25,26,27]. The blue-gray-red-yellow color scale of the MTsat maps covered a range from − 0.1 pu (light blue; lower limit of the CSF mode) to 1.2 pu (gray, GM) to 2.5 pu (yellow; WM). The upper threshold was chosen post hoc below the MTsat values of myelinated WM of controls (3 pu) to increase the sensitivity in the range observed with severe hypomyelination.
Mice
Plp1 transgenic Plp1tg-72 [51] mice harbor three copies of the murine Plp1 gene. The substrain of Plp1tg-72 mice (Plp1tgB) spontaneously developed after infection with helminthes and worms followed by standard treatment. While severity of histopathology differs between Plp1tg mouse strains, Plp1 overexpression in spinal cord remained unchanged at about 1.2 fold normalized to wild type mice (Online Resource Supplemental Fig. 13). Wild type C57BL6 mice were obtained from Charles River. Only male mice were used in this study. Animals were allocated to cages of 4 animals per cage by block randomization for adaptation to the new environment for 2 weeks before cages were assigned to the different treatment groups.
Diets
Mice were fed normal chow (ssniff V1124 containing 11% fat, 36% protein, 53% carbohydrates (5% sugar) metabolizable energy), high-fat/low-carbohydrate ketogenic diet (ssniff E15149-30 containing 94% fat, 5% protein, 1% carbohydrates/sugar), medium-chain triglyceride diet (ssniff modified Surwit diet containing 70% fat, 15% protein, 15% carbohydrates (1% sugar)) or a normal chow supplemented with 0.5–5% w/w cholesterol.
Cuprizone
Adult male C57BL/6N mice (8–10 weeks of age) were randomly assigned to an experimental group. Mice were fed 0.2% w/w cuprizone (bis-cyclohexanone oxaldihydrazone, Sigma-Aldrich) in powdered chow (ssniff V1120) for 4 weeks (acute paradigm) or 12 weeks (chronic paradigm), followed by cuprizone withdrawal and feeding mice SD or KD ad libitum.
Motor coordination
For the elevated beam test, mice were put on a beam (width 1.5 cm) and allowed to run toward a hiding box. The number of slips in a defined 55 cm distance was assessed as a mean of two repeats per time point. For rotarod analysis, mice were placed on the rotarod starting with a speed of 4 rpm. Speed was increased with 10 rpm/min until 26 rpm was reached and held for 10 s. Maximum trial duration was 150 s and the latency to fall was recorded.
Antibodies
The following antibodies were used: actin (Sigma A3853), ACO2 (Sigma, HPA001097), APP (Chemicon MAB348), ATF6 (Abcam 40256), CAII (Said Ghandour), GFAP (Chemicon MAB3402), Iba1 (Wako 019-19741), MAC3 (Pharmigen 01781D), NF200 (Sigma N4142), Olig2 (Charles Stiles/John Alberta, DF308), OXCT1 (Proteintech Europe 12175-1AP), SMI31 (Covance SMI-31P), VDAC (Rockland), isolectin IB4 coupled to Alexa 594 (Vectorlab). For generation of MCT1 (SLC16a1) antisera, rabbits were immunized with the intracellular peptide 221–236 of mouse MCT1 (CDANTDLIGGSPKGEKL). Anti-MCT1 antibodies were purified by affinity chromatography.
Histochemistry
Mice were sacrificed by cervical dislocation and immersion fixed for 48 h or perfused with 4% formaldehyde (PFA). Brain samples of cuprizone treated animals were cut at Bregma 1.58 for comparable pathology. Tissue embedded in paraffin and cut into 5 µm sections (HMP 110, MICROM). Gallyas silver impregnation was done as described [6]. For immunohistological analyses, sections were deparaffinized followed by antigen-retrieval in sodium citrate buffer (0.01 M, pH 6.0). For immunofluorescence, sections were blocked with serum free protein block (Dako). Primary antibodies were diluted in 2% bovine serum albumin (BSA)/PBS and incubated for 48 h followed by fluorophor coupled secondary antibodies.
For volume analysis of mitochondria, oversampled images suitable for subsequent deconvolution were acquired with a Zeiss LSM 510 Meta confocal laser-scanning microscope (CLSM). A 63× (NA = 1.25) oil immersion objective was used to record images composed of 48 nm pixels, with z-planes being acquired every 190 nm. Signal-to-noise, resolution signal and sample geometry of the resulting z-stacks were corrected by deconvolution using the Huygens Professional software package (version 18.04, Scientific Volume imaging B.V., Hilversum, Netherlands). An image stack volume of 49.05 × 49.05 × 5.5 µm was used to generate 3D models of mitochondria by the Imaris software package (version 9.2.1, Bitplane AG, Zurich, Switzerland) of which the volumetric information in the range of 0.02–1 µm3 was extracted.
For immunohistochemistry, endogenous peroxidase activity was blocked with 3% hydrogen peroxide. Sections were then blocked (20% goat serum in BSA/PBS) and incubated with primary antibodies. Detection was done with the LSAB2 kit (Dako, Hamburg, Germany) or the Vector Elite ABC kit (Vector Labs). HRP substrate 3,3′-Diaminobenzidine was applied by using the DAB Zytomed Kit (Zytomed Systems GmbH). Haematoxylin stain was done to label nuclei. Sections were dehydrated prior to mounting (Eukitt). Specimens were analyzed on an Axio Imager.Z1 (Zeiss) equipped with an AxioCam MRc3, 0.63 × Camera Adapter and the ZEN 2012 blue edition software using 10× objective (Plan Apochromat 10×/0.45 M27) or 20× objective (Plan-Apochromat 20×/0.8) and evaluated with Image J software. Quantification of areas (Gallyas, GFAP, MAC3) were done by applying semi-automated Fiji software [60] macro to threshold (variable threshold in case of Gallyas and fixed threshold for antibody staining) and color deconvolute the images of the spinal cord or corpus callosum above the fornix. Three to five sections per animal were analyzed for quantification.
Electron microscopic analysis was done as previously described [6, 57]. Briefly, tissue was fixed in 4% PFA, 2.5% glutaraldehyde, 0.1 M phosphate buffer. Sagittal brain sections were cut on a vibratome (Leica VT1200, 300 µm) and the corpus callosum with adjacent tissue (-0.04 mm lateral) was punched with a 2 mm diameter punching tool. Tissue punches or spinal cord sections were embedded in epon (LynxII, EMS). At least 15 digital pictures using an on-axis 2048 × 2048-CCD camera (12,000 × magnification, TRS, Moorenweis) of uranyl acetate contrasted ultrathin sections were taken with the LEO912 electron microscope (Zeiss, Oberkochen). For quantification of mitochondrial sizes, the area at least 60 mitochondrial profiles per animal were measured. Only mitochondria of cross sectioned axons were considered. For electron microscopic analysis on high pressure frozen samples, optic nerves of P23 old mice were freshly prepared and cryo-immobilized by high pressure freezing using a HPM100 instrument (Leica, Vienna) and 20% PVP in PBS as filler. Freeze substitution was carried out as described [42].
Protein analysis
Tissue samples were lysed in sucrose buffer (18% sucrose, 10 mM Tris/HCl pH 7.4, 1 mM sodium bicarbonate, 1 mM magnesium chloride, 0.1% Triton, 0.2% lithiumdodecyl sulphate, 0.025% sodium deoxycholate) with protease inhibition (Roche) using a Precellys 24 homogenizer (Bertin technologies). SDS gel electrophoresis, semi-dry blotting on PVDF membranes (Hybond P, Biosciences) and antibody incubations were done using standard procedures. Detection of immunolabeled proteins was performed with Western-Lighting Plus-ECL Reagent (Perkin Elmer) using ChemoCam Imager (Intas).
Expression analysis
Expression analyses were done as described [6]. RNA was extracted from dissected tissue using QIAshredder and RNeasy protocols (Qiagen). Concentration and quality of RNA was evaluated using a NanoDrop spectrophotometer and RNA Nano (Agilent). cDNA was synthesized with Superscript III (Invitrogen) and quantitative PCRs were done in triplicates with the GoTaq qPCR Master Mix (Promega) on a 7500 Fast Real-Time PCR System (Applied Biosystems). Expression values were normalized to the mean of two housekeeping genes, Hprt (hypoxanthin-phosphoribosyl-transferase 1) and Rplp0 (60S acidic ribosomal protein P). Quantification was done by applying the ΔΔCt method, normalized to age matched untreated controls (set to 1). All primers were intron-spanning. Expression of the following genes was measured: Acat1 (acetyl-Coenzyme A acetyltransferase 1), Aif1 (allograft inflammatory factor 1), Alox5 (arachidonate 5-lipoxygenase), Atf4 (activating transcription factor 4), Atf6, Bdh1 (3-hydroxybutyrate dehydrogenase 1), Hspa5 (heat shock protein 5) also termed Bip or Grp78, Car2 (carbonic anhydrase 2), Cat (catalase), Cox1 (cyclooxygenase 1), Ddit3 (DNA-damage inducible transcript 3) also termed Chop (C/EBP homoologous protein), Dmn1 l (dynamin 1 like), Gfap (glial fibrillary acidic protein), Slc16a1 (solute carrier family 16 member 1, also termed monocarboxylate transporter 1 MCT1), Mct2/Slc16a7, Olig2 (oligodendrocyte lineage transcription factor 2), Pdgfa (platelet derived growth factor alpha), Ppargc1a (peroxisome proliferative activated receptor, gamma, coactivator 1 alpha), also termed Pgc1a (PPAR gamma coactivator-1), Plp1 (proteolipid protein 1), Sod1 (superoxide dismutase 1), Sod2, Tfam (transcription factor A, mitochondrial), Vdac (voltage dependent anion channel 1), Xbp1 (X-box binding protein 1). All primer sequences are listed in Table S1.
Electrophysiological recordings
Electrophysiological recordings were done as described [54, 69]. Optic nerves were excised from decapitated mice, placed into an interface perfusion chamber (Harvard Apparatus, Holliston, MA) and continuously superfused with artificial cerebrospinal fluid (aCSF). The perfusion chamber was continuously aerated by a humidified gas mixture of 95% O2/5% CO2 and experiments were performed at 37 °C. Custom-made suction electrodes back-filled with aCSF were used for stimulation and recording as described [54, 65]. The stimulating electrode, connected to a battery (Stimulus Isolator 385; WPI, Berlin, Germany) delivered a supramaximal stimulus of 0.75 mA to the nerve evoking compound action potentials (CAP). The recording electrode was connected to an EPC9 amplifier (Heka Elektronik, Lambrecht/Pfalz, Germany). The signal was amplified 500 times, filtered at 30 kHz, and acquired at 100 kHz. The CAP, elicited by the maximum stimulation of 0.75 mA, was recorded at baseline stimulation frequency at 0.1 Hz in the presence of 10 mM glucose and 3 mM lactate with or without bath application of 50 µM 4-aminopyridine (4-AP). To determine the energetic capability of axons to sustain repeated firing, we depleted optic nerves of endogenous energy stores for 5 min (no substrate) followed by incubation in 3 mM lactate in the presence of continuous 5 Hz stimulation which was applied consisting of 100 stimuli, separated by 460 ms, during which the CAP was recorded. After 75 min, stimulation settings were switched back to baseline conditions and the CAP recovery was assessed. For I–V measurements, the evoked response of the optic nerve was elicited every 10 s, by subsequent 0.1 mA stimulation intensity steps, ranging from 0 to 1 mA.
Tracer studies
Bodipy-cholesterol (Topflour, Avanti Polar Lipids) was injected i.p. (5 µg/g body weight). After 1 week, mice were perfused with PBS to remove tracer from the circulatory system. Evans Blue (50 mg/g body weight) was i.v. injected and mice were flushed after 4 h incubation as described [5]. Brains were dissected and immediately frozen on dry ice, weighed and stored at − 80 °C for further processing. Tissue was lyophilized (Christ LMC-1 BETA 1-16) at −36 °C for 24 h under vacuum of 0.2 mBar. For tracer extraction, hemispheres were incubated shaking in 10 µl formamide per mg brain at 57 °C for 24 h. Integrated density of tracer fluorescence was determined in triplicates on a fluorescent microscope (Observer Z2, Zeiss, Germany), equipped with an AxioCam MRc3, 1× Camera Adapter and the ZEN 2012 blue edition software recorded at 10× magnification (Plan-Apochromat 10×/0.45 M27). Tracer concentration was calculated using a standard curve and normalized to matched controls (set to 1).
Statistical analysis
Statistical evaluation was done using GraphPad Prism (GraphPad Inc.), applying either unpaired two-sided Student’s t test for pairwise comparisons, ANOVA for comparisons of more than two groups or the Kolmogorov–Smirnov test for non-parametric comparisons, as stated in the figure legends. ANOVA was combined with a post test to evaluate individual groups. For all statistical tests, significance was measured against an alpha value of 0.05. Sample sizes are given in the figure legends. All error bars show SEM. P values are shown as *P < 0.05; **P < 0.01; ***P < 0.001. No statistical methods were used to predetermine sample sizes, but our sample sizes are similar to those reported in previous publications [6, 57]. Data analysis was performed blind to the experimental groups.
Change history
03 September 2019
The original article was published
References
Ahmed RM, Ke YD, Vucic S, Ittner LM, Seeley W, Hodges JR et al (2018) Physiological changes in neurodegeneration—mechanistic insights and clinical utility. Nat Rev Neurol 14:259–271. https://doi.org/10.1038/nrneurol.2018.23
Anchisi L, Dessi S, Pani A, Mandas A (2012) Cholesterol homeostasis: a key to prevent or slow down neurodegeneration. Front Physiol 3:486. https://doi.org/10.3389/fphys.2012.00486
Augustin K, Khabbush A, Williams S, Eaton S, Orford M, Cross JH et al (2018) Mechanisms of action for the medium-chain triglyceride ketogenic diet in neurological and metabolic disorders. Lancet Neurol 17:84–93. https://doi.org/10.1016/S1474-4422(17)30408-8
Bagchi B, Al-Sabi A, Kaza S, Scholz D, O’Leary VB, Dolly JO et al (2014) Disruption of myelin leads to ectopic expression of K(V)1.1 channels with abnormal conductivity of optic nerve axons in a cuprizone-induced model of demyelination. PLoS ONE 9:87736. https://doi.org/10.1371/journal.pone.0087736
Berghoff SA, Düking T, Spieth L, Winchenbach J, Stumpf SK, Gerndt N et al (2017) Blood–brain barrier hyperpermeability precedes demyelination in the cuprizone model. Acta Neuropathol Commun 5:94. https://doi.org/10.1186/s40478-017-0497-6
Berghoff SA, Gerndt N, Winchenbach J, Stumpf SK, Hosang L, Odoardi F et al (2017) Dietary cholesterol promotes repair of demyelinated lesions in the adult brain. Nat Commun 8:14241. https://doi.org/10.1038/ncomms14241
Björkhem I, Meaney S (2004) Brain cholesterol: long secret life behind a barrier. Arterioscler Thromb Vasc Biol 24:806–815. https://doi.org/10.1161/01.ATV.0000120374.59826.1b
Bock M, Karber M, Kuhn H (2018) Ketogenic diets attenuate cyclooxygenase and lipoxygenase gene expression in multiple sclerosis. EBioMedicine 36:293–303. https://doi.org/10.1016/j.ebiom.2018.08.057
Boespflug-Tanguy O, Bertini E, Baethmann M, Gärtner J, Hanefeld F, Kohlschütter A et al (2004) Pelizaeus–Merzbacher disease and “hypomyelinated” leukodystrophies. In: Uziel G, Taroni F (eds) Hereditary leukoencephalopathies and demyelinating neuropathies in children, vol 12. John Libbey Eurotext, Montrouge, pp 37–42
Brown AM, Wender R, Ransom BR (2001) Metabolic substrates other than glucose support axon function in central white matter. J Neurosci Res 66:839–843. https://doi.org/10.1002/jnr.10081
Cailloux F, Gauthier-Barichard F, Mimault C, Isabelle V, Courtois V, Giraud G et al (2000) Genotype-phenotype correlation in inherited brain myelination defects due to proteolipid protein gene mutations. Clinical European Network on Brain Dysmyelinating Disease. Eur J Hum Genet 8:837–845. https://doi.org/10.1038/sj.ejhg.5200537
Choi IY, Piccio L, Childress P, Bollman B, Ghosh A, Brandhorst S et al (2016) A diet mimicking fasting promotes regeneration and reduces autoimmunity and multiple sclerosis symptoms. Cell Rep 15:2136–2146. https://doi.org/10.1016/j.celrep.2016.05.009
Chrast R, Saher G, Nave KA, Verheijen MH (2011) Lipid metabolism in myelinating glial cells: lessons from human inherited disorders and mouse models. J Lipid Res 52:419–434. https://doi.org/10.1194/jlr.R009761
Courtney R, Landreth GE (2016) LXR regulation of brain cholesterol: from development to disease. Trends Endocrinol Metab 27:404–414. https://doi.org/10.1016/j.tem.2016.03.018
Criste G, Trapp B, Dutta R (2014) Axonal loss in multiple sclerosis: causes and mechanisms. Handb Clin Neurol 122:101–113. https://doi.org/10.1016/B978-0-444-52001-2.00005-4
Dahlin M, Martin DA, Hedlund Z, Jonsson M, von Dobeln U, Wedell A (2015) The ketogenic diet compensates for AGC1 deficiency and improves myelination. Epilepsia 56:e176–e181. https://doi.org/10.1111/epi.13193
Dathan-Stumpf A, Vogel M, Hiemisch A, Thiery J, Burkhardt R, Kratzsch J et al (2016) Pediatric reference data of serum lipids and prevalence of dyslipidemia: results from a population-based cohort in Germany. Clin Biochem 49:740–749. https://doi.org/10.1016/j.clinbiochem.2016.02.010
DeBarber AE, Eroglu Y, Merkens LS, Pappu AS, Steiner RD (2011) Smith–Lemli–Opitz syndrome. Expert Rev Mol Med 13:e24. https://doi.org/10.1017/S146239941100189X
Dreha-Kulaczewski SF, Brockmann K, Henneke M, Dechent P, Wilken B, Gärtner J et al (2012) Assessment of myelination in hypomyelinating disorders by quantitative MRI. J Magn Reson Imaging 36:1329–1338. https://doi.org/10.1002/jmri.23774
Edgar JM, McCulloch MC, Montague P, Brown AM, Thilemann S, Pratola L et al (2010) Demyelination and axonal preservation in a transgenic mouse model of Pelizaeus–Merzbacher disease. EMBO Mol Med 2:42–50. https://doi.org/10.1002/emmm.200900057
Fledrich R, Abdelaal T, Rasch L, Bansal V, Schutza V, Brügger B et al (2018) Targeting myelin lipid metabolism as a potential therapeutic strategy in a model of CMT1A neuropathy. Nat Commun 9:3025. https://doi.org/10.1038/s41467-018-05420-0
Freeman J, Veggiotti P, Lanzi G, Tagliabue A, Perucca E, Institute of Neurology ICMF (2006) The ketogenic diet: from molecular mechanisms to clinical effects. Epilepsy Res 68:145–180
Fünfschilling U, Supplie LM, Mahad D, Boretius S, Saab AS, Edgar J et al (2012) Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature 485:517–521. https://doi.org/10.1038/nature11007
Gruenenfelder FI, Thomson G, Penderis J, Edgar JM (2011) Axon-glial interaction in the CNS: what we have learned from mouse models of Pelizaeus–Merzbacher disease. J Anat 219:33–43. https://doi.org/10.1111/j.1469-7580.2011.01363.x
Helms G, Dathe H, Dechent P (2008) Quantitative FLASH MRI at 3T using a rational approximation of the Ernst equation. Magn Reson Med 59:667–672. https://doi.org/10.1002/mrm.21542
Helms G, Dathe H, Dechent P (2010) Modeling the influence of TR and excitation flip angle on the magnetization transfer ratio (MTR) in human brain obtained from 3D spoiled gradient echo MRI. Magn Reson Med 64:177–185. https://doi.org/10.1002/mrm.22379
Helms G, Dathe H, Kallenberg K, Dechent P (2008) High-resolution maps of magnetization transfer with inherent correction for RF inhomogeneity and T1 relaxation obtained from 3D FLASH MRI. Magn Reson Med 60:1396–1407. https://doi.org/10.1002/mrm.21732
Hogan V, White K, Edgar J, McGill A, Karim S, McLaughlin M et al (2009) Increase in mitochondrial density within axons and supporting cells in response to demyelination in the Plp1 mouse model. J Neurosci Res 87:452–459. https://doi.org/10.1002/jnr.21867
Iyoda K, Tanaka J, Suzuki Y, Nagao Y, Ohtahara S (1988) Histopathologic and biochemical analysis of classic Pelizaeus–Merzbacher disease. Pediatr Neurol 4:252–254
Keys A, Mickelsen O, Ev M, Chapman CB (1950) The relation in man between cholesterol levels in the diet and in the blood. Science 112:79–81
Kippert A, Trajkovic K, Rajendran L, Ries J, Simons M (2007) Rho regulates membrane transport in the endocytic pathway to control plasma membrane specialization in oligodendroglial cells. J Neurosci 27:3560–3570. https://doi.org/10.1523/JNEUROSCI.4926-06.2007
Koper JW, Lopes-Cardozo M, Van Golde LM (1981) Preferential utilization of ketone bodies for the synthesis of myelin cholesterol in vivo. Biochim Biophys Acta 666:411–417
Kornek B, Storch MK, Weissert R, Wallstroem E, Stefferl A, Olsson T et al (2000) Multiple sclerosis and chronic autoimmune encephalomyelitis: a comparative quantitative study of axonal injury in active, inactive, and remyelinated lesions. Am J Pathol 157:267–276. https://doi.org/10.1016/S0002-9440(10)64537-3
Kuhlmann T, Lingfeld G, Bitsch A, Schuchardt J, Brück W (2002) Acute axonal damage in multiple sclerosis is most extensive in early disease stages and decreases over time. Brain 125:2202–2212
Laukka JJ, Kamholz J, Bessert D, Skoff RP (2016) Novel pathologic findings in patients with Pelizaeus–Merzbacher disease. Neurosci Lett 627:222–232. https://doi.org/10.1016/j.neulet.2016.05.028
Lee Y, Morrison BM, Li Y, Lengacher S, Farah MH, Hoffman PN et al (2012) Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature 487:443–448. https://doi.org/10.1038/nature11314
Leino RL, Gerhart DZ, Duelli R, Enerson BE, Drewes LR (2001) Diet-induced ketosis increases monocarboxylate transporter (MCT1) levels in rat brain. Neurochem Int 38:519–527. https://doi.org/10.1016/S0197-0186(00)00102-9
Liemburg-Apers DC, Willems PH, Koopman WJ, Grefte S (2015) Interactions between mitochondrial reactive oxygen species and cellular glucose metabolism. Arch Toxicol 89:1209–1226. https://doi.org/10.1007/s00204-015-1520-y
Matsushima GK, Morell P (2001) The neurotoxicant, cuprizone, as a model to study demyelination and remyelination in the central nervous system. Brain Pathol 11:107–116
Mayer JA, Griffiths IR, Goldman JE, Smith CM, Cooksey E, Radcliff AB et al (2015) Modeling the natural history of Pelizaeus–Merzbacher disease. Neurobiol Dis 75:115–130. https://doi.org/10.1016/j.nbd.2014.12.023
Meyer N, Richter N, Fan Z, Siemonsmeier G, Pivneva T, Jordan P et al (2018) Oligodendrocytes in the mouse corpus callosum maintain axonal function by delivery of glucose. Cell Rep 22:2383–2394. https://doi.org/10.1016/j.celrep.2018.02.022
Möbius W, Cooper B, Kaufmann WA, Imig C, Ruhwedel T, Snaidero N et al (2010) Electron microscopy of the mouse central nervous system. Methods Cell Biol 96:475–512. https://doi.org/10.1016/S0091-679X(10)96020-2
Naseebullah Siddiqui M, Saeed S, Chaudary Y (2014) A rare case of Palizaeus Merzbacher disease in a female patient diagnosed radiologically. J Pak Med Assoc 64:1313–1314
Neishabouri AM, Faisal AA (2011) The metabolic efficiency of myelinated vs unmyelinated axons. BMC Neurosci 12:P100. https://doi.org/10.1186/1471-2202-12-S1-P100
Nguyen HB, Sui Y, Thai TQ, Ikenaka K, Oda T, Ohno N (2018) Decreased number and increased volume with mitochondrial enlargement of cerebellar synaptic terminals in a mouse model of chronic demyelination. Med Mol Morphol. https://doi.org/10.1007/s00795-018-0193-z
Olson CA, Vuong HE, Yano JM, Liang QY, Nusbaum DJ, Hsiao EY (2018) The gut microbiota mediates the anti-seizure effects of the ketogenic diet. Cell 173(1728–1741):e1713. https://doi.org/10.1016/j.cell.2018.04.027
Palumbo S, Bosetti F (2013) Alterations of brain eicosanoid synthetic pathway in multiple sclerosis and in animal models of demyelination: role of cyclooxygenase-2. Prostaglandins Leukot Essent Fatty Acids 89:273–278. https://doi.org/10.1016/j.plefa.2013.08.008
Picard M, Turnbull DM (2013) Linking the metabolic state and mitochondrial DNA in chronic disease, health, and aging. Diabetes 62:672–678. https://doi.org/10.2337/db12-1203
Praet J, Guglielmetti C, Berneman Z, Van der Linden A, Ponsaerts P (2014) Cellular and molecular neuropathology of the cuprizone mouse model: clinical relevance for multiple sclerosis. Neurosci Biobehav Rev 47:485–505. https://doi.org/10.1016/j.neubiorev.2014.10.004
Puchalska P, Crawford PA (2017) Multi-dimensional roles of ketone bodies in fuel metabolism, signaling, and therapeutics. Cell Metab 25:262–284. https://doi.org/10.1016/j.cmet.2016.12.022
Readhead C, Schneider A, Griffiths I, Nave KA (1994) Premature arrest of myelin formation in transgenic mice with increased proteolipid protein gene dosage. Neuron 12:583–595. https://doi.org/10.1016/0896-6273(94)90214-3
Reiber H (2003) Proteins in cerebrospinal fluid and blood: barriers, CSF flow rate and source-related dynamics. Restor Neurol Neurosci 21:79–96
Ruiz M, Begou M, Launay N, Ranea-Robles P, Bianchi P, Lopez-Erauskin J et al (2017) Oxidative stress and mitochondrial dynamics malfunction are linked in Pelizaeus–Merzbacher disease. Brain Pathol. https://doi.org/10.1111/bpa.12571
Saab AS, Tzvetavona ID, Trevisiol A, Baltan S, Dibaj P, Kusch K et al (2016) Oligodendroglial NMDA receptors regulate glucose import and axonal energy metabolism. Neuron 91:119–132. https://doi.org/10.1016/j.neuron.2016.05.016
Saher G, Brügger B, Lappe-Siefke C, Möbius W, Tozawa R, Wehr MC et al (2005) High cholesterol level is essential for myelin membrane growth. Nat Neurosci 8:468–475. https://doi.org/10.1038/nn1426
Saher G, Quintes S, Möbius W, Wehr MC, Krämer-Albers EM, Brügger B et al (2009) Cholesterol regulates the endoplasmic reticulum exit of the major membrane protein P0 required for peripheral myelin compaction. J Neurosci 29:6094–6104. https://doi.org/10.1523/JNEUROSCI.0686-09.2009
Saher G, Rudolphi F, Corthals K, Ruhwedel T, Schmidt KF, Löwel S et al (2012) Therapy of Pelizaeus–Merzbacher disease in mice by feeding a cholesterol-enriched diet. Nat Med. https://doi.org/10.1038/nm.2833
Sarret C, Lemaire JJ, Sontheimer A, Coste J, Savy N, Pereira B et al (2018) Brain diffusion imaging and tractography to distinguish clinical severity of human PLP1-related disorders. Dev Neurosci. https://doi.org/10.1159/000492218
Sarret C, Lemaire JJ, Tonduti D, Sontheimer A, Coste J, Pereira B et al (2016) Time-course of myelination and atrophy on cerebral imaging in 35 patients with PLP1-related disorders. Dev Med Child Neurol 58:706–713. https://doi.org/10.1111/dmcn.13025
Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T et al (2012) Fiji: an open-source platform for biological-image analysis. Nat Methods 9:676–682. https://doi.org/10.1038/nmeth.2019
Simeone TA, Simeone KA, Stafstrom CE, Rho JM (2018) Do ketone bodies mediate the anti-seizure effects of the ketogenic diet? Neuropharmacology 133:233–241. https://doi.org/10.1016/j.neuropharm.2018.01.011
Simons M, Krämer EM, Macchi P, Rathke-Hartlieb S, Trotter J, Nave KA et al (2002) Overexpression of the myelin proteolipid protein leads to accumulation of cholesterol and proteolipid protein in endosomes/lysosomes: implications for Pelizaeus–Merzbacher disease. J Cell Biol 157:327–336. https://doi.org/10.1083/jcb.200110138
Simons M, Krämer EM, Thiele C, Stoffel W, Trotter J (2000) Assembly of myelin by association of proteolipid protein with cholesterol- and galactosylceramide-rich membrane domains. J Cell Biol 151:143–154
Sinha K, Karimi-Abdolrezaee S, Velumian AA, Fehlings MG (2006) Functional changes in genetically dysmyelinated spinal cord axons of shiverer mice: role of juxtaparanodal Kv1 family K + channels. J Neurophysiol 95:1683–1695. https://doi.org/10.1152/jn.00899.2005
Stys PK, Ransom BR, Waxman SG (1991) Compound action potential of nerve recorded by suction electrode: a theoretical and experimental analysis. Brain Res 546:18–32
Sumida K, Inoue K, Takanashi J, Sasaki M, Watanabe K, Suzuki M et al (2016) The magnetic resonance imaging spectrum of Pelizaeus–Merzbacher disease: a multicenter study of 19 patients. Brain Dev 38:571–580. https://doi.org/10.1016/j.braindev.2015.12.007
Torii T, Miyamoto Y, Yamauchi J, Tanoue A (2014) Pelizaeus–Merzbacher disease: cellular pathogenesis and pharmacologic therapy. Pediatr Int 56:659–666. https://doi.org/10.1111/ped.12450
Tracey TJ, Steyn FJ, Wolvetang EJ, Ngo ST (2018) Neuronal lipid metabolism: multiple pathways driving functional outcomes in health and disease. Front Mol Neurosci 11:10. https://doi.org/10.3389/fnmol.2018.00010
Trevisiol A, Saab AS, Winkler U, Marx G, Imamura H, Möbius W et al (2017) Monitoring ATP dynamics in electrically active white matter tracts. Elife. https://doi.org/10.7554/elife.24241
van Berkel AA, IJff DM, Verkuyl JM (2018) Cognitive benefits of the ketogenic diet in patients with epilepsy: a systematic overview. Epilepsy Behav 87:69–77. https://doi.org/10.1016/j.yebeh.2018.06.004
van der Knaap MS, Valk J, Barkhof F (2005) Magnetic resonance of myelination and myelin disorders, 3rd edn. Springer, Berlin
W. H. O. Multicentre Growth Reference Study Group (2006) WHO Motor Development Study: windows of achievement for six gross motor development milestones. Acta Paediatr Suppl 450:86–95
Wood PL, Smith T, Pelzer L, Goodenowe DB (2011) Targeted metabolomic analyses of cellular models of Pelizaeus–Merzbacher disease reveal plasmalogen and myo-inositol solute carrier dysfunction. Lipids Health Dis 10:102. https://doi.org/10.1186/1476-511X-10-102
Woodward KJ (2008) The molecular and cellular defects underlying Pelizaeus–Merzbacher disease. Expert Rev Mol Med 10:e14. https://doi.org/10.1017/S1462399408000677
Yoshikawa K, Palumbo S, Toscano CD, Bosetti F (2011) Inhibition of 5-lipoxygenase activity in mice during cuprizone-induced demyelination attenuates neuroinflammation, motor dysfunction and axonal damage. Prostaglandins Leukot Essent Fatty Acids 85:43–52. https://doi.org/10.1016/j.plefa.2011.04.022
Acknowledgements
Open access funding provided by Max Planck Society. We thank Annette Fahrenholz, Silvia Thüne, and Tanja Pawels for excellent technical support. Funding: This study is supported by the European Leukodystrophy Association and the Deutsche Forschungsgemeinschaft (to GS). WM is supported by Deutsche Forschungsgemeinschaft Cluster of Excellence Research Center Nanoscale Microscopy and Molecular Physiology of the Brain. KAN is supported by a European Research Council Advanced Grant. AB is supported by “Freunde der Kinderklinik des UK Eppendorf eV”.
Author information
Authors and Affiliations
Contributions
SKS and SAB designed the study and were involved in all experiments, AT did electrophysiological recordings. LS, TD, LVS, LS, and DB contributed to data acquisition. SDK, AB, JD, PG, and JG performed PMD studies. KK made the MCT1 antibody, TR and WM performed electron microscopic analysis. HR and MM performed volume analysis of mitochondria. KAN edited the manuscript, and Gesine Saher designed the study, analyzed data and wrote the manuscript. All authors approved the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors have nothing to declare.
Animal studies
All animal studies were performed in compliance with the animal policies of the Max Planck Institute of Experimental Medicine, and were approved by the German Federal State of Lower Saxony.
Human studies
The cholesterol supplementation in PMD patients was started as an individual trial following the general guidelines and ethical standards established by the Universities Medicine Göttingen and Hamburg. Research based MRI studies were approved by the local Ethics Committees of the Universities Medicine Gttingen and Hamburg. Informed consents were obtained from the parents.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Stumpf, S.K., Berghoff, S.A., Trevisiol, A. et al. Ketogenic diet ameliorates axonal defects and promotes myelination in Pelizaeus–Merzbacher disease. Acta Neuropathol 138, 147–161 (2019). https://doi.org/10.1007/s00401-019-01985-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-019-01985-2