
Abstract
Nodding syndrome is an epidemic neurologic disorder of unknown cause that affects children in the subsistence-farming communities of East Africa. We report the neuropathologic findings in five fatal cases (13–18 years of age at death) of nodding syndrome from the Acholi people in northern Uganda. Neuropathologic examination revealed tau-immunoreactive neuronal neurofibrillary tangles, pre-tangles, neuropil threads, and dot-like lesions involving the cerebral cortex, subcortical nuclei and brainstem. There was preferential involvement of the frontal and temporal lobes in a patchy distribution, mostly involving the crests of gyri and the superficial cortical lamina. The mesencephalopontine tegmental nuclei, substantia nigra, and locus coeruleus revealed globose neurofibrillary tangles and threads. We conclude that nodding syndrome is a tauopathy and may represent a newly recognized neurodegenerative disease.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Nodding syndrome (NS) is a neurologic disorder of children in East Africa. NS is characterized by stereotypical head dropping movements, cognitive impairment, impaired growth, and seizures [2, 4, 5, 7, 16, 19, 23, 24]. The age of onset is usually 5–15 years of age [5]. Although NS has been documented in the Republic of South Sudan [2, 20, 23, 24] and Tanzania [19], it is currently a major health problem in the subsistence-farming villages of the Acholi people in northern Uganda [5, 7, 16]. NS emerged as an epidemic in the Kitgum district of Uganda in 1998 [5, 11] during internal armed conflict and displacement of children into camps. Over 3000 NS-affected children have been documented in northern Uganda [11]. Clinical descriptions of NS have focused on the seizures that are found in the disorder [2, 4, 7, 16, 19]. However, relentless neurologic deterioration [19] and death are often reported.
The cause of NS is not known. Different causal theories have been proposed including: onchocerciasis [4, 5, 20, 24], neurotropic virus [18], war-induced post-traumatic stress disorder [15], and environmental and nutritional factors [18, 20]. The current leading and controversial theory is that NS may be due to an autoimmune reaction to leiomodin-1 on the basis of a shared epitope in Onchocerca volvulus and the human brain [8].
No clinicopathologic studies have been published on NS. The aim of the current study was to investigate the neuropathologic basis of NS. On this basis, we now report the neuropathologic and immunohistochemical hallmarks of NS in five cases. We present histologic evidence the NS is a novel tauopathy.
Methods
Subjects
Five cases of NS who died in their villages in northern Uganda, or in a local hospital, in 2014 and 2017 were used for this study. All cases fulfilled the consensus-based diagnostic criteria for probable or confirmed NS [4]. Clinical history was obtained at the time of autopsy, or retrospective interview of the family (Table 1). Permission to conduct autopsies and publish the results was obtained from the affected families.
Histologic preparation
Histologic sections were obtained from formalin-fixed brains. In some cases, the brains had already been sectioned and blocks had already been sampled. The cerebrum was cut in the coronal plane, the brainstem sectioned in the horizontal plane, and the cerebellum sectioned sagittally and para-sagittally. Standardized blocks for paraffin embedding were obtained, if available, from the following areas: frontopolar cortex (Brodmann area 10); cingulate gyrus at the level of the head of the caudate nucleus; superior and middle frontal gyri at the level of the anterior striatum; superior and middle temporal gyrus at the lateral geniculate body; amygdala at the level of the anterior commissure; hippocampus and hippocampal gyrus; anterior striatum; inferior parietal lobule; calcarine cortex (Brodmann area 17); thalamus including subthalamic nucleus; midbrain at the level of the red nucleus; pons at the level of the locus coeruleus; medulla at the level of the olive; caudal medulla at the level of the nucleus cuneatus and gracilis; and cerebellum including the dentate nucleus. Sections were prepared for routine staining with Luxol fast blue with hematoxylin and eosin. Selected sections were stained with the Bielschowsky method.
Immunohistochemistry
All brain areas were immunohistochemically studied with an antibody to abnormally phosphorylated tau (AT8, Invitrogen), using the immunoperoxidase technique. Selected brains areas including the cerebral cortex, brainstem and cerebellum were also immunostained with antibodies to α-synuclein (LB509, Novex), α-amyloid (6F/3D, Dako), and phosphorylated TDP-43 (MABN14, Milliepore). Sections of cerebral cortex from cases 2 were immunostained with antibodies to 3-repeat tau (RD3, Milliepore) and 4-repeat tau (RD4, Milliepore). Brain tissue from a case of Alzheimer’s disease, Lewy body-type Parkinson’s disease and frontotemporal degeneration with TDP-43 positive-inclusions were used as positive controls for immunoreactivity for the appropriate antibody. The primary antibody was omitted from the immunoperoxidase method for the negative control. The severity of AT8-immunoreactivity was graded on a four point scale: 0—none; +—mild; ++—moderate; and +++—marked.
Results
Clinical characteristics
All five cases derived from northern Uganda from the three districts inhabited by the Acholi people: Kitgum, Pader and Gulu (Fig. 1). The cases ranged from 13 to 18 years of age at death with three males and two females (Table 1). In all cases, the clinical symptoms were classical for NS with head dropping spells (nodding), cognitive impairment, seizures, and neurologic deterioration resulting in terminal wasting in four cases, and sudden death in one case. None of patients had chronic neurologic symptoms or signs before the onset of the symptoms of NS. All patients were treated with the anti-epileptic drugs carbamazepine and valproic acid. All patients developed symptoms following the height of the recent insurgency in northern Uganda and resided in camps for internally displaced people [11] at some time in the course of their illness. Three of the affected children had multiple injuries complicating seizure-related falls. Four of the children were malnourished and wasted due to the inability to swallow at the end stage of the illness. One child apparently died of aspiration of gastric contents secondary to a seizure (case 2), whereas the others died of a lung abscess or malnutrition.

Three districts in northern Uganda affected by nodding syndrome [11]
Neuropathologic findings
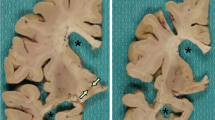
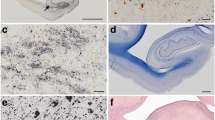
All five cases showed the same pathologic findings in the same anatomical distribution with some variation in severity (Table 2). Macroscopically, there was mild frontotemporal cortical atrophy. On routine staining, the cerebral cortex showed superficial microvacuolation with neuronal neurofibrillary tangles, in all cases, with patchy frontal cortical status spongiosus (cases 2–5). The tangles ranged from argentophilic cytoplasmic wisps to classical flame-shaped tangles. Immunostaining for phosphorylated tau revealed tangles, pre-tangles (diffuse perikaryal and dendritic immunoreactivity), dot-like immunoreactivity in the neuropil, and neuropil threads (Fig. 2). The tangles, pre-tangles, dot-like lesions and threads were found mostly in the upper cortical lamina and tended to cluster. These changes were extensive but multifocally distributed in discontinuous bands, chiefly involving gyral crowns and less frequently in the depths of sulci. There was no perivascular predilection. Ghost tangles were often found in the superficial lamina. In areas of neocortex with the most tangles, all cortical lamina contained tangles with extensive involvement of pyramidal cells. Tangles, pre-tangles, dot-like lesions and threads were differentially distributed in the cortex. The most severe changes were present in the prefrontal cortex, and the superior and middle frontal gyri. The superior and middle temporal gyri and focally the parietal cortex were extensively involved. The occipital lobe was relatively spared. There was complete sparing of the hippocampus with minimal involvement of the cingulate gyrus, amygdala and hippocampal gyrus.

Histologic findings in nodding syndrome. a Tau-immunoreactive in frontal cortex, mostly in gryal crowns (AT8, scale bar: 1000 μm). b Cortical neurofibrillary tangles (Bielschowsky stain, scale bar: 100 μm). c Neurofibrillary tangles, dystrophic neurites and dot-like immunoreactivity containing phosphorylated tau in cerebral cortex (AT8, scale bar: 200 μm). d Neurofibrillary tangles and dystrophic neurites in neurons in the base of pons (AT8, scale bar: 75 μm)
Globose tangles, pre-tangles and threads were found in various subcortical and brainstem sites (Fig. 2). The tegmental nuclei of the upper brainstem, including the Edinger–Westphal nucleus, substantia nigra, locus coeruleus and the nuclei of the pontine base were preferentially affected. Less extensive formation of tangles, pre-tangles and threads was found in the striatum, lentiform nuclei thalamus and medulla. There was relatively minimal or no involvement of the olives or dentate nuclei. Patchy to extensive Purkinje cell loss with Bergman gliosis and empty baskets in the cerebellum were found in three cases (cases 2, 3 and 5). There was moderate diffuse pallor of myelin staining in one case with preferential involvement of subcortical white matter and the pontocerebellar fibres (case 2). In severely involved areas of neocortex, there were tau-immunoreactive threads in subcortical white matter. Glial cellular inclusions were not prominent in any brain; however, occasional fuzzy/granular astrocytes were found in the locus coeruleus in case 4.
The tangles were immunoreactive with antibodies to 3-repeat and 4-repeat tau. No immunostaining was found with antibodies to α-synuclein, TDP-43, or β-amyloid. Other pertinent negative observations include no ballooned neurons, no Pick bodies, no Lewy bodies, no neuritic plaques, and no spongiform change. The pathologic features of subacute sclerosing panencephalitis, or other viral or autoimmune encephalitis were not present.
Discussion
The hallmarks of NS include widespread formation of tangles, pre-tangles, neuropil threads and dot-like tau deposits in the cerebral cortex and brainstem with lesser involvement of the basal ganglia. The cortical involvement shows a frontotemporal predilection but parietal lobe involvement is also present. The prefrontal cortex and the anterior frontal lobe are the most severely affected. The brainstem involvement is principally centred on the substantia nigra, locus coeruleus, tegmental nuclei and the nuclei of the pontine base. These findings indicate that NS is a tauopathy. Based on the progressive clinical course and the nature of the histologic findings, NS may be a newly recognized neurodegenerative disease. Cerebellar degeneration was also present in three of the five cases, and could be explained by four different mechanisms: the direct effect of the NS, hypoxia secondary to seizures, anti-epileptic medication, or a combination of mechanisms. However, the cerebellar degeneration is likely related to the anti-epileptic medication because the severity of the tau-burden did not correlate with presence of cerebellar degeneration, (e.g., case 4 had a substantial tau-burden, but lacked cerebellar degeneration) and cerebellar ataxia is not a documented clinical feature of NS.
It is interesting to compare and contrast NS with other tauopathies. The distribution and cellular pathology in NS does not precisely match any other known tauopathy. The patchy cortical involvement in NS is similar to chronic traumatic encephalopathy [13]. However, the distribution of pathology within the cortex is different. In NS, the crests of gyri, rather than the depths of sulci, are preferentially involved and there is no perivascular predilection [12]. In addition, there is some overlap between the cortical deposition of tau in NS and temporal lobe epilepsy [21]. However, numerous cortical and subcortical tangles are not reported to occur in epilepsy, but these are distinctive findings in NS. Furthermore, a detailed mapping of tangles and tau-immunoreactivity, beyond the temporal lobe, in cases of epilepsy has found a propensity for a limbic distribution [22]. There is dissimilarity in the distribution of pathology of our five cases of NS. Thus, it is unclear if repeated seizures can explain the widespread tauopathy in NS. One possibility is that the cortical tauopathy form epileptogenic foci that cause the ongoing seizures in NS. However, if repeated seizures can cause widespread cortical and subcortical tangles and other tau-immunoreactive lesions, then this could be an alternate explanation.
NS also shares some pathological features with progressive supranuclear palsy due to a similar distribution of tangles [3]. However, the glial pathology in this series of NS cases is limited and was represented by granular/fuzzy astrocytes in the locus coeruleus in case 4. In addition, the tau deposits had both 3-repeat and 4-repeat tau isoforms, rather than selective enrichment of 4-repeat tau that occurs in progressive supranuclear palsy [1]. Case 4 was the oldest child in the series and had the longest duration of disease. The presence of granular/fuzzy astrocytes is similar to that described in ageing-related tau astrogliopathy [10]. Further characterization of the potential spectrum of astrogliopathy in a larger number of NS cases, at different stages of disease, is indicated. In particular, it will be interesting to determine if granular/fuzzy astrocytes develop into tufted astrocytes [9].
The clinical descriptions of NS have described cognitive impairment [16] and Parkinsonism [19]. But, no studies have described the late stages of disease progression in NS. Although the tegmentum, locus coeruleus and substantia nigra contain tangles and threads, neuronal loss and gliosis were not prominent features in five cases reported herein. Further histologic studies of the brainstem of in a larger cases series of NS with clinicopathologic correlation will be informative. Specifically, neurologic assessment for supranuclear palsy and extra-pyramidal signs in late-stage cases of NS with correlation with brainstem lesions will be important.
There are both similarities and differences between NS and the classical disease described by Hirano on Guam [6], and that was also later recognized in other Western Pacific foci [17]. The main similarities are that both disorders emerged as epidemics in indigenous people at a particular time and place, and both diseases have abundant tangles. In addition, both diseases are fading epidemics of unknown cause. The main differences are that NS appears to be purely a tauopathy, whereas the Guam disease is a polyproteinopathy [14], and the age of onset and clinical presentations are different. In addition, the Guam disease has extensive limbic involvement [6], whereas NS does not.
The cause of NS is not apparent from our neuropathological findings. However, there was no histologic evidence of viral or autoimmune encephalitis, or cerebral parasitism. The overall pathological process was neurodegeneration with tau-deposition. Therefore, if NS is caused by infection with a virus, or other infectious agent, then the nexus is unlikely through the pathogenic mechanisms that cause a post-encephalitic state, as occurs in measles infections leading to subacute sclerosing panencephalitis.
In conclusion, we have shown that the epidemic neglected tropical disease known as NS is a tauopathy. On this basis, NS may be a newly recognized neurodegenerative disease with a regional cluster in East Africa. The discovery that NS is a tauopathy may facilitate determining the cause of NS, because it opens up new lines of inquiry that were unavailable prior to our neuropathologic studies.
References
Arai T, Ikeda K, Akiyama H, Shikamoto Y, Tsuchiya K, Yagishita S, Beach T, Rogers J, Schwab C, McGeer PL (2001) Distinct isoforms of tau aggregated in neurons and glial cells in brains of patients with Pick’s disease, corticobasal degeneration and progressive supranuclear palsy. Acta Neuropathol 101:167–173
de Polo G, Romaniello R, Otim A, Benjamin K, Bonanni P, Borgatti R (2015) Neurophysiological and clinical findings on Nodding Syndrome in 21 South Sudanese children and a review of the literature. Seizure 31:64–71
Dickson DW, Kouri N, Murray ME, Josephs KA (2011) Neuropathology of frontotemporal lobar degeneration-tau (FTLD-tau). J Mol Neurosci 45:384–389
Dowell SF, Sejvar JJ, Riek L, Vandemaele KA, Lamunu M, Kuesel AC, Schmutzhard E, Matuja W, Bunga S, Foltz J, Nutman TB, Winkler AS, Mbonye AK (2013) Nodding syndrome. Emerg Infect Dis 19:1374–1384
Foltz JL, Makumbi I, Sejvar JJ, Malimbo M, Ndyomugyenyi R, Atai-Omoruto AD, Alexander LN, Abang B, Melstrom P, Kakooza AM, Olara D, Downing RG, Nutman TB, Dowell SF, Lwamafa DK (2013) An epidemiologic investigation of potential risk factors for Nodding Syndrome in Kitgum District. PLoS one, Uganda. https://doi.org/10.1371/journal.pone.0066419
Hirano A, Malamud N, Kurland LT (1961) Parkinsonism-dementia complex, an endemic disease on the island of Guam. II. Pathological features. Brain 84:662–679
Idro R, Opoka RO, Aanyu HT, Kakooza-Mwesige A, Piloya-Were T, Namusoke H, Musoke SB, Nalugya J, Bangirana P, Mwaka AD, White S, Chong K, Atai-Omoruto AD, Mworozi E, Nankunda J, Kiguli S, Aceng JR, Tumwine JK (2013) Nodding syndrome in Ugandan children–clinical features, brain imaging and complications: a case series. BMJ Open. https://doi.org/10.1136/bmjopen-2012-002540
Johnson TP, Tyagi R, Lee PR, Lee MH, Johnson KR, Kowalak J, Elkahloun A, Medynets M, Hategan A, Kubofcik J, Sejvar J, Ratto J, Bunga S, Makumbi I, Aceng JR, Nutman TB, Dowell SF, Nath A (2017) Nodding syndrome may be an autoimmune reaction to the parasitic worm Onchocerca volvulus. Sci Transl Med. https://doi.org/10.1126/scitranslmed.aaf6953
Kovacs GG, Robinson JL, Xie SX, Lee EB, Grossman M et al (2017) Evaluating the patterns of aging-related tau astrogliopathy unravels novel insights into brain aging and neurodegenerative diseases. J Neuropathol Exp Neurol 76:270–288
Kovacs GG, Xie SX, Lee EB, Robinson JL, Caswell C et al (2017) Multisite assessment of aging-related tau astrogliopathy (ARTAG). J Neuropathol Exp Neurol 76:605–619
Landis JL, Palmer VS, Spencer PS (2014) Nodding syndrome in Kitgum District, Uganda: association with conflict and internal displacement. BMJ Open. https://doi.org/10.1136/bmjopen-2014-006195
McKee AC, Cairns NJ, Dickson DW, Folkerth RD, Keene CD et al (2015) The first NINDS/NIBIB consensus meeting to define neuropathological criteria for the diagnosis of chronic traumatic encephalopathy. Acta Neuropathol 131:75–86
McKee AC, Cantu RC, Nowinski CJ, Hedley-Whyte ET, Gavett BE, Budson AE, Santini VE, Lee HS, Kubilus CA, Stern RA (2009) Chronic traumatic encephalopathy in athletes: progressive tauopathy after repetitive head injury. J Neuropathol Exp Neurol 68:709–735
Mimuro M, Yoshida M, Kuzuhara S, Kokubo Y (2018) Amyotrophic lateral sclerosis and parkinsonism-dementia complex of the Hohara focus of the Kii Peninsula: a multiple proteinopathy? Neuropathology 38:98–107
Musisi S, Akena D, Nakimuli-Mpungu E, Abbo C, Okello J (2013) Neuropsychiatric perspectives on nodding syndrome in northern Uganda: a case series study and a review of the literature. Afr Health Sci. https://doi.org/10.4314/ahs.v13i2.3
Sejvar JJ, Kakooza AM, Foltz JL, Makumbi I, Atai-Omoruto AD, Malimbo M, Ndyomugyenyi R, Alexander LN, Abang B, Downing RG, Ehrenberg A, Guilliams K, Helmers S, Melstrom P, Olara D, Perlman S, Ratto J, Trevathan E, Winkler AS, Dowell SF, Lwamafa D (2013) Clinical, neurological, and electrophysiological features of nodding syndrome in Kitgum, Uganda: an observational case series. Lancet Neurol 12:166–174
Spencer PS, Kisby GE, Ludolph AC (1991) Slow toxins, biologic markers, and long-latency neurodegenerative disease in the western Pacific region. Neurology 41:62–66
Spencer PS, Mazumder R, Palmer VS, Lasarev MR, Stadnik RC et al (2016) Environmental, dietary and case-control study of nodding syndrome in Uganda: a post-measles brain disorder triggered by malnutrition? J Neurol Sci 15:191–203
Spencer PS, Palmer VS, Jilek-Aall L (2013) Nodding syndrome: origins and natural history of a longstanding epileptic disorder in sub-Saharan Africa. Afr Health Sci 13:176–182
Spencer PS, Vandemaele K, Richer M, Palmer VS, Chungong S, Anker M, Ayana Y, Opoka ML, Klaucke BN, Quarello A, Tumwine JK (2013) Nodding syndrome in Mundri county, South Sudan: environmental, nutritional and infectious factors. Afr Health Sci 13:183–204
Tai XY, Koepp M, Duncan JS, Fox N, Thompson P, Baxendale S, Liu JY, Reeves C, Michalak Z, Thom M (2016) Hyperphosphorylated tau in patients with refractory epilepsy correlates with cognitive decline: a study of temporal lobe resections. Brain 139:2441–2455
Thom M, Liu JY, Thompson P, Phadke R, Narkiewicz M, Martinian L, Marsdon D, Koepp M, Caboclo L, Catarino CB, Sisodiya SM (2011) Neurofibrillary tangle pathology and Braak staging in chronic epilepsy in relation to traumatic brain injury and hippocampal sclerosis: a post-mortem study. Brain 134:2969–2981
Tumwine JK, Vandemaele K, Chungong S, Richer M, Anker M, Ayana Y, Opoka ML, Klaucke DN, Quarello A, Spencer PS (2012) Clinical and epidemiologic characteristics of nodding syndrome in Mundri County, southern Sudan. Afr Health Sci 12:242–248
Winkler AS, Friedrich K, König R, Meindl M, Helbok R, Unterberger I, Gotwald T, Dharsee J, Velicheti S, Kidunda A, Jilek-Aall L, Matuja W, Schmutzhard E (2008) The head nodding syndrome–clinical classification and possible causes. Epilepsia 49:2008–2015
Acknowledgements
This project was funded by the Raymond Chang Foundation. We thank Drs. P. Spencer and R. Mazumder for helpful discussions. We acknowledge the Centre for Disease and Control and Prevention for returning nodding syndrome brains to Uganda for neuropathologic examination.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Pollanen, M.S., Onzivua, S., Robertson, J. et al. Nodding syndrome in Uganda is a tauopathy. Acta Neuropathol 136, 691–697 (2018). https://doi.org/10.1007/s00401-018-1909-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-018-1909-9