
Abstract
Mitochondrial complex I deficiency occurs in the substantia nigra of individuals with Parkinson’s disease. It is generally believed that this phenomenon is caused by accumulating mitochondrial DNA damage in neurons and that it contributes to the process of neurodegeneration. We hypothesized that if these theories are correct, complex I deficiency should extend beyond the substantia nigra to other affected brain regions in Parkinson’s disease and correlate tightly with neuronal mitochondrial DNA damage. To test our hypothesis, we employed a combination of semiquantitative immunohistochemical analyses, Western blot and activity measurements, to assess complex I quantity and function in multiple brain regions from an extensively characterized population-based cohort of idiopathic Parkinson’s disease (n = 18) and gender and age matched healthy controls (n = 11). Mitochondrial DNA was assessed in single neurons from the same areas by real-time PCR. Immunohistochemistry showed that neuronal complex I deficiency occurs throughout the Parkinson’s disease brain, including areas spared by the neurodegenerative process such as the cerebellum. Activity measurements in brain homogenate confirmed a moderate decrease of complex I function, whereas Western blot was less sensitive, detecting only a mild reduction, which did not reach statistical significance at the group level. With the exception of the substantia nigra, neuronal complex I loss showed no correlation with the load of somatic mitochondrial DNA damage. Interestingly, α-synuclein aggregation was less common in complex I deficient neurons in the substantia nigra. We show that neuronal complex I deficiency is a widespread phenomenon in the Parkinson’s disease brain which, contrary to mainstream theory, does not follow the anatomical distribution of neurodegeneration and is not associated with the neuronal load of mitochondrial DNA mutation. Our findings suggest that complex I deficiency in Parkinson’s disease can occur independently of mitochondrial DNA damage and may not have a pathogenic role in the neurodegenerative process.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Parkinson’s disease (PD) is the second most common neurodegenerative disorder affecting 1.8% of the population above the age of 65 years [11]. It is characterized by progressive motor dysfunction including bradykinesia, rigidity, resting tremor and postural instability. In addition, a number of non-motor symptoms occur, including autonomic dysfunction, dementia, depression and anxiety. The pathological hallmark of PD is the degeneration of the dopaminergic neurons in the pars compacta of the substantia nigra (SNc) in the presence of α-synuclein immunoreactive neuronal inclusions collectively termed Lewy pathology. Other affected areas comprise multiple brainstem and cholinergic nuclei, the hippocampus and the neocortex. Neuropathologically spared regions include the striatum, cerebellum, thalamus, red nucleus and the pontine nuclei [12].
Although the etiology of PD is unknown, mitochondrial dysfunction has been associated with both familial and idiopathic forms of the disease. In the early 1980s, it was discovered that substances inhibiting complex I of the mitochondrial respiratory chain (MRC) cause nigrostriatal degeneration and parkinsonism [6, 22, 30]. Subsequently, evidence of complex I deficiency was shown in the SNc of individuals with idiopathic PD [25, 35]. While this phenomenon has been consistently reproduced by multiple studies [17, 18, 23, 24, 34] its significance remains undetermined. It is unclear whether complex I deficiency in PD is a primary pathogenic event compromising neuronal integrity, an adaptive response to disease-related stress, or merely a symptom of end-stage dysfunction in terminally ill neurons. The SNc, where this phenomenon has been best characterized, has already lost at least 60% of its dopaminergic neuronal population at symptom onset [9] and suffers end-stage neurodegeneration in the vast majority of autopsy-derived samples. Studies of other brain areas are both limited in number and show conflicting results; while some experiments have found decreased complex I activity in frontal cortex homogenate from individuals with PD [19, 27] others showed no difference between individuals with PD and controls [18, 36]. One recent study found that complex I activity was decreased in prefrontal cortex homogenate from individuals with Parkinson’s disease dementia (PDD), but within normal range in non-demented PD individuals [14]. Complex I activity measurements and/or Western blot quantification in homogenized samples of the cerebellum and striatum have shown normal findings [18, 24, 25, 36]. Other brain areas remain unexplored.
We hypothesized that if complex I deficiency is actively involved in the neurodegenerative process of PD, it should not be limited to the SNc, but extend to other regions of the brain involved in the disease. To address this question, we performed comprehensive, quantitative and functional studies of the MRC in multiple brain regions from a population-based cohort of individuals with validated idiopathic PD and healthy controls. Our findings show that neuronal complex I quantity and function are decreased throughout the PD brain, including mildly affected areas and areas unaffected by neurodegeneration. Intriguingly, we find no correlation between complex I deficiency and mitochondrial DNA (mtDNA) damage, suggesting an alternative etiology.
Materials and methods
Patient cohorts and tissue samples
Brain tissue was available from individuals with idiopathic PD (n = 18) and neurologically healthy controls (Ctrl n = 11). The individuals with PD were obtained from the Norwegian Park West study, a prospective population-based cohort that has been described in detail [2]. Individuals with PD fulfilled the National Institute of Neurological Disorders and Stroke [15] and the UK Parkinson’s disease Society Brain Bank [43] diagnostic criteria for the disease at their final visit. Whole-exome sequencing had been performed on all patients using Roche-NimbleGen Sequence Capture EZ Exome v3 kit and paired-end 100nt sequencing on the Illumina HiSeq platform (unpublished material). To exclude monogenic cases, which could potentially confound our study, genes associated with familial parkinsonism (SNCA, LRRK2, VPS35, EIF4G1, DCTN1, CHCHD2, PARK2, PINK1, PARK7, ATP13A2, PLA2G6, FBXO7, COQ2, SLC6A3) were extracted and screened with no evidence of known pathogenic mutations. In addition, coverage analysis of the SNCA exons showed no evidence of copy number variation. Mitochondrial disease due to known nuclear gene mutations (e.g. POLG, TWNK, OPA1 or nuclear encoded respiratory subunits) was also excluded from the exome data. None of the study participants had clinical signs of mitochondrial disease. Controls had no known neurological disease and were matched for age and gender. Individuals with PD and controls had no history of mitochondrial disorder or use of medication known to influence mitochondrial function (Supplementary Table 1).
Brains were collected at autopsy and split sagittally along the corpus callosum. One hemisphere was fixed whole in formaldehyde and the other coronally sectioned and snap-frozen in liquid nitrogen. All samples were collected using a standard technique and fixation time of ~ 2 weeks. There was no significant difference in post-mortem (PM) time (independent t test, P = 0.06), age (independent t test, P = 0.17) or gender (Chi-square test P > 0.96) between PD subjects and controls. Subject demographics and tissue availability are summarized in Table 1.
Immunohistochemistry
Immunohistochemistry (IHC) for mitochondrial markers was carried out on formalin-fixed, paraffin-embedded brain tissue from the following areas of individuals with PD and neurologically healthy controls: prefrontal cortex, Brodmann areas 8–9 (PD n = 14, Ctrl n = 6), hippocampus (PD n = 12, Ctrl n = 5), putamen (PD n = 14, Ctrl n = 5), cerebellum (PD n = 14, Ctrl n = 7), mesencephalon at the level of the third cranial nerve (PD n = 8, Ctrl n = 6) and basal pons at the level of the locus coeruleus (PD n = 12, Ctrl n = 5). Serial 3 μm thick sections were stained with primary antibodies (all from Abcam) against complex I (NDUFB8, ab110242, dilution 1:300), complex II (anti-SDHA, ab14715, dilution 1:4000), complex III (anti-UQCRC2, ab14745, dilution 1:10,000), complex IV (COX-I, ab14705, dilution 1:10,000), complex V (ATP-synthase, ab14748, dilution 1:10,000), and the mitochondrial membrane marker porin (VDAC1, ab14734, dilution 1:10,000). We chose the NDUFB8 subunit because it is central to complex I assembly and function [10, 39, 46] and has been widely used as a marker of the integrity of the complex [16, 40, 41]. Sections were deparaffinised in Histoclear and rehydrated in graded ethanol. Antigen retrieval was performed in 1 mM EDTA at pH 8. The sections were blocked in 3% hydrogen peroxide in TBS (tris-buffered saline) and permeabilized in TBS containing 0.1% Tween 20 (TBST). Incubation with primary antibody diluted in TBST was executed at 4 °C overnight. MACH4 Universal HRP (horseradish peroxidase)-polymer (Biocare M4U534) and DAB (3′3-diaminobenzidine) chromogen kit (Biocare DB801) were used for visualization.
Double fluorescent immunohistochemistry was performed for α-synuclein (clone KM51, NCL-L-ASYN, Leica, dilution 1:20) and complex I (NDUFS1, ab169540, Abcam, dilution 1:100) in the SNc (PD n = 4, Ctrl n = 1). Anti-NDUFS1 was chosen here instead of anti-NDUFB8 because it produced better results (stronger signal) in immunofluorescent staining. NDUFS1 is a core subunit considered essential for complex I function [5]. Sections were deparaffinised in Histoclear and rehydrated in graded ethanol. Antigen retrieval was performed in 1 mM EDTA at pH 8. Sections were permeabilized in 1% Triton X-100 in TBS, and blocked in 3% normal goat serum in TBST. Incubation with primary antibody diluted in 3% normal goat serum in TBST was executed at 4 °C overnight. Secondary antibodies (goat anti-rabbit IgG (Alexa Fluor 488, A11008), goat anti-mouse IgG1 (Alexa Fluor 594, A21125) and goat anti-mouse IgG2b (Alexa Fluor 647, A21242); all from Invitrogen) were diluted in 3% normal goat serum in TBST for 1 h at room temperature. DAPI (Invitrogen) was used to visualize the nucleus, and Sudan Black (Sigma Aldrich) was used to quench auto-fluorescence.
Immunohistochemistry analysis
IHC of sections stained with HRP-conjugated secondary antibodies was evaluated visually and quantitatively. First, high-resolution microphotographs were taken at 400× magnification from randomly chosen locations of the following brain regions (the number of photographs taken per region and individual is given in parenthesis): prefrontal cortex (15), hippocampus (12; comprising three from each of CA4, CA3, CA2 and CA1), putamen (5), SNc (five from the pars compacta region), red nucleus (3), dentate nucleus (5), cerebellar cortex (6), basis pontis (3). Analysis of the locus coeruleus was not possible due to severe degeneration in PD. All photographs were taken using a light microscope (Leica) equipped with a Zeiss AxioCam MRc5 camera and Zen 2011 software (Carl Zeiss MicroImaging GmbH, Jena, Germany) under standardized conditions.
For visual assessment, photographed areas were evaluated by three investigators with expertise in mitochondrial staining and neuropathology (IF, CT, ML). For the quantitative analyses, all neurons with a visible nucleolus were cut out from each of the photographed areas, and stacked on a uniform black background using Fiji v2.0.0 [37]. Next, the nucleus was cut out, and photomontages were made for each area including one unstained neuron from a negative control without primary antibody. The colour threshold of each montage was calibrated using the negative control to define absence of staining. Subsequently the number of positive cytoplasmic pixels (i.e. pixels showing colour saturation above the level of the negative control) was counted and a ratio was made between the number of positive and total pixels of neuronal cytoplasm. Thus, the fraction of positively stained cytoplasm of each neuron (i.e. positively stained cytoplasmic area/total cytoplasmic area) was calculated. We termed this positive fractional cytoplasmic area (PFCA) (Fig. 1a–f). To ensure adequate sample size, we analysed a total of 1197 control and 2569 PD neurons. The number of neurons analysed from each individual and region are shown in Table 2.

Determination of the positive fractional cytoplasmic area for semiquantitative IHC analysis. The positive fractional cytoplasmic area (PFCA), i.e. the area percentage of positive cytoplasmic staining, was determined as measure for expression of mitochondrial proteins as follows: Multiple microphotographs were taken at ×400 magnification from each region. As an example, a single photograph from prefrontal cortex is shown (a). All neurons with a visible nucleolus (arrowheads in a) were cut out (b) and the background and nuclei removed leaving only the cytoplasm (c). Neurons from each area were combined into a single photomontage (d) including an unstained negative control as calibrator (*). Subsequently, the total cytoplasmic area (coloured red in e) and positively stained cytoplasmic area compared to the unstained calibrator (coloured red in f) were measured and the ratio of positive to total cytoplasm was used to calculate the PFCA. Due to interference of neuromelanin, the substantia nigra was analysed visually from a montage including two negative (*) and two positive (**) controls (g). Dopaminergic neurons in the substantia nigra were categorised as negative, intermediate or positive. Scale bar: 50 µm
The entire SNc was photographed at 200× magnification and photomontages of individual neurons were constructed as described above. Due to the presence of neuromelanin interfering with automated measurement procedures, the photomontages were evaluated visually by IHF, CD and KH. Investigators were blinded for disease group. A mean of the measurements from the three evaluators was used to determine the percentage of positive, intermediate and negative neurons (Fig. 1g). A total of 374 SNc dopaminergic neurons were evaluated in PD and 767 in controls.
Fluorescent immunohistochemistry for complex I and α-synuclein in the SNc was assessed using a Leica DMRBE fluorescence microscope and a Zeiss AxioCam MRm camera and Zen 2011 software (Carl Zeiss MicroImaging GmbH, Jena, Germany). A total of 123 dopaminergic SNc neurons were assessed from 13 microphotographs, taken at 400× magnification, from each of 4 PD individuals. All neuromelanin containing neurons in the photographs were classified as complex I positive or negative, and Lewy pathology positive or negative. Evaluation was performed visually by IHF, CD and CT.
Complex I enzyme activity measurement
Approximately 100 mg of brain tissue from prefrontal cortex (PD n = 7, Ctrl n = 6), striatum (PD n = 8, Ctrl n = 8), and cerebellar cortex (PD n = 9, Ctrl n = 7) were cut in small pieces and manually homogenized in five volumes of Medium A (0.32 M Sucrose, 10 mM Tris–HCl pH 7.4, 1 mM EDTA). The homogenate was centrifuged at 800g for 5 min at 4 °C. The pellet was discarded and the supernatant containing the enriched mitochondrial fraction was collected. NADH:Coenzyme Q1 oxidoreductase activity was measured in a 96-well plate reader by measuring the decrease in absorbance at 340 nm wave-length, in the absence or presence of rotenone [20, 38]. Citrate synthase activity, an indicator of mitochondrial volume, was measured for normalization [20, 38].
Blue-native gel electrophoresis and immunoblotting
1–1.5 mg of mitochondrial protein from prefrontal cortex (PD n = 11, Ctrl n = 11), striatum (PD n = 6, Ctrl n = 6), and cerebellar cortex (PD n = 8, Ctrl n = 2) were solubilized using 1.6 mg dodecyl-maltoside/mg protein to analyse complex I by blue-native gel electrophoresis and Western blot as described [26, 33, 44]. 50 μg of mitochondrial protein was separated by electrophoresis and complexes I and II were immuno-visualised using anti-NDUFB8 (Abcam ab110242, dilution 1:1000) and anti-SDHB (Abcam ab14714, dilution 1:1000), respectively.
Single neuron laser-microdissection studies
Qualitative and quantitative mtDNA studies were performed in a total of 1189 single laser-microdissected neurons from 20 µm thick sections of fresh-frozen tissue, which had been stained with cresyl violet. The data from the SNc, cerebellum and frontal cortex have been described in detail in our previously published work using consistent subject ID’s [13]. To allow for a more complete correlation with our complex I data, we expanded the experiment to include pyramidal neurons from the CA2 and CA3 area of the hippocampus (47 neurons from nine individuals with PD and 34 neurons from four controls) and principal neurons of the putamen (72 neurons from nine individuals with PD and 65 neurons from eight controls). Total mtDNA copy number and the fraction of major arc deletion were determined using a duplex real-time PCR assay as previously described [13] in 7–12 single neurons per brain region from each PD individual or control.
Statistical analysis
Comparison of neuronal staining levels (PFCA) between PD and controls was performed using linear mixed models analyses, to account for intraindividual random variability and interindividual variation in the number of analysed neurons per region. Subjects were specified as random effects and multiple mixed models were produced to assess the effect of disease state, sex, age of death, and PM-time on neuronal staining levels. These parameters were assessed in our models as fixed effects both individually and in combinations. The error distribution was assumed normal, and solution to the mixed models was obtained using the maximum likelihood estimation method to determine if there were significant differences between the models. The likelihood ratio of different mixed models was sequentially tested using ANOVA. Model selection was based on the results from the ANOVA of model comparison together with visual inspection of the Akaike information criterion (AIC). The mixed-effect model analyses were implemented using the “Linear and Nonlinear Mixed Effects Models” package in R (v3.3.0).
Complex I levels in the SNc were analysed by independent t test to compare the fraction of negative/positive or intermediate neurons in controls vs PD. The association between Lewy pathology and complex I state in the SNc was assessed by Pearson Chi-square test. Densitometric blot measurements in the prefrontal cortex and striatum were compared by Mann–Whitney U test as these were not normally distributed. Statistical analysis of blot data from the cerebellum was not performed due to a small control sample size (n = 2). mtDNA data comparison was performed by linear mixed models. These analyses were carried out in SPSS version 22.0.0 and Prism version 7.0a. Data is presented as mean ± SD. A P < 0.05 was considered statistically significant in all analyses. Correlations with clinical parameters were not undertaken as the sample size did not provide sufficient power to draw definite conclusions.
Results
Widespread neuronal complex I deficiency in the PD brain
The demographic and clinical characteristics of the individuals with PD are summarized in Table 1. To characterize the type and distribution of MRC dysfunction in the PD, we first performed a pilot study of IHC on a randomly selected subset of individuals with PD (n = 3) and controls (n = 3) from our cohorts. Sections from prefrontal cortex, hippocampus, putamen, pons, and SNc were stained for complexes I–V. Visual evaluation of the sections suggested that complex I was decreased in all areas in PD compared to the controls. No differences were detected for complexes II, III, IV and V, in any of the areas with the exception of the SNc where a mild complex IV deficiency was found (Fig. 2a).

Selective complex I deficiency in PD neurons. a MRC complex IHC in the prefrontal cortex of a representative individual with PD and control (magnification ×400, scale bar 50 µm). Staining intensity is representative for all areas examined. b Mean neuronal staining levels per brain region for each MRC complex, expressed as PFCA (bars show SD). c Visual evaluation of the SNc. The fraction (%) of positively stained neurons for each complex is shown. Individuals with PD have a higher fraction of complex I negative neurons. There is also a mild complex IV deficiency with increased levels of intermediate neurons, although negative neurons were not detected. Staining of the neuropil occurs with all MRC complexes as expected. Bars show SD. For columns with no bars, SD = 0, i.e. all neurons were assessed as positive
To allow for accurate comparison between brain regions and individuals, and correlation with clinical variables in our cohort, we applied a standardized semiquantification method to calculate the positive fractional cytoplasmic area (PFCA, details in methods; Fig. 1). Quantification confirmed that individuals with PD had decreased neuronal complex I staining in all areas, whereas complexes II–V stained similar to healthy controls (Fig. 2b).
To confirm and further characterize the anatomical distribution of neuronal complex I changes in a larger sample we performed quantitative analyses of complex I staining in our entire cohort of individuals with PD (n = 17) and controls (n = 7). Linear mixed model analyses showed no significant impact for sex, age of death or PM-time on staining levels. Individuals with PD had significantly decreased neuronal complex I staining in pyramidal neurons of the prefrontal cortex (PD 53.9 ± 32.9%, Ctrl 84.0 ± 13.9%, P = 0.023), pyramidal neurons of the hippocampus CA region (PD 56.6 ± 26.0%, Ctrl 83.8 ± 16.3%, P = 0.018), neurons of the putamen (PD 51.5 ± 29.0%, Ctrl 85.4 ± 9.9%, P = 0.012), Purkinje cells of cerebellar cortex (PD 44.0 ± 23.8%, Ctrl 89.2 ± 14.5%, P < 0.001), and principal neurons of the dentate nucleus (PD 44.0 ± 23.8%, Ctrl 94.6 ± 9.9%, P = 0.021) (Figs. 3 and 4a–e). A trend for lower complex I staining in PD was also seen in the principal neurons of the red nucleus (PD 58.6 ± 33.2%, Ctrl 91.7 ± 11.7%, P = 0.051) and the basis pontis (PD 66.7 ± 36.6%, Ctrl 96.1 ± 3.8%, P = 0.088), although it did not reach statistical significance (Figs. 3 and 4f–g). In the SNc, there was a significantly higher number of complex I negative (PD 29.5 ± 23.6%, Ctrl 7.0 ± 6.1%, P = 0.036) and intermediate (PD 29.1 ± 10.8%, Ctrl 16.0 ± 6.68%, P = 0.019) neurons in PD compared to controls (Figs. 3 and 4h). Individual results of the complex I analysis are summarized in Tables 2 and 3.

Widespread complex I deficiency in the PD brain. Representative photographs of complex I IHC in individuals with PD and controls; a prefrontal cortex, b hippocampus, c putamen, d cerebellar cortex, e dentate nucleus, f pons, g red nucleus, h SNc. Complex I deficiency is visually evident in all areas (except the basal pontine nuclei). Of note, decreased complex I staining in PD was observed not only in the neuronal somata, but also in the surrounding neuropil. a–e Magnification ×400, scale bar 50 µm. h Magnification ×200, scale bar 100 µm

Semiquantitative analyses of complex I deficiency in PD and control neurons. Individuals with PD show significantly decreased neuronal complex I PFCA in the prefrontal cortex (P = 0.023) (a), hippocampus (P = 0.018) (b), putamen (P = 0.012) (c), cerebellar cortex (P < 0.001) (d) and the dentate nucleus (P = 0.021) (e). There is also a clear trend towards lower complex I levels in the basal pontine nuclei (P = 0.088) (f) and the red nucleus (P = 0.051) (g), although not reaching statistical significance. The substantia nigra (h) was analysed visually. There is a significantly higher number of complex I negative (P = 0.036) and intermediate (P = 0.019) neurons and lower number of complex I positive neurons (P = 0.008) in PD. Bars show mean with SD. PFCA positive fractional cytoplasmic area. ns non-significant
Decreased complex I staining was observed also diffusely in the neuropil, the dense network of neuronal processes (dendrites, axons, synapses) and glial cells surrounding neuronal somata. However, our method did not allow us to resolve and differentiate specific components of the neuropil for complex I quantification.
To confirm that the observed complex I deficiency was not due to an overall reduction of mitochondrial mass, we quantified the PFCA for complex II (PD 96.6 ± 6.0%, n = 355, Ctrl 98.0 ± 2.3%, n = 136), and porin (PD 90.3 ± 10.1%, n = 232, Ctrl 87.3 ± 14.2%, n = 119) in sequential sections of the prefrontal cortex from all samples. PD and controls showed similar neuronal staining for complex II (P = 0.32) and porin (P = 0.41).
To further assess the functional impact of neuronal complex I reduction in the PD brain, we performed kinetic measurements of complex I enzymatic activity using fresh frozen brain tissue from the prefrontal cortex, putamen and cerebellum. Individuals with PD had a decreased ratio of complex I to citrate synthase activity in the prefrontal cortex (Ctrl 27.8 ± 7.6, PD 14.4 ± 4.9, P = 0.003) and putamen (Ctrl 12.2 ± 4.1, PD 7.6 ± 3.4, P = 0.036). While the decrease was not significant at the group level in the cerebellum (Ctrl 4.1 ± 2.4, PD 3.2 ± 1.9, P = 0.43), there was high interindividual variation with five of nine PD samples showing substantially lower activity compared to controls (Fig. 5a–c).

Reduced complex I activity in the PD brain. Complex I activity in homogenate from prefrontal cortex (a), striatum (b) and cerebellum (c). Each individual with PD is shown against the mean of controls (Ctrl) with SD. There is a significant decrease of complex I activity in the frontal cortex (P = 0.003) and striatum (P = 0.036) of individuals with PD. The cerebellum shows no significant difference (P = 0.43), but with high variability in the PD group. Individuals with PD 7, 10, 12, 13 and 14 shows substantially lower complex I activity compared to controls. d–k Tissue homogenates from prefrontal cortex (d, f), striatum (e) and cerebellum (g) were separated by blue-native gel electrophoresis, followed by Western blotting and probing for complex I (CI) and complex II (CII). Prefrontal cortex was analysed in two batches (prefrontal I, II) to accommodate gel size. Densitometric analysis showed no significant difference between controls and PD at the group level in any of the examined regions. Prefrontal cortex batch I (P = 0.252) (h), prefrontal cortex batch II (P = 0.429) (i), striatum (P = 0.24) (j). Statistical analysis was not performed in the cerebellum due to a low number of controls (k)
Native Western blot analysis in homogenized tissue also showed lower complex I quantity, both visually and densitometrically in most PD samples. This difference was not statistically significant at the group level, however. Notably, large interindividual variability was seen in the PD group: prefrontal cortex batch 1 (85 ± 8% of Ctrl P = 0.252), prefrontal cortex batch II (87 ± 14% of Ctrl P = 0.429), striatum (95 ± 41% of Ctrl P = 0.24) (Fig. 5d–k).
Complex I deficiency occurs independently of mtDNA changes
While all areas we examined showed neuronal complex I deficiency, only dopaminergic neurons of the SNc harbored significant mtDNA changes in PD in the form of higher deletion levels and wild-type mtDNA depletion [13]. The remaining neuronal populations showed no significant mtDNA differences between PD and controls. Results from the hippocampus and putamen are shown in Fig. 6. Data from the SNc, frontal cortex and cerebellum from the same individuals have been reported previously [13].

Neuronal complex I deficiency does not correlate with mtDNA damage in hippocampus and putamen. Scatter plot showing mtDNA copy number (a, b) and deletion levels (c, d) in single neurons from the putamen and hippocampus. There is no significant difference between controls and individuals with PD for mtDNA copy number or deletion levels in the putamen (Ctrl 15,204 ± 6736, PD 15,927 ± 7453 mtDNA copies/neuron, P = 0.551; Ctrl 19.40 ± 5.80%, PD 17.87 ± 7.28% deleted mtDNA, P = 0.178), or hippocampus (Ctrl 14,770 ± 6118, PD 16,990 ± 7079 mtDNA copies/neuron, P = 0.140; Ctrl 14.69 ± 5.52%, PD 12.91 ± 4.31% deleted mtDNA, P = 0.107). Bars show mean and SD
Lewy pathology is less prevalent in complex I negative neurons
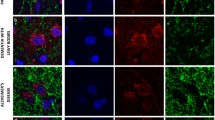
To test for a potential association between Lewy pathology and complex I deficiency, we performed double fluorescent immunohistochemistry for complex I and α-synuclein in the SNc (Fig. 7). Lewy pathology, including Lewy bodies, pale bodies and Lewy neurites, was found throughout the SNc of individuals with PD and was not present in the control. Lewy pathology was significantly less prevalent in complex I negative compared to complex I positive dopaminergic SNc neurons (P = 0.005, OR 0.24, 95% CI 0.09–0.69). The neuronal counts are shown in Table 4.

Lewy pathology is less common in complex I negative neurons. Representative neurons from the SNc of individuals with PD: complex I positive without Lewy pathology (a); complex I negative without Lewy pathology (b); complex I positive with Lewy pathology (c); complex I negative with Lewy pathology (d). Sections have been stained for complex I subunit NDUFS1 (green), alpha-synuclein (red) and the nuclear marker DAPI (blue). Nuclei (#) and somata have been outlined (dotted line) in the merged images. Neuromelanin (*) and Lewy pathology (arrowhead) have been labelled. Scale bar: 15 µm
Discussion
We show that mitochondrial complex I deficiency is not limited to the SNc and other areas affected by neurodegeneration, but is a widespread phenomenon affecting neurons throughout the brain of individuals with PD. Neuronal complex I is decreased both in areas universally affected by neurodegeneration, such as the SNc, areas with mild and/or variable neuronal loss, like the hippocampus and prefrontal cortex, and even areas believed to remain unaffected such as the cerebellar cortex and dentate nucleus [12]. Neuronal complex I deficiency appeared most pronounced in the SNc, putamen and cerebellar cortex, and mildest in the basal pontine nuclei and red nucleus, but regional differences were generally minor and all eight brain regions we examined showed decreased complex I staining. While our measurements in the basal pontine nuclei and red nucleus did not reach statistical significance at the group level, mean complex I PFCA was substantially lower in PD and the lack of significance was attributed to interneuronal and interindividual variation. The remaining respiratory complexes and porin showed no difference between PD and controls suggesting a selective reduction of complex I, rather than a general respiratory chain defect or decrease of total mitochondrial mass.
When interpreting our findings, it is important to be aware of the methodological limitations of IHC as a semiquantitative method. Due to the fact that antigen–antibody reactions are non-stoichiometric and DAB-based staining intensity does not follow the Lambert–Beer law of light absorbance [42], the relationship between staining intensity and antigen quantity is nonlinear. Therefore, direct measurement of staining intensity can lead to over- or under-estimation of the actual difference in neuronal complex I content between samples. To address this limitation, we measured instead the fraction of cytoplasmic immunoreactivity for complex I, i.e. how much of the neuronal cytoplasm is at all stained for complex I (PFCA). The advantage of this approach is that it does not rely on direct quantification of pixel intensity. Instead, each cytoplasmic pixel is binarized into one of two possible states, stained or unstained, and the cytoplasmic fraction that stains positive compared to a negative control is calculated. Thus, while still semiquantitative, our method of PFCA quantification allows an objective and more precise relative quantification of neuronal complex I content between brain regions and individuals. A similar approach has been successfully used to assess the endosomal–lysosomal system in prion disease [21].
The NDUFB8 subunit is essential for the assembly, structure stability and function of the complex [39, 46] and its knockdown decreases complex I activity in animals [10]. We, therefore, reasoned that NDUFB8 deficiency should reflect the entire complex. This was corroborated by activity measurements showing widespread functional complex I deficiency in the PD brain. While most individuals with PD had lower complex I activity in the cerebellum compared to controls, this difference was not significant at the group level due to large interindividual variation. This may be real biological variation, or a technical artefact reflecting the tissue architecture of the cerebellum. The ratio of Purkinje cells, which are the main complex I source in the cerebellar cortex, to other cell types and underlying white matter is susceptible to inter-sample variability due to the highly convoluted structure of the cerebellar surface and small number of Purkinje cells compared to other cell populations.
Western blot analysis showed a trend for lower complex I levels in PD, but this did not reach statistical significance at the group level. We believe this to be due to a combination of overall lower sensitivity of Western blotting, which only allows robust changes in protein quantity to be reliably quantified and large interindividual variation. This discrepancy between Western blotting and other techniques of assessing the respiratory chain in PD has been reported previously [32]. Our IHC analyses suggest that neuronal complex I deficiency is generally mild in PD and shows large interneuronal variability, which may be masked by studies in homogenized tissue. Thus, single cell analysis is a more reliable method for assessing complex I deficiency in PD.
Our findings appear to contradict studies showing normal complex I activity in homogenized tissue of the cerebellum, striatum [18, 24, 25, 36] and frontal cortex [18, 36] in PD. This difference may be partly due to lower sensitivity of analyses in brain homogenate. We show that complex I levels in PD can vary greatly between neighboring neurons in the same individual (Fig. 4). This heterogeneity is not detectable in homogenized tissue and may create sampling bias contributing to the variable and contradictory results of previous studies. Our method of PFCA measurement circumvents this problem by assessing complex I at the level of individual neurons. Nevertheless, in our experiments, we detected lower complex I activity and a mild quantitative reduction also in homogenized tissue. Therefore, while PFCA measurements appear to be more sensitive, this is not sufficient to explain the negative findings of other studies. Sample heterogeneity may also play a role. Previous studies were based on material from brain banks, which may be subject to selection bias (i.e. not entirely population-based) and are commonly not accompanied by systematically collected prospective clinical information. Pathology alone is not always sufficient to distinguish PD from other synucleinopathies and previous research suggests that complex I may be intact in other parkinsonisms [36]. Our sample is from an entirely population-based, longitudinally followed cohort, which has been extensively characterized clinically, pathologically and genetically, and is, therefore, by current standards as close as possible to true idiopathic PD.
Our findings do not corroborate the current hypothesis that complex I deficiency in PD may be the result of accumulating deletions and quantitative depletion of the mtDNA [4, 16]. Moreover, ultra-deep-sequencing studies in single neurons and brain homogenates have shown no difference in the point mutational burden of mtDNA-encoded complex I genes between PD individuals and controls, suggesting that mtDNA point mutations do not contribute to the observed complex I deficiency [8, 13]. Here, we show that complex I deficiency occurs in neurons from at least eight different brain areas in PD, whereas levels of mtDNA damage only exceed those of controls in the SNc. In fact, we found substantial complex I deficiency even in areas with very low levels of mtDNA damage such as the prefrontal cortex and cerebellum. Moreover, complex I deficiency in PD has been reported in tissues with minimal or no mtDNA damage burden such as skeletal muscle, platelets and lymphocytes [3, 7, 27]. Collectively, these findings suggest that, contrary to dominant theory, neuronal complex I deficiency in PD can occur independently of mtDNA damage.
Interestingly, we found that Lewy pathology is significantly less prevalent in complex I negative neurons of the SNc. This is in line with a previous report showing α-synuclein pathology only in neurons with high levels of mitochondrial respiratory subunits [31]. These results do not support a causal link between complex I deficiency and α-synuclein pathology. The reason for the inverse association of these two phenomena remains, however, unknown. One possibility is that neurons containing both pathologies (i.e. complex I deficiency and Lewy pathology) suffer a double-insult and have a short lifespan, thus being unlikely to detect by pathology at end-stage. Alternatively, it could be speculated that intact complex I activity is required for Lewy body formation, at least in the initial phase of aggregation.
In spite of its widespread distribution, neuronal complex I deficiency does not follow the anatomical distribution of neurodegeneration or mitochondrial DNA damage. These findings do not support a direct link between complex I dysfunction and neuronal loss in PD. It is plausible that complex I deficiency is only one of multiple hits required to trigger neuronal death in PD. Another, more intriguing possibility is that complex I is actively downregulated as a compensatory response to moderate excess generation of reactive oxygen species (ROS). Evidence of increased oxidative damage has been consistently found in the PD brain [1, 45] and it has been shown that, under certain conditions, complex I inhibition [28] or modulation [29] can substantially decrease ROS generation. Irrespective of whether it is a pathogenic event or compensatory response, the extent of neuronal complex I deficiency in PD suggests that it is an important and inherent part of the disorder. We, therefore, believe that further elucidating this phenomenon will be key to understanding the pathogenesis of PD.
Abbreviations
- PD:
-
Parkinson’s disease
- PDD:
-
Parkinson’s disease dementia
- Ctrl:
-
Control
- CVLT 2:
-
California Verbal Learning Test 2
- DAB:
-
3′3-Diaminobenzidine
- IHC:
-
Immunohistochemistry
- MMSE:
-
Mini mental status examination
- MRC:
-
Mitochondrial respiratory chain
- mtDNA:
-
Mitochondrial DNA
- PFCA:
-
Positive fractional cytoplasmic area
- SNc:
-
Substantia nigra pars compacta
- UPRDS III:
-
Unified Parkinson’s disease rating scale III (motor symptoms)
References
Alam ZI, Jenner A, Daniel SE, Lees AJ, Cairns N, Marsden CD, Jenner P, Halliwell B (1997) Oxidative DNA damage in the parkinsonian brain: an apparent selective increase in 8-hydroxyguanine levels in substantia nigra. J Neurochem 69:1196–1203
Alves G, Muller B, Herlofson K, HogenEsch I, Telstad W, Aarsland D, Tysnes OB, Larsen JP, Norwegian ParkWest study g (2009) Incidence of Parkinson’s disease in Norway: the Norwegian ParkWest study. J Neurol Neurosurg Psychiatry 80:851–857. https://doi.org/10.1136/jnnp.2008.168211
Barroso N, Campos Y, Huertas R, Esteban J, Molina JA, Alonso A, Gutierrez-Rivas E, Arenas J (1993) Respiratory chain enzyme activities in lymphocytes from untreated patients with Parkinson disease. Clin Chem 39:667–669
Bender A, Krishnan KJ, Morris CM, Taylor GA, Reeve AK, Perry RH, Jaros E, Hersheson JS, Betts J, Klopstock T, Taylor RW, Turnbull DM (2006) High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat Genet 38:515–517. https://doi.org/10.1038/ng1769
Benit P, Chretien D, Kadhom N, de Lonlay-Debeney P, Cormier-Daire V, Cabral A, Peudenier S, Rustin P, Munnich A, Rotig A (2001) Large-scale deletion and point mutations of the nuclear NDUFV1 and NDUFS1 genes in mitochondrial complex I deficiency. Am J Hum Genet 68:1344–1352. https://doi.org/10.1086/320603
Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT (2000) Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat Neurosci 3:1301–1306. https://doi.org/10.1038/81834
Bindoff LA, Birch-Machin MA, Cartlidge NE, Parker WD Jr, Turnbull DM (1991) Respiratory chain abnormalities in skeletal muscle from patients with Parkinson’s disease. J Neurol Sci 104:203–208
Coxhead J, Kurzawa-Akanbi M, Hussain R, Pyle A, Chinnery P, Hudson G (2016) Somatic mtDNA variation is an important component of Parkinson’s disease. Neurobiol Aging. https://doi.org/10.1016/j.neurobiolaging.2015.10.036
Damier P, Hirsch EC, Agid Y, Graybiel AM (1999) The substantia nigra of the human brain. II. Patterns of loss of dopamine-containing neurons in Parkinson’s disease. Brain 122(Pt 8):1437–1448
Davis CW, Hawkins BJ, Ramasamy S, Irrinki KM, Cameron BA, Islam K, Daswani VP, Doonan PJ, Manevich Y, Madesh M (2010) Nitration of the mitochondrial complex I subunit NDUFB8 elicits RIP1- and RIP3-mediated necrosis. Free Radic Biol Med 48:306–317. https://doi.org/10.1016/j.freeradbiomed.2009.11.001
de Rijk MC, Launer LJ, Berger K, Breteler MM, Dartigues JF, Baldereschi M, Fratiglioni L, Lobo A, Martinez-Lage J, Trenkwalder C, Hofman A (2000) Prevalence of Parkinson’s disease in Europe: a collaborative study of population-based cohorts. Neurologic Diseases in the Elderly Research Group. Neurology 54:S21–S23
Dickson DW (2012) Parkinson’s disease and parkinsonism: neuropathology. Cold Spring Harb Perspect Med. https://doi.org/10.1101/cshperspect.a009258
Dölle C, Flones I, Nido GS, Miletic H, Osuagwu N, Kristoffersen S, Lilleng PK, Larsen JP, Tysnes OB, Haugarvoll K, Bindoff LA, Tzoulis C (2016) Defective mitochondrial DNA homeostasis in the substantia nigra in Parkinson disease. Nat Commun 7:13548. https://doi.org/10.1038/ncomms13548
Gatt AP, Duncan OF, Attems J, Francis PT, Ballard CG, Bateman JM (2016) Dementia in Parkinson’s disease is associated with enhanced mitochondrial complex I deficiency. Mov Disord 31:352–359. https://doi.org/10.1002/mds.26513
Gelb DJ, Oliver E, Gilman S (1999) Diagnostic criteria for Parkinson disease. Arch Neurol 56:33–39
Grunewald A, Rygiel KA, Hepplewhite PD, Morris CM, Picard M, Turnbull DM (2016) Mitochondrial DNA depletion in respiratory chain-deficient Parkinson disease neurons. Ann Neurol 79:366–378. https://doi.org/10.1002/ana.24571
Hattori N, Tanaka M, Ozawa T, Mizuno Y (1991) Immunohistochemical studies on complexes I, II, III, and IV of mitochondria in Parkinson’s disease. Ann Neurol 30:563–571. https://doi.org/10.1002/ana.410300409
Janetzky B, Hauck S, Youdim MB, Riederer P, Jellinger K, Pantucek F, Zochling R, Boissl KW, Reichmann H (1994) Unaltered aconitase activity, but decreased complex I activity in substantia nigra pars compacta of patients with Parkinson’s disease. Neurosci Lett 169:126–128
Keeney PM, Xie J, Capaldi RA, Bennett JP Jr (2006) Parkinson’s disease brain mitochondrial complex I has oxidatively damaged subunits and is functionally impaired and misassembled. J Neurosci 26:5256–5264. https://doi.org/10.1523/JNEUROSCI.0984-06.2006
Kirby DM, Thorburn DR, Turnbull DM, Taylor RW (2007) Biochemical assays of respiratory chain complex activity. Methods Cell Biol 80:93–119. https://doi.org/10.1016/S0091-679X(06)80004-X
Kovacs GG, Gelpi E, Strobel T, Ricken G, Nyengaard JR, Bernheimer H, Budka H (2007) Involvement of the endosomal-lysosomal system correlates with regional pathology in Creutzfeldt-Jakob disease. J Neuropathol Exp Neurol 66:628–636. https://doi.org/10.1097/nen.0b013e318093ecc7
Langston JW, Ballard P, Tetrud JW, Irwin I (1983) Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 219:979–980
Mann VM, Cooper JM, Daniel SE, Srai K, Jenner P, Marsden CD, Schapira AH (1994) Complex I, iron, and ferritin in Parkinson’s disease substantia nigra. Ann Neurol 36:876–881. https://doi.org/10.1002/ana.410360612
Mann VM, Cooper JM, Krige D, Daniel SE, Schapira AH, Marsden CD (1992) Brain, skeletal muscle and platelet homogenate mitochondrial function in Parkinson’s disease. Brain 115(Pt 2):333–342
Mizuno Y, Ohta S, Tanaka M, Takamiya S, Suzuki K, Sato T, Oya H, Ozawa T, Kagawa Y (1989) Deficiencies in complex I subunits of the respiratory chain in Parkinson’s disease. Biochem Biophys Res Commun 163:1450–1455
Nijtmans LG, Henderson NS, Holt IJ (2002) Blue Native electrophoresis to study mitochondrial and other protein complexes. Methods 26:327–334. https://doi.org/10.1016/S1046-2023(02)00038-5
Parker WD Jr, Boyson SJ, Parks JK (1989) Abnormalities of the electron transport chain in idiopathic Parkinson’s disease. Ann Neurol 26:719–723. https://doi.org/10.1002/ana.410260606
Pryde KR, Hirst J (2011) Superoxide is produced by the reduced flavin in mitochondrial complex I: a single, unified mechanism that applies during both forward and reverse electron transfer. J Biol Chem 286:18056–18065. https://doi.org/10.1074/jbc.M110.186841
Pryde KR, Taanman JW, Schapira AH (2016) A LON-ClpP proteolytic axis degrades complex I to extinguish ROS production in depolarized mitochondria. Cell Rep 17:2522–2531. https://doi.org/10.1016/j.celrep.2016.11.027
Ramsay RR, Salach JI, Singer TP (1986) Uptake of the neurotoxin 1-methyl-4-phenylpyridine (MPP+) by mitochondria and its relation to the inhibition of the mitochondrial oxidation of NAD+-linked substrates by MPP+. Biochem Biophys Res Commun 134:743–748
Reeve AK, Park TK, Jaros E, Campbell GR, Lax NZ, Hepplewhite PD, Krishnan KJ, Elson JL, Morris CM, McKeith IG, Turnbull DM (2012) Relationship between mitochondria and alpha-synuclein: a study of single substantia nigra neurons. Arch Neurol 69:385–393. https://doi.org/10.1001/archneurol.2011.2675
Schagger H (1995) Quantification of oxidative phosphorylation enzymes after blue native electrophoresis and two-dimensional resolution: normal complex I protein amounts in Parkinson’s disease conflict with reduced catalytic activities. Electrophoresis 16:763–770
Schagger H (1996) Electrophoretic techniques for isolation and quantification of oxidative phosphorylation complexes from human tissues. Methods Enzymol 264:555–566
Schapira AH, Cooper JM, Dexter D, Clark JB, Jenner P, Marsden CD (1990) Mitochondrial complex I deficiency in Parkinson’s disease. J Neurochem 54:823–827
Schapira AH, Cooper JM, Dexter D, Jenner P, Clark JB, Marsden CD (1989) Mitochondrial complex I deficiency in Parkinson’s disease. Lancet 1:1269
Schapira AH, Mann VM, Cooper JM, Dexter D, Daniel SE, Jenner P, Clark JB, Marsden CD (1990) Anatomic and disease specificity of NADH CoQ1 reductase (complex I) deficiency in Parkinson’s disease. J Neurochem 55:2142–2145
Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A (2012) Fiji: an open-source platform for biological-image analysis. Nat Methods 9:676–682. https://doi.org/10.1038/nmeth.2019
Spinazzi M, Casarin A, Pertegato V, Salviati L, Angelini C (2012) Assessment of mitochondrial respiratory chain enzymatic activities on tissues and cultured cells. Nat Protoc 7:1235–1246. https://doi.org/10.1038/nprot.2012.058
Stroud DA, Surgenor EE, Formosa LE, Reljic B, Frazier AE, Dibley MG, Osellame LD, Stait T, Beilharz TH, Thorburn DR, Salim A, Ryan MT (2016) Accessory subunits are integral for assembly and function of human mitochondrial complex I. Nature 538:123–126. https://doi.org/10.1038/nature19754
Talasila KM, Rosland GV, Hagland HR, Eskilsson E, Flones IH, Fritah S, Azuaje F, Atai N, Harter PN, Mittelbronn M, Andersen M, Joseph JV, Hossain JA, Vallar L, Noorden CJ, Niclou SP, Thorsen F, Tronstad KJ, Tzoulis C, Bjerkvig R, Miletic H (2017) The angiogenic switch leads to a metabolic shift in human glioblastoma. Neuro Oncol 19:383–393. https://doi.org/10.1093/neuonc/now175
Tzoulis C, Tran GT, Coxhead J, Bertelsen B, Lilleng PK, Balafkan N, Payne B, Miletic H, Chinnery PF, Bindoff LA (2014) Molecular pathogenesis of polymerase gamma-related neurodegeneration. Ann Neurol 76:66–81. https://doi.org/10.1002/ana.24185
van der Loos CM (2008) Multiple immunoenzyme staining: methods and visualizations for the observation with spectral imaging. J Histochem Cytochem 56:313–328. https://doi.org/10.1369/jhc.2007.950170
Ward CD, Gibb WR (1990) Research diagnostic criteria for Parkinson’s disease. Adv Neurol 53:245–249
Wittig I, Braun HP, Schagger H (2006) Blue native PAGE. Nat Protoc 1:418–428. https://doi.org/10.1038/nprot.2006.62
Yoritaka A, Hattori N, Uchida K, Tanaka M, Stadtman ER, Mizuno Y (1996) Immunohistochemical detection of 4-hydroxynonenal protein adducts in Parkinson disease. Proc Natl Acad Sci USA 93:2696–2701
Zhu J, Vinothkumar KR, Hirst J (2016) Structure of mammalian respiratory complex I. Nature 536:354–358. https://doi.org/10.1038/nature19095
Acknowledgements
We are grateful to the patients and their families for participating in this study. We thank Prof. Gabor Kovacs for the helpful discussions regarding the immunohistochemical analyses.
Funding
This work was supported by grants from the Regional Health Authority of Western Norway (Grant no. 911903 and 911988) and the Research Council of Norway (Grant no. 240369/F20). We thank patients and family for contributing to this study.
Author information
Authors and Affiliations
Contributions
Conception, design and funding: CT. Acquisition and analysis of data: IHF, EFV, ML, CD, KH, GOS, HM, PKL, GA, OBT, CT, MZ. Drafting the manuscript: IHF, CT. Statistical analysis: IHF, BB, GOS. Critical revision of the manuscript: KH, HM, PKL, GA, OBT, MZ.
Corresponding author
Ethics declarations
Ethical approval
These studies were approved by the Regional Committee for Medical and Health Research Ethics, Western Norway (REK 131/04 and REK 2010/23).
Informed consent
Informed consent was obtained from all subjects.
Reseach involving human/animal participants
All procedures performed in the studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Flønes, I.H., Fernandez-Vizarra, E., Lykouri, M. et al. Neuronal complex I deficiency occurs throughout the Parkinson’s disease brain, but is not associated with neurodegeneration or mitochondrial DNA damage. Acta Neuropathol 135, 409–425 (2018). https://doi.org/10.1007/s00401-017-1794-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-017-1794-7