
Abstract
Studies in pediatric high-grade astrocytomas (HGA) by our group and others have uncovered recurrent somatic mutations affecting highly conserved residues in histone 3 (H3) variants. One of these mutations leads to analogous p.Lys27Met (K27M) mutations in both H3.3 and H3.1 variants, is associated with rapid fatal outcome, and occurs specifically in HGA of the midline in children and young adults. This includes diffuse intrinsic pontine gliomas (80 %) and thalamic or spinal HGA (>90 %), which are surgically challenging locations with often limited tumor material available and critical need for specific histopathological markers. Here, we analyzed formalin-fixed paraffin-embedded tissues from 143 pediatric HGA and 297 other primary brain tumors or normal brain. Immunohistochemical staining for H3K27M was compared to tumor genotype, and also compared to H3 tri-methylated lysine 27 (H3K27me3) staining, previously shown to be drastically decreased in samples carrying this mutation. There was a 100 % concordance between genotype and immunohistochemical analysis of H3K27M in tumor samples. Mutant H3K27M was expressed in the majority of tumor cells, indicating limited intra-tumor heterogeneity for this specific mutation within the limits of our dataset. Both H3.1 and H3.3K27M mutants were recognized by this antibody while non-neoplastic elements, such as endothelial and vascular smooth muscle cells or lymphocytes, did not stain. H3K27me3 immunoreactivity was largely mutually exclusive with H3K27M positivity. These results demonstrate that mutant H3K27M can be specifically identified with high specificity and sensitivity using an H3K27M antibody and immunohistochemistry. Use of this antibody in the clinical setting will prove very useful for diagnosis, especially in the context of small biopsies in challenging midline tumors and will help orient care in the context of the extremely poor prognosis associated with this mutation.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Recent years have seen an explosion of genomic data across many types of human cancer including high-grade gliomas, through the increasing use of next generation sequencing (NGS) technologies and efforts of independent laboratories, as well as large consortia such as The Cancer Genome Atlas (TCGA) and the International Cancer Genome Consortium (ICGC). In children and young adults, critical epigenetic alterations were discovered in subgroups of these tumors. Somatic recurrent mutations resulting in amino acid substitutions at lysine 27 (p.Lys27Met, K27M) and glycine 34 (p.Gly34Arg/Val, G34R/V) in H3F3A, which encodes histone 3 (H3) variant 3 (H3.3), were shown by our group and others to characterize about one-third of pediatric high-grade astrocytomas (HGA) [20, 24]. These mutations resulted in somatic heterozygous lysine 27 to methionine (p.Lys27Met, K27M) and glycine 34 to arginine or valine (p.Gly34Arg/Val, G34R/V) amino acid substitutions at these positions within the H3.3 tail [20, 24]. A large cohort of 784 gliomas of all ages and histopathological grades (WHO I–IV) confirmed the prevalence of H3F3A mutations in pediatric and young adult HGA [20] as did additional studies performed by other groups [9, 26]. Genomic analysis of HGA and correlation of results with age and tumor site showed that H3.3G34R/V mutations characterize HGA located within the cerebral hemispheres, mainly temporo-parietal regions in adolescents and young adults with this tumor [12, 21]. K27M mutations in H3.3 and histone variant H3.1 (encoded by HIST1H3B) were also shown to be present in 71–78 % of diffuse intrinsic pontine gliomas (DIPGs), which are HGA of the pons [12, 24]. Moreover, H3F3A K27M mutations also characterize pediatric [8, 20, 21] and younger adult HGA [1] arising in the thalamus, the spinal cord or the cerebellum [8, 21] thus confirming preponderance of H3K27M mutations in HGA in the midline [12, 20, 21, 24]. These brain regions are notoriously difficult areas for surgical resection and stereotactic biopsy and the frequency and specificity of these mutations in histones strongly support their use in diagnosis and as biomarkers for these HGA tumors in children and younger patients [3, 4, 8, 22, 25].
The current routine procedure for assessing H3 status is through DNA sequencing. This is a time-consuming and laborious process which requires extraction of nucleic acids and relatively elaborate laboratory equipment that may not be available in every pathology department. In contrast, immunohistochemistry (IHC) is routinely performed in pathology laboratories. Critical insight into the effects of the K- to M change in H3 tails was provided by the Allis group who showed that this mutation acted as a gain-of-function through inhibition of the methyltransferase activity of EZH2 [14]. This leads to decreased levels of K27me3 because the Polycomb repressive complex 2 which contains EZH2 is unable to mediate this activity in the presence of H3K27M [14]. Decreased H3K27me3 levels in H3K27M-mutant HGA samples were shown by several groups further confirming this effect on tumor tissues [2, 23]. Therefore, use of decreased H3K27me3 levels by IHC was proposed as a surrogate marker to diagnose H3.3K27M mutation in the clinical setting [23]. However, the normal pattern of H3K27me3 in control brain and other brain tumors is not clearly established, and the availability of a mutation-specific antibody that specifically recognizes the mutation similar to the one derived for the most common isocitrate dehydrogenase (IDH) mutation in gliomas (IDH1R132H) would greatly help clinical management [5]. This is especially true in the context of the very poor outcome established for this specific mutation compared to H3.3 G34R/V mutations, IDH mutations, or tumors that are wild type for these genes [7, 12, 21]. We show here on a cohort of HGA enriched for midline and pediatric tumors that a commercially available rabbit polyclonal antibody directed against K27M mutant H3 variants can detect these mutations in fixed tissues using standard immunohistochemistry with high sensitivity and specificity.
Materials and methods
Patient samples and pathological review
All samples were obtained with informed consent after approval of the Institutional Review Board of the respective hospitals they were treated in, and were independently reviewed by pediatric neuropathologists (SA, BE, TP, AK, KL) according to the WHO guidelines. Samples were obtained from the Montreal Children’s Hospital (Montreal, McGill University Health Centre), University of Bonn (Bonn, Germany), Boston Children’s Hospital (Boston, Harvard University), Sainte-Justine Hospital (Montreal, Canada), University of Heidelberg (Heidelberg, Germany), and the Brain Tumor Toronto Bank (BTTB, Toronto). All samples were from formalin-fixed paraffin-embedded material. All midline pediatric HGA samples were from needle biopsies (pons) or partial resections (thalamus, spine, cerebellum) and most were obtained prior radio- and/or chemotherapy. Slides were available from individual tumor samples or from previously reported tissue microarrays (TMA) [6, 13, 20]. The pediatric HGA cohort (mean age 11 years) included tumor tissue from 124 WHO Grade IV astrocytomas (glioblastoma, GBM), 14 GBM recurrences, and 11 WHO Grade III astrocytomas (anaplastic astrocytoma, AA) (Table 1 ). A range of other brain tumors was also investigated and included other pediatric gliomas (n = 6), control brain (n = 8) and three TMAs containing, respectively, 97 medulloblastomas (MB), 71 primitive neuroectodermal tumors (PNET) samples, or 115 WHO Grade I (pilocytic astrocytomas, PA) (Supplementary Table 1). Clinical characteristics of patients with HGA are summarized in Table 1. TMAs comprised an average of three tumor cores from the same sample with a mean diameter of 1.5 mm for each core. Cores were selected from the original tumor sample and oriented on the TMA by the neuropathologist and controlled for adequate tumor representation by hematoxylin/eosin staining.
Automated immunohistochemistry
Tissue samples were cut at 5 µm, placed on SuperFrost/Plus slides (Fisher) and dried overnight at 37 °C. The slides were then loaded onto a Discovery XT Autostainer (Ventana Medical System). All solutions used for automated immunohistochemistry were from Ventana Medical System unless otherwise specified. Slides underwent de-paraffinization and heat-induced epitope retrieval (CC1 pre-diluted solution Cat# 950-124) following standard protocol. Immunostaining for H3K27M mutant and H3K27me3 were performed using a heat protocol. Briefly, rabbit polyclonal anti-H3K27M (#ABE419 Millipore, 1:500), or rabbit monoclonal anti-H3K27me3 (C36B11, #9733 Cell Signalling, diluted at 1:75) diluted in the antibody diluent (Cat# 251-018) were applied manually for 32 min at 37 °C and then incubated using the appropriate detection kit (OmniMap anti-Rabbit-HRP, Cat# 760-4311) for 8 min, followed by ChromoMap-DAB Cat# 760-159). Omission of the primary antibody was used as negative control. Slides were then counterstained with hematoxylin for 4 min, blued with Bluing Reagent for 4 min, removed from the autostainer, washed in warm soapy water, dehydrated through graded alcohols, cleared in xylene, and mounted with Permount. Sections were analyzed by conventional light microscopy. Immunohistochemistry for INI1 (clone 25/BAF47, 1:400, BD Biosciences) and CD45 (clone T29/33, pre-diluted, Dako) was performed according to the manufacturer’s instructions on a Dako autostainer. Slides were scanned using the Aperio system and independently scored for H3K27M and H3K27me3-positive staining by three independent individuals including two neuropathologists (SA, BE) blinded to the tumor genotype. Results were merged and consensus scoring was obtained as previously described [15, 20]. Briefly, samples were considered as positive for H3K27M staining if tumor cells showed nuclear staining and the core included a control blood vessel or normal brain with negative staining for the antibody. H3K27me3 staining was performed as previously described [23]. If no H3K27me3 or H3K27M staining were observed, we performed a control staining against a nuclear protein INI1 used as control for nuclear staining and tissue/fixation quality. Immunohistochemistry for INI1 (clone 25/BAF47, 1:400, BD Biosciences) and double labeling for H3K27me3 (DAB brown chromogen) and CD45 (clone T29/33, pre-diluted, Dako, AP red chromogen) was performed according to the manufacturer’s instructions on a Dako autostainer. Three HGA samples from our initial dataset were thus excluded based on negative INI1 staining. Notably, archival material fixed with Bouin’s solution showed a very strong background using the H3K27M antibody and poor H3K27me3 staining, and were therefore excluded from further analysis (n = 7, Supplementary Fig. 1a). Also, autopsy samples showed a high background when stained using the anti-H3K27M antibody and were not included in the dataset (Supplementary Fig. 1b).
DNA sequencing of H3 variants
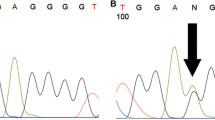
For HGA samples not previously screened using whole exome/Sanger sequencing [8, 12, 20] high-resolution melting (HRM) or pyrosequencing were used as previously described [20]. Briefly, H3F3A and HIST1H3B mutation screening using HRM was performed on a Light Cycler 480 using triplicates for each sample. Each reaction contained 10 μl of LC480 High Resolution Master Mix 2× (Roche), 0.2 μM of each primer, 2.5 μM MgCl2, 5–20 ng of genomic DNA and water to a final volume of 20 μl. PCR conditions were as follows: 95 °C for 10 min followed by 45–50 cycles of 95 °C for 10 s, a touchdown of 68–58 °C for 15 s (1 °C/cycle) and 72 °C for 15 s. Primers were HPLC-purified (Integrated DNA Technologies). MgCl2 concentration was optimized for each reaction to give low C t values and a high plateau phase. Primer sequences are H3F3A-F: 5′-GTACAAAGCAGACTGCCCGCAAAT-3′, H3F3A-R: 5′-GTGGATACATAC AAGAGAGACTTTGTCCC-3′, and Hist1H3B-F: 5′-ACAGACGTCTCTGCAGGCAAGC-3′ and Hist1H3B-R: 5′-GGCGGTAACGGTGAGGCTTT-3′. After amplification, the PCR product was denatured at 95 °C for 1 min, cooled down to 40 °C to allow duplex formation, then high-resolution melting data were acquired when the temperature was increased from 70 to 95 °C (1 °C/cycle—25 data acquisition per °C). Analysis was performed with the Light Cycler 480 Gene scanning software. Melting curves were first normalized, which were then shifted along the temperature, and finally a difference plot was generated. The grouping method chosen was the “In-run” standards where the software applies grouping on melting standard samples included in the run.
Results and discussion
We performed targeted sequencing using HRM or pyrosequencing to analyze the genotype of tumor and control brain samples included in this study for which information on H3.1 (HIST31B and HIST31C) and H3.3 (H3F3A) mutational status at position K27 was not available. We identified a K27M substitution in 48 samples, 5 HGA carrying H3.1K27M (4 on HIST1B and 1 on HIST1C) and 43 HGA with H3.3K27M. Six HGA cases had no available material to perform genotype analysis and were excluded from further analysis.
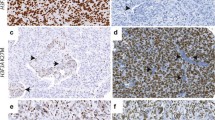
We then assessed H3K27M expression by IHC on the 143 genotyped HGA (118 GBM, 14 GBM recurrences, 11 AA, Table 1). Results were compared to H3K27me3 staining on consecutive sections and to genomic data. Anti-H3K27 M antibody showed a strong nuclear staining of most tumor cells (>80 %) in all 48 samples with the H3K27M genotype (Fig. 1; Tables 1, 2). None of the H3K27M wild-type samples showed nuclear staining (Table 2; Fig. 1; Supplementary Fig. 2). Importantly, the antibody was able to recognize both H3.3 and H3.1 K27M mutant tumors (Fig. 2), as expected given that the peptide it was raised against is identical in both mutant H3 variants. The anti-H3K27M antibody did not stain any of the H3.3G34R/V HGA included in this dataset (n = 7). It did not recognize endothelial cells or other vascular structures within H3K27M mutant HGA which still expressed H3K27me3 (Fig. 1; Supplementary Fig. 2).

Immunohistochemical (IHC) staining of pediatric high-grade astrocytomas (HGA) using the anti-H3K27M antibody correlates with tumor genotype and decreased H3K27Me3 in tumors. Representative IHC of pediatric HGA using anti-H3K27M (a, c, e, g) or anti-H3K27me3 (b, d, f, h) antibodies and counterstained with hematoxylin. H3K27M shows strong nuclear positivity in tumor cells, but no staining in the nuclei of endothelial and smooth muscle cells in blood vessels in K27M mutant tumors (a, c). Tumors wild type for H3K27 show no nuclear staining with the anti-H3K27M antibody (e, g). Corresponding H3K27me3 staining on the same samples shows global decrease of the expression of this histone mark in H3K27M mutant tumors (b, d) compared to tumors wild type for this mutation (f, h). Notably, positivity for H3K27me3 was mainly seen in tumor vessels (b, d) even though a degree of intra-tumor staining was also seen in H3K27M mutant samples (d)

Immunohistochemical (IHC) staining of H3.1K27M and H3.3K27M mutant high-grade astrocytomas (HGA) shows similar results using the anti-H3K27M and anti-H3K27me3 antibodies. Representative sections from HGAs (WHO Grade III) carrying H3.3K27M (a, b) and H3.1K27M (c, d, e, f) immunostained with anti-H3K27M (a, c, e) and anti-H3K27me3 (b, d, f) antibodies. Two areas from the same H3.1K27M WHO Grade III DIPG sample are shown to further illustrate that the K27M mutation is also present in seemingly lower grade tumor sections (c)
H3K27me3 staining was negative or strongly decreased in K27M mutant samples as previously described [2, 14, 23], while tumor cells in samples wild type for H3K27M showed high expression of this post-translational histone modification (Fig. 1). Out of the 14 recurrent GBM, 3 were genotyped to be H3K27M mutants. All three GBM showed strong H3K27M expression across most tumor cells. Conversely, recurrent samples wild type for this mutation showed no staining. This further confirms that when H3K27M is identified at diagnosis it is maintained by most tumor cells at relapse and is not acquired at the time of tumor recurrence similar to IDH-mutant gliomas [10]. To further assess the specificity of H3K27M antibody, we screened TMAs containing PA, MB and PNET samples and a set of other pediatric brain tumors or normal brain (Supplementary Table 1). As expected, none of these samples had positive H3K27M staining (Supplementary Table 1, Supplementary Fig. 3 and data not shown).
A small number of samples (n = 6) wild type for H3K27M showed scattered cells with pale, nonspecific-appearing cytoplasmic staining with the anti-H3K27M antibody, mostly in areas of necrosis or inflammation, but these cases were not viewed as equivocal and were not interpreted as positive by scoring pathologists blinded to the genotype. As for the anti-H3K27me3 antibody, some strong extravascular nuclear staining was seen in ~10 % of H3K27M mutant samples (which generally do not stain except in blood vessels). In some cases the staining appeared mainly to be from mononuclear inflammatory cells including microglia, which we confirmed by immunostaining and co-immunostaining with anti-CD45, a marker that recognizes lymphocytes, on consecutive slides from the same tumors (Fig. 3). However, nuclear staining for H3K27me3 in H3K27M samples could not be solely accounted for by CD45-positive cells, which highlights the fact that loss of H3K27me3 is not reliable to predict H3K27M genotype (Fig. 3b; Supplementary Fig. 4). In addition, based on the limits of IHC at the single cell level, we cannot exclude that a small subset of tumor cells is negative for both H3K27M and H3K27me3 in H3K27M tumor samples (Supplementary Fig. 4).

H3K27M staining and H3K27me3/CD45 co-immunostaining on consecutive slides of an H3K27M mutant high-grade astrocytoma. a anti-H3K27M staining (nuclear); b co-immunostaining using a CD45 antibody (cytoplasmic, purple) and an H3K27me3 antibody (nuclear, dark brown); results indicate that only a proportion of cells positive for H3K27me3 staining in H3K27M mutant high-grade astrocytomas are positive for CD45 staining and represent a lymphocytic infiltrate (light blue arrows). Endothelial and smooth muscle vascular cells show uptake of the H3K27me3 antibody alone (asterisk) while few cells within the tumor remain positive for H3K27me3 (black arrow)
Our results indicate that the rabbit monoclonal antibody directed against H3K27M mutations specifically recognizes mutant proteins using simple IHC. Nuclear immunostaining was restricted to tumor cells in samples carrying the mutant H3K27M genotype, both in H3.1 and H3.3. This staining is more specific than H3K27me3 investigation as it specifically targets the mutation and not one of its downstream effects. Other pediatric posterior fossa tumors including ependymomas [16] and different sub-groups of medulloblastomas [11, 17–19] have strong H3K27me3 positive staining based on their own biology. In addition, based on the scattered H3K27me3-positive staining in several mutant H3K27M HGA, interpretation of the results may prove challenging when using H3K27me3 expression levels as a surrogate marker of H3K27M. In summary, our data show that the H3K27M antibody is highly useful for tumor diagnosis in the clinical setting where IHC is routine procedure in pathology laboratories. This is especially true in the context of midline and hindbrain tumors where surgical material is limited. The concurrent use of this antibody with other known prognostic pediatric HGA markers including TP53 will help orient patients to optimal clinical care and management based on the very poor prognosis and limited survival seen in the context of this mutation.
References
Aihara K, Mukasa A, Gotoh K, Saito K, Nagae G, Tsuji S, Tatsuno K, Yamamoto S, Takayanagi S, Narita Y, Shibui S, Aburatani H, Saito N (2014) H3F3A K27M mutations in thalamic gliomas from young adult patients. Neuro-oncology 16(1):140–146. doi:10.1093/neuonc/not144
Bender S, Tang Y, Lindroth AM, Hovestadt V, Jones DT, Kool M, Zapatka M, Northcott PA, Sturm D, Wang W, Radlwimmer B, Hojfeldt JW, Truffaux N, Castel D, Schubert S, Ryzhova M, Seker-Cin H, Gronych J, Johann PD, Stark S, Meyer J, Milde T, Schuhmann M, Ebinger M, Monoranu CM, Ponnuswami A, Chen S, Jones C, Witt O, Collins VP, von Deimling A, Jabado N, Puget S, Grill J, Helin K, Korshunov A, Lichter P, Monje M, Plass C, Cho YJ, Pfister SM (2013) Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high-grade gliomas. Cancer Cell 24(5):660–672. doi:10.1016/j.ccr.2013.10.006
Buczkowicz P, Bartels U, Bouffet E, Becher O, Hawkins C (2014) Histopathological spectrum of paediatric diffuse intrinsic pontine glioma: diagnostic and therapeutic implications. Acta Neuropathol. doi:10.1007/s00401-014-1319-6
Buczkowicz P, Hoeman C, Rakopoulos P, Pajovic S, Letourneau L, Dzamba M, Morrison A, Lewis P, Bouffet E, Bartels U, Zuccaro J, Agnihotri S, Ryall S, Barszczyk M, Chornenkyy Y, Bourgey M, Bourque G, Montpetit A, Cordero F, Castelo-Branco P, Mangerel J, Tabori U, Ho KC, Huang A, Taylor KR, Mackay A, Bendel AE, Nazarian J, Fangusaro JR, Karajannis MA, Zagzag D, Foreman NK, Donson A, Hegert JV, Smith A, Chan J, Lafay-Cousin L, Dunn S, Hukin J, Dunham C, Scheinemann K, Michaud J, Zelcer S, Ramsay D, Cain J, Brennan C, Souweidane MM, Jones C, Allis CD, Brudno M, Becher O, Hawkins C (2014) Genomic analysis of diffuse intrinsic pontine gliomas identifies three molecular subgroups and recurrent activating ACVR1 mutations. Nat Genet 46(5):451–456. doi:10.1038/ng.2936
Capper D, Zentgraf H, Balss J, Hartmann C, von Deimling A (2009) Monoclonal antibody specific for IDH1 R132H mutation. Acta Neuropathol 118(5):599–601. doi:10.1007/s00401-009-0595-z
Dubuc AM, Remke M, Korshunov A, Northcott PA, Zhan SH, Mendez-Lago M, Kool M, Jones DT, Unterberger A, Morrissy AS, Shih D, Peacock J, Ramaswamy V, Rolider A, Wang X, Witt H, Hielscher T, Hawkins C, Vibhakar R, Croul S, Rutka JT, Weiss WA, Jones SJ, Eberhart CG, Marra MA, Pfister SM, Taylor MD (2013) Aberrant patterns of H3K4 and H3K27 histone lysine methylation occur across subgroups in medulloblastoma. Acta Neuropathol 125(3):373–384. doi:10.1007/s00401-012-1070-9
Fontebasso AM, Liu XY, Sturm D, Jabado N (2013) Chromatin remodeling defects in pediatric and young adult glioblastoma: a tale of a variant histone 3 tail. Brain Pathol 23(2):210–216. doi:10.1111/bpa.12023
Fontebasso AM, Papillon-Cavanagh S, Schwartzentruber J, Nikbakht H, Gerges N, Fiset PO, Bechet D, Faury D, De Jay N, Ramkissoon LA, Corcoran A, Jones DT, Sturm D, Johann P, Tomita T, Goldman S, Nagib M, Bendel A, Goumnerova L, Bowers DC, Leonard JR, Rubin JB, Alden T, Browd S, Geyer JR, Leary S, Jallo G, Cohen K, Gupta N, Prados MD, Carret AS, Ellezam B, Crevier L, Klekner A, Bognar L, Hauser P, Garami M, Myseros J, Dong Z, Siegel PM, Malkin H, Ligon AH, Albrecht S, Pfister SM, Ligon KL, Majewski J, Jabado N, Kieran MW (2014) Recurrent somatic mutations in ACVR1 in pediatric midline high-grade astrocytoma. Nat Genet 46(5):462–466. doi:10.1038/ng.2950
Gielen GH, Gessi M, Hammes J, Kramm CM, Waha A, Pietsch T (2013) H3F3A K27M mutation in pediatric CNS tumors: a marker for diffuse high-grade astrocytomas. Am J Clin Pathol 139(3):345–349. doi:10.1309/AJCPABOHBC33FVMO
Johnson BE, Mazor T, Hong C, Barnes M, Aihara K, McLean CY, Fouse SD, Yamamoto S, Ueda H, Tatsuno K, Asthana S, Jalbert LE, Nelson SJ, Bollen AW, Gustafson WC, Charron E, Weiss WA, Smirnov IV, Song JS, Olshen AB, Cha S, Zhao Y, Moore RA, Mungall AJ, Jones SJ, Hirst M, Marra MA, Saito N, Aburatani H, Mukasa A, Berger MS, Chang SM, Taylor BS, Costello JF (2014) Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science 343(6167):189–193. doi:10.1126/science.1239947
Jones DT, Jager N, Kool M, Zichner T, Hutter B, Sultan M, Cho YJ, Pugh TJ, Hovestadt V, Stutz AM, Rausch T, Warnatz HJ, Ryzhova M, Bender S, Sturm D, Pleier S, Cin H, Pfaff E, Sieber L, Wittmann A, Remke M, Witt H, Hutter S, Tzaridis T, Weischenfeldt J, Raeder B, Avci M, Amstislavskiy V, Zapatka M, Weber UD, Wang Q, Lasitschka B, Bartholomae CC, Schmidt M, von Kalle C, Ast V, Lawerenz C, Eils J, Kabbe R, Benes V, van Sluis P, Koster J, Volckmann R, Shih D, Betts MJ, Russell RB, Coco S, Tonini GP, Schuller U, Hans V, Graf N, Kim YJ, Monoranu C, Roggendorf W, Unterberg A, Herold-Mende C, Milde T, Kulozik AE, von Deimling A, Witt O, Maass E, Rossler J, Ebinger M, Schuhmann MU, Fruhwald MC, Hasselblatt M, Jabado N, Rutkowski S, von Bueren AO, Williamson D, Clifford SC, McCabe MG, Collins VP, Wolf S, Wiemann S, Lehrach H, Brors B, Scheurlen W, Felsberg J, Reifenberger G, Northcott PA, Taylor MD, Meyerson M, Pomeroy SL, Yaspo ML, Korbel JO, Korshunov A, Eils R, Pfister SM, Lichter P (2012) Dissecting the genomic complexity underlying medulloblastoma. Nature 488(7409):100–105. doi:10.1038/nature11284
Khuong-Quang DA, Buczkowicz P, Rakopoulos P, Liu XY, Fontebasso AM, Bouffet E, Bartels U, Albrecht S, Schwartzentruber J, Letourneau L, Bourgey M, Bourque G, Montpetit A, Bourret G, Lepage P, Fleming A, Lichter P, Kool M, von Deimling A, Sturm D, Korshunov A, Faury D, Jones DT, Majewski J, Pfister SM, Jabado N, Hawkins C (2012) K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol 124(3):439–447. doi:10.1007/s00401-012-0998-0
Korshunov A, Sturm D, Ryzhova M, Hovestadt V, Gessi M, Jones DT, Remke M, Northcott P, Perry A, Picard D, Rosenblum M, Antonelli M, Aronica E, Schuller U, Hasselblatt M, Woehrer A, Zheludkova O, Kumirova E, Puget S, Taylor MD, Giangaspero F, Peter Collins V, von Deimling A, Lichter P, Huang A, Pietsch T, Pfister SM, Kool M (2014) Embryonal tumor with abundant neuropil and true rosettes (ETANTR), ependymoblastoma, and medulloepithelioma share molecular similarity and comprise a single clinicopathological entity. Acta Neuropathol 128(2):279–289. doi:10.1007/s00401-013-1228-0
Lewis PW, Muller MM, Koletsky MS, Cordero F, Lin S, Banaszynski LA, Garcia BA, Muir TW, Becher OJ, Allis CD (2013) Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 340(6134):857–861. doi:10.1126/science.1232245
Liu XY, Gerges N, Korshunov A, Sabha N, Khuong-Quang DA, Fontebasso AM, Fleming A, Hadjadj D, Schwartzentruber J, Majewski J, Dong Z, Siegel P, Albrecht S, Croul S, Jones DT, Kool M, Tonjes M, Reifenberger G, Faury D, Zadeh G, Pfister S, Jabado N (2012) Frequent ATRX mutations and loss of expression in adult diffuse astrocytic tumors carrying IDH1/IDH2 and TP53 mutations. Acta Neuropathol 124(5):615–625. doi:10.1007/s00401-012-1031-3
Mack SC, Witt H, Piro RM, Gu L, Zuyderduyn S, Stutz AM, Wang X, Gallo M, Garzia L, Zayne K, Zhang X, Ramaswamy V, Jager N, Jones DT, Sill M, Pugh TJ, Ryzhova M, Wani KM, Shih DJ, Head R, Remke M, Bailey SD, Zichner T, Faria CC, Barszczyk M, Stark S, Seker-Cin H, Hutter S, Johann P, Bender S, Hovestadt V, Tzaridis T, Dubuc AM, Northcott PA, Peacock J, Bertrand KC, Agnihotri S, Cavalli FM, Clarke I, Nethery-Brokx K, Creasy CL, Verma SK, Koster J, Wu X, Yao Y, Milde T, Sin-Chan P, Zuccaro J, Lau L, Pereira S, Castelo-Branco P, Hirst M, Marra MA, Roberts SS, Fults D, Massimi L, Cho YJ, Van Meter T, Grajkowska W, Lach B, Kulozik AE, von Deimling A, Witt O, Scherer SW, Fan X, Muraszko KM, Kool M, Pomeroy SL, Gupta N, Phillips J, Huang A, Tabori U, Hawkins C, Malkin D, Kongkham PN, Weiss WA, Jabado N, Rutka JT, Bouffet E, Korbel JO, Lupien M, Aldape KD, Bader GD, Eils R, Lichter P, Dirks PB, Pfister SM, Korshunov A, Taylor MD (2014) Epigenomic alterations define lethal CIMP-positive ependymomas of infancy. Nature 506(7489):445–450. doi:10.1038/nature13108
Northcott PA, Shih DJ, Peacock J, Garzia L, Morrissy AS, Zichner T, Stutz AM, Korshunov A, Reimand J, Schumacher SE, Beroukhim R, Ellison DW, Marshall CR, Lionel AC, Mack S, Dubuc A, Yao Y, Ramaswamy V, Luu B, Rolider A, Cavalli FM, Wang X, Remke M, Wu X, Chiu RY, Chu A, Chuah E, Corbett RD, Hoad GR, Jackman SD, Li Y, Lo A, Mungall KL, Nip KM, Qian JQ, Raymond AG, Thiessen NT, Varhol RJ, Birol I, Moore RA, Mungall AJ, Holt R, Kawauchi D, Roussel MF, Kool M, Jones DT, Witt H, Fernandez LA, Kenney AM, Wechsler-Reya RJ, Dirks P, Aviv T, Grajkowska WA, Perek-Polnik M, Haberler CC, Delattre O, Reynaud SS, Doz FF, Pernet-Fattet SS, Cho BK, Kim SK, Wang KC, Scheurlen W, Eberhart CG, Fevre-Montange M, Jouvet A, Pollack IF, Fan X, Muraszko KM, Gillespie GY, Di Rocco C, Massimi L, Michiels EM, Kloosterhof NK, French PJ, Kros JM, Olson JM, Ellenbogen RG, Zitterbart K, Kren L, Thompson RC, Cooper MK, Lach B, McLendon RE, Bigner DD, Fontebasso A, Albrecht S, Jabado N, Lindsey JC, Bailey S, Gupta N, Weiss WA, Bognar L, Klekner A, Van Meter TE, Kumabe T, Tominaga T, Elbabaa SK, Leonard JR, Rubin JB, Liau LM, Van Meir EG, Fouladi M, Nakamura H, Cinalli G, Garami M, Hauser P, Saad AG, Iolascon A, Jung S, Carlotti CG, Vibhakar R, Ra YS, Robinson S, Zollo M, Faria CC, Chan JA, Levy ML, Sorensen PH, Meyerson M, Pomeroy SL, Cho YJ, Bader GD, Tabori U, Hawkins CE, Bouffet E, Scherer SW, Rutka JT, Malkin D, Clifford SC, Jones SJ, Korbel JO, Pfister SM, Marra MA, Taylor MD (2012) Subgroup-specific structural variation across 1,000 medulloblastoma genomes. Nature 488(7409):49–56. doi:10.1038/nature11327
Pugh TJ, Weeraratne SD, Archer TC, Pomeranz Krummel DA, Auclair D, Bochicchio J, Carneiro MO, Carter SL, Cibulskis K, Erlich RL, Greulich H, Lawrence MS, Lennon NJ, McKenna A, Meldrim J, Ramos AH, Ross MG, Russ C, Shefler E, Sivachenko A, Sogoloff B, Stojanov P, Tamayo P, Mesirov JP, Amani V, Teider N, Sengupta S, Francois JP, Northcott PA, Taylor MD, Yu F, Crabtree GR, Kautzman AG, Gabriel SB, Getz G, Jager N, Jones DT, Lichter P, Pfister SM, Roberts TM, Meyerson M, Pomeroy SL, Cho YJ (2012) Medulloblastoma exome sequencing uncovers subtype-specific somatic mutations. Nature 488(7409):106–110. doi:10.1038/nature11329
Robinson G, Parker M, Kranenburg TA, Lu C, Chen X, Ding L, Phoenix TN, Hedlund E, Wei L, Zhu X, Chalhoub N, Baker SJ, Huether R, Kriwacki R, Curley N, Thiruvenkatam R, Wang J, Wu G, Rusch M, Hong X, Becksfort J, Gupta P, Ma J, Easton J, Vadodaria B, Onar-Thomas A, Lin T, Li S, Pounds S, Paugh S, Zhao D, Kawauchi D, Roussel MF, Finkelstein D, Ellison DW, Lau CC, Bouffet E, Hassall T, Gururangan S, Cohn R, Fulton RS, Fulton LL, Dooling DJ, Ochoa K, Gajjar A, Mardis ER, Wilson RK, Downing JR, Zhang J, Gilbertson RJ (2012) Novel mutations target distinct subgroups of medulloblastoma. Nature 488(7409):43–48. doi:10.1038/nature11213
Schwartzentruber J, Korshunov A, Liu XY, Jones DT, Pfaff E, Jacob K, Sturm D, Fontebasso AM, Quang DA, Tonjes M, Hovestadt V, Albrecht S, Kool M, Nantel A, Konermann C, Lindroth A, Jager N, Rausch T, Ryzhova M, Korbel JO, Hielscher T, Hauser P, Garami M, Klekner A, Bognar L, Ebinger M, Schuhmann MU, Scheurlen W, Pekrun A, Fruhwald MC, Roggendorf W, Kramm C, Durken M, Atkinson J, Lepage P, Montpetit A, Zakrzewska M, Zakrzewski K, Liberski PP, Dong Z, Siegel P, Kulozik AE, Zapatka M, Guha A, Malkin D, Felsberg J, Reifenberger G, von Deimling A, Ichimura K, Collins VP, Witt H, Milde T, Witt O, Zhang C, Castelo-Branco P, Lichter P, Faury D, Tabori U, Plass C, Majewski J, Pfister SM, Jabado N (2012) Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 482(7384):226–231. doi:10.1038/nature10833
Sturm D, Witt H, Hovestadt V, Khuong-Quang DA, Jones DT, Konermann C, Pfaff E, Tonjes M, Sill M, Bender S, Kool M, Zapatka M, Becker N, Zucknick M, Hielscher T, Liu XY, Fontebasso AM, Ryzhova M, Albrecht S, Jacob K, Wolter M, Ebinger M, Schuhmann MU, van Meter T, Fruhwald MC, Hauch H, Pekrun A, Radlwimmer B, Niehues T, von Komorowski G, Durken M, Kulozik AE, Madden J, Donson A, Foreman NK, Drissi R, Fouladi M, Scheurlen W, von Deimling A, Monoranu C, Roggendorf W, Herold-Mende C, Unterberg A, Kramm CM, Felsberg J, Hartmann C, Wiestler B, Wick W, Milde T, Witt O, Lindroth AM, Schwartzentruber J, Faury D, Fleming A, Zakrzewska M, Liberski PP, Zakrzewski K, Hauser P, Garami M, Klekner A, Bognar L, Morrissy S, Cavalli F, Taylor MD, van Sluis P, Koster J, Versteeg R, Volckmann R, Mikkelsen T, Aldape K, Reifenberger G, Collins VP, Majewski J, Korshunov A, Lichter P, Plass C, Jabado N, Pfister SM (2012) Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 22(4):425–437. doi:10.1016/j.ccr.2012.08.024
Taylor KR, Mackay A, Truffaux N, Butterfield YS, Morozova O, Philippe C, Castel D, Grasso CS, Vinci M, Carvalho D, Carcaboso AM, de Torres C, Cruz O, Mora J, Entz-Werle N, Ingram WJ, Monje M, Hargrave D, Bullock AN, Puget S, Yip S, Jones C, Grill J (2014) Recurrent activating ACVR1 mutations in diffuse intrinsic pontine glioma. Nat Genet 46(5):457–461. doi:10.1038/ng.2925
Venneti S, Garimella MT, Sullivan LM, Martinez D, Huse JT, Heguy A, Santi M, Thompson CB, Judkins AR (2013) Evaluation of histone 3 lysine 27 trimethylation (H3K27me3) and enhancer of Zest 2 (EZH2) in pediatric glial and glioneuronal tumors shows decreased H3K27me3 in H3F3A K27M mutant glioblastomas. Brain Pathol 23(5):558–564. doi:10.1111/bpa.12042
Wu G, Broniscer A, McEachron TA, Lu C, Paugh BS, Becksfort J, Qu C, Ding L, Huether R, Parker M, Zhang J, Gajjar A, Dyer MA, Mullighan CG, Gilbertson RJ, Mardis ER, Wilson RK, Downing JR, Ellison DW, Zhang J, Baker SJ (2012) Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet 44(3):251–253. doi:10.1038/ng.1102
Wu G, Diaz AK, Paugh BS, Rankin SL, Ju B, Li Y, Zhu X, Qu C, Chen X, Zhang J, Easton J, Edmonson M, Ma X, Lu C, Nagahawatte P, Hedlund E, Rusch M, Pounds S, Lin T, Onar-Thomas A, Huether R, Kriwacki R, Parker M, Gupta P, Becksfort J, Wei L, Mulder HL, Boggs K, Vadodaria B, Yergeau D, Russell JC, Ochoa K, Fulton RS, Fulton LL, Jones C, Boop FA, Broniscer A, Wetmore C, Gajjar A, Ding L, Mardis ER, Wilson RK, Taylor MR, Downing JR, Ellison DW, Zhang J, Baker SJ, St. Jude Children’s Research Hospital-Washington University Pediatric Cancer Genome P (2014) The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat Genet 46(5):444–450. doi:10.1038/ng.2938
Zhang J, Wu G, Miller CP, Tatevossian RG, Dalton JD, Tang B, Orisme W, Punchihewa C, Parker M, Qaddoumi I, Boop FA, Lu C, Kandoth C, Ding L, Lee R, Huether R, Chen X, Hedlund E, Nagahawatte P, Rusch M, Boggs K, Cheng J, Becksfort J, Ma J, Song G, Li Y, Wei L, Wang J, Shurtleff S, Easton J, Zhao D, Fulton RS, Fulton LL, Dooling DJ, Vadodaria B, Mulder HL, Tang C, Ochoa K, Mullighan CG, Gajjar A, Kriwacki R, Sheer D, Gilbertson RJ, Mardis ER, Wilson RK, Downing JR, Baker SJ, Ellison DW, St. Jude Children’s Research Hospital-Washington University Pediatric Cancer Genome P (2013) Whole-genome sequencing identifies genetic alterations in pediatric low-grade gliomas. Nat Genet 45(6):602–612. doi:10.1038/ng.2611
Acknowledgments
This work was performed within the context of the I-CHANGE consortium (International Childhood Astrocytoma iNntegrated Genomics and Epigenomics consortium) and supported by funding from Genome Canada, Genome Quebec, The Institute for Cancer Research of the Canadian Institutes for Health Research (CIHR) McGill University and the Montreal Children’s Hospital Foundation. N. Jabado is a member of the Penny Cole lab and the recipient of a Chercheur Clinicien Senior Award. D.B. is supported by a studentship from the T.D. Trust/Montreal Children’s Hospital Foundation. The authors would like to thank Leonie Michael for critical reading of the manuscript.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Bechet, D., Gielen, G.G.H., Korshunov, A. et al. Specific detection of methionine 27 mutation in histone 3 variants (H3K27M) in fixed tissue from high-grade astrocytomas. Acta Neuropathol 128, 733–741 (2014). https://doi.org/10.1007/s00401-014-1337-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-014-1337-4