
Abstract
The GGGGCC (G4C2) repeat expansion in C9ORF72 is the most common cause of familial amyotrophic lateral sclerosis (ALS), frontotemporal lobar dementia (FTLD) and ALS–FTLD, as well as contributing to sporadic forms of these diseases. Screening of large cohorts of ALS and FTLD cohorts has identified that C9ORF72-ALS is represented throughout the clinical spectrum of ALS phenotypes, though in comparison with other genetic subtypes, C9ORF72 carriers have a higher incidence of bulbar onset disease. In contrast, C9ORF72-FTLD is predominantly associated with behavioural variant FTD, which often presents with psychosis, most commonly in the form of hallucinations and delusions. However, C9ORF72 expansions are not restricted to these clinical phenotypes. There is a higher than expected incidence of parkinsonism in ALS patients with C9ORF72 expansions, and the G4C2 repeat has also been reported in other motor phenotypes, such as primary lateral sclerosis, progressive muscular atrophy, corticobasal syndrome and Huntington-like disorders. In addition, the expansion has been identified in non-motor phenotypes including Alzheimer’s disease and Lewy body dementia. It is not currently understood what is the basis of the clinical variation seen with the G4C2 repeat expansion. One potential explanation is repeat length. Sizing of the expansion by Southern blotting has established that there is somatic heterogeneity, with different expansion lengths in different tissues, even within the brain. To date, no correlation with expansion size and clinical phenotype has been established in ALS, whilst in FTLD only repeat size in the cerebellum was found to correlate with disease duration. Somatic heterogeneity suggests there is a degree of instability within the repeat and evidence of anticipation has been reported with reducing age of onset in subsequent generations. This variability/instability in expansion length, along with its interactions with environmental and genetic modifiers, such as TMEM106B, may be the basis of the differing clinical phenotypes arising from the mutation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
One of the most interesting features of hexanucleotide repeat expansions of C9ORF72 is that the associated phenotype is extremely variable and indeed, except in certain pedigrees, the expansion does not appear to be 100 % penetrant [1]. Although it was discovered in patients with amyotrophic lateral sclerosis (ALS) and/or frontotemporal lobar degeneration (FTLD) [2, 3] many of the original studies of cohorts of C9ORF72-related patients noted the presence of other clinical phenotypes in probands and family members, at higher frequencies than would be expected by chance [4]. For the purposes of this review, clinical presentations will be divided into motor and non-motor phenotypes within which various recognised symptom complexes will be discussed (Fig. 1). The variability in, and indeed within, clinical presentations of C9ORF72-related disease [4] is a source of hope as well as a challenge as it suggests that multiple disease modifiers are at work, each of which is a potential therapeutic target. Understanding what these potential modifiers might be is at an early stage and indeed the number of likely modifiers means that large numbers of cases are going to be needed to illustrate any single effect. The most obvious potential modifier is expansion size, but as yet a clear relationship has not been established in ALS, although there is more indication of a correlation with phenotype in FTLD.

Clinical phenotypes associated with G4C2 repeat expansion of C9ORF72: both motor and non-motor phenotypes are associated with C9ORF72 expansions. The primary phenotype in each case is amyotrophic lateral sclerosis (ALS) and frontotemporal lobar dementia (FTLD). Other phenotypes have been noted in very small numbers of patients: motor phenotypes in this group are parkinsonism, olivopontocerebellar degeneration, corticobasal syndrome and Huntington’s disease phenocopies. The only non-motor phenotype identified in more than a few individuals is Alzheimer’s disease
The penetrance of C9ORF72-related disease does not appear to differ between ALS and FTLD pedigrees. Identification of repeat expansions in the general population during mutation screening of neurologically normal controls [1, 4] suggests that C9ORF72 expansions may actually be as common in “controls” as in specific clinical phenotypes, such as corticobasal syndrome (CBS) and sporadic Creutzfeldt–Jakob disease [1, 5]. It has been suggested that these C9ORF72 “controls” might represent pre-symptomatic disease as estimates suggest that C9ORF72 expansions are non-penetrant at <35 years, 50 % penetrant by 58 years and fully penetrant at 80 years [5, 6]. Alternatively, the penetrance of the expansion may not reach 100 % even at 80 years, as others have reported C9ORF72 expansion carriers aged 80 years and over without a clinical phenotype [7]. This implies that the expansions in Alzheimer’s disease cases may be chance occurrences. Based on estimates of prevalence of ALS and FTLD in the UK population being 0.3 and 0.07 %, respectively [8, 9] and the proportion of cases with C9ORF72 expansions being 10 % for ALS and 13.6 % for FTLD [5], we have calculated the lifetime risk of C9ORF72-related disease to be approximately 0.04 %. There is, therefore, a 15-fold difference between the estimated frequency of C9ORF72 repeat expansions in controls of up to 0.6 % [4], and the lifetime risk of C9ORF72-related disease. This difference is perhaps too large to be explained by premature death of expansion carriers from other causes, although this remains to be verified. The variable penetrance also raises the possibility that C9ORF72 expansions may be a risk factor for disease and may not be capable of producing disease in isolation. There is a higher than expected coincidence of repeat expansions found in individuals carrying other genetic variants of ALS, so-called oligogenic inheritance [10], suggesting that another ‘hit’ may be necessary for clinical disease. Oligogenic inheritance has also been reported in FTLD, with the C9ORF72 expansion being identified in patients carrying GRN mutations [11, 12].
C9ORF72 in ALS and other motor phenotypes
Epidemiology
Whilst the majority of cases of ALS are sporadic, data show that 5–10 % of ALS cases show clear autosomal dominant inheritance [13], whilst twin studies estimate that the genetic heritability of disease is between 0.38 and 0.78 [14]. Over 20 loci have been identified, with the genes identified at 17, the most common causes being mutations of C9ORF72, SOD1, TARDBP and FUS [15].
Screening for the C9ORF72 expansion in ALS cohorts worldwide has identified this mutation as the most common genetic cause of ALS in Caucasians, and the proportion of patients affected is significant in this complex neurodegenerative disease. In the Northern England population, C9ORF72 expansions are present in 43 % of ALS cases with an identifiable family history and in 7 % of apparently sporadic cases [4]. However, this frequency varies globally: C9ORF72 expansions were found in 61 % of familial ALS and 19 % of sporadic ALS patients in Finland [5], 22 % of familial ALS cases in Germany [5] and only 3.4 % in Japan [16]. The apparent correlation of expansion frequency with distance from Scandinavia is consistent with a suggested common founder rather than repeated de novo occurrence. Indeed the C9ORF72 expansion was identified through study of a risk haplotype at the 9p21 locus [17]. Following the screening of numerous populations for the C9ORF72 expansion, it has been shown to occur in the presence of the same haplotype in all populations considered, or with the A-allele of rs389942, which is associated with the haplotype [18], including populations which might be considered relatively genetically different from Scandinavia such as Japan [16]. Data analysis of a 42-SNP haplotype from carriers of the expansion in Europe, USA and Australia led to the hypothesis that there was a common founder carrying the pathogenic mutation approximately 100 generations ago in Northern Europe and this expansion then spread across the world [5]. A European study using an 82-SNP haplotype suggests the expansion is much older, with the common founder arose over 251 generations ago [19]. As the disease has a late age of onset, this means it is not subject to reproductive pressure. However, other studies have contradicted the founder effect, showing no evidence of recent shared ancestry in cases identified within the South-east region of the UK and the presence of the expansion in a patient homozygous for the non-risk G-allele [1, 20].
The origin of the expansion becomes even more intriguing if it is considered whether it is the expansion itself or a propensity for the region to expand that is inherited. One of the initial studies of C9ORF72 noted that the risk haplotype was associated with an increased number of repeats even in controls [3]. It was determined, in a group of controls, that longer repeat lengths were associated with the rs389942 A-allele and that over 10 repeats was associated with inter-generational changes in repeat length [1].
C9ORF72-related ALS phenotypes
ALS is a neurodegenerative disease defined clinically as the loss of upper and/or lower motor neurons. It is a disease of ageing: peak age of onset is between 50 and 70 years [21]. Progression is relentless and death usually results from respiratory failure, between 3 and 5 years after onset of symptoms [22]. However, the ALS phenotype is notably variable; approximately 20 % of patients survive longer than 5 years [23] and cases have been reported in teenagers and the very elderly. The disease usually starts in one area and spreads in an anatomically contiguous fashion throughout the motor system [24]. Typically this involves insidious progression of weakness from one limb or the bulbar muscles; rarely does the disease simultaneously start in multiple areas or in the respiratory muscles.
Broadly, the ALS phenotype associated with G4C2 repeat expansion of C9ORF72 is representative of the whole clinical spectrum of ALS [4, 25, 26]. However, within the broad range of phenotypes, bulbar onset has been found to be more frequently associated with C9ORF72-related ALS, than with non-C9ORF72 ALS [4, 27–29]; and as would be expected from the association of C9ORF72 with FTLD, there is a greater incidence of dementia or a family history of dementia in C9ORF72-related ALS, than in non-C9ORF72 ALS [4, 25, 29–32]. In addition, there is evidence of an earlier age of onset in C9ORF72-related ALS compared to non-C9ORF72 ALS cases [30, 31] though not all cohorts reached significance [4, 31]. However, in a Belgian cohort, where specifically familial cases with C9ORF72-related ALS were compared to non-C9ORF72 familial ALS cases, the age of onset was later in C9ORF72-related familial ALS [27]. In contrast, there were no differences between C9ORF72-related and non-C9ORF72 sporadic ALS cases. Finally, evidence suggests that C9ORF72-related ALS has a shorter survival than non-C9ORF72 ALS [4, 27–32]. Some of the discrepancies between these reports may be explained by different cohorts being used in the comparisons (e.g. all C9ORF72-related ALS v all non-C9ORF72 ALS as opposed to familial C9ORF72-related ALS v familial non-C9ORF72 ALS) and due to the extensive variability in the non-C9ORF72 ALS phenotype. However, gender may also play a role as male C9ORF72-related ALS cases have been reported to have a younger age of onset than non-C9ORF72 ALS cases [33].
To address the variability of non-C9ORF72 ALS, one study compared a large cohort of C9ORF72-related ALS cases directly with patients carrying other genetic variants [34]. This showed that C9ORF72-related cases had a significantly higher incidence of bulbar onset compared to ALS cases with mutations in SOD1, TARDBP, FUS and other familial ALS cases, as well as a greater association with FTLD. Disease duration of C9ORF72-related ALS was significantly shorter than in patients with mutations in SOD1, TARDBP or other familial ALS cases, but did not differ from FUS cases, whilst there was an older age of onset in C9ORF72-related ALS compared to SOD1 and FUS-related ALS, but not when compared with TARDBP and other familial ALS cases.
Other motor phenotypes
Parkinsonism
What is most clear about the C9ORF72-related ALS phenotype compared to other ALS cases, whether familial or sporadic, is an association with other neurological phenotypes both motor and extramotor [4, 34]. Most prominently this includes FTLD and a number of other non-motor phenotypes (which will be discussed further in the section on “FTLD and other dementia and psychosis-related phenotypes”). In addition to FTLD, we and others also noted an association with other motor phenotypes including parkinsonism; by which we refer to the clinical presentation rather than confirmed Parkinson’s disease (PD) with α-synucleinopathy. It is clear that parkinsonism is over-represented compared to chance in C9ORF72-related ALS cases and their families [3, 4, 35, 36]. Despite this, a number of studies have failed to find C9ORF72 expansions in patients with a clinical diagnosis of pure PD [37–39] or only very rarely and often in patients with a family history of ALS/FTLD [40, 41] perhaps suggesting a pathogenesis distinct from classical idiopathic PD. Focusing on the neuropathology of these cases, added some clarity to this point. Whilst there was no evidence for over-representation of C9ORF72 expansions in a cohort of PD patients with confirmed α-synucleinopathy, neurodegeneration was shown in the substantia nigra of C9ORF72-related ALS cases [40]. Therefore, it is suggested that whilst C9ORF72 expansions are not a cause of classical PD, the neuropathology associated with C9ORF72-related ALS can affect the substantia nigra and cause parkinsonism. Thus, C9ORF72 expansions can affect multiple brain areas which lead to different clinical consequences.
Multiple sclerosis
Another phenotype associated with C9ORF72-related ALS was demyelination [4]. To further investigate this, a cohort of multiple sclerosis (MS) cases was screened but no C9ORF72 expansions were identified. However, in a small number of prospectively identified cases with MS who subsequently developed ALS, there was a significantly higher than expected number of C9ORF72 expansions [42]. Rather than C9ORF72 expansions causing MS it is suggested MS increased the penetrance of the C9ORF72 expansion, and this was supported by the fact that C9ORF72-related ALS was more rapidly progressive in the patients with a previous history of MS.
Other motor phenotypes screened or associated with for C9ORF72 expansions
With the widening clinical phenotypes associated with ALS, several other neurodegenerative disease cohorts have been screened to determine if they are associated with the C9ORF72 repeat expansion. Whilst ALS is defined as the loss of both upper and lower motor neurons, primary lateral sclerosis (PLS) is characterised by the degeneration of only upper motor neurons whilst the loss of only lower motor neurons is defined as progressive muscular atrophy (PMA). These three disorders are considered to form part of a spectrum of disease. However, screening for the C9ORF72 expansion found the expanded repeats at relatively low frequencies in the PMA and PLS cohorts, at 1.6 and 0.9 %, respectively [43], and in a smaller cohort of PMA, PLS and progressive bulbar palsy patients, no expansions were found [44]. Thus, C9ORF72 expansions appear to be more commonly associated with an ALS phenotype within this spectrum of disease.
Hereditary spastic paraplegia (HSP) describes a group of over 50 genetically heterogeneous diseases which are characterised by a progressive dying back of the distal axons of upper motor neurons, leading to lower limb spasticity, pyramidal distribution weakness in the lower limbs and brisk reflexes. To establish if C9ORF72 might be a modifier of the HSP phenotype, a Danish cohort of 182 HSP cases was screened for the repeat expansion [45]. However, no mutations were identified. The mutation was not associated with any cases of corticobasal syndrome or progressive supranuclear palsy (PSP) in Italy or the United States [46, 47] but was identified in a Danish patient with CBS [48]. In addition, where ALS + CBS and ALS + PSP were the clinical diagnosis, C9ORF72 expansions were identified [49]. Finally, a pathological characterisation of C9ORF72-FTLD cases identified an individual with FTLD-tau and corticobasal degeneration [50].
In the Danish cohort of dementia cases, which included the C9ORF72-related CBS case, C9ORF72 expansions were also found in one patient clinically diagnosed with olivopontocerebellar degeneration (OPCD) (where his father had ALS) and one patient with atypical Parkinsonian syndrome (APS) [48]. The range of motor phenotypes associated with C9ORF72 expansions was widened further with expansions found in multiple cases of a cohort of Huntington disease-like syndromes and “other neurodegenerative diseases” (excluding ALS and FTLD) [1]. The association of C9ORF72 with HD-like symptoms was supported by 1.95 % cases within a cohort of HD phenocopies being positive for the expansion [51]. Thus, the C9ORF72 expansion is associated with additional motor phenotypes, outside of the ALS spectrum, albeit at low frequencies.
C9ORF72 in FTLD and other dementia and psychosis-related phenotypes
Epidemiology
Whilst the majority of cases of FTLD are sporadic, a body of evidence shows that approximately 13 % of FTLD cases show clear autosomal dominant inheritance. However, up to 40 % of cases have an additional member of the family with the disease, though whether this is a contribution of genes or environment or both remains to be established [52].
Mutations in MAPT, CHMP2B, VCP, GRN, TARDBP and FUS have been previously published as genetic causes of autosomal dominant FTLD. However, with the identification of the C9ORF72 repeat expansion as a cause of FTLD and ALS–FTLD [2, 3], many cohorts of FTLD cases have been screened to establish the frequency of this gene in FTLD. A consistent finding is that the C9ORF72 expansion is the most frequent mutation associated with familial cases, accounting for 25.1 % of familial FTLD [5]. In addition, the C9ORF72 mutation is also found in 6 % of sporadic FTLD cases, though there is a wide variation in frequency across different populations, with the highest frequencies in geographically isolated populations such as Finland (21 %) [53] and the lowest frequency found in Holland (2.2 %) [5]. The mutation is most frequently found in Caucasian populations, although the expansion has recently been shown in two Chinese cases, one with sporadic FTLD and another with familial ALS–FTLD [54]. This is in contrast to ALS, where C9ORF72 carriers are seen in Native American, Hispanic, Middle Eastern and Asian populations [5].
As discussed earlier, the expansion is suggested to have arisen over 100 generations ago in Northern Europe, based upon analysis that suggested expansion carriers all carried the same risk haplotype [5]. However, as with ALS, this is not always the case [11]. Ferrari and colleagues report four cases which carried the surrogate risk haplotype marker rs3849942 A-allele, but did not carry the complete 42 SNP risk haplotype. This finding is most likely to be due to the ethnically diverse background of these patients from the USA and the effect of recombination events throughout the preceding generations.
In addition, what is particularly interesting about the chromosome 9p21 region is that if expansion carriers are excluded, the locus still shows significant linkage to the ALS phenotype, suggesting a further genetic risk factor may be associated with the same region. This may perhaps point to the presence of another expansion [55]. Further sequencing and analysis of the region will be required to elucidate the basis of this linkage finding.
C9ORF72-related FTLD phenotypes
FTLD is the second most common form of degenerative dementia after Alzheimer’s disease to occur in individuals younger than 65 years old, and accounts for 5–15 % of all dementia cases. Survival is on average 7 years from onset. It can be subdivided according to the presenting features into behavioural variant FTLD (bvFTLD), where there is a change in the patients behaviour associated with a progressive deterioration of personality, usually beginning with hallucinations, delusions, disordered thinking or paranoia, or primary progressive aphasia (PPA). PPA can be further subdivided into progressive non-fluent aphasia (PNFA) where patients have difficulties with word retrieval, non-fluent speech patterns and a progressive loss of speech, and semantic dementia (SD), where there is a loss of memory regarding the understanding of words and objects. In addition, FTLD can be associated with extrapyramidal movement disorders, such as parkinsonism or corticobasal syndrome as well as amyotrophic lateral sclerosis/motor neurone disease (FTLD–ALS/FTLD–MND), which will be discussed later.
The predominant FTLD phenotype associated with C9ORF72 mutations is that of bvFTLD. The high incidence of this variant in the C9ORF72 expansion carriers was reported in the initial publications [2, 3]. Subsequent screening of FTLD cohorts across the UK, Europe, America and Australia identified bvFTLD as consistently more prevalent in C9ORF72-related FTLD than non-C9ORF72 FTLD cases [6, 11, 20, 25, 26, 35, 46, 47, 53, 56–60]. The percentage of bvFTLD within C9ORF72-related FTLD varied within the cohorts, but it was always higher by 12–19 % [6, 53, 59]. Within the C9ORF72 bvFTLD cases there were a significant number who presented with psychosis, most commonly hallucinations and delusions [47, 59]. This is exemplified in a UK study where a third of the C9ORF72-related FTLD cases presented with psychosis, compared to only 4 % in the non-C9ORF72 cases [59]. Whilst bvFTLD was the most common FTLD variant associated with C9ORF72 expansion, PNFA was the second most common FTLD variant associated with C9ORF72 expansions, although the frequency of PNFA did not differ between carriers and non-carriers of the expansion. SD has been seen more rarely associated with C9ORF72 expansions [58, 59, 61].
The clinical phenotype of C9ORF72-related FTLD has also been compared to FTLD cases carrying mutations in GRN and MAPT. In an early study, no clinical differences were seen between C9ORF72, GRN and MAPT cases, although neuroimaging showed predominant frontal atrophy was more common in the C9ORF72-related cases. A subsequent study showed that the C9ORF72-related FTLD cases showed an earlier age of onset than GRN mutation carriers, whilst survival was similar, and whereas bvFTLD was most commonly associated with C9ORF72 expansions, PNFA was more commonly associated with GRN mutations [60]. Within a cohort of bvFTLD the relative frequencies of these three genetic variants showed that in familial cases, the frequency of C9ORF72 was similar to MAPT mutations (14.7 and 12.7 %, respectively) but GRN mutations were of lower frequency (6.8 %) [35]. In sporadic bvFTLD cases, however, the frequency of these three mutations in the cohort was similar, at 3–4 %.
Other dementia and psychosis-related phenotypes
Alzheimer’s disease
Initial screening of Alzheimer’s disease (AD) patients either failed to find any C9ORF72 expansions or suggested that the cases were misdiagnosed FTLD cases [5, 47, 62]. Indeed, pathological studies of these initial Alzheimer’s disease cases identified none of the p62-positive inclusions in hippocampus and cerebellum which are considered pathognomonic for C9ORF72 disease [41, 63]. However, subsequent screening of AD cohorts has identified several cases with C9ORF72 expansions, including individuals with autopsy confirmed AD [64–66]. The cases were also shown to carry the risk A-allele at rs389942. In addition, it was found that these C9ORF72-related AD cases have a later age of onset, at 77.8 years, compared to the rest of the cohort of over 1,000 definite or probable AD cases [66, 67]. However, no association of C9ORF72 with the APOE ε4 status was identified [65, 66]. Therefore, the C9ORF72 expansion is associated with AD at low frequencies (<1 %), but notably more often than in healthy controls [64–66].
Other dementia-related disorders screened or associated with for C9ORF72 expansions
Expansions of C9ORF72 have also been identified in single cases of dementia with Lewy bodies [68] and sporadic Creutzfeldt–Jakob disease [1], although the former patient is proposed to suffer from a FTLD TDP-proteinopathy that mimics Lewy body dementia. Interestingly, whilst psychosis is highly associated with C9ORF72-related bvFTLD, no C9ORF72 expansions were seen in a cohort of 192 schizophrenia cases [69].
ALS–FTLD
ALS and FTLD have been linked both through families presenting with ALS, FTLD or both diseases. It was through studying these families that the Chr9p21 locus and C9ORF72 as a genetic cause of ALS and FTLD was identified [2, 3]. Several other genes associated with ALS have subsequently been found, albeit less commonly, in FTLD (TARDBP, FUS) and vice versa (CHMP2B). However, C9ORF72 provides the strongest link between the two disorders. Whilst previously familial ALS or familial FTLD was classified if there was a close family member with the disease, the incidence of familial disease increases if both ALS and dementia are included [4]. Screening of ALS–FTLD cases has shown that C9ORF72 expansion is the most common cause of familial ALS–FTLD, accounting for the majority (sometimes all) of the ALS–FTLD cases screened [25, 26, 56]. Individuals can present with ALS or FTLD first and then go on to develop the other disease manifestation later. Clinically and pathologically patients possess both classical ALS and FTLD symptoms and neuropathology. The molecular basis for this disease specification is currently unknown, although work is beginning to elucidate the effect of the expansion length and other genetic and environmental modifiers on C9ORF72 disease.
Clinical phenotype specification: role of expansion length
In a repeat expansion-related disorder the most obvious candidate for a genetic modifier is repeat number. Measurement of the G4C2 repeat length initially proved technically challenging as it is not amenable to PCR-based sequencing. However, a number of groups have now optimised Southern hybridisation-based techniques [1, 70, 71] which allow sizing of the expansion. So far, in a pure ALS group, no aspect of phenotype has been shown to significantly correlate with the length of the expansion regardless of the tissue tested [71, 72]. However, in FTLD, van Blitterswijk et al. [71] revealed a direct correlation between repeat size in the frontal cortex and age of onset, and a threshold repeat size in the cerebellum associated with reduced survival. The work also established that the repeat length in the cerebellum was shorter than in other CNS areas (mean of approximately 1,667 repeat units compared to approximately 5,250 repeat units in the frontal cortex). It is hypothesised that repeat expansions can increase in size through a human lifetime resulting in significant somatic heterogeneity [73] and therefore, perhaps the minimum repeat length in the CNS is more reflective of the germline repeat number. If so, the expansion length in the cerebellum may best represent the repeat length which initiated the disease pathogenesis; and the correlation with age of onset in frontal cortex may simply reflect the patient’s age. In this context it is interesting, but not conclusive, to note that the same study found smaller repeat lengths in blood from two out of three unaffected expansion carriers compared to their affected relatives. A smaller study identified a positive correlation between repeat length and age of onset in C9ORF72-related patients with a variety of neurodegenerative phenotypes including FTLD, ALS and Alzheimer’s disease [1] but as described above, this may simply reflect the individual’s age at the time of sampling. It has also been shown that the length of the non-expanded allele is not a disease modifier in C9ORF72-related or non-C9ORF72 ALS or FTLD [71, 74, 75].
One of the significant questions is whether there is evidence of anticipation in C9ORF72-related disease. Benussi and colleagues [6] reported on C9ORF72-related FTLD families which showed evidence of anticipation with a mean difference in age of onset between the parent and offspring of 9.8 years; This was also seen by Chio et al. [76], with age of onset 7 years earlier in the subsequent generation in Italian ALS cases. However, whether this related to expansion size is still to be determined, perhaps through Southern blotting analysis of parents and offspring DNA.
In summary, a clear relationship between C9ORF72 expansion size and disease severity has not yet been identified, particularly in ALS. Comparison with the literature on myotonic dystrophy type I (DM1) is informative on this point. Similar to C9ORF72-related disease, DM1 is caused by a repeat expansion in a non-coding region of DMPK. In DM1, correlation between repeat size and phenotype is apparent, but only when a large number of cases (>100) are considered [77]. The reason for this is thought to be the presence of other disease modifiers, the effect of which must be averaged out before a genotype–phenotype correlation is identifiable. Clinically this is relevant because it means that measurement of repeat length could never serve as a prognostic biomarker. As yet the studies of expansion length in C9ORF72-ALS have only included relatively small numbers of patients and so there is still a case to answer. Work on the pathogenic mechanism in C9ORF72 disease appears to be settling on a gain-of-function toxicity mediated either by RNA foci formed from the repeat sequence [2] or dipeptide repeat protein formed by repeat associated non-ATG (RAN) translation [78, 79]. Numbers of RNA foci have been correlated with pathogenic severity in cell models [80, 81], and in tissue from FTLD cases [82]. RAN-translated dipeptide repeat protein appears to be toxic in a cell model [83], but levels of the aberrantly translated protein observed do not correlate with neurodegeneration in autopsy material [34]. If, as postulated, pathogenesis is mediated in a gain-of-function manner then expansion size would be expected to modify the disease phenotype (Fig. 2).

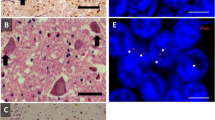
Proposed pathogenic mechanisms and modifiers in C9ORF72 disease. Three prominent mechanisms of pathogenesis have been proposed in C9ORF72 disease: (1) Haploinsufficiency, (2) RAN translation of the expansion to form dipeptide repeats and (3) the formation of toxic RNA foci. Poly-(Gly-Ala)- dipeptide repeat protein is shown (stained in green, arrowed) aggregated in the cytoplasm of a cerebellar granule cell from a C9ORF72-ALS patient. RNA foci containing the G4C2 repeat sequence are shown (stained red, arrowed) within a fibroblast obtained from a C9ORF72-ALS patient. There is some evidence that the repeat length is a modifier of the disease phenotype which would be consistent with mechanisms (2) and (3). However, case reports suggest that disease phenotype is not directly proportional to the number of affected alleles which goes against mechanism (1). The function of the C9ORF72 protein is unknown, but a role in membrane trafficking has been proposed which is consistent with the modifier effect of the TMEM106B genotype: both C9ORF72 and TMEM106B are postulated to be involved in lysosome function
Other than disease severity it is also interesting to ask whether expansion size has an effect on perhaps the most noticeable source of variability in the C9ORF72 disease phenotypes: the primary group of neurons affected. Currently, work has been limited to comparing C9ORF72-ALS and C9ORF72-FTLD cases. One study suggested that longer repeat sizes in blood are associated with ALS as opposed to FTLD [72], but others suggested that there is no difference between the two subtypes [1, 71]. However, the largest group size with a pure phenotype in either of these studies was 38 patients; larger numbers will be required to verify either conclusion.
Finally, the G4C2 expansions have been described as pathogenic if they are over 30 repeats. However, relatively small repeat sizes are reportedly pathogenic in FTLD [84] and the clinical phenotype of ALS cases with 20–30 repeats are similar to those with over 30 repeats [85]. Therefore, the minimum number of repeats required for pathogenicity may be smaller than the currently adopted cut-off size of 30 repeats. If true, this has an impact on another hypothesised pathogenic mechanism in C9ORF72 disease: haploinsufficiency. Reduced expression of C9ORF72 mRNA is seen in the presence of the expansion [2]. However, it has been demonstrated that small expansions of approximately 50 repeats do not reduce transcription [86] possibly because smaller expansions do not lead to hypermethylation of a CpG island 5′ to the repeat sequence [87]. If smaller repeat lengths are pathogenic, then haploinsufficiency is not the responsible mechanism. Another observation not consistent with haploinsufficiency comes from two patients with expansions of both C9ORF72 loci; one a homozygote [16] and the other a compound heterozygote [86]. Both cases suffered FTLD but neither phenotype was outside the usual phenotypic spectrum. This is not consistent with a pure haploinsufficiency model which would predict a disease severity in proportion to the number of involved alleles. It remains to be seen whether haploinsufficiency is a disease modifier, in particular we await the arrival of a specific antibody to accurately detect protein levels of C9ORF72, but evidence is growing that it cannot be the sole or even the primary mechanism in C9ORF72 disease (Fig. 2).
Clinical phenotype specification: other potential disease modifiers
The striking variability in the phenotype associated with the C9ORF72 expansion suggests that multiple modifiers may exist, which may be genetic or environmental. Each identified modifier is a potential therapeutic target and, therefore, understanding factors influencing the phenotype of C9ORF72 disease is likely to be an important avenue of research in the near future. In addition to the expansion length (described above), a number of other modifiers have been investigated, including TMEM106B genotype.
TMEM106B genotype
A genome-wide association study initially identified single nucleotide polymorphisms (SNPs) in TMEM106B as risk factors for FTLD with TDP-43 positive pathology (which is found in C9ORF72-related FTLD cases amongst other subtypes) [88]. The protein product of TMEM106B is localised to the lysosome. Its function is as yet only beginning to be understood, but it has been suggested that the protective isoform is expressed at a lower level because of increased protein degradation mediated via altered glycosylation [89].
The haplotype associated with higher risk of FTLD-TDP, more particularly the major, or T, allele of rs1990622, has recently been investigated in the context of C9ORF72-related disease [90]. The major allele is significantly associated with FTLD presentations of C9ORF72-related disease and is associated with an earlier age of onset in GRN-related FTLD. However, in C9ORF72-FTLD, the major allele is associated with a later age of onset and death. There is no effect on the phenotype in GRN-negative non-C9ORF72 FTLD-TDP patients. This fascinating complexity suggests an interaction between the C9ORF72 expansion and TMEM106B genotype and suggests both proteins may have similar function. Interestingly, it has been suggested that the C9ORF72 protein is structurally associated with the DENN family of proteins [91] which are implicated in the regulation of membrane trafficking required for lysosome function (Fig. 2).
Interestingly, in contrast to the C9ORF72-FTLD findings; it has been shown that neither TMEM106B allele is significantly associated with a C9ORF72-related ALS presentation [92]. Why the TMEM106B genotype modifies the risk of one phenotype and not the other is unknown, but this suggests that the mechanism of neurotoxicity may be different in each case.
Other modifiers
As mentioned above, we have suggested that multiple sclerosis influences the penetrance and phenotype of C9ORF72-related ALS patients. Moreover, we showed that, in contrast to non-C9ORF72 ALS cases, C9ORF72-related ALS cases showed a reduction in CSF levels of the cytokine CXCL10 [42] which is reportedly neuroprotective in ALS [93]. Neuroinflammation and activation of microglia have been detected pathologically [94] and in imaging studies [95] of ALS patients. It is thought that the activity of non-neuronal cells may be key determinants of disease propagation through the CNS [96], if not in disease initiation directly.
Conclusion
Identification of the C9ORF72 repeat expansion has established the most common cause of ALS, FTLD and ALS–FTLD. Recent screening of related clinical phenotypes has extended the disease spectrum in which the C9ORF72 expansion is found, including Alzheimer’s disease and parkinsonism. As Southern blotting is used to more accurately size the repeat in larger cohorts of cases, and in multiple tissues, correlations between the expansion length and clinical characteristics may be revealed. Finally, given the frequency of the expansion in both ALS and FTLD, this population will provide an ideal group to elucidate the role of genetic and environmental modifiers contributing to the heterogeneity of disease.
References
Beck J, Poulter M, Hensman D, Rohrer JD, Mahoney CJ, Adamson G, Campbell T, Uphill J, Borg A, Fratta P, Orrell RW, Malaspina A, Rowe J, Brown J, Hodges J et al (2013) Large C9orf72 hexanucleotide repeat expansions are seen in multiple neurodegenerative syndromes and are more frequent than expected in the UK population. Am J Hum Genet 92(3):345–353. doi:10.1016/j.ajhg.2013.01.011
DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J, Kouri N, Wojtas A, Sengdy P, Hsiung GY, Karydas A et al (2011) Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72(2):245–256. doi:10.1016/j.neuron.2011.09.011
Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L, Kalimo H, Paetau A, Abramzon Y, Remes AM, Kaganovich A et al (2011) A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72(2):257–268. doi:10.1016/j.neuron.2011.09.010
Cooper-Knock J, Hewitt C, Highley JR, Brockington A, Milano A, Man S, Martindale J, Hartley J, Walsh T, Gelsthorpe C, Baxter L, Forster G, Fox M, Bury J, Mok K et al (2012) Clinico-pathological features in amyotrophic lateral sclerosis with expansions in C9ORF72. Brain 135(Pt 3):751–764. doi:10.1093/brain/awr365
Majounie E, Renton AE, Mok K, Dopper EG, Waite A, Rollinson S, Chio A, Restagno G, Nicolaou N, Simon-Sanchez J, van Swieten JC, Abramzon Y, Johnson JO, Sendtner M, Pamphlett R et al (2012) Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol 11(4):323–330. doi:10.1016/S1474-4422(12)70043-1
Benussi L, Rossi G, Glionna M, Tonoli E, Piccoli E, Fostinelli S, Paterlini A, Flocco R, Albani D, Pantieri R, Cereda C, Forloni G, Tagliavini F, Binetti G, Ghidoni R (2013) C9ORF72 Hexanucleotide Repeat Number in Frontotemporal Lobar Degeneration: a Genotype-Phenotype Correlation Study. J Alzheimers Dis. doi:10.3233/JAD-131028
Galimberti D, Arosio B, Fenoglio C, Serpente M, Cioffi SM, Bonsi R, Rossi P, Abbate C, Mari D, Scarpini E (2013) Incomplete Penetrance of the C9ORF72 Hexanucleotide Repeat Expansions: frequency in a Cohort of Geriatric Non-Demented Subjects. J Alzheimers Dis. doi:10.3233/JAD-131172
Johnston CA, Stanton BR, Turner MR, Gray R, Blunt AH, Butt D, Ampong MA, Shaw CE, Leigh PN, Al-Chalabi A (2006) Amyotrophic lateral sclerosis in an urban setting: a population based study of inner city London. J Neurol 253(12):1642–1643. doi:10.1007/s00415-006-0195-y
Mercy L, Hodges JR, Dawson K, Barker RA, Brayne C (2008) Incidence of early-onset dementias in Cambridgeshire, United Kingdom. Neurology 71(19):1496–1499. doi:10.1212/01.wnl.0000334277.16896.fa
van Blitterswijk M, van Es MA, Hennekam EA, Dooijes D, van Rheenen W, Medic J, Bourque PR, Schelhaas HJ, van der Kooi AJ, de Visser M, de Bakker PI, Veldink JH, van den Berg LH (2012) Evidence for an oligogenic basis of amyotrophic lateral sclerosis. Hum Mol Genet 21(17):3776–3784. doi:10.1093/hmg/dds199
Ferrari R, Mok K, Moreno JH, Cosentino S, Goldman J, Pietrini P, Mayeux R, Tierney MC, Kapogiannis D, Jicha GA, Murrell JR, Ghetti B, Wassermann EM, Grafman J, Hardy J et al (2012) Screening for C9ORF72 repeat expansion in FTLD. Neurobiol Aging 33(8):1850.e1–1850.e11. doi:10.1016/j.neurobiolaging.2012.02.017
Lashley T, Rohrer JD, Mahoney C, Gordon E, Beck J, Mead S, Warren J, Rossor M, Revesz T (2013) A pathogenic progranulin mutation and C9orf72 repeat expansion in a family with frontotemporal dementia. Neuropathol Appl Neurobiol. doi:10.1111/nan.12100
Byrne S, Walsh C, Lynch C, Bede P, Elamin M, Kenna K, McLaughlin R, Hardiman O (2011) Rate of familial amyotrophic lateral sclerosis: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry 82(6):623–627. doi:10.1136/jnnp.2010.224501
Al-Chalabi A, Fang F, Hanby MF, Leigh PN, Shaw CE, Ye W, Rijsdijk F (2010) An estimate of amyotrophic lateral sclerosis heritability using twin data. J Neurol Neurosurg Psychiatry 81(12):1324–1326. doi:10.1136/jnnp.2010.207464
Goodall EF, Bury JJ, Cooper-Knock J, Shaw PJ, Kirby J (2012) Genetics of familial amyotrophic lateral sclerosis. In: Maurer MH (ed) Amyotrophic lateral sclerosis. Intech, Rijeka, pp 517–536
Konno T, Shiga A, Tsujino A, Sugai A, Kato T, Kanai K, Yokoseki A, Eguchi H, Kuwabara S, Nishizawa M, Takahashi H, Onodera O (2013) Japanese amyotrophic lateral sclerosis patients with GGGGCC hexanucleotide repeat expansion in C9ORF72. J Neurol Neurosurg Psychiatry 84(4):398–401. doi:10.1136/jnnp-2012-302272
Shatunov A, Mok K, Newhouse S, Weale ME, Smith B, Vance C, Johnson L, Veldink JH, van Es MA, van den Berg LH, Robberecht W, Van Damme P, Hardiman O, Farmer AE, Lewis CM et al (2010) Chromosome 9p21 in sporadic amyotrophic lateral sclerosis in the UK and seven other countries: a genome-wide association study. Lancet Neurol 9(10):986–994. doi:10.1016/S1474-4422(10)70197-6
Mok K, Traynor BJ, Schymick J, Tienari PJ, Laaksovirta H, Peuralinna T, Myllykangas L, Chiò A, Shatunov A, Boeve BF, Boxer AL, DeJesus-Hernandez M, Mackenzie IR, Waite A, Williams N et al (2012) Chromosome 9 ALS and FTD locus is probably derived from a single founder. Neurobiol Aging 33(1):209.e3–209.e8. doi:10.1016/j.neurobiolaging.2011.08.005
Smith BN, Newhouse S, Shatunov A, Vance C, Topp S, Johnson L, Miller J, Lee Y, Troakes C, Scott KM, Jones A, Gray I, Wright J, Hortobagyi T, Al-Sarraj S et al (2013) The C9ORF72 expansion mutation is a common cause of ALS ± FTD in Europe and has a single founder. Eur J Hum Genet 21(1):102–108. doi:10.1038/ejhg.2012.98
Dobson-Stone C, Hallupp M, Bartley L, Shepherd CE, Halliday GM, Schofield PR, Hodges JR, Kwok JB (2012) C9ORF72 repeat expansion in clinical and neuropathologic frontotemporal dementia cohorts. Neurology 79(10):995–1001. doi:10.1212/WNL.0b013e3182684634
del Aguila MA, Longstreth WT Jr, McGuire V, Koepsell TD, van Belle G (2003) Prognosis in amyotrophic lateral sclerosis: a population-based study. Neurology 60(5):813–819
McDermott CJ, Shaw PJ (2008) Diagnosis and management of motor neurone disease. BMJ 336(7645):658–662. doi:10.1136/bmj.39493.511759.BE
Preux PM, Couratier P, Boutros-Toni F, Salle JY, Tabaraud F, Bernet-Bernady P, Vallat JM, Dumas M (1996) Survival prediction in sporadic amyotrophic lateral sclerosis. Age and clinical form at onset are independent risk factors. Neuroepidemiology 15(3):153–160
Cooper-Knock J, Jenkins T, Shaw PJ (2013) Clinical and Molecular Aspects of Motor Neuron Disease. Colloq Ser Genomic Mol Med 2(2):1–60
Gijselinck I, Van Langenhove T, van der Zee J, Sleegers K, Philtjens S, Kleinberger G, Janssens J, Bettens K, Van Cauwenberghe C, Pereson S, Engelborghs S, Sieben A, De Jonghe P, Vandenberghe R, Santens P et al (2012) A C9orf72 promoter repeat expansion in a Flanders-Belgian cohort with disorders of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum: a gene identification study. Lancet Neurol 11(1):54–65. doi:10.1016/S1474-4422(11)70261-7
Murray ME, DeJesus-Hernandez M, Rutherford NJ, Baker M, Duara R, Graff-Radford NR, Wszolek ZK, Ferman TJ, Josephs KA, Boylan KB, Rademakers R, Dickson DW (2011) Clinical and neuropathologic heterogeneity of c9FTD/ALS associated with hexanucleotide repeat expansion in C9ORF72. Acta Neuropathol 122(6):673–690. doi:10.1007/s00401-011-0907-y
Debray S, Race V, Crabbé V, Herdewyn S, Matthijs G, Goris A, Dubois B, Thijs V, Robberecht W, Van Damme P (2013) Frequency of C9orf72 repeat expansions in amyotrophic lateral sclerosis: a Belgian cohort study. Neurobiol Aging 34(12):2890.e7–2890.e12. doi:10.1016/j.neurobiolaging.2013.06.009
Ratti A, Corrado L, Castellotti B, Del Bo R, Fogh I, Cereda C, Tiloca C, D’Ascenzo C, Bagarotti A, Pensato V, Ranieri M, Gagliardi S, Calini D, Mazzini L, Taroni F et al (2012) C9ORF72 repeat expansion in a large Italian ALS cohort: evidence of a founder effect. Neurobiol Aging 33(10):2528.e7–2528.e14. doi:10.1016/j.neurobiolaging.2012.06.008
Stewart H, Rutherford NJ, Briemberg H, Krieger C, Cashman N, Fabros M, Baker M, Fok A, DeJesus-Hernandez M, Eisen A, Rademakers R, Mackenzie IR (2012) Clinical and pathological features of amyotrophic lateral sclerosis caused by mutation in the C9ORF72 gene on chromosome 9p. Acta Neuropathol 123(3):409–417. doi:10.1007/s00401-011-0937-5
Byrne S, Elamin M, Bede P, Shatunov A, Walsh C, Corr B, Heverin M, Jordan N, Kenna K, Lynch C, McLaughlin RL, Iyer PM, O’Brien C, Phukan J, Wynne B et al (2012) Cognitive and clinical characteristics of patients with amyotrophic lateral sclerosis carrying a C9orf72 repeat expansion: a population-based cohort study. Lancet Neurol 11(3):232–240. doi:10.1016/S1474-4422(12)70014-5
Garcia-Redondo A, Dols-Icardo O, Rojas-Garcia R, Esteban-Perez J, Cordero-Vazquez P, Munoz-Blanco JL, Catalina I, Gonzalez-Munoz M, Varona L, Sarasola E, Povedano M, Sevilla T, Guerrero A, Pardo J, Lopez de Munain A et al (2013) Analysis of the C9orf72 gene in patients with amyotrophic lateral sclerosis in Spain and different populations worldwide. Hum Mutat 34(1):79–82. doi:10.1002/humu.22211
Sabatelli M, Conforti FL, Zollino M, Mora G, Monsurrò MR, Volanti P, Marinou K, Salvi F, Corbo M, Giannini F, Battistini S, Penco S, Lunetta C, Quattrone A, Gambardella A et al (2012) C9ORF72 hexanucleotide repeat expansions in the Italian sporadic ALS population. Neurobiol Aging 33(8):1848.e15–1848.e20. doi:10.1016/j.neurobiolaging.2012.02.011
Williams KL, Fifita JA, Vucic S, Durnall JC, Kiernan MC, Blair IP, Nicholson GA (2013) Pathophysiological insights into ALS with C9ORF72 expansions. J Neurol Neurosurg Psychiatry 84(8):931–935. doi:10.1136/jnnp-2012-304529
Millecamps S, Boillee S, Le Ber I, Seilhean D, Teyssou E, Giraudeau M, Moigneu C, Vandenberghe N, Danel-Brunaud V, Corcia P, Pradat PF, Le Forestier N, Lacomblez L, Bruneteau G, Camu W et al (2012) Phenotype difference between ALS patients with expanded repeats in C9ORF72 and patients with mutations in other ALS-related genes. J Med Genet 49(4):258–263. doi:10.1136/jmedgenet-2011-100699
Boeve BF, Boylan KB, Graff-Radford NR, DeJesus-Hernandez M, Knopman DS, Pedraza O, Vemuri P, Jones D, Lowe V, Murray ME, Dickson DW, Josephs KA, Rush BK, Machulda MM, Fields JA et al (2012) Characterization of frontotemporal dementia and/or amyotrophic lateral sclerosis associated with the GGGGCC repeat expansion in C9ORF72. Brain 135(Pt 3):765–783. doi:10.1093/brain/aws004
O’Dowd S, Curtin D, Waite AJ, Roberts K, Pender N, Reid V, O’Connell M, Williams NM, Morris HR, Traynor BJ, Lynch T (2012) C9ORF72 expansion in amyotrophic lateral sclerosis/frontotemporal dementia also causes parkinsonism. Mov Disord 27(8):1072–1074. doi:10.1002/mds.25022
Akimoto C, Forsgren L, Linder J, Birve A, Backlund I, Andersson J, Nilsson AC, Alstermark H, Andersen PM (2013) No GGGGCC-hexanucleotide repeat expansion in C9ORF72 in parkinsonism patients in Sweden. Amyotroph Lateral Scler Frontotemporal Degener 14(1):26–29. doi:10.3109/17482968.2012.725415
Harms MB, Neumann D, Benitez BA, Cooper B, Carrell D, Racette BA, Perlmutter JS, Goate A, Cruchaga C (2013) Parkinson disease is not associated with C9ORF72 repeat expansions. Neurobiol Aging 34(5):1519.e1–1519.e12. doi:10.1016/j.neurobiolaging.2012.10.001
Majounie E, Abramzon Y, Renton AE, Keller MF, Traynor BJ, Singleton AB (2012) Large C9orf72 repeat expansions are not a common cause of Parkinson’s disease. Neurobiol Aging 33(10):2527.e1–2527.e2. doi:10.1016/j.neurobiolaging.2012.05.007
Cooper-Knock J, Frolov A, Highley JR, Charlesworth G, Kirby J, Milano A, Hartley J, Ince PG, McDermott CJ, Lashley T, Revesz T, Shaw PJ, Wood NW, Bandmann O (2013) C9ORF72 expansions, parkinsonism, and Parkinson disease: a clinicopathologic study. Neurology. doi:10.1212/WNL.0b013e3182a2cc38
Lesage S, Le Ber I, Condroyer C, Broussolle E, Gabelle A, Thobois S, Pasquier F, Mondon K, Dion PA, Rochefort D, Rouleau GA, Durr A, Brice A, French Parkinson’s Disease Genetics Study G (2013) C9orf72 repeat expansions are a rare genetic cause of parkinsonism. Brain 136(Pt 2):385–391. doi:10.1093/brain/aws357
Ismail A, Cooper-Knock J, Highley JR, Milano A, Kirby J, Goodall E, Lowe J, Scott I, Constantinescu CS, Walters SJ, Price S, McDermott CJ, Sawcer S, Compston DA, Sharrack B et al (2013) Concurrence of multiple sclerosis and amyotrophic lateral sclerosis in patients with hexanucleotide repeat expansions of C9ORF72. J Neurol Neurosurg Psychiatry 84(1):79–87. doi:10.1136/jnnp-2012-303326
van Rheenen W, van Blitterswijk M, Huisman MH, Vlam L, van Doormaal PT, Seelen M, Medic J, Dooijes D, de Visser M, van der Kooi AJ, Raaphorst J, Schelhaas HJ, van der Pol WL, Veldink JH, van den Berg LH (2012) Hexanucleotide repeat expansions in C9ORF72 in the spectrum of motor neuron diseases. Neurology 79(9):878–882. doi:10.1212/WNL.0b013e3182661d14
Rutherford NJ, DeJesus-Hernandez M, Baker MC, Kryston TB, Brown PE, Lomen-Hoerth C, Boylan K, Wszolek ZK, Rademakers R (2012) C9ORF72 hexanucleotide repeat expansions in patients with ALS from the Coriell Cell Repository. Neurology 79(5):482–483. doi:10.1212/WNL.0b013e31826170f1
Nielsen TT, Svenstrup K, Duno M, Nielsen JE (2013) Hereditary spastic paraplegia is not associated with C9ORF72 repeat expansions in a Danish cohort. Spinal Cord. doi:10.1038/sc.2013.116
Galimberti D, Fenoglio C, Serpente M, Villa C, Bonsi R, Arighi A, Fumagalli GG, Del Bo R, Bruni AC, Anfossi M, Clodomiro A, Cupidi C, Nacmias B, Sorbi S, Piaceri I et al (2013) Autosomal dominant frontotemporal lobar degeneration due to the C9ORF72 hexanucleotide repeat expansion: late-onset psychotic clinical presentation. Biol Psychiatry 74(5):384–391. doi:10.1016/j.biopsych.2013.01.031
Sha SJ, Takada LT, Rankin KP, Yokoyama JS, Rutherford NJ, Fong JC, Khan B, Karydas A, Baker MC, DeJesus-Hernandez M, Pribadi M, Coppola G, Geschwind DH, Rademakers R, Lee SE et al (2012) Frontotemporal dementia due to C9ORF72 mutations: clinical and imaging features. Neurology 79(10):1002–1011. doi:10.1212/WNL.0b013e318268452e
Lindquist SG, Duno M, Batbayli M, Puschmann A, Braendgaard H, Mardosiene S, Svenstrup K, Pinborg LH, Vestergaard K, Hjermind LE, Stokholm J, Andersen BB, Johannsen P, Nielsen JE (2013) Corticobasal and ataxia syndromes widen the spectrum of C9ORF72 hexanucleotide expansion disease. Clin Genet 83(3):279–283. doi:10.1111/j.1399-0004.2012.01903.x
Ticozzi N, Tiloca C, Calini D, Gagliardi S, Altieri A, Colombrita C, Cereda C, Ratti A, Pezzoli G, Borroni B, Goldwurm S, Padovani A, Silani V (2013) C9orf72 repeat expansions are restricted to the ALS-FTD spectrum. Neurobiol Aging. doi:10.1016/j.neurobiolaging.2013.09.037
Mann DM, Rollinson S, Robinson A, Bennion CJ, Thompson JC, Snowden JS, Gendron T, Petrucelli L, Masuda-Suzukake M, Hasegawa M, Davidson Y, Pickering-Brown S (2013) Dipeptide repeat proteins are present in the p62 positive inclusions in patients with frontotemporal lobar degeneration and motor neurone disease associated with expansions in C9ORF72. Acta Neuropathol Commun 1(1):68. doi:10.1186/2051-5960-1-68
Hensman Moss DJ, Poulter M, Beck J, Hehir J, Polke JM, Campbell T, Adamson G, Mudanohwo E, McColgan P, Haworth A, Wild EJ, Sweeney MG, Houlden H, Mead S, Tabrizi SJ (2013) C9orf72 expansions are the most common genetic cause of Huntington disease phenocopies. Neurology. doi:10.1212/WNL.0000000000000061
Liu Y, Yu JT, Sun FR, Ou JR, Qu SB, Tan L (2013) The clinical and pathological phenotypes of frontotemporal dementia with C9ORF72 mutations. J Neurol Sci 335(1–2):26–35. doi:10.1016/j.jns.2013.09.013
Kaivorinne AL, Bode MK, Paavola L, Tuominen H, Kallio M, Renton AE, Traynor BJ, Moilanen V, Remes AM (2013) Clinical Characteristics of C9ORF72-Linked Frontotemporal Lobar Degeneration. Dement Geriatr Cogn Dis Extra 3(1):251–262. doi:10.1159/000351859
Jiao B, Tang B, Liu X, Yan X, Zhou L, Yang Y, Wang J, Xia K, Shen L (2013) Identification of C9orf72 repeat expansions in patients with amyotrophic lateral sclerosis and frontotemporal dementia in mainland China. Neurobiol Aging. doi:10.1016/j.neurobiolaging.2013.10.001
Jones AR, Woollacott I, Shatunov A, Cooper-Knock J, Buchman V, Sproviero W, Smith B, Scott KM, Balendra R, Abel O, McGuffin P, Ellis CM, Shaw PJ, Morrison KE, Farmer A et al (2013) Residual association at C9orf72 suggests an alternative amyotrophic lateral sclerosis-causing hexanucleotide repeat. Neurobiol Aging 34(9):2234.e17–2234.e7. doi:10.1016/j.neurobiolaging.2013.03.003
Hsiung GY, DeJesus-Hernandez M, Feldman HH, Sengdy P, Bouchard-Kerr P, Dwosh E, Butler R, Leung B, Fok A, Rutherford NJ, Baker M, Rademakers R, Mackenzie IR (2012) Clinical and pathological features of familial frontotemporal dementia caused by C9ORF72 mutation on chromosome 9p. Brain 135(Pt 3):709–722. doi:10.1093/brain/awr354
Khan BK, Yokoyama JS, Takada LT, Sha SJ, Rutherford NJ, Fong JC, Karydas AM, Wu T, Ketelle RS, Baker MC, Hernandez MD, Coppola G, Geschwind DH, Rademakers R, Lee SE et al (2012) Atypical, slowly progressive behavioural variant frontotemporal dementia associated with C9ORF72 hexanucleotide expansion. J Neurol Neurosurg Psychiatry 83(4):358–364. doi:10.1136/jnnp-2011-301883
Simon-Sanchez J, Dopper EG, Cohn-Hokke PE, Hukema RK, Nicolaou N, Seelaar H, de Graaf JR, de Koning I, van Schoor NM, Deeg DJ, Smits M, Raaphorst J, van den Berg LH, Schelhaas HJ, De Die-Smulders CE et al (2012) The clinical and pathological phenotype of C9ORF72 hexanucleotide repeat expansions. Brain 135(Pt 3):723–735. doi:10.1093/brain/awr353
Snowden JS, Rollinson S, Thompson JC, Harris JM, Stopford CL, Richardson AM, Jones M, Gerhard A, Davidson YS, Robinson A, Gibbons L, Hu Q, DuPlessis D, Neary D, Mann DM et al (2012) Distinct clinical and pathological characteristics of frontotemporal dementia associated with C9ORF72 mutations. Brain 135(Pt 3):693–708. doi:10.1093/brain/awr355
Van Langenhove T, van der Zee J, Gijselinck I, Engelborghs S, Vandenberghe R, Vandenbulcke M, De Bleecker J, Sieben A, Versijpt J, Ivanoiu A, Deryck O, Willems C, Dillen L, Philtjens S, Maes G et al (2013) Distinct clinical characteristics of C9orf72 expansion carriers compared with GRN, MAPT, and nonmutation carriers in a Flanders-Belgian FTLD cohort. JAMA Neurol 70(3):365–373. doi:10.1001/2013.jamaneurol.181
Cerami C, Marcone A, Galimberti D, Zamboni M, Fenoglio C, Serpente M, Scarpini E, Cappa SF (2013) Novel evidence of phenotypical variability in the hexanucleotide repeat expansion in chromosome 9. J Alzheimers Dis 35(3):455–462. doi:10.3233/JAD-122302
Rollinson S, Halliwell N, Young K, Callister JB, Toulson G, Gibbons L, Davidson YS, Robinson AC, Gerhard A, Richardson A, Neary D, Snowden J, Mann DM (1846) Pickering-Brown SM (2012) Analysis of the hexanucleotide repeat in C9ORF72 in Alzheimer’s disease. Neurobiol Aging 33(8):e1845–e1846. doi:10.1016/j.neurobiolaging.2012.01.109
Davidson YS, Robinson AC, Snowden JS, Mann DM (2013) Pathological assessments for the presence of hexanucleotide repeat expansions in C9ORF72 in Alzheimer’s disease. Acta Neuropathol Commun 1(1):50. doi:10.1186/2051-5960-1-50
Cacace R, Van Cauwenberghe C, Bettens K, Gijselinck I, van der Zee J, Engelborghs S, Vandenbulcke M, Van Dongen J, Bäumer V, Dillen L, Mattheijssens M, Peeters K, Cruts M, Vandenberghe R, De Deyn PP et al (2013) C9orf72 G4C2 repeat expansions in Alzheimer’s disease and mild cognitive impairment. Neurobiol Aging 34(6):1712.e1–1712.e7. doi:10.1016/j.neurobiolaging.2012.12.019
Harms M, Benitez BA, Cairns N, Cooper B, Cooper P, Mayo K, Carrell D, Faber K, Williamson J, Bird T, Diaz-Arrastia R, Foroud TM, Boeve BF, Graff-Radford NR, Mayeux R et al (2013) C9orf72 hexanucleotide repeat expansions in clinical Alzheimer disease. JAMA Neurol 70(6):736–741. doi:10.1001/2013.jamaneurol.537
Kohli MA, John-Williams K, Rajbhandary R, Naj A, Whitehead P, Hamilton K, Carney RM, Wright C, Crocco E, Gwirtzman HE, Lang R, Beecham G, Martin ER, Gilbert J, Benatar M et al (2013) Repeat expansions in the C9ORF72 gene contribute to Alzheimer’s disease in Caucasians. Neurobiol Aging 34(5):1519.e5–1519.e12. doi:10.1016/j.neurobiolaging.2012.10.003
Wojtas A, Heggeli KA, Finch N, Baker M, Dejesus-Hernandez M, Younkin SG, Dickson DW, Graff-Radford NR, Rademakers R (2012) C9ORF72 repeat expansions and other FTD gene mutations in a clinical AD patient series from Mayo Clinic. Am J Neurodegener Dis 1(1):107–118
Snowden JS, Rollinson S, Lafon C, Harris J, Thompson J, Richardson AM, Jones M, Gerhard A, Neary D, Mann DM, Pickering-Brown S (2012) Psychosis, C9ORF72 and dementia with Lewy bodies. J Neurol Neurosurg Psychiatry 83(10):1031–1032. doi:10.1136/jnnp-2012-303032
Huey ED, Nagy PL, Rodriguez-Murillo L, Manoochehri M, Goldman J, Lieberman J, Karayiorgou M, Mayeux R (2013) C9ORF72 repeat expansions not detected in a group of patients with schizophrenia. Neurobiol Aging 34(4):1309.e9–1309.e10. doi:10.1016/j.neurobiolaging.2012.08.011
Buchman VL, Cooper-Knock J, Connor-Robson N, Higginbottom A, Kirby J, Razinskaya OD, Ninkina N, Shaw PJ (2013) Simultaneous and independent detection of C9ORF72 alleles with low and high number of GGGGCC repeats using an optimised protocol of Southern blot hybridisation. Mol Neurodegener 8:12. doi:10.1186/1750-1326-8-12
van Blitterswijk M, DeJesus-Hernandez M, Niemantsverdriet E, Murray ME, Heckman MG, Diehl NN, Brown PH, Baker MC, Finch NA, Bauer PO, Serrano G, Beach TG, Josephs KA, Knopman DS, Petersen RC et al (2013) Association between repeat sizes and clinical and pathological characteristics in carriers of C9ORF72 repeat expansions (Xpansize-72): a cross-sectional cohort study. Lancet Neurol 12(10):978–988. doi:10.1016/S1474-4422(13)70210-2
Dols-Icardo O, Garcia-Redondo A, Rojas-Garcia R, Sanchez-Valle R, Noguera A, Gomez-Tortosa E, Pastor P, Hernandez I, Esteban-Perez J, Suarez-Calvet M, Anton-Aguirre S, Amer G, Ortega-Cubero S, Blesa R, Fortea J et al (2013) Characterization of the repeat expansion size in C9orf72 in amyotrophic lateral sclerosis and frontotemporal dementia. Hum Mol Genet. doi:10.1093/hmg/ddt460
Clark RM, De Biase I, Malykhina AP, Al-Mahdawi S, Pook M, Bidichandani SI (2007) The GAA triplet-repeat is unstable in the context of the human FXN locus and displays age-dependent expansions in cerebellum and DRG in a transgenic mouse model. Hum Genet 120(5):633–640. doi:10.1007/s00439-006-0249-3
Dobson-Stone C, Hallupp M, Loy CT, Thompson EM, Haan E, Sue CM, Panegyres PK, Razquin C, Seijo-Martinez M, Rene R, Gascon J, Campdelacreu J, Schmoll B, Volk AE, Brooks WS et al (2013) C9ORF72 repeat expansion in Australian and Spanish frontotemporal dementia patients. PLoS One 8(2):e56899. doi:10.1371/journal.pone.0056899
Rutherford NJ, Heckman MG, Dejesus-Hernandez M, Baker MC, Soto-Ortolaza AI, Rayaprolu S, Stewart H, Finger E, Volkening K, Seeley WW, Hatanpaa KJ, Lomen-Hoerth C, Kertesz A, Bigio EH, Lippa C et al (2012) Length of normal alleles of C9ORF72 GGGGCC repeat do not influence disease phenotype. Neurobiol Aging 33(12):2950.e5–2950.e7. doi:10.1016/j.neurobiolaging.2012.07.005
Chio A, Borghero G, Restagno G, Mora G, Drepper C, Traynor BJ, Sendtner M, Brunetti M, Ossola I, Calvo A, Pugliatti M, Sotgiu MA, Murru MR, Marrosu MG, Marrosu F et al (2012) Clinical characteristics of patients with familial amyotrophic lateral sclerosis carrying the pathogenic GGGGCC hexanucleotide repeat expansion of C9ORF72. Brain 135(Pt 3):784–793. doi:10.1093/brain/awr366
Radvansky J, Kadasi L (2010) The expanding world of myotonic dystrophies: how can they be detected? Genet Test Mol Biomarkers 14(6):733–741. doi:10.1089/gtmb 2010.0073
Ash PE, Bieniek KF, Gendron TF, Caulfield T, Lin WL, Dejesus-Hernandez M, van Blitterswijk MM, Jansen-West K, Paul JW 3rd, Rademakers R, Boylan KB, Dickson DW, Petrucelli L (2013) Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 77(4):639–646. doi:10.1016/j.neuron.2013.02.004
Mori K, Weng SM, Arzberger T, May S, Rentzsch K, Kremmer E, Schmid B, Kretzschmar HA, Cruts M, Van Broeckhoven C, Haass C, Edbauer D (2013) The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 339(6125):1335–1338. doi:10.1126/science.1232927
Donnelly CJ, Zhang PW, Pham JT, Heusler AR, Mistry NA, Vidensky S, Daley EL, Poth EM, Hoover B, Fines DM, Maragakis N, Tienari PJ, Petrucelli L, Traynor BJ, Wang J et al (2013) RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron 80(2):415–428. doi:10.1016/j.neuron.2013.10.015
Sareen D, O’Rourke JG, Meera P, Muhammad AK, Grant S, Simpkinson M, Bell S, Carmona S, Ornelas L, Sahabian A, Gendron T, Petrucelli L, Baughn M, Ravits J, Harms MB et al (2013) Targeting RNA foci in iPSC-Derived Motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci Transl Med 5(208):208ra149. doi:10.1126/scitranslmed.3007529
Lagier-Tourenne C, Baughn M, Rigo F, Sun S, Liu P, Li HR, Jiang J, Watt AT, Chun S, Katz M, Qiu J, Sun Y, Ling SC, Zhu Q, Polymenidou M et al (2013) Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc Natl Acad Sci USA. doi:10.1073/pnas.1318835110
Zu T, Liu Y, Banez-Coronel M, Reid T, Pletnikova O, Lewis J, Miller TM, Harms MB, Falchook AE, Subramony SH, Ostrow LW, Rothstein JD, Troncoso JC, Ranum LP (2013) RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc Natl Acad Sci USA. doi:10.1073/pnas.1315438110
Gomez-Tortosa E, Gallego J, Guerrero-Lopez R, Marcos A, Gil-Neciga E, Sainz MJ, Diaz A, Franco-Macias E, Trujillo-Tiebas MJ, Ayuso C, Perez-Perez J (2013) C9ORF72 hexanucleotide expansions of 20-22 repeats are associated with frontotemporal deterioration. Neurology 80(4):366–370. doi:10.1212/WNL.0b013e31827f08ea
Byrne S, Heverin M, Elamin M, Walsh C, Hardiman O (2013) Intermediate repeat expansion length in C9orf72 may be pathological in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. doi:10.3109/21678421.2013.838586
Cooper-Knock J, Higginbottom A, Connor-Robson N, Bayatti N, Bury JJ, Kirby J, Ninkina N, Buchman VL, Shaw PJ (2013) C9ORF72 transcription in a frontotemporal dementia case with two expanded alleles. Neurology. doi:10.1212/01.wnl.0000435295.41974.2e
Xi Z, Zinman L, Moreno D, Schymick J, Liang Y, Sato C, Zheng Y, Ghani M, Dib S, Keith J, Robertson J, Rogaeva E (2013) Hypermethylation of the CpG island near the G4C2 repeat in ALS with a C9orf72 expansion. Am J Hum Genet 92(6):981–989. doi:10.1016/j.ajhg.2013.04.017
Van Deerlin VM, Sleiman PM, Martinez-Lage M, Chen-Plotkin A, Wang LS, Graff-Radford NR, Dickson DW, Rademakers R, Boeve BF, Grossman M, Arnold SE, Mann DM, Pickering-Brown SM, Seelaar H, Heutink P et al (2010) Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions. Nat Genet 42(3):234–239. doi:10.1038/ng.536
Nicholson AM, Finch NA, Wojtas A, Baker MC, Perkerson RB 3rd, Castanedes-Casey M, Rousseau L, Benussi L, Binetti G, Ghidoni R, Hsiung GY, Mackenzie IR, Finger E, Boeve BF, Ertekin-Taner N et al (2013) TMEM106B p. T185S regulates TMEM106B protein levels: implications for frontotemporal dementia. J Neurochem 126(6):781–791. doi:10.1111/jnc.12329
Gallagher MD, Suh E, Grossman M, Elman L, McCluskey L, Van Swieten JC, Al-Sarraj S, Neumann M, Gelpi E, Ghetti B, Rohrer JD, Halliday G, Van Broeckhoven C, Seilhean D, Shaw PJ et al (2013) TMEM106B is a genetic modifier of frontotemporal lobar degeneration with C9orf72 hexanucleotide repeat expansions. Acta Neuropathol [Epub ahead of print]
Levine TP, Daniels RD, Gatta AT, Wong LH, Hayes MJ (2013) The product of C9orf72, a gene strongly implicated in neurodegeneration, is structurally related to DENN Rab-GEFs. Bioinformatics 29(4):499–503. doi:10.1093/bioinformatics/bts725
van Blitterswijk M, Mullen B, Nicholson AM, Bieniek KF, Heckman MG, Baker MC, Dejesus-Hernandez M, Finch NA, Brown PH, Murray ME, Hsiung GY, Stewart H, Karydas AM, Finger E, Kertesz A et al (2014) TMEM106B protects C9ORF72 expansion carriers against frontotemporal dementia. Acta Neuropathol. doi:10.1007/s00401-013-1240-4
Tateishi T, Yamasaki R, Tanaka M, Matsushita T, Kikuchi H, Isobe N, Ohyagi Y, Kira J (2010) CSF chemokine alterations related to the clinical course of amyotrophic lateral sclerosis. J Neuroimmunol 222(1–2):76–81. doi:10.1016/j.jneuroim.2010.03.004
Kawamata T, Akiyama H, Yamada T, McGeer PL (1992) Immunologic reactions in amyotrophic lateral sclerosis brain and spinal cord tissue. Am J Pathol 140(3):691–707
Turner MR, Cagnin A, Turkheimer FE, Miller CC, Shaw CE, Brooks DJ, Leigh PN, Banati RB (2004) Evidence of widespread cerebral microglial activation in amyotrophic lateral sclerosis: an [11C](R)-PK11195 positron emission tomography study. Neurobiol Dis 15(3):601–609. doi:10.1016/j.nbd.2003.12.012
Henkel JS, Beers DR, Wen S, Rivera AL, Toennis KM, Appel JE, Zhao W, Moore DH, Powell SZ, Appel SH (2013) Regulatory T-lymphocytes mediate amyotrophic lateral sclerosis progression and survival. EMBO Mol Med 5(1):64–79. doi:10.1002/emmm.201201544
Acknowledgments
JCK is funded by a Motor Neurone Disease Association/Medical Research Council Lady Edith Wolfson Fellowship and PJS is supported as an NIHR Senior Investigator. PJS and JK are supported by the MNDA, a European Community’s Seventh Framework Programme (FP7/2007-2013) under the Euro-MOTOR project (Grant agreement No.: 259867) and by funding from the European Union Joint Programme–Neurodegenerative Disease Research (JPND), Sampling and biomarker OPtimization and Harmonization In ALS and other motor neuron diseases (SOPHIA). This is an EU Joint Programme–Neurodegenerative Disease Research (JPND) project. The project is supported through the following funding organisations under the aegis of JPND–www.jpnd.eu: France, Agence Nationale de la Recherche (ANR); Germany, Bundesministerium für Bildung und Forschung (BMBF); Ireland, Health Research Board (HRB); Italy, Ministero della Salute; The Netherlands, The Netherlands Organisation for Health Research and Development (ZonMw); Poland, Narodowe Centrum Badań i Rozwoju; Portugal, Fundação a Ciência e a Tecnologia; Spain, Ministerio de Ciencia e Innovación; Switzerland, Schweizerischer Nationalfonds zur Förderung der wissenschaftlichen Forschung (SNF); Turkey, Tübitak; United Kingdom, Medical Research Council (MRC).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Cooper-Knock, J., Shaw, P.J. & Kirby, J. The widening spectrum of C9ORF72-related disease; genotype/phenotype correlations and potential modifiers of clinical phenotype. Acta Neuropathol 127, 333–345 (2014). https://doi.org/10.1007/s00401-014-1251-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-014-1251-9