
Abstract
Inflammatory lesions in the central nervous system of patients with neuromyelitis optica are characterized by infiltration of T cells and deposition of aquaporin-4-specific antibodies and complement on astrocytes at the glia limitans. Although the contribution of aquaporin-4-specific autoantibodies to the disease process has been recently elucidated, a potential role of aquaporin-4-specific T cells in lesion formation is unresolved. To address this issue, we raised aquaporin-4-specific T cell lines in Lewis rats and characterized their pathogenic potential in the presence and absence of aquaporin-4-specific autoantibodies of neuromyelitis optica patients. We show that aquaporin-4-specific T cells induce brain inflammation with particular targeting of the astrocytic glia limitans and permit the entry of pathogenic anti-aquaporin-4-specific antibodies to induce NMO-like lesions in spinal cord and brain. In addition, transfer of aquaporin-4-specific T cells provoked mild (subclinical) myositis and interstitial nephritis. We further show that the expression of the conformational epitope, recognized by NMO patient-derived aquaporin-4-specific antibodies is induced in kidney cells by the pro-inflammatory cytokine gamma-interferon. Our data provide further support for the view that NMO lesions may be induced by a complex interplay of T cell mediated and humoral immune responses against aquaporin-4.
Similar content being viewed by others


Introduction
Inflammatory lesions in the central nervous system (CNS) of patients with neuromyelitis optica (NMO) are characterized by infiltration of T cells and deposition of aquaporin-4 (AQP-4)-specific antibodies and complement on astrocytes at the superficial and perivascular glia limitans. Several lines of evidence suggest that T cells are not just innocent bystanders of lesion pathogenesis, but that they are actively involved in this process:
First, AQP-4-specific antibodies and complement components are unable to cross through the endothelial blood–brain barrier in the absence of activated, CNS antigen-specific T cells [5], but they can cause tissue damage if the blood–brain barrier is by-passed by direct injection of these molecules into the CNS parenchyma [32]. Secondly, the leaky phenotype of the blood–brain barrier in immature rats is not enough to allow AQP-4-specific antibodies to initiate complement-mediated tissue damage in the absence of encephalitogenic T cells [5]. Thirdly, even in circumventricular organs characterized by a lack of tight junctions between endothelial cells, these molecules seem to be unable to enter in sufficiently high concentrations to cause tissue damage in the absence of inflammation [5].
Further evidence for a critical role of T cells in NMO comes from observations in Japanese NMO patients, where T cell receptors using the Vβ1 or Vβ13 chain were clonally expanded [38]. T cell recognize “their” specific antigen in context with major histocompatibility complex (MHC) class I or II products, which show an association with NMO in some patient cohorts. For example, in AQP-4-antibody-positive Japanese patients with opticospinal multiple sclerosis [23], which also belong to the group of NMO spectrum diseases, the phenotypic frequency of the human leukocyte antigen (HLA) class II allele HLA-DPB1*0501 is significantly increased [23], while in Brazilian Mulatto NMO patients, the HLA-DRB1*03 allele group is overrepresented [6].
All these different observations suggest that T cells may play a crucial role in the initiation and/or progression of NMO. However, the antigen specificity of these cells is unknown: on the one hand, it was hypothesized that AQP-4-specific T cells might be present and activated in NMO patients [8], on the other hand, it was assumed that tolerance induction due to the presence of AQP-4 in peripheral organs might interfere with the activation and expansion of encephalitogenic T cell responses against this antigen, or that AQP-4-specific T cells might cause inflammation in peripheral organs as well. In this study, we specifically addressed this issue, using the Lewis rat as a model system to search for AQP-4-specific T cells and to test their encephalitogenic potential.
Materials and methods
Animals
Lewis rats were obtained from Charles River Wiga (Sulzfeld, Germany) and used at an age of 8 weeks (~170 g body weight). They were housed in the Decentral Facilities of the Institute for Biomedical Research (Medical University Vienna) under standardized conditions. The experiments were approved by the Ethics Commission of the Medical University Vienna and performed with the license of the Austrian Ministry for Science and Research.
Sources and characterization of patient-derived immunoglobulin preparations
All immunoglobulin preparations from NMO, MS, or control patients have been described before [5], and are used here according to the former nomenclature. Unless otherwise indicated, experiments were performed with the anti-AQP-4 antibody containing human NMO-IgG derived from patient J0 [5]. The use of the patients’ plasma for this study was approved by the Ethics Committee of Tohoku University School of Medicine (No. 2007-327).
Antigens
For immunization and T cell isolation/propagation, the following peptides and fusion proteins were used: (1) AQP-4207–232 (YTGASMNPARSFGPAVIMGNWENHWI containing the putative RT1.BL-binding epitope AQP-4220–228 (PAVIMGNWE)), which was synthesized by Centic Biotech (Weimar, Germany), (2) a thioredoxin fusion protein containing the C-terminal, intracellular human AQP-4 peptide AQP-4277–322 (MEVEDNRSQVETDDLILKPGVVHVIDVDRGEEKKGKDQSGEVLSSV) expressed in E. coli using the pBAD/TOPO ThioFusion Expression System (Invitrogen, Carlsbad, CA, USA). The expression of the AQP-4277–322 thioredoxin fusion protein was induced by arabinose and the recombinant protein was purified by affinity chromatography on Ni–NTA agarose columns (Qiagen, Hilden, Germany) under denaturing conditions (8 M urea, 100 mM NaH2PO4, 10 mM Tris–HCl). This fusion protein includes the epitope hAQP-4296–304 (GVVHVIDVD), (3) hAQP-4296–304 (GVVHVIDVD), synthesized by Centic Biotech, (4) guinea pig myelin basic protein (MBP; Sigma, Vienna, Austria), (5) ovalbumin (OVA, Sigma).
Immunization and T cell line preparation
Animals were subcutaneously immunized with 100 μl of a 1:1 mixture of the relevant antigen (stock 2 mg/ml) in Freund′s incomplete adjuvants supplemented with 4 mg/ml mycobacterium tuberculosis H37Ra. After 9–11 days of immunization, the animals were killed. At this point, they were all clinically healthy and did not show any evidence for inflammation of the CNS or of peripheral organs. The lymph nodes draining the immunization site were removed, and peptide-specific T cell lines were established as described [2]. These lines represent a mixed population of antigen-specific CD3+/CD4+ T lymphocytes which belonged to the T-helper 1 subset of T cells, as indicated by strong expression of IL-2 and IFN-γ which was similar in AQP-4277–322- and AQP-4207–232-specific cells (data not shown).
EAE induction and tissue preparation
Experimental autoimmune encephalomyelitis (EAE) was induced in Lewis rats by intraperitoneal transfer of 3.5–7 × 107 activated T cells. On day 4 after T cell transfer, some animals were left untreated, while others received an additional, intraperitoneal injection with 1-ml phosphate buffered saline (PBS) and 10 mg anti-AQP-4 antibody containing NMO-IgG or 10 mg/ml commercially available human IgG (Subcuvia®, Baxter, Vienna, Austria). This amount of IgG transferred is well tolerated by rats [5, 34] 24 h later, the animals were killed with CO2. Afterwards, they were perfused with 4% phosphate buffered paraformaldehyde (PFA). Brain, spinal cord, and other tissues were dissected and immersed for another 18 h in PFA. The PFA-fixed material was routinely embedded in paraffin. Tissue sections were stained with hematoxylin and eosin (HE), and with Luxol fast blue.
Immunohistochemistry
All tissue blocks were cut in serial sections, and all staining were done essentially as described [1], using the following antibodies: CD3 (T cells; rabbit polyclonal; NeoMarkers, Fremont, USA); ED 1 (macrophages, microglia; mouse monoclonal; Serotec, Germany); OX 6 (MHC class II; mouse monoclonal, Serotec, Germany); commercial anti-AQP4 (rabbit polyclonal; Sigma, Germany); anti-AQP4-antibody containing NMO-IgG; anti-GFAP (rabbit polyclonal; Dako, Denmark, or mouse monoclonal; NeoMarkers); anti-human Ig (biotinylated donkey; polyclonal; GE Healthcare/Amersham, Vienna, Austria), anti-complement C9 (rabbit polyclonal [28]). Immunohistochemistry was completed using biotinylated secondary antibodies (sheep anti-mouse, donkey anti-rabbit, or donkey anti-sheep/goat, all from GE Healthcare/Amersham), and subsequent incubation of the sections with peroxidase labeled avidin (Sigma).
When anti-AQP4-antibody containing NMO-IgG in a stock concentration of 10 mg/ml was used for immunohistochemistry on frozen kidney sections, it was first diluted 1:10 in 10%FCS/PBS, mixed with 40 mg rabbit liver powder (RLP) (Sigma), and incubated for 1 h at room temperature. Afterwards, the antibody preparation was centrifuged (10 min, 13,000 rpm, room temperature). Subsequently, the supernatant was mixed with 25 mg RLP, incubated for 1 h at room temperature and centrifuged (10 min, 13,000 rpm, room temperature). Then, the supernatant was mixed with 20 mg RLP, incubated over night at 4°C, then for 1 h at room temperature, and centrifuged (10 min, 13,000 rpm, room temperature). Finally, the supernatant was diluted with 10% FCS/PBS to a final concentration of 1:200–1:400 and applied onto the tissue sections. Immunohistochemistry was completed with biotinylated sheep anti-human immunoglobulin and streptavidin-Cy2.
Quantitative evaluation of immunostained sections and statistic evaluation
Quantification was done by manual counting. We used whole renal cross sections to count collecting ducts which had at least 75% of their surface covered by T cells or macrophages. In addition, we counted collecting ducts which were invaded by T cells or macrophages. Statistics was calculated with the Statgraphics Plus program. Mann–Whitney (Wilcoxon) W test (comparison of medians) was used in all cases.
PCR analysis of connective tissue, T cells and microglia cells/perivascular macrophages to determine mRNA expression of AQP-4
We isolated RNA from the omentum, from established T cell lines, and from microglia cells/perivascular macrophages and astrocytes derived from the fetal brain, using the RNeasy kit (Qiagen, Hilden, Germany) according to the instructions of the manufacturer. This RNA was transcribed to cDNA and then subjected to PCR analysis, using primers specific for AQP4 (forward 5′-ACTGGGAAAACCATGGATATATTG-3′; reverse 5′-AGATAATACCTCTCCAGACGAGTCC-3′), and β-actin (forward 5′-ATGAAGTGTGACGTTGACATCC-3′; reverse 5′-CAAAAATGAAGTATTAAGGCGG-3′) or GAPDH (forward 5′-GGCATTGCTCTCAATGACACC-3′; reverse 5′-TGAGGGTGCAGCGAACTTTAT-3′).
For PCR reactions, 5 μl 10× PCR buffer (200 mM Tris–HCl, pH 8.4, 500 mM KCl), 1 μl 10 mM dNTP mix, 1 μl forward primer (100 pmol/μl), 1 μl reverse primer (100 pmol/μl), 0.4 μl polymerase (5 U/μl), 1 μl cDNA and 40.6 μl H2O were mixed together. The reaction mix was heated for 11 min at 95°C (denaturation), and then subjected to 40 cycles of denaturation (30 s, 95°C), annealing (30 s, 55°C for AQP-4; 53°C for β-actin) and elongation (30 s, 72°C). The final extension was made for 10 min at 72°C. The size of the resulting PCR products (287 bp for AQP4, 303 bp for β-actin, and 311 bp for GAPDH) was determined by agarose gel electrophoresis. Our PCR reactions and subsequent sequencing analysis of the amplified products revealed the presence of AQP-4 message in astrocytes, and its absence in the omentum, T cells, and microglia cells/perivascular macrophages (data not shown).
Production of astrocyte cultures
0–24 h-old Lewis rats were killed, their brains dissected and transferred to RPMI 1640/10% fetal calf serum (FCS). After stripping of the meninges, the brains were mechanically dissociated by gentle pipetting. The resulting single cell suspensions were cultured for 5–7 days in poly-l-lysine-coated culture flasks, using RPMI 1650/10% FCS, changing the medium every other day. After this time period, the mixed glial cell cultures consisted of a monolayer of astrocytes and fibroblasts. On top of this monolayer, loosely adherent, ramified microglial cells and glial progenitor cells were found. Confluent mixed glial cultures were agitated for 12–15 h (180 rpm, 37°C). Then, the supernatant was discarded, and the firmly adherent cells (mostly astrocytes and fibroblasts) were rinsed with PBS, trypsinized, re-seeded onto poly-l-lysine-coated cover slips (for studies on MHC class II and AQP-4 expression by immunocytochemistry) or 96 well plates (for usage as antigen-presenting cells), and cultured for an additional 1 day in RPMI 1640/10% FCS prior to their treatment. Based on GFAP staining, at least 95% of all cells in these cultures were astrocytes, the remaining cells mostly meningeal fibroblasts.
IFN-γ treatment and characterization of astrocytes
For the treatment, each cover slip was transferred to a separate well of a 12-well plate and inverted. Small dots of paraffin served as spacers between the cell layer of the cover slip and the bottom of the 12-well plate. The astrocytes growing on the cover slip were either left untreated as a control, or received IFN-γ (100 ng/ml) for 72 h. Afterwards, the cover slips were fixed with 4% PFA for 15 min (room temperature), washed with 0.05 M Tris-buffered saline (TBS) (pH 7.4) and permeabilized with TBS containing 0.5% Triton X-100, 5% bovine serum albumin (BSA, Sigma) and 5% donkey serum (Sigma) for 30 min at room temperature. The cells were then washed twice with TBS and incubated overnight at 4°C with either OX6 (murine monoclonal against MHC class II, Serotec) or with commercial rabbit anti-AQP-4 antibody and mouse anti-GFAP antibody (both diluted 1:100 in TBS/1% donkey serum). Afterwards, the cells were washed three times with TBS and incubated for 1 h at room temperature with donkey anti-mouse Cy2 (for OX6) or with donkey anti-mouse Cy3 (Jackson Immuno Research Laboratories, West Grove, PA, USA, for AQP-4) and donkey anti-rabbit Cy2 (Jackson Immuno Research Laboratories for GFAP), all diluted 1:100 in TBS/1% donkey serum). The cells were washed with TBS and mounted with gallate/geltol.
Antigen presentation by IFN-γ-treated astrocytes
The ability of astrocytes cultured in the absence or presence of 100 ng/ml IFN-γ for 72 h prior to the experiment to act as antigen-presenting cells of endogenous AQP-4 or exogenously added AQP-4 peptides was determined using these astrocytes seeded into flat bottomed 96-well plates at a density of 1 × 104 cells/well. Prior to their use as antigen-presenting cells, the astrocytes were irradiated with 30 Gray. 5 × 105 T cells were added to give a final volume of 200 μl/well. Then, the cells were cultured in the presence or absence of antigen, and T cell proliferation was determined essentially as described [11].
IFN-γ treatment and characterization of collecting duct epithelial cells
Madin–Darby canine kidney (MDCK) cells were cultured in the presence or absence of 50 ng/ml recombinant canine IFN-γ (R&D Systems) for the last 72 h. Then, the cells were detached with accutase (PAA Laboratories GmbH, Pansching, Austria) and incubated for 30 min at 4°C with human NMO-IgG (stock 10 mg/ml) or with Subcuvia as human control IgG. All antibody preparations were used at a dilution of 1:100 in stain buffer (BD Pharmingen, San Diego, USA). After incubation, the cells were washed in stain buffer and incubated for 30 min at 4°C with a biotinylated sheep anti-human antibody (GE Healthcare/Amersham; 1:100). Then, the cells were washed in stain buffer and incubated for 30 min at 4°C with an allophycocyanin (APC)-labeled anti-biotin antibody (MACS Miltenyi Biotec, Bergisch-Gladbach, Germany). Then, the cells were analyzed by flow cytometry.
Analysis of parameters of kidney injury in blood and urine
Experimental autoimmune encephalomyelitis (EAE) was induced in Lewis rats by intraperitoneal transfer of 5 × 107 activated AQP4207–232 spec. T cells. On day 4 after T cell transfer, the animals received an intraperitoneal injection with 10 mg anti-AQP-4 antibody containing NMO-IgG or 10 mg/ml commercially available human IgG (Subcuvia®). Other animals received either 10 mg anti-AQP-4 antibody containing NMO-IgG or 10 mg/ml commercially available human IgG (Subcuvia®) without prior T cell injection. 10 h after antibody injection, the animals were placed for 14 h in metabolic cages for urine collection, and killed with CO2. Then, blood was taken from these animals for serum analysis. We also included untreated animals in this study.
The urine samples were analyzed for the following parameters: volume/hour, sediment composition, and content (glucose, leukocytes, erythrocytes, nitrite and proteins, as analyzed with the Combur5 Test® HC (Roche Diagnostics, Mannheim, Germany). In the serum, blood urea nitrogen and creatinine was determined. All these parameters were similar in all different experimental groups, indicating that the inflammatory reactions at the renal cortico-medullary junction took a subclinical course.
Results
AQP-4 contains epitopes suitable for binding to Lewis rat MHC class II (RT1.BL)
Informations about possible T cell epitopes on rat AQP-4 were not yet available. However, autoimmune-associated T cell responses in Lewis rats are MHC class II RT1.BL restricted, we used the defined extended RT1.BL-peptide-binding motif to predict possible T cell epitopes on AQP-4 [39]. According to these predictions, positions 4 and 5 do not play an important role in RT1.BL binding. Hence, these positions were neglected in peptide search.
We found two perfect matches: the first optimal binder, AQP-4220–228, is contained in the extracellular loop E of AQP-4, which is 100% identical between rats and humans, and which has the sequence PAVIMGNWE [8]. This sequence contains glutamic acid (E), a negatively charged residue at position 9, which is a prerequisite for RT1.BL binding. In addition to this anchor at position 9, it also contains the auxiliary anchors proline (P), valine (V), asparagine (N), and tryptophan (W) at positions 1, 3, 7, and 8, respectively (Fig. 1).

Identification of RT1.BL-peptide-binding motifs of rat AQP-4. a List of amino acids enhancing or decreasing binding to RT1.BL at peptide positions 1–9 [39]. Please note that positions 4 and 5 do not play an important role in RT1.BL binding [39] and were neglected in peptide search. b Amino acid sequences of two rat AQP-4 nonamers showing a 100% match to the binding requirements at positions 1,3,7,8, and 9 (red), and of seven rat aquaporin 4 nonamers showing 1 mismatch (green) to the optimal binding motif at these positions. c Alignment of the completely matching epitopes with rat aquaporin 4
The second epitope, rAQP-4296–304, is contained in the intracellular C-terminal part of AQP-4 and had the sequence GVVHVIDID. This sequence contains aspartic acid (D), a negatively charged residue at position 9, which is a prerequisite for RT1.BL binding. In addition to this anchor at position 9, it also contains the auxiliary anchors glycine (G), valine (V), aspartic acid (D), and isoleucine (I, rat) at positions 1, 3, 7, and 8, respectively (Fig. 1).
Because it is easier to obtain rat encephalitogenic T cell responses with “foreign” CNS antigens, and since in Lewis rats, T cells specific for guinea pig MBP and bovine S100β induce a more severe EAE than T cells specific for the autologous proteins, we used the human counterpart of the identified epitope, hAQP-4296–304 (GVVHVIDVD) with its amino acid exchange at position 8. The valine at position 8 of the human sequence is as suitable to bind to RT1.BL as the isoleucine at position 8 of the rat sequence.
Lewis rats can mount encephalitogenic T cell responses against AQP-4
When we immunized experimental animals with AQP-4 peptides containing the identified epitopes, we did not see any evidence for clinical or histologically evident CNS disease (data not shown). However, we were able to raise antigen-specific T cell lines.
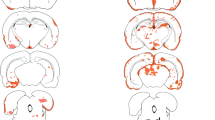
Transfer of AQP-4207–232-specific T cells did not cause any clinical signs of EAE (data not shown), although they induced inflammatory lesions along the entire neuraxis (Fig. 2), and were highly specific for their cognate antigen (Fig. 3). In the brain, these lesions were observed in cerebral and cerebellar cortex, the basal hypothalamus and the brain stem. Predilection sites for these lesions were the AQP-4 rich glial lamellae of the basal/lateral hypothalamus and the meninges, where the inflammatory cells formed multilayered, densely packed foci, which were either located on top of a seemingly intact subpial glia limitans, or infiltrated the parenchyma from this site (Fig. 2). The inflammatory lesions were dominated by T lymphocytes (T cells: macrophages/activated microglia cell ratio = 4.4). We did not detect any inflammatory lesions in the optic system. In the spinal cord, inflammatory lesions were found at the thoracic as well as at the lumbar/sacral level and frequently involved the dorsal horns (Fig. 2). Both in brain and spinal cord, astrocytes remained intact, and there was no evidence for myelin/oligodendrocyte injury. When AQP-4207–232-specific T cells were transferred together with anti-AQP-4 antibody containing NMO-IgG, lesions were more numerous and larger. The parenchymal infiltration by T cells (Fig. 4) and the numbers of macrophages/activated microglia cells was more pronounced (T cell: macrophages/activated microglia cell ratio = 1.3). AQP-4 loss was readily seen. In the brain, areas of AQP-4 loss involved the periventricular pons, subpial cortical areas of the cerebral cortex, and perivascular sites of cerebellar deep white matter and medulla (Fig. 4). In the spinal cord, AQP-4 loss mostly involved the perivascular glia limitans of the gray matter, and was much less pronounced at the white matter perivascular or subpial glia limitans (Fig. 4). Myelin and oligodendrocytes remained intact.

CNS inflammation caused by AQP-4207–232-specific T cells. a Distribution of densely packed inflammatory lesions along the neuraxis, using schemes provided by Paxinos and Watson [27] as guide lines. Brain and spinal cord [cervical (C1–7), thoracal (T1–10) and lumbar/sacral (L1–S4)] sections of eight animals were analyzed, and the location of each lesion was projected in red color into the relevant scheme. 2/8 animals had been injected with AQP-4207–232-specific T cells only; 3/8 animals had been injected with AQP-4207–232-specific T cells and Subcuvia, and 3/8 animals had been challenged with AQP-4207–232-specific T cells and NMO-IgG. The data shown here were obtained from 48 brain sections and 120 spinal cord sections. Inflammation of the glial lamellae of the basal hypothalamus (b) and the orbital cortex (c, d). e Inflammatory cells overlaying and infiltrating the dorsal horn of the spinal cord. f–g Massive build-up of inflammatory cells on top of an essentially intact subpial glia limitans of the spinal cord (d–g). Tissue sections were stained with antibodies against AQP-4 (b, d, e, f) or GFAP (c, g), the nuclei were counterstained with hematoxylin

Antigen specificity of AQP-4207–232-specific T cells. 5 × 105 AQP-4207–232-specific T cells were cocultured in triplicates with 1 × 106 thymic antigen-presenting cells in the absence of externally added antigens (0), or in the presence of myelin basic protein (MBP, as irrelevant CNS antigen), AQP-4207–232 representing AQP-4 loop E (E), myelin oligodendrocyte glycoprotein (MOG, as second irrelevant CNS antigen) or concanavalin A (ConA, as positive control) (upper panel). In a second set of experiments, 5 × 105 AQP4207–232-specific T cells were cocultured in triplicates with 1 × 106 thymic antigen-presenting cells in the absence of externally added antigen (0), or in the presence of AQP-454–71 representing AQP-4 loop A (a), AQP-4137–157 representing AQP-4 loop C (C), AQP-4207–232 representing AQP-4 loop E (E), AQP-4277–322 representing the C-terminal, intracellular domain of AQP-4 (ic), ovalbumin (OVA, as an irrelevant antigen), or ConA as positive control. T cell activation, as evidenced by incorporation of 3H thymidine, and measured in counts per minute (cpm) was only detected upon exposure to AQP-4207–232 and in response to ConA. Shown here are the mean values of cpm ± standard deviation

AQP-4 loss caused by the combined action of AQP-4207–232-specific T cells and NMO-IgG. Subpial (a) and perivascular (b) loss of AQP-4 reactivity. Consecutive sections of lumbar spinal cord. Note the presence of large numbers of inflammatory cells (c), the dominance of granulocytes in the infiltrate (d), the presence of macrophages with degradation products (red e), and the massive loss of AQP-4 reactivity in the tissue (f). The inflammatory lesion is associated with the presence of human IgG (g) and with the deposition of complement (h). Tissue sections were stained with antibodies against AQP-4 (brown a, b, f) analyzed by hematoxylin/eosin (c, d) or Luxol fast blue staining (myelin blue, degradation products red e), or anti-human IgG (brown g) or anti-C9neo (red h). The nuclei were counterstained with hematoxylin
Transfer of hAQP-4296–304-specific T cells provoked a subclinical EAE with few inflammatory lesions in the CNS (Fig. 5). The CNS lesions presented mostly as small foci of meningeal inflammation, from which T cells entered the CNS parenchyma via the subpial glia limitans (Fig. 5). The optic system was not affected, and astrocytes as well as oligodendrocytes remained intact. Co-transfer of hAQP-4296–304-specific T cells with anti-AQP-4 antibody containing NMO-IgG did not increase the lesion load in the CNS, did not increase the parenchymal infiltration of the CNS by T cells, and was not associated with noticeable loss of AQP-4 reactivity in the CNS (data not shown). The low degree of CNS inflammation induced by hAQP-4296–304-specific T cells coincided with an accumulation of these cells in the omentum (supplementary fig. 1), although PCR analysis did not show evidence for AQP-4 expression at this site (data not shown). The inability of large numbers of these cells to leave the peritoneal cavity stood in marked contrast to the behavior of AQP-4207–232-specific T cells or of any other T cells injected at this site [5, 7, 13] and was not further analyzed.

CNS inflammation caused by hAQP-4277–322- and hAQP-4296–304-specific T cells. a Distribution of densely packed inflammatory lesions along the neuraxis, using schemes provided by Paxinos and Watson [27] as guide lines. Brain and spinal cord [cervical (C1–7), thoracal (T1–10) and lumbar/sacral (L1–S4)] sections of seven animals were analyzed, and the location of each lesion was projected in red color into the relevant scheme. The data shown here were obtained from 42 brain sections and 140 spinal cord sections. Inflammatory foci at the subpial glia limitans of the brain (b–g) and spinal cord (h, i) are shown. Please note the ongoing parenchymal infiltration in b and f, and the diffuse distribution of T cells in the dorsal horns (h). Tissue sections were stained with anti-CD3 antibodies to identify T cells (b, e–i), with the antibody ED1 to identify macrophages/activated microglia cells, c or with anti-AQP-4 antibody (d), the nuclei were counterstained with hematoxylin
The infiltration of AQP-4207–232- and AQP-4296–304-specific T cells from the subpial glia limitans into the CNS parenchyma raised the question whether astrocytes could trigger some T cell reactivation at the blood–brain barrier. To address this question, we first made immunohistochemical analysis of the inflamed CNS, but we were unable to find any evidence for MHC class II expression on GFAP+ astrocytes (data not shown). However, this still left us with the possibility that astrocytes in the lesions carry MHC class II products on the surface, which are below the limit of detection by conventional immunohistochemistry, but would nevertheless exert biological functions. Therefore, we addressed this possibility in in vitro studies. We treated astrocytes with IFN-γ to induce MHC class II products and used these cells in T cell activation assays (Fig. 6). We used AQP4207–232-specific T cells in our assay, since these cells showed a higher encephalitogenic potential than their AQP-4296–304-specific counterparts, and because they also showed a deeper infiltration into the parenchyma. We observed that MHC class II+ astrocytes were able to activate AQP-4207–232-specific T cells only in the presence of the exogeneously added peptide AQP-4207–232, but not in its absence (Fig. 6). This observation suggested that astrocytes were unable to present fragments of endogenous AQP-4 to AQP-4207–232-specific T cells.

Astrocytes as antigen-presenting cells. Astrocytes cultured in vitro for 72 h in the absence (a, c) or presence of IFN-γ (b, d) maintain GFAP (red) and AQP-4 (green) expression. None of the untreated (c) and approximately 10% of the IFN-γ treated astrocytes (d) express MHC class II products (OX-6 staining red). e Presentation of exogenous antigens by IFN-γ treated astrocytes. When these cells were cocultured with AQP-4207–232-specific T cells in the absence of externally added antigens (0), or in the presence of AQP-454–71 representing AQP-4 loop A (A), AQP-4137–157 representing AQP-4 loop C (C), AQP-4207–232 representing AQP-4 loop E (E), AQP-4277–322 representing the C-terminal, intracellular domain of AQP-4 (ic), myelin basic protein (MBP) as an irrelevant CNS antigen, or concanavalin A (ConA), T cell activation was only detected upon exposure to AQP-4207–232 and in response to ConA. Shown in this graph are differences in the average counts per minute (Δcpm) obtained from triplicate cultures of T cells challenged with antigens and IFN-γ treated or untreated astrocytes, given as mean ± SD. The graph is representative of two experiments. f No presentation of endogenous AQP-4 peptides by IFN-γ-treated astrocytes. The average cpm (mean ± SD) seen when astrocytes were cocultured in triplicates with AQP-4207–232-specific T cells in the absence of AQP-4207–232 (273.3 ± 110.0) is in the range of the sum of the cpm (mean ± SD) of astrocytes alone (131.7 ± 42.7) plus AQP-4207–232-specific T cells cultured with AQP-4207–232 only (137.5 ± 27.6; to reveal carryover of antigen-presenting cells in the T cell preparation used) or AQP-4207–232-specific T cells cultured without antigen (64.3 ± 9.1). The graph is representative of two experiments
We also observed inflammatory lesions in muscle (Fig. 7) and kidney (Fig. 8). Because these organs not only express AQP-4, but also many other AQPs, we first analyzed whether these other AQPs share sequence homology to AQP-4220–228- and hAQP-4296–304. This was not the case (Fig. 9). Hence, these inflammatory lesions must be triggered by the local availability of AQP-4 and appropriate antigen-presenting cells.

Muscle inflammation upon transfer of hAQP-4296–304-specific T cells (HE staining). Eosinophilic granulocytes are labeled with white arrowheads, neutrophilic granulocytes with a black arrow

Renal pathology following the transfer of AQP-4296–304-specific T cells in combination with AQP-4 antibody containing NMO-IgG. In the intact kidney (a–c), AQP-4 positive collecting ducts (brown) are essentially absent in the cortex, appear at the cortico-medullary junction (a), and are found in high numbers in the renal papilla (a). Intact collecting duct cells show a basolateral staining pattern (b, c), which is much stronger, when the commercial anti-AQP-4 antibody was used (b brown) than it was, when anti-AQP-4 antibody containing NMO-IgG was utilized (c red). d–e Peri-ductal inflammation at the renal cortico-medullary junction following transfer of AQP-4296–304-specific T cells in combination with AQP-4 antibody containing NMO-IgG. Consecutive sections of the cortico-medullary junction, stained with HE (d) and commercial anti-AQP-4 (e brown). Inflammation was not seen in the renal papilla, although collecting ducts at this site expressed high levels of AQP-4 (f brown). g Flow cytometry analysis revealed that binding of AQP-4 antibody containing NMO-IgG (gray) to the surface of collecting duct cells (MDCK cells) was below the limit of detection in the absence of IFN-γ. In the presence of IFN-γ, the availability of the antibody-reactive AQP-4 epitope increased, and AQP-4 antibody containing NMO-IgG could readily bind. In both cases, binding of Subcuvia (red) served as negative control

Alignment of the amino acid sequence of RT1.BL binding T cell epitopes of rat AQP-4 with other known AQPs. The following accession numbers were used to align sequences: AQP-1 (EDL88090.1), AQP-2 (NP_037041.2), AQP-3 (NP_113891.1), AQP-4 (NP_001135838.1), AQP-5 (NP_036911.1), AQP6 (NP_071517.1), AQP-8 (NP_062031.1), AQP-9 (NP_075249.1), AQP-12 (NP_001102479.1). a Alignment of AQP-4220–228. The amino acid sequence of AQP-4220–228 (red) is unique among all AQPs studied. All other AQP sequences have either amino acid mismatches (black) or insertions (small letters) at this site. b Alignment of AQP-4296–304. The amino acid sequence of AQP-4296–304 (red) is unique among all AQPs studied. All other sequences have either no significant similarity (−), or just an arbitrary match of four amino acid within their sequence (AQP-3)
Muscle lesions were observed in 2/6 animals injected with hAQP-4296–304-specific T cells. The inflammatory cell population was located in the connective tissue compartment and contained neutrophils and eosinophils. The muscle fibers themselves were not invaded (Fig. 7). The real extent of muscle involvement in the AQP-4-specific T cell injected animals is probably higher, since we only sampled and analyzed the m. glutaeus.
We also observed kidney inflammation in all animals injected with AQP-4207–232- and hAQP-4296–304-specific T cells. This finding was subject of more detailed analysis.
AQP-4207–232- and hAQP-4296–304-specific T cells profoundly enhance the anti-AQP-4 antibody containing NMO-IgG induced peri-ductal inflammation at the renal cortico-medullary junction
The presence of inflammatory cells surrounding and even infiltrating collecting ducts at the renal cortico-medullary junction was a common feature of all animals injected with anti-AQP-4 antibody containing NMO-IgG (Table 1; Fig. 8). It was not associated with clinically overt kidney disease or with noticeable changes of the urine (data not shown). Periductal renal inflammation could be reproduced using anti-AQP-4 antibody containing NMO-IgG from an additional AQP-4 antibody+ NMO patient tested (patient J1 of [5]). It was not observed with IgG preparations obtained from AQP-4 antibody-negative NMO patients (patients J3 and J4 of [5]), MS patients (patients J6 and J7 of [5]), or controls (patients J8 and J9 of [5], and was also not evident after transfer of Subcuvia (Table 1).
In the presence of AQP-4207–232- and AQP-4296–304-specific T cells, the AQP-4 antibody-induced peri-ductal inflammation at the renal cortico-medullary junction was exacerbated, while only low numbers of inflammatory cells were observed at this site when AQP-4207–232- and AQP-4296–304-specific T cells had been injected into the recipient animals (Fig. 8). T cells were essentially undetectable at the cortico-medullary renal junction upon transfer of OVA- or MBP-specific T lymphocytes (Table 1). The peri-ductal inflammation provoked by anti-AQP-4 antibody containing NMO-IgG remained detectable upon transfer of OVA-specific T cells, but was completely abolished in the presence of MBP-specific T cells (Table 1). This finding cannot be explained by a selective depletion of antibodies by astrocytes in the course of severe CNS inflammation, since animals injected with NMO-IgG only and animals injected with MBP-specific T cells and NMO-IgG had comparable antibody titers in the serum (both in the range of 1:512–1:1,024).
In collecting ducts of the intact kidney, AQP-4 could be readily detected with the commercial antibody directed against an intracellular epitope of this molecule, while the binding efficiency of the pathogenic anti-AQP-4 antibodies contained in our NMO-IgG preparations was very low (Fig. 8). Moreover, in the presence of these pathogenic antibodies alone, the degree of peri-ductal inflammation at the renal cortico-medullary junction was lower than the one observed in the presence of both AQP-4-specific T cells and AQP-4-specific autoantibodies. These two observations raised the question whether an inflammatory milieu provoked by kidney-infiltrating T cells could lead to an enhanced exposure/availability of AQP-4 epitopes recognized by the pathogenic anti-AQP-4 antibodies from human NMO patients. To specifically address this question, we used the canine collecting duct cell line MDCK [10] for further studies. This was possible since the three AQP4 extracellular loops A, C, and E participate in the formation of the NMO-IgG epitope [29], and since these loops are highly conserved in human, rat, and canine AQP-4. We cultured MDCK cells in absence and presence of IFN-γ, and studied the expression of AQP-4 on these cells by flow cytometry. In the absence of IFN-γ, binding of anti-AQP-4 antibody containing NMO-IgG to the surface of MDCK cells was below the limit of detection. However, binding of this antibody was observed when the cells had been cultured in the presence of IFN-γ (Fig. 8).
Discussion
Our study provides insights, which may be relevant for the pathogenesis of NMO. It first shows that AQP-4-specific T cells are part of the normal immune repertoire of rats and, following expansion and activation can induce T cell-mediated brain inflammation, which is sufficient for AQP-4-specific antibodies to reach the brain and to initiate NMO-like lesions in the CNS. Secondly, it provides evidence that organs with high AQP-4 expression other than the CNS, such as kidney and muscle, may be affected by the disease process.
Inflammatory lesions in the CNS of NMO patients are characterized by infiltration of T lymphocytes and deposition of AQP-4-specific antibodies and complement on astrocytes at the glia limitans [20, 31]. The formation of antibodies depends on T cell help, but whether or not AQP-4-specific T cells can also contribute to the inflammatory process remains an open issue. We show here, that the normal immune repertoire of Lewis rats harbors AQP-4-specific T cells, just as the murine immune repertoire does [12, 24], that AQP-4-specific T cells have encephalitogenic potential, and that they are able to cooperate with anti-AQP-4 antibodies to produce astrocyte-destructive lesions. Myelin/oligodendrocyte loss was not observed, due to the acute nature of our experiments [5]. The encephalitogenic potential of our AQP-4-specific T cell lines was low and, thus, despite the transfer of high numbers of T cells no clinical deficits were present in the recipient animals. One possible reason for this observation could be that the natural processing of AQP4 might preferentially generate peptides different from those used by us for the isolation of AQP-4-specific T cells, and this point has to be addressed in future studies. Nevertheless, our AQP-4 peptide-specific T cells induced subclinical inflammation in the brain and spinal cord with a pattern of inflammation unique to this T cell population. In contrast to S100β-specific T cells, which recognize another astrocytic protein [15], there was only very little inflammatory response in the CNS parenchyma. Instead, the glia limitans was selectively targeted, leading to characteristic T cell infiltrates between the astrocytic layers. Moreover, on a background of diffuse T cell infiltration, S100β T cell transferred animals showed ~13% of their spinal cord lesions in the dorsal horns, while AQP4207–232-specific T cell transferred animals had ~53% of spinal cord lesions at this site (data not shown). It has been shown before in cotransfer models of MOG reactive T cells and demyelinating anti-MOG antibodies [21] as well as in transfer experiments with human NMO-IgG [3, 5] that pathogenic antibodies induce lesions, when reaching the CNS at sites of T cell mediated inflammation. T cell inflammation leads to opening of the blood–brain barrier, allowing antibodies and complement to reach the target tissue [22] and also to the activation of macrophages or microglia which induce tissue injury through antibody-dependent cellular cytotoxicity. In the absence of T cell-mediated inflammation this can be achieved by local injection of demyelinating antibodies together with IFN-γ [37]. To initiate antibody-mediated damage in the CNS only a very mild, subclinical T cell mediated inflammation is required [18], which is also seen in our studies. Our results, thus, indicate that a T cell-mediated immune response may be instrumental in the induction of NMO lesions, even when such a T cell response alone is insufficient to induce disease. Whether NMO patients mount a T cell response against AQP-4 and whether such a T cell response is associated with relapses of the disease has to be shown in future studies.
Moreover, additional animal models of NMO are needed, preferentially involving T cell- and B cell-specific immune responses against AQP-4 and such studies are warranted by recent observations in an animal model of spontaneous opticospinal encephalomyelitis involving MOG-reactive T and B cells which suggest that autoimmune T and B cells may interact on several levels to create disease [16, 30]: Antigen-specific T cells could recruit endogenous antigen-specific B cells [30], drive their differentiation to Ig-producing plasma cells or long-lived memory B cells [16], and could even control Ig affinity maturation and isotype switching [16], while antigen-specific B cells could capture even highly diluted protein antigens and present them to specific T cells [17].
AQP-4 is highly expressed also in other tissues than the CNS, such as muscle and kidney. However, so far NMO is regarded as a CNS-specific disease, leaving other organs unaffected. This may have one major reason. As we show in this study, there is only a very low or even absent expression and/or accessibility of the pathogenic NMO epitope, recognized by the patient’s autoantibodies in kidney and muscle of normal animals. Since the pathogenic autoantibodies of NMO patients recognize a conformational epitope of AQP-4, which depends on clustering of the protein in orthogonal arrays [25], expression of AQP-4 alone not necessarily implies that the respective cells are a target in the disease. Here, however, we show a binding of AQP-4 autoantibodies to kidney collecting duct cells stimulated with IFN-γ. Hence, T cell-mediated inflammation may lead to a slight induction of expression of AQP-4 targets on the cell surface, or to a loss of surface proteins interfering with the binding of pathogenic autoantibodies to AQP-4.
We observed an accumulation of inflammatory cells in the muscle of 2/6 animals injected with hAQP-4296–304-specific cells. Transient myositis or rhabdomyolysis could also be a pathological feature in NMO, because elevated levels of creatine kinase (CK) as a marker for muscle injury were recently described in some patients [35], in line with AQP-4 expression in fast-twitch skeletal muscle fibers [14].
In the kidney, AQP-4 is expressed at the basolateral surface of collecting duct epithelial cells [8, 36]. These ducts are much more frequent in the renal papilla than at the cortico-medullary junction and yet, peri-ductal inflammation was almost exclusively confined to the renal cortico-medullary junction. This is probably due to the facts that the affinity of antibody-antigen interactions is lowered under high salt conditions [26], and that collecting ducts of the papilla have to deal with much higher pericellular salt concentrations than their cortico-medullary counterparts [19]. This could also provide a rationale for the absence of peri-ductal inflammation at the renal cortico-medullary junction in animals injected with MBP-specific T cells and anti-AQP-4-specific antibody containing NMO-IgG, and for its presence in animals injected with the same NMO-IgG preparation in the absence of T cell transfer: the EAE induced by MBP-specific T cells was accompanied by a pronounced weight loss of ~10% body weight within 24 h of disease onset (data not shown), and most of this weight loss can be attributed to dehydration, which might further enhance the pericellular salt concentrations in collecting ducts. Alternatively, dehydration might also directly downregulate renal AQP-4 expression [33].
Renal disease in NMO patients has not been described so far. In addition, in our present experiment animals, no clinically overt renal disease was seen and urinary analysis did not show pathological alterations, indicating that AQP-4-specific T cells with or without NMO autoantibodies induced a mild and subclinical interstitial nephritis. Interestingly, in humans, interstitial nephritis can take an asymptomatic course [9], and could escape from diagnosis using conventional markers of kidney injury [4]. Thus, kidney inflammation may represent a pathological feature of NMO previously overlooked in NMO patients and inflammatory changes in the urine of such patients may not simply represent urinary tract infection in such patients with severe neurological disability. Alternatively, we cannot exclude that T cells specific for different AQP-4 epitopes differ in their tropism to CNS, muscle, and kidney, or, that T cells with specificities unrelated with AQP4 prepare the field for NMO-IgG induced CNS pathology in NMO patients.
References
Aboul-Enein F, Bauer J, Klein M, Schubart A, Flugel A, Ritter T et al (2004) Selective and antigen-dependent effects of myelin degeneration on central nervous system inflammation. J Neuropathol Exp Neurol 63:1284–1296
Ben Nun A, Wekerle H, Cohen IR (1981) The rapid isolation of clonable antigen-specific T lymphocyte lines capable of mediating autoimmune encephalomyelitis. Eur J Immunol 11:195–199
Bennett JL, Lam C, Kalluri SR, Saikali P, Bautista K, Dupree C et al (2009) Intrathecal pathogenic anti-aquaporin-4 antibodies in early neuromyelitis optica. Ann Neurol 66:617–629
Bonventre JV, Vaidya VS, Schmouder R, Feig P, Dieterle F (2010) Next-generation biomarkers for detecting kidney toxicity. Nat Biotech 28:436–440
Bradl M, Misu T, Takahashi T, Watanabe M, Mader S, Reindl M et al (2009) Neuromyelitis optica: pathogenicity of patient immunoglobulin in vivo. Ann Neurol 66:630–643
Brum DG, Barreira AA, dos Santos AC, Kaimen-Maciel DR, Matiello M, Costa RM et al (2010) HLA-DRB association in neuromyelitis optica is different from that observed in multiple sclerosis. Mult Scler 16:21–29
Flugel A, Berkowicz T, Ritter T, Labeur M, Jenne DE, Li Z et al (2001) Migratory activity and functional changes of green fluorescent effector cells before and during experimental autoimmune encephalomyelitis. Immunity 14:547–560
Graber DJ, Levy M, Kerr D, Wade WF (2008) Neuromyelitis optica pathogenesis and aquaporin 4. J Neuroinflamm 5:22–43
Halbritter J, Mayer C, Rasche FM, Amann K, Lindner TH (2009) Interstitielle nephritis. Der Intern 50:1111–1126
Herzlinger DA, Easton TG, Ojakian GK (1982) The MDCK epithelial cell line expresses a cell surface antigen of the kidney distal tubule. J Cell Biol 93:269–277
Hochmeister S, Zeitelhofer M, Bauer J, Nicolussi E-M, Fischer M-T, Heinke B et al (2008) After injection into the striatum, in vitro-differentiated microglia- and bone marrow-derived dendritic cells can leave the central nervous system via the blood stream. Am J Pathol 173:1669–1681
Kalluri SR, Rothhammer V, Staszewski O, Srivastava R, Petermann F, Prinz M, Hemmer B, Korn T (2011) Functional characterization of aquaporin-4 specific T cells: towards a model for neuromyelitis optica. PLoS One 6:e16083
Kawakami N, Lassmann S, Li Z, Odoardi F, Ritter T, Ziemssen T et al (2004) The activation status of neuroantigen-specific T cells in the target organ determines the clinical outcome of autoimmune encephalomyelitis. J Exp Med 199:185–197
King LS, Kozono D, Agre P (2004) From structure to disease: the evolving tale of aquaporin biology. Nat Rev Mol Cell Biol 5:687–698
Kojima K, Berger T, Lassmann H, Hinze-Selch D, Zhang Y, Gehrmann J et al (1994) Experimental autoimmune panencephalitis and uveoretinitis transferred to the Lewis rat by T lymphocytes specific for the S100ß molecule, a calcium binding protein of astroglia. J Exp Med 180:817–829
Krishnamoorthy G, Lassmann H, Wekerle H, Holz A (2006) Spontaneous opticospinal encephalomyelitis in a double-transgenic mouse model of autoimmune T cell/B cell cooperation. J Clin Invest 116:2385–2392
Lanzavecchia A (1990) Receptor-mediated antigen uptake and its effects on antigen presentation to class II-restricted T lymphocytes. Ann. Rev. Immunol. 8:773–794
Lassmann H, Brunner C, Bradl M, Linington C (1988) Experimental allergic encephalomyelitis: the balance between encephalitogenic T lymphocytes and demyelinating antibodies determines size and structure of demyelinated lesions. Acta Neuropathol (Berl) 75:566–576
Lee WK, Jang SB, Cha SH, Lee JH, Lee KH, Kim J et al (2002) Different sensitivity to nephrotoxic agents and osmotic stress in proximal tubular and collecting duct cell lines derived from transgenic mice. Toxicol In Vitro 16:55–62
Lennon VA, Wingerchuck DN, Kryzer TJ, Pittock SJ, Lucchinetti CF, Fujihara K et al (2004) A serum autoantibody marker of neuromyelitis potica: distinction from multiple sclerosis. Lancet 264:2106–2112
Linington C, Bradl M, Lassmann H, Brunner C, Vass K (1988) Augmentation of demyelination in rat acute allergic encephalomyelitis by circulating mouse monoclonal antibodies directed against a myelin/oligodendrocyte glycoprotein. Am J Pathol 130:443–454
Linington C, Morgan BP, Scolding NJ, Wilkins P, Piddlesden S, Compston DA (1989) The role of complement in the pathogenesis of experimental allergic encephalomyelitis. Brain 112:895–911
Matsushita T, Matsuoka T, Isobe N, Kawano Y, Minohara M, Shi N et al (2009) Association of the HLA-DPB1*0501 allele with anti-aquaporin-4 antibody positivity in Japanese patients with idiopathic central nervous system demyelinating disorders. Tissue Antigens 73:171–176
Nelson PA, Khodadoust M, Prodhomme T, Spencer C, Patarroyo JC, Varrin-Doyer M, Ho JD, Stroud RM, Zamvil SS (2010) Immunodominant T cell determinants of aquaporin-4, the autoantigen associated with neuromyelitis optica. PLoS One 5:e15050
Nicchia GP, Mastrototaro M, Rossi A, Pisani F, Tortorella C, Ruggieri M, Lia A, Trojano M, Frigeri A, Svelto M (2009) Aquaporin-4 orthogonal arrays of particles are the target for neuromyelitis optica autoantibodies. Glia 57:1363–1373
Paul WE (2008) Fundamental immunology, 6th edn. Lippincott Williams & Wilkins, Philadelphia
Paxinos G, Watson C (1997) The rat brain in stereotaxic coordinates, 3rd edn. Academic Press, San Diego
Piddlesden SJ, Lassmann H, Zimprich F et al (1993) The demyelinating potential of antibodies to myelin oligodendrocyte glycoprotein is related to their ability to fix complement. Am J Pathol 143:555–564
Pisani F, Mastrototaro M, Rossi A, Nicchia GP, Tortorella C, Ruggieri M et al (2011) Identification of two major conformational AQP4 epitopes for neuromyelitis optica autoantibodies binding. J Biol Chem. doi:10.1074/jbc.M110.123000
Pöllinger B, Krishnamoorthy G, Berer K, Lassmann H, Bösl MR, Dunn R, Domingues HS, Holz A, Kurschus FC, Wekerle H (2009) Spontaneous relapsing-remitting EAE in the SJL/J mouse: MOG-reactive transgenic T cells recruit endogenous MOG-specific B cells. J Exp Med 206:1303–1316
Roemer SF, Parisi JE, Lennon VA, Mandler R, Weinshenker BG, Benarroch E et al (2007) Distinct pattern of aquaporin-4 expression in neuromyelitis optica lesions. Brain 130:1194–1205
Saadoun S, Waters P, Bell BA, Vincent A, Verkman AS, Papadopoulos MC (2010) Intra-cerebral injection of neuromyelitis optica immunoglobulin G and human complement produces neuromyelitis optica lesions in mice. Brain 133:349–361
Saito N, Ikegami H, Shimada K (2005) Effect of water deprivation on aquaporin 4 (AQP4) mRNA expression in chickens (Gallus domesticus). Brain Res Mol Brain Res 30:193–197
Sommer C, Weishaupt A, Brinkhoff J, Biko L, Wessig C, Gold R et al (2005) Paraneoplastic stiff-person syndrome: passive transfer to rats by means of IgG antibodies to amphiphysin. Lancet 365:1406–1411
Suzuki N, Takahashi T, Aoki M, Misu T, Konohana S, Okumura T et al (2010) Neuromyelitis optica preceded by hyperCKemia episode. Neurology 11:1543–1545
Terris J, Ecelbarger CA, Marples D, Knepper MA, Nielsen S (1995) Distribution of aquaporin-4 water channel expression within rat kidney. Am J Physiol 269:F775–F785
Vass K, Heininger K, Schäfer B, Linington C, Lassmann H (1992) Interferon-gamma potentiates antibody mediated demyelination in vivo. Ann Neurol 32:189–206
Warabi Y, Yagi K, Hayashi H, Matsumoto Y (2006) Characterization of the T cell receptor repertoire in the Japanese neuromyelitis optica: T cell activity is upregulated compared to multiple sclerosis. J Neurol Sci 249:145–152
Wauben MHM, van der Kraan M, Grosfeld-Stulemeyer MC, Joosten I (1997) Definition of an extended MHC class II-peptide binding motif for the autoimmune disease-associated Lewis rat RT1.BL molecule. Int Immunol 9:281–290
Acknowledgments
We thank Marianne Leisser, Ulrike Köck and Angela Kury for excellent technical assistance. This work was supported by the European Union (Grant number LSHM-CT-2005-018637), the Austrian Science Fund (Grant number P21581-B09 to MB), the PhD programme Cell Communication in Health and Disease (CCHD, co-funded by the Austrian Science Fund and the Medical University Vienna), and a research grant (number 2007104) of the interdisciplinary center for research and treatment (IFTZ) of Innsbruck Medical University. It was supported in parts by grants-in-aid for Scientific Research of the Ministry of Education, Culture, Sports, Science and Technology (19209032, 20390241, 22229008), and the Health and Labour Sciences Research Grant for Neuroimmunological Diseases of the Ministry of Health, Labor and Welfare of Japan.
Conflict of interest
Nothing to declare.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
401_2011_824_MOESM1_ESM.tif
Following the intraperitoneal injection of activated hAQP-4296-304-specific T cells, cell accumulations were most frequently observed on the peritoneal coating of the liver. Predilection site for this perihepatitis was an area just underneath the diaphragm in close vicinity to the pars abdominalis of the oesophagus. Similar cellular aggregates were not observed following the intraperitoneal injection of activated AQP-4207-232-specific T cells (TIFF 2735 kb)
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Pohl, M., Fischer, MT., Mader, S. et al. Pathogenic T cell responses against aquaporin 4. Acta Neuropathol 122, 21–34 (2011). https://doi.org/10.1007/s00401-011-0824-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-011-0824-0