
Abstract
Primary loss and dysfunction of astrocytes may trigger demyelination, as seen in neuromyelitis optica, an inflammatory disease of the central nervous system. In most patients affected by this disease, injury to astrocytes is initiated by the action of autoantibodies targeting aquaporin 4 (AQP-4), a water channel on astrocytes. We show here that damage of astrocytes and subsequent demyelination can also occur in the absence of autoantibody-mediated mechanisms. Following injection of lipopolysaccharide into the white matter initial microglia activation is followed by a functional disturbance of astrocytes, mainly reflected by retraction of astrocytic foot processes at the glia limitans and loss of AQP-4 and connexins, which are involved in the formation of gap junctions between astrocytes and oligodendrocytes. Demyelination and oligodendrocyte degeneration in this model follows astrocyte pathology. Similar structural abnormalities were also seen in a subset of active lesions in multiple sclerosis. Our studies suggest that astrocyte injury may be an important early step in the cascade of lesion formation in brain inflammation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Tissue injury in inflammatory demyelinating diseases, such as multiple sclerosis (MS), is generally believed to be induced by primarily targeting myelin sheaths and/or oligodendrocytes [29]. However, demyelinating diseases, such as neuromyelitis optica (NMO) reveal that a direct targeting of myelin or oligodendrocytes is not essential. In NMO, specific autoantibodies are directed against the astrocytic antigen aquaporin 4 (AQP-4) [20, 21] and these antibodies can initiate tissue injury in the central nervous system when they reach their target in animals with T-cell-mediated brain inflammation [6, 7]. In this experimental paradigm [7], as well as in patients with NMO [34], initial destruction of astrocytes is followed by oligodendrocyte degeneration and primary demyelination. These studies indicate that oligodendrocyte injury and demyelination may in some instances be secondary consequences of astrocyte dysfunction or degeneration.
Recently, a new model of spinal cord demyelination has been introduced, resulting from the focal injection of lipopolysaccharide (LPS) into the spinal white matter [11]. Using this model, it was suggested that microglia, activated through mechanisms of innate immunity produce toxic molecules which affect myelin sheaths and oligodendrocytes and thereby induce primary demyelination with partial axonal preservation [11, 26]. However, local microglial activation is an early response to LPS injection [26], but demyelination only starts 5–6 days later. This time lag between microglial activation and the onset of demyelination argues against a direct toxicity of products of activated microglia in the induction of demyelination. In light of the new findings in NMO and its experimental models, we have analyzed the astrocytic response to LPS both in vitro and in vivo. Our results show that structural and functional changes in astrocytes precede demyelination in a similar way as seen in experimental NMO lesions raising the possibility that the astrocytes are causally involved in the demyelination. We also provide evidence that astrocyte dysfunction may be involved in the pathogenesis of a subset of active MS lesions.
Materials and methods
Experimental design
Astrocyte pathology was analyzed in four different models of brain inflammation in rats (Table 1). T-cell-mediated inflammation was induced by passive transfer of myelin basic protein (MBP) reactive encephalitogenic T-cell lines. Specific targeting of astrocytes was obtained by co-transfer of human AQP-4 autoantibody containing immunoglobulins into animals with T-cell-mediated autoimmune encephalomyelitis (EAE). For controls, demyelination and oligodendrocyte destruction was induced in a similar way with co-transfer EAE, using monoclonal antibodies against myelin oligodendrocyte glycoprotein (MOG). In another model, inflammatory demyelination was triggered by innate immunity induced by focal injection of LPS into the spinal cord. Finally, we analyzed astrocyte pathology in different active MS lesions from patients with acute, relapsing and progressive MS.
T-cell-mediated EAE
The MBP-specific T cells used in this study were derived from Lewis rats according to the standard protocols [5]. The Lewis rats were intraperitoneally injected with 1 × 105 activated MBP-specific T cells or with saline (controls). Afterwards, the rats were monitored daily for evidence of weight loss or clinical symptoms of EAE. 3, 6, 9, 12 and 15 days after the induction of EAE, the animals were killed with an overdose of CO2, and perfused transcardially with 4% paraformaldehyde (PFA) in 0.1 M phosphate buffered saline (PBS, pH 7.4). The spinal cords were dissected-free, post-fixed overnight in 4% PFA, washed with PBS and embedded in paraffin.
Co-transfer of encephalitogenic T cells with specific autoantibodies
Lewis rats challenged with MBP-specific T cells, as described above received 10 mg human immunoglobulin from NMO patients or alternatively 10 mg normal human IgG (Subcuvia® [7]). Another set of animals with T-cell-mediated EAE received 1 mg of the monoclonal antibody Z12, directed against the conformational demyelinating epitope of MOG as described in detail before [23, 31]. Antibodies were injected systemically at day 4 after T-cell transfer, when T-cell-mediated inflammation in the central nervous system is initiated. Animals were killed 1 day after antibody transfer, the brains and spinal cord were dissected and routinely embedded in paraffin. To study later time points in lesion development, Lewis rats were actively sensitized with 100 µg of MBP in complete Freund’s adjuvant at the base of the tail. At the onset of clinical disease (day 10 after immunization), the animals received an intraperitoneal injection of NMO IgG or subcuvia. This was repeated 24 h later. Analysis of lesions in the central nervous system was performed on day 12 after immunization.
The LPS model
LPS-mediated lesions were induced in rat spinal cord as described in detail by Felts et al. [11]. Under isoflurane anesthesia, a quarter laminectomy was performed at the T12 vertebral level and a small hole was made in the dura. A drawn glass micropipette of an external tip diameter of 25 μm was then inserted into the dorsal funiculus and 0.5 μl of LPS (100 ng/μl in saline) was injected at two adjacent sites, longitudinally 1 mm apart, at depths of 0.7 and 0.4 mm (2 μl in total). LPS from Salmonella abortus equi (Sigma, Poole, UK) was used without further purification. Control animals received injections of saline alone. The injection site was marked by placing a small amount of sterile powered charcoal on the adjacent dura.
At different time points after the injections (8 h and 1, 3, 5, 8, 12 and 15 days), the animals were anesthetized with isoflurane and were then perfused transcardially with 4% PFA in 0.15 M PBS. The spinal cord was dissected-free and tissue blocks from the injection site as well as from a region 1 cm rostral and caudal to the injection site were embedded in paraffin.
All animal tissues used in this study were retrieved from the archives of the Center for Brain Research and the respective studies have been published earlier [7, 26, 23].
Human autopsy tissue
This study was performed on autopsy brains of MS patients and control cases from paraffin blocks archived in the Center of Brain Research, Medical University of Vienna, Austria (Table 1). Hemispheric or double hemispheric sections of these autopsy brains were characterized and classified to identify acute pattern II, pattern III [26], slowly expanding lesions of progressive MS, and chronic inactive lesions [12]. As a control, we included autopsy tissues from eight patients without neurological disease and without any CNS lesions.
Immunohistochemistry
Immunohistochemical investigations were performed on 3–5-μm thick paraffin sections of both human and rat material. Sections were de-paraffinized twice with xylol substitute (XEM) (Fluka analytical, Germany) for 20 min each, rinsed twice in 96% EtOH, treated with hydrogen peroxide in methanol for 30 min to block endogenous peroxidase, rehydrated in a descending series of EtOH (96 >70 > 50 > 30%) and further incubated for 1 h in phosphate buffered saline containing 10% fetal calf serum (PBS/CSF) to block non-specific antibody binding. Antigen retrieval was performed by heating the sections for 60–90 min in EDTA (1 mM EDTA, 10 mM Tris, pH 8.5 or 9) or 0.1 mM citrate buffer (pH 6) in a household food steamer device. The primary antibodies (listed in Table 2) were applied overnight in PBS/FCS. Afterwards, the slides were washed 3–4 times in PBS. Then, the slides were incubated with biotinylated secondary antibodies (sheep anti-mouse, donkey anti-rabbit, donkey anti-goat; all from Amersham or Jackson Immuno Research) for 1 h at room temperature. After washing 3–4 times in PBS, the sections were treated with avidin peroxidase (diluted 1:100 in 10% FCS/PBS), and incubated for 1 h at room temperature. For visualization of the bound antibodies, diaminobenzidine was used as chromogen as described previously [18].
Confocal laser microscopy
Areas of collagen IV (col IV) deposition and of loss of astrocytic markers in the EAE and LPS models were analyzed in serial sections. Fluorescence immunohistochemistry was performed on 3–5-μm thick paraffin sections of both human and rat as described earlier with few modifications. The sections were deparaffinized as described above, treated with hydrogen peroxide in methanol to block endogenous peroxidase, and incubated for 20 min in DAKO diluent to block nonspecific antibody reactions. For confocal fluorescent double labeling, glial fibrillary acidic protein (GFAP) immunoreactivity was visualized using either a mouse monoclonal or a rabbit polyclonal antibody. As second marker, we applied mouse monoclonal α-col IV, anti-α-dystroglycan antibodies, polyclonal α-connexin 43, or α-AQP-4 antibodies.
The next day, slides were washed twice with PBS. Then, either fluorochrome-conjugated or biotinylated secondary antibodies were applied simultaneously for 1 h at room temperature. When the secondary antibodies were biotinylated, the labeling was finished by application of streptavidin Cy2/Cy3 complexes (Jackson ImmunoResearch, 1:75) for 1 h at room temperature in the dark. The slides were mounted with cover slips using Geltol/Gellate. Once slides were dried (after 24 h at 4°C), they were analyzed by confocal microscopy. A Leica TCS SP5 LASAF microscope (Leica microsystems, CMS-GmbH, Germany) was used, equipped with an argon-ion laser (488 nm excitation) and two HeNe lasers (543 and 633 nm excitations). Fluorescence signals were collected simultaneously in the green (Cy2) and red (Cy3) channels.
Quantitative analysis
Quantitative analysis of demyelination, density of GFAP-reactive astrocytes and of the expression of astrocytic antigens within spinal cord cross-sections was performed on sections stained with LFB or immunocytochemistry, respectively. Sections were overlaid with a morphometric grid and the area of myelin loss and loss of astrocytic antigens was manually determined by counting the grid points over the respective areas. In addition, we determined the area of extracellular collagen IV (col IV) deposits within the CNS parenchyma as well as the number of astrocytes with cytoplasmic expression of col IV, and the number of blood vessels with split basal laminae. Areas of col IV deposition and of loss of astrocytic markers in the EAE and LPS models were analyzed in serial sections, using a microscopic field of 1 mm2. All values are expressed as cell counts and/or area per mm2.
Characterization of LPS effects on microglial cells and astrocytes
Production of mixed glial cultures as a source of microglia and astrocytes
0-24-h-old Lewis rats were killed and their brains dissected and transferred to RPMI 1640/10% fetal calf serum (FCS). After stripping of the meninges, the brains were mechanically dissociated by gentle pipetting. The resulting single cell suspensions were cultured for 5–7 days in poly-l-lysine-coated culture flasks, using RPMI1650/10% FCS, changing the medium every other day. After this time period, the mixed glial cell cultures consisted of a monolayer of astrocytes and fibroblasts. On top of this monolayer, loosely adherent, ramified microglial cells and glial progenitor cells were found.
Microglia cultures
Confluent mixed glial cultures were agitated for 12–15 h (180 rpm, 37°C). The supernatant was centrifuged (1,400 rpm, RT). Afterwards, the cell pellet was re-suspended in RPMI 10% FBS, seeded into uncoated Petri dishes and incubated for 10 min at 37°C, 5% CO2. During this incubation period, only microglial cells became adherent and stuck to the floor of the Petri dish, while other contaminating cells—mostly glial progenitor cells—remained in the supernatant. The supernatant was removed and the adherent microglial cells were washed 2–3 times with PBS at room temperature. These microglial cultures had a purity of ~99%.
Astrocyte-enriched cultures
Confluent mixed glial cultures were agitated for 12–15 h (180 rpm, 37°C). Then, the supernatant was discarded, and the firmly adherent cells (mostly astrocytes and fibroblasts) were rinsed with PBS, trypsinized, re-seeded onto poly-l-lysine-coated cover slips, and cultured for an additional 1–2 days in RPMI 1640/10% FCS prior to their treatment. About 75% of all cells in these cultures were astrocytes-based on GFAP staining.
Stimulation of astrocyte-enriched cultures
For the treatment, each cover slip was transferred to a separate well of a 12-well-plate and inverted. Small dots of paraffin served as spacers between the cell layer of the cover slip and the bottom of the 12-well-plate. The astrocytes growing on the cover slip were either left untreated as a control (1), received LPS (100 ng/ml) for 12 h (2), or were co-cultured with microglia cells growing on the bottom of the plate (1:1 ratio of astrocytes:microglia cells) in the absence (3) or presence (4) of LPS (100 ng/ml) for 12 h. Afterwards, the cover slips were washed twice with PBS and used for immunocytochemical analysis.
Immunocytochemical analysis
The differentially treated cells were fixed with 4% PFA for 15 min (room temperature), washed with 0.05 M Tris-buffered saline (TBS) (pH 7.4) and permeabilized with TBS containing 0.5% Triton, 5% bovine serum albumin (BSA, Sigma) and 5% donkey serum (Sigma) for 30 min at room temperature. The cells were then washed twice with TBS and incubated overnight at 4°C with commercial rabbit anti-AQP-4 antibody and mouse anti-GFAP antibody (both diluted 1:100 in TBS/1% donkey serum). Afterwards, the cells were washed three times with TBS and incubated for 1 h at room temperature with donkey anti-rabbit Cy3 (Jackson Immuno Research Laboratories, West Grove, PA, USA) to visualize AQP-4 (red) and donkey anti-mouse Cy2 (Jackson Immuno Research Laboratories) to identify astrocytic GFAP (green) (both diluted 1:100 in TBS/1% donkey serum). The cells were washed with TBS and mounted with gallate/geltol.
Statistical analysis
Quantitative data were analyzed by statistical evaluation using non-parametric tests. Descriptive analysis included mean values and standard error. Owing to sample size and variability of the data, we used non-parametric group tests (Kruskal–Wallis). SPSS 14.0 statistical software system (SPSS Inc., Chicago) was used for calculations.
Results
Astrocyte pathology in T-cell-mediated autoimmune encephalomyelitis amplified by NMO patient-derived circulating AQP-4 antibodies
As described before [7] profound loss of AQP-4 and astrocyte damage is the earliest pathological alteration in the lesions in the central nervous system of animals with T-cell-mediated EAE, which received additional systemic injections of AQP-4 antibody containing immunoglobulin from patients with NMO (Table 1). One day after antibody injection, a profound loss of AQP-4 (Fig. 1b) and GFAP was seen (Fig. 1c) in the absence of demyelination (Fig. 1a). This was associated with a major disturbance of the perivascular glia limitans, as seen in sections stained for α-dystroglycan (ADG) (Fig. 1d), while the vascular basement membranes only showed some splitting in the area of inflammation (Fig. 1n). Astrocyte pathology in these lesions was associated with profound immunoglobulin and complement deposition (Fig. 1h). Three days after injection of AQP-4 antibody containing immunoglobulin in animals with actively induced EAE similar alterations of astrocytes were present (Fig. 1f–h), but at this time point demyelination was observed within the lesions. These data show that in the experimental model for NMO, where the specific antibody response is directed against the astrocytic antigen AQP-4, demyelination follows astrocyte injury. Comparable astrocyte alterations and demyelination were absent in rats with T-cell-mediated EAE, which received control immunoglobulin (Table 3; Fig. 1i–l, o–p).

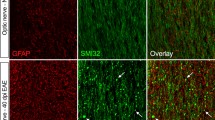
Astrocyte pathology and demyelination in different experimental models of autoimmune encephalomyelitis: immunocytochemistry for the different markers was performed on serial sections of lesions. T-cell-mediated EAE with co-transfer of human immunoglobulin containing aquaporin 4 antibody. 24 h after antibody transfer, there is no demyelination (Luxol fast blue staining a), loss of aquaporin 4 (b), GFAP (c) and α-dystroglycan (d) in the lesions. 72 h after antibody transfer, the loss of aquaporin 4 (f) and GFAP (g) is associated with focal demyelination (Luxol fast blue staining e) and deposition of complement C9neo antigen (h). T-cell-mediated EAE with co-transfer of control immunoglobulin. There is no demyelination (Luxol fast blue staining i), no loss of aquaporin 4 (j) GFAP (k) or α-dystroglycan (l). T-cell-mediated EAE with co-transfer of human immunoglobulin containing aquaporin 4 antibody, 24 h after antibody transfer. m Connexin 43 immunoreactivity is lost in areas of aquaporin 4 loss (see Fig. 1b). n Staining for collagen IV shows some splitting of basement membranes in inflamed vessels. T-cell-mediated EAE with co-transfer of control immunoglobulin; no loss of connexin 43 is seen (o); staining for collagen IV shows some splitting of basement membranes in inflamed vessels (p). T-cell-mediated EAE with co-transfer of anti-MOG antibodies. There is perivascular demyelination (luxol fast blue staining q), but no change in aquaporin 4 expression (r), and no GFAP loss (s). However, connexin 43 immunoreactivity is profoundly reduced within the lesion in comparison with the adjacent normal white matter (t). The insert shows the punctate connexin 43 reactivity associated with the surface of glial cells (right), which is lost in the lesion (left). Bar 100 µm
As an additional control, we used spinal cord sections of animals, in which EAE has been induced with MBP-specific T cells and demyelinating antibodies directed against MOG. In these animals, profound perivascular demyelination was already present 24 h after the antibody injection. However, no astrocyte pathology, comparable to that seen in the experimental NMO model, was present (Fig. 1q–s).
Early loss of AQP-4 in astrocytes in the LPS model
Focal injection of saline into the spinal cord white matter did not induce demyelination or astrocyte injury (Fig. 2a–d). By studying the time course of lesion development in the LPS model, we found loss of AQP-4 immunoreactivity at the injection site already on the first day after LPS injection, and the extent of AQP-4 loss increased with time (Table 3; Fig. 2e–l). In the early stages after LPS injection GFAP-positive astrocytes still appeared normal (Figs. 2g, 3), but retraction of astrocytic processes was already seen (Figs. 2a, 3). The reduction in astrocyte numbers was observed at later stages after LPS injection (Fig. 3k). Similarly, earliest signs of demyelination and nuclear changes of oligodendrocytes, resembling apoptosis were seen at day 5 and the size of demyelinated lesions increased thereafter, reaching a peak 12 days after LPS injection (Fig. 2e, i).

Astrocyte pathology and demyelination in LPS-induced spinal cord lesions: a–d no pathological alterations of astrocytes and no demyelination are seen in animals injected with saline into the spinal cord white matter. 3 days after injection of LPS, there is no demyelination (Luxol fast blue staining e); staining for aquaporin 4 is reduced (f), but astrocytes are still preserved (g). In contrast, there is a profound loss of α-dystroglycan reactivity at the vascular glia limitans (h). At day 12 after LPS injection, a focal plaque of demyelination is visible (Luxol fast blue staining (i), associated with complete loss of aquaporin 4 (j) and α-dystroglycan reactivity (l) and a profound reduction in astrocytes and their processes (k). Connexin 30 immunoreactivity is progressively lost with time in LPS lesions (n–p) in comparison with saline-injected animals (m). q–t Connexin 43 immunoreactivity shows similar changes in comparison with that for connexin 30. u–x LPS lesions show an increasing amount of collagen IV deposited in the spinal cord extracellular space with time. Bar 100 µm

Immunocytochemical double staining for GFAP and other markers visualized by confocal laser microscopy. First column normal white matter, second column day 3 after LPS injection, third column 12 days after LPS injection, lesion edge, fourth column 12 days after LPS injection, lesion center. In comparison with the control white matter, there is a progressive loss of astrocytes and their processes with time after LPS injection. In contrast to the results shown in Fig. 2, confocal microscopy reveals more pronounced changes by day 3 after LPS injection, due to the very thin optical section thickness and the much higher resolution of this technique. Note that in w and x most of the remaining astrocytes show collagen 4 immunoreactivity within their cytoplasm (yellow color), suggesting synthesis of this antigen within astrocytes. a–e, g–j, n–p, u–x Bar 25 µm. f, k, l, m, q–t Bar 10 µm
Loss of AQP-4 is associated with structural disturbance of the glia limitans
AQP-4 is concentrated in astrocytic foot processes at the glia limitans and loss of AQP-4 in experimental NMO lesions occurs concurrently with a retraction or destruction of perivascular astrocytic foot processes at the glia limitans. To analyze whether this is also the case after LPS injection, we used two markers which allow analysis of the structural organization of the glia limitans: ADG and col IV. In the LPS lesions ADG reactive glial end feet were lost in parallel with the loss of AQP-4 (Figs. 2h, i, 3e–l). Col IV is present in the basement membranes of cerebral endothelial cells and glial end feet, but no col IV is seen in the extracellular space of the CNS parenchyma (Fig. 2n). Damage of the perivascular glia limitans in LPS lesions, however, resulted in the presence of col IV in the extracellular matrix of the spinal cord tissue, which was accentuated around the cell bodies of reactive astrocytes (Figs. 2v–x, 3v–x). Furthermore, col IV immunoreactivity was also seen within the cytoplasm of astrocytes, suggesting its synthesis within the cells (Fig. 3w, x). No alterations of ADG or col IV expression were seen in control animals injected with saline (Figs. 2d, u, 3i, u).
Molecules which metabolically connect astrocytes with oligodendrocytes are lost in lesions with both, primary astrocyte or oligodendrocyte damage
Because demyelination in the LPS and the NMO model follows astrocyte dysfunction, we analyzed additional astrocytic molecules which play a role in oligodendrocyte homeostasis. Oligodendrocytes in the CNS are linked to astrocytes by heterotypic gap junctions composed of connexin 32 (Cx32) and connexin 47 (Cx47) in oligodendrocytes, and of connexin 30 (Cx30) and connexin 43 (Cx43) in astrocytes, with Cx32/Cx30 and Cx47/Cx43 as coupling partners [22, 30]. These gap junctions allow the trafficking of metabolites and ions between astrocytes and oligodendrocytes [3, 36]. The expression of both Cx30 and Cx43 was profoundly reduced in both, the LPS lesions (Fig. 3m–t) as well as the experimental NMO lesions (Fig. 1m). The time course of loss of Cx 43 and 30 in the LPS model closely followed that described above for AQP-4. Interestingly, however, a similar loss of Cx 30 and 43 immunoreactivity was also seen in primary demyelinating lesions induced by anti-MOG antibodies (Fig. 1t).
Microglia cells are the primary target of LPS
LPS is recognized by toll-like receptor 4 (TLR4), which acts in concert with the cluster of differentiation 14 (CD14) protein. Astrocytes in vivo express none of these molecules [9]. However, in marked contrast, microglia cells in vivo readily express both TLR4 and CD14 [32]. Therefore, the LPS effects on astrocytes following injections into the dorsal funiculus seemed to be driven by microglia-derived factors. In a next set of experiments, we therefore co-cultured astrocytes with microglial cells with and without LPS treatment, and analyzed astrocytic changes in morphology and AQP-4 expression. Normally, the astrocyte cultures were a mixture of both protoplasmic and stellate cells (Fig. 4a, e). AQP-4 expression was very weak on the stellate cells, and much stronger on the protoplasmic astrocytes. In both cases, AQP-4 reactivity was uniformly distributed over the cell membrane of astrocytes, with some accentuation on membrane ruffles (Fig. 4i, m, q). This picture did not change following the exposure of enriched astrocyte cultures for 24 h with LPS (Fig. 4j, n, r). However, upon co-culture with microglial cells, many astrocytes acquired a reactive phenotype characterized by the retraction of cell processes (Fig. 4c, g). Stellate astrocytes were less frequently observed, and the number of astrocytes with a rounded-up appearance increased. Although AQP-4 reactivity was still distributed over the entire astrocytic cell surface, an aggregation of AQP-4 molecules was readily detectable, which was strongly suggestive for internalization of AQP-4 by endocytosis (Fig. 4k, o, s) [17]. The effect of microglial cells on astrocytes in vitro can readily be explained, since these microglia have an activated phenotype, even in the absence of additional stimuli. However, when LPS was added to the microglia–astrocyte co-cultures, the changes in astrocyte morphology and AQP-4 reactivity became more pronounced: stellate astrocytes were almost completely absent from the cultures, the numbers of reactive and rounded-up astrocytes increased (Fig. 4d, h), and aggregates of AQP-4 reactivity were more frequently detected (Fig. 4p, t).

Consequences of co-culture with activated microglial cells on the morphology of, and the expression of aquaporin 4 in, astrocytes in vitro. a–h Changes in morphology, as revealed by GFAP immunoreactivity (green). Astrocyte cultures incubated without microglial cells in the absence of LPS (a higher magnification in e) and in the presence of 100 ng/ml LPS (b higher magnification in f) contain protoplasmic and stellate astrocytes. Upon incubation with microglial cells, the numbers of astrocytes with large cellular processes decreases and the astrocytes start to round up (c higher magnification in g). This process is even more pronounced upon incubation of astrocytes with microglial cells in the presence of 100 ng/ml LPS (d higher magnification in h). i–t Changes in aquaporin 4 expression. The astrocytes were double stained with GFAP (green i–l, overlay q–t) and AQP-4 (red m–p, overlay q–t). Astrocytes cultured without microglial cells, both in the absence (i, m, q) or presence of 100 ng/ml LPS (j, n, r), reveal a uniform, punctate staining pattern of AQP-4, which is accentuated at membrane ruffles and cellular processes. Astrocytes cultured in the presence of microglial cells (k, o, s) also show punctate AQP-4 immunoreactivity on the cell surface. However, in addition to this, a large number of astrocytes also show vesicle-like AQP-4 aggregates. This is also seen in astrocytes cultured with microglia cells in the presence of 100 ng/ml LPS (l, p, t). The data shown are representative of two independently performed experiments. Bar 100 µm
Astrocyte pathology is seen in lesions of a subset of MS patients with fulminant disease exacerbations
Having established that astrocyte dysfunction and damage may be an important component in the pathogenesis of inflammatory demyelinating lesions triggered by innate immunity, we analyzed whether similar astrocytic changes can also be seen in lesions of MS patients. We screened MS autopsies, which included patients who died from acute MS, or who had relapsing-remitting MS (patterns II and III cases, Table 4 [24]), or secondary progressive or primary progressive MS, and studied the lesions for evidence of disturbance of the astrocytic glia limitans. To do so, we used antibodies to AQP-4, ADG, col IV and GFAP. MS lesions were classified as classical active lesions, slowly expanding lesions or inactive lesions as described before [12]. In the majority of MS lesions, regardless of activity or disease stage, we did not see evidence for a disruption of the perivascular glia limitans (Fig. 5h–m). Only in a subset of active lesions mainly following pattern III demyelination [24], which were present in patients with acute MS or fulminant attacks of chronic MS, loss of perivascular astrocytic foot processes was associated with col IV deposition in the parenchymal extracellular space (Fig. 5a–g). Furthermore, col IV expression was also seen within the cytoplasm of highly reactive astrocytes, and deposition of precipitated col IV was present on the extracellular surface of reactive astrocyte cell bodies and processes, together with massive surface expression of AQP-4 (Fig. 5d, f, g). Split basement membranes, reflecting the widening of the perivascular space due to inflammation, were seen in similar extent in active lesions following pattern II and III demyelination (Table 4).

Astrocyte pathology in multiple sclerosis lesions: a–g Fulminant active lesions of acute multiple sclerosis following pattern III demyelination [21]. Myelin staining reveals an initial lesion (I) with myelin pallor and a more advanced lesion with complete demyelination (DM a). In the area of the initial lesion, there are already macrophages with myelin degradation products within their cytoplasm (b). Within the lesion, there is abundant expression of GFAP, but the perivascular glia limitans is disrupted (c). Staining for aquaporin 4 shows patchy loss of immunoreactivity with numerous highly reactive protoplasmatic astrocytes with intense aquaporin 4 immunoreactivity on their surface (d insert). α-Dystroglycan immunoreactivity is only present on the glia limitans of some vessels and there the intensity of expression is uneven and weak (e). Collagen IV immunoreactivity is seen within the cytoplasm of reactive astrocytes, which have lost their processes forming the glia limitans (f, g). In addition, there is profound precipitation of collagen IV on the surface of reactive astrocytes (g). h–n Astrocytes in a chronic expanding lesion following pattern II demyelination [24]. Edge of an active lesion with a broad rim of macrophages with early myelin degradation products (h, i). In these lesions, profound astrocytic gliosis is seen and the glia limitans is still preserved in GFAP stained lesions (j). Aquaporin 4 is densely expressed on astrocytes between the macrophages, containing myelin debris (k). α-Dystroglycan expression in the normal white matter (l) and within the lesion (m) shows comparable expression patterns at the glia limitans. Collagen IV staining shows profound splitting of the vascular basement membranes within the lesions, but no immunoreactivity in astrocytes and no collagen deposition within the parenchyma (n). Bar 100 µm; f bar 50 µm
Discussion
Our current study shows that astrocyte dysfunction precedes demyelination in two experimental models of inflammatory demyelination with fundamentally different pathogenesis. In NMO lesions specific autoantibodies target the astrocytic membrane protein AQP-4 and destroy astrocytes through either activated complement, through a mechanism of antibody-dependent cellular cytotoxicity, or a mixture of both mechanisms [6, 7]. In the LPS model, autoantibodies against AQP-4 do not play a role. Autoantibodies against AQP-4 have not been seen in normal rats [7] and the time between LPS injection and the appearance of AQP-4 loss within the lesions (1 day) is too short for the induction of an autoantibody response. Furthermore, we did not find immunoglobulin and complement deposition in the lesions. In contrast, we saw that supernatants of LPS-activated microglia can induce astrocyte dystrophy in vitro. It has been described that LPS-activated microglial cells produce a large number of molecules, among them interleukin-1, tumor necrosis factor-alpha, and nitric oxide, which can impair astrocyte function in vitro [33]. Despite the completely different modes of the induction of astrocyte dystrophy, the consequences for astrocyte function and lesion formation in vivo are essentially similar. In both conditions, astrocytic foot processes at the perivascular glia limitans are first and most severely affected. This is reflected by the loss of ADG, which is an important anchor protein, linking the astrocyte processes with the molecules of the extracellular matrix of the perivascular space, and which is also involved in AQP-4 clustering at the glia limitans. It is well established that ADG is selectively cleaved at the parenchymal basement membrane at sites of leukocyte extravasation in EAE [2]. However, in simple EAE models, this cleavage does not seem to translate to a complete loss of ADG reactivity in the lesions. Col IV, too, is at least in part a marker for the intactness of the perivascular glia limitans. It is also produced by astrocytes and under normal conditions deposited in a polarized manner on the astrocytic surface of the glia limitans. We show here that col IV is expressed in the cytoplasm of reactive astrocytes and excreted into the parenchymal extracellular space of the CNS tissue, when the structure of the glia limitans is impaired. In addition, it decorates the surface of reactive astrocytes within the lesions, which may further disturb astrocyte function.
The time course of events seen in our experiments suggests that in both the NMO and the LPS lesions demyelination may be a secondary consequence of astrocytic dysfunction. This may reflect several different underlying mechanisms. Astrocyte dysfunction may impair blood–brain barrier function, leading to perivascular precipitation of fibrin. Precipitated fibrin is present at the onset of demyelination in the LPS lesions [26] and is known to be an additional trigger for microglia activation in inflammatory conditions [1]. In addition, we have tested the expression of molecules that may in part be involved in oligodendrocyte injury. The Cx30 and Cx43 form heteromeric gap junctions between astrocytes and oligodendrocytes [30], and are thought to play a vital role for the delivery, removal and general flow of ions, osmotic water and metabolites that sustain the normal functioning of oligodendrocytes [16]. When this connection is interrupted, oligodendrocytes may undergo a state of energy deprivation. A previously reported alternative candidate for oligodendrocyte injury is the excitatory amino acid transporter EAAT2 [38 15]. It is expressed in astrocytes and it is involved in the clearance of glutamate from the extracellular space and, thus, it contributes to extracellular glutamate homeostasis. Because oligodendrocytes are sensitive to glutamate receptor-mediated injury [27], decreased extracellular glutamate buffering may be involved in oligodendrocyte degeneration and demyelination. It has been concluded previously from in vitro data that this mechanism may be important for the pathogenesis of demyelination in NMO.
The link between astrocyte injury and demyelination has also been observed in earlier studies. For example, ablation of astrocytes lead to severe demyelination and neuronal as well as oligodendrocyte death after spinal cord injury [10], and to an increased spread of inflammation and tissue damage in EAE [37]. Similar results were obtained when signal transducer and activator of transcription 3 (STAT3)-positive astrocytes were selectively killed [14]. Moreover, depletion of GFAP-positive astrocytes after forebrain stab injury lead to substantial neuronal degeneration that could be attenuated by chronic glutamate receptor blockade [8]; and finally, a primary loss of astrocytes followed by extensive demyelination was also observed after injection of human complement and NMO IgG into the murine cerebrum [35].
Although the profound structural impairment of astrocytes is well documented in the lesions of patients with NMO, potential pathological alterations of astrocytes in MS lesions have not received major attention. Reactive protoplasmatic gliosis and fibrillar gliosis have been well described in active and chronic MS lesions [19]. Here, we show that at least in a subset of MS lesions astrocytic changes are present which bear similarities to those in NMO and LPS lesions. However, there is a major difference between MS and NMO lesions. In NMO lesions, astrocytes are widely destroyed leading to large areas devoid of astrocytes [28]. In contrast in MS astrocyte pathology is mainly affecting the cell processes and destruction or loss of astrocytes is minor. The functional significance of these changes is currently unknown, but in analogy to NMO one may speculate that they too may be involved in the pathogenesis of myelin and oligodendrocyte damage. Interestingly, those fulminant pattern III lesions, where we predominantly found astrocyte dystrophy in our study, show a rather low extent of T-cell infiltration [4, 13, 26] and in many respects are similar to the lesions induced by focal LPS injection into the spinal cord [11, 26]. It was thus suggested before that in these lesions innate immunity may play a prominent pathogenetic role.
Finally, our current observations have consequences for diagnostic neuropathology. Diagnosis of NMO is based on the detection of circulating AQP-4 autoantibodies [20, 21]. However, there is a subset of patients, which is variable in different cohorts and tests negative for AQP-4. We show here an alternative mechanism of astrocytic injury and AQP-4 loss, which does not depend on the presence of AQP-4 autoantibodies. In addition, AQP-4 autoantibody titers are frequently unavailable, when neuropathologists are confronted with an autopsy or biopsy specimen with suspected NMO. Currently, immunocytochemical detection of AQP-4 loss in the lesions is regarded as a very good diagnostic marker for NMO lesions. In the light of our current results, this assumption has to be qualified. AQP-4 loss can be seen in lesions with a completely different immunopathogenesis than AQP-4 antibody-driven NMO. However, in contrast to NMO lesions [28, 34], this AQP-4 loss is patchy and associated with profound overexpression of this molecule on the cell surface of reactive astrocytes. In addition, complete loss and destruction of astrocytes is minor as compared to that seen in NMO lesions [25, 28, 34] and specific deposition of immunoglobulin and complement at the site of astrocyte damage is absent.
References
Adams RA, Bauer J, Flick MJ, Sikorski SL, Nuriel T, Lassmann H, Degen JL, Akassoglou K (2007) The fibrin-derived gamma377–395 peptide inhibits microglia activation and suppresses relapsing paralysis in central nervous system autoimmune disease. J Exp Med 204:571–582
Agrawal S, Anderson P, Durbeej M, van Rooijen N, Ivars F, Opdenakker G, Sorokin LM (2006) Dystroglycan is selectively cleaved at the parenchymal basement membrane at sites of leukocyte extravasation in experimental autoimmune encephalomyelitis. J Exp Med 203:1007–1019
Ball KK, Gandhi GK, Thrash J, Cruz NF, Dienel GA (2007) Astrocytic connexin distributions and rapid, extensive dye transfer via gap junctions in the inferior colliculus: implications for [(14)C]glucose metabolite trafficking. J Neurosci Res 85:3267–3283
Barnett MH, Prineas JW (2004) Relapsing and remitting multiple sclerosis: pathology of the newly forming lesion. Ann Neurol 55:458–468. doi:10.1002/ana.20016
Ben-Nun A, Cohen IR (1981) Vaccination against autoimmune encephalomyelitis (EAE): attenuated autoimmune T lymphocytes confer resistance to induction of active EAE but not to EAE mediated by the intact T lymphocyte line. Eur J Immunol 11:949–952
Bennett JL, Lam C, Kalluri SR, Saikali P, Bautista K, Dupree C, Glogowska M, Case D, Antel JP, Owens GP, Gilden D, Nessler S, Stadelmann C, Hemmer B (2009) Intrathecal pathogenic anti-aquaporin-4 antibodies in early neuromyelitis optica. Ann Neurol 66:617–629
Bradl M, Misu T, Takahashi T, Watanabe M, Mader S, Reindl M, Adzemovic M, Bauer J, Berger T, Fujihara K, Itoyama Y, Lassmann H (2009) Neuromyelitis optica: pathogenicity of patient immunoglobulin in vivo. Ann Neurol 66:630–643
Bush TG, Puvanachandra N, Horner CH, Polito A, Ostenfeld T, Svendsen CN, Mucke L, Johnson MH, Sofroniew MV (1999) Leukocyte infiltration, neuronal degeneration, and neurite outgrowth after ablation of scar-forming, reactive astrocytes in adult transgenic mice. Neuron 23:297–308
Farina C, Aloisi F, Meinl E (2007) Astrocytes are active players in cerebral innate immunity. Trends Immunol 28:138–145
Faukner JR, Herrmann JE, Woo MJ, Tansey KE, Doan NB, Sofroniew MV (2004) Reactive astrocytes protect tissue and preserve function after spinal cord injury. J Neurosci 24:2143–2155
Felts PA, Woolston AM, Fernando HB, Asquith S, Gregson NA, Mizzi OJ, Smith KJ (2005) Inflammation and primary demyelination induced by the intraspinal injection of lipopolysaccharide. Brain 128:1649–1666
Frischer JM, Bramow S, Dal-Bianco A, Lucchinetti CF, Rauschka H, Schmidbauer M, Laursen H, Sorensen PS, Lassmann H (2009) The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain 132:1175–1189
Henderson AP, Barnett MH, Parratt JD, Prineas JW (2009) Multiple sclerosis: distribution of inflammatory cells in newly forming lesions. Ann Neurol 66:739–753
Herrmann JE, Imura T, Song B, Qi J, Ao Y, Nguyen TK, Korsak RA, Takeda K, Akira S, Sofroniew MV (2008) STAT3 is a critical regulator of astrogliosis and scar formation after spinal cord injury. J Neurosci 28:7231–7243
Hinson SR, Roemer SF, Lucchinetti CF, Fryer JP, Chamberlain JL, Howe CL, Pittock SJ, Lennon VA (2008) Aquaporin-4-binding autoantibodies in patients with neuromyelitis optica impair glutamate transport by down-regulating EAAT2. J Exp Med 205:2473–2481
Kamasawa N, Sik A, Morita M, Yasumura T, Davidson KG, Nagy JI, Rash JE (2005) Connexin-47 and connexin-32 in gap junctions of oligodendrocyte somata, myelin sheaths, paranodal loops and Schmidt–Lanterman incisures: implications for ionic homeostasis and potassium siphoning. Neuroscience 136:65–86
Kamsteeg EJ, Hendriks G, Boone M, Konings IB, Oorschot V, van der Sluijs P, Klumperman J, Deen PM (2006) Short-chain ubiquitination mediates the regulated endocytosis of the aquaporin-2 water channel. Proc Natl Acad Sci USA 103:18344–18349
Kutzelnigg A, Lucchinetti CF, Stadelmann C, Bruck W, Rauschka H, Bergmann M, Schmidbauer M, Parisi JE, Lassmann H (2005) Cortical demyelination and diffuse white matter injury in multiple sclerosis. Brain 128:2705–2712
Lassmann H, Bruck W, Lucchinetti CF (2007) The immunopathology of multiple sclerosis: an overview. Brain Pathol 17:210–218
Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SR (2005) IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med 202:473–477
Lennon VA, Wingerchuk DM, Kryzer TJ, Pittock SJ, Lucchinetti CF, Fujihara K, Nakashima I, Weinshenker BG (2004) A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet 364:2106–2112
Li X, Penes M, Odermatt B, Willecke K, Nagy JI (2008) Ablation of Cx47 in transgenic mice leads to the loss of MUPP1, ZONAB and multiple connexins at oligodendrocyte-astrocyte gap junctions. Eur J Neurosci 28:1503–1517
Linington C, Bradl M, Lassmann H, Brunner C, Vass K (1988) Augmentation of demyelination in rat acute allergic encephalomyelitis by circulating mouse monoclonal antibodies directed against a myelin/oligodendrocyte glycoprotein. Am J Pathol 130:443–454
Lucchinetti C, Bruck W, Parisi J, Scheithauer B, Rodriguez M, Lassmann H (2000) Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol 47:707–717
Lucchinetti CF, Mandler RN, McGavern D, Bruck W, Gleich G, Ransohoff RM, Trebst C, Weinshenker B, Wingerchuk D, Parisi JE, Lassmann H (2002) A role for humoral mechanisms in the pathogenesis of Devic’s neuromyelitis optica. Brain 125:1450–1461
Marik C, Felts PA, Bauer J, Lassmann H, Smith KJ (2007) Lesion genesis in a subset of patients with multiple sclerosis: a role for innate immunity? Brain 130:2800–2815
Matute C (2007) Interaction between glutamate signalling and immune attack in damaging oligodendrocytes. Neuron Glia Biol 3:281–285
Misu T, Fujihara K, Kakita A, Konno H, Nakamura M, Watanabe S, Takahashi T, Nakashima I, Takahashi H, Itoyama Y (2007) Loss of aquaporin 4 in lesions of neuromyelitis optica: distinction from multiple sclerosis. Brain 130:1224–1234
Noseworthy JH, Lucchinetti C, Rodriguez M, Weinshenker BG (2000) Multiple sclerosis. N Engl J Med 343:938–952
Orthmann-Murphy JL, Freidin M, Fischer E, Scherer SS, Abrams CK (2007) Two distinct heterotypic channels mediate gap junction coupling between astrocyte and oligodendrocyte connexins. J Neurosci 27:13949–13957
Piddlesden SJ, Lassmann H, Zimprich F, Morgan BP, Linington C (1993) The demyelinating potential of antibodies to myelin oligodendrocyte glycoprotein is related to their ability to fix complement. Am J Pathol 143:555–564
Reed-Geaghan EG, Savage JC, Hise AG, Landreth GE (2009) CD14 and toll-like receptors 2 and 4 are required for fibrillar A{beta}-stimulated microglial activation. J Neurosci 29:11982–11992
Retamal MA, Froger N, Palacios-Prado N, Ezan P, Saez PJ, Saez JC, Giaume C (2007) Cx43 hemichannels and gap junction channels in astrocytes are regulated oppositely by proinflammatory cytokines released from activated microglia. J Neurosci 27:13781–13792
Roemer SF, Parisi JE, Lennon VA, Benarroch EE, Lassmann H, Bruck W, Mandler RN, Weinshenker BG, Pittock SJ, Wingerchuk DM, Lucchinetti CF (2007) Pattern-specific loss of aquaporin-4 immunoreactivity distinguishes neuromyelitis optica from multiple sclerosis. Brain 130:1194–1205
Saadoun S, Waters P, Bell BA, Vincent A, Verkman AS, Papdopoulos MC (2010) Intra-cerebral injection of neuromyelitis optica immunoglobulin G and human complement produces neuromyelitis optica lesions in mice. Brain 133:349–361
Sánchez-Abarca LI, Tabernero A, Medina JM (2001) Oligodendrocytes use lactate as a source of energy and as a precursor of lipids. Glia 36:321–329
Voskuhl RR, Peterson RS, Song B, Ao Y, Morales LB, Tiwari-Woodruff S, Sofroniew MV (2009) Reactive astrocytes form scar-like perivascular barriers to leukocytes during adaptive immune inflammation of the CNS. J Neurosci 29:11511–11522
Zeng XN, Sun XL, Gao L, Fan Y, Ding JH, Hu G (2007) Aquaporin-4 deficiency down-regulates glutamate uptake and GLT-1 expression in astrocytes. Mol Cell Neurosci 34:34–39
Acknowledgments
We thank Ulrike Köck, Angela Kury and Marianne Leisser for expert technical assistance. This study was funded by the PhD programme Cell Communication in Health and Disease (CCHD, co-funded by the Austrian Science Fund and the Medical University Vienna), by the Austrian Science Fund (FWF, Project P19854-B02 to HL and P21581-B09 to MB), by the European Union (LSHM-CT-2005-018637), by the UK MRC to KJS, the Multiple Sclerosis Society of GB and NI to KJS, and the Brain Research Trust to KJS.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Sharma, R., Fischer, MT., Bauer, J. et al. Inflammation induced by innate immunity in the central nervous system leads to primary astrocyte dysfunction followed by demyelination. Acta Neuropathol 120, 223–236 (2010). https://doi.org/10.1007/s00401-010-0704-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-010-0704-z