
Abstract
Six subtypes of sporadic Creutzfeldt–Jakob disease with distinctive clinico-pathological features have been identified largely based on two types of the abnormal prion protein, PrPSc, and the methionine (M)/valine (V) polymorphic codon 129 of the prion protein. The existence of affected subjects showing mixed phenotypic features and concurrent PrPSc types has been reported but with inconsistencies among studies in both results and their interpretation. The issue currently complicates diagnosis and classification of cases and also has implications for disease pathogenesis. To explore the issue in depth, we carried out a systematic regional study in a large series of 225 cases. PrPSc types 1 and 2 concurrence was detected in 35% of cases and was higher in MM than in MV or VV subjects. The deposition of either type 1 or 2, when concurrent, was not random and always characterized by the coexistence of phenotypic features previously described in the pure subtypes. PrPSc type 1 accumulation and related pathology predominated in MM and MV cases, while the type 2 phenotype prevailed in VVs. Neuropathological examination best identified the mixed types 1 and 2 features in MMs and most MVs, and also uniquely revealed the co-occurrence of pathological variants sharing PrPSc type 2. In contrast, molecular typing best detected the concurrent PrPSc types in VV subjects and MV cases with kuru plaques. The present data provide an updated disease classification and are of importance for future epidemiologic and transmission studies aimed to identify etiology and extent of strain variation in sporadic Creutzfeldt–Jakob disease.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Transmissible spongiform encephalopathies (TSEs) or prion diseases are invariably fatal neurodegenerative disorders affecting humans and other mammals such as sheep, elk, and cattle [36]. These diseases are characterized clinically by a rapidly progressive neurological dysfunction and neuropathologically by spongiform degeneration, neuronal loss, gliosis, and sometimes by amyloid plaques, which combine in varying degrees of severity and regional distribution.
Biochemically, prion diseases are characterized by the conversion of a constitutively expressed cellular protein, the prion protein (PrPC) into an abnormally folded, insoluble, partially protease-resistant isoform (PrPSc) [4, 8, 27]. Incubation of a brain homogenate from prion-infected individuals with proteinase K (PK) under conditions leading to a complete degradation of PrPC generates an N-terminal truncated form of PrPSc, commonly referred to as PrP27-30, an established molecular marker for the disease [5, 38].
Sporadic Creutzfeldt–Jakob disease (sCJD), the commonest form of the human prion diseases, comprises a broad spectrum of clinico-pathological variants [30]. Prion strains are believed to be the main cause of TSE phenotypic diversity. Prion strains are defined as isolates that show distinct disease phenotypes upon transmission to syngenic animals, persisting on serial transmission [6, 15, 34]. In addition, the host genotype variability in the gene encoding PrPC (PRNP), as determined by polymorphisms and mutations, is another recognized causal factor for phenotypic heterogeneity [2, 12]. The hypothesis that PrPSc itself may encode strain-specific properties has gained support over the years by the demonstration of distinct physico-chemical subtypes of PrPSc that include differences in the level of glycosylation and in the size of proteinase K-digested PrPSc fragments, possibly reflecting distinct protein conformations [1, 3, 9, 10, 14, 22, 28, 29, 32, 39, 41, 42]. Although the final proof that conformational variants of PrPSc represent the biological basis of prion strains is lacking, the described physicochemical differences among PrPSc types are increasingly used as surrogate markers of strain properties in a given host. Based on differences in gel mobility and N-terminal sequence of the core fragments (i.e. PrP27-30) generated by PK digestion, Parchi et al. [28, 29, 32] originally identified two major human PrPSc types: type 1 with a relative molecular mass of 21 kDa and the primary cleavage site at residue 82, and type 2 with relative molecular mass of 19 kDa and the primary cleavage site at residue 97. The two PrPSc types, in conjunction with the codon 129 genotype, significantly explained sCJD phenotypic variability and provided a molecular basis for disease classification [7, 28, 30]. Six subtypes of sCJD with distinctive phenotypic features were identified, each resulting from a specific codon 129 genotype/PrPSc type combination with two exceptions. MM1 and MV1 cases were phenotypically indistinguishable and, therefore, merged in one subtype (MM/MV 1), whereas, on the contrary, MM2 subjects were associated with 2 subtypes with distinctive histopathological features in the cerebral cortex and in the thalamus and designated accordingly (MM2-cortical or MM 2C and MM2-thalamic or MM 2T) [30]. By showing that at least some of these sCJD subtypes correspond to specific agent strain-host genotype combinations, preliminary transmission studies seem to confirm the biological basis of this classification [16, 17, 23, 42].
An important aspect of sCJD phenotypic variability, which has not yet been investigated thoroughly, concerns the existence of cases presenting more than one PrPSc type and mixed histopathological features. This issue still represents a potential drawback for the widespread application of the current disease classification for diagnostic and epidemiologic purposes. Since the original report of this phenomenon [30], based on the analyses of a few samples for each brain, the co-occurrence of PrPSc type 1 and 2 in a percentage varying between 12 and 44% of sCJD cases analyzed has been confirmed by a number of subsequent studies [13, 18, 20, 37, 40, 43]. In many of them, the question of how far the proportion of cases with concurrent PrPSc types is a function of the sampling protocol employed was raised. More recently, two studies using novel antibodies recognizing an epitope between residues 82 and 96 (i.e. not detecting type 2) found that all type 2 samples show at least some associated protein migrating in the 21 kDa molecular weight range and concluded that PrPSc type 1 coexists in all type 2 sCJD cases and that the number of cases with concurrent PrPSc types is likely larger than previously thought [35, 45]. It was later shown, however, that the “type 1 selective” antibodies fail to discriminate properly between a bona fide PK-resistant PrPSc core and the partially cleaved fragments generated during digestion due to the several PK cleavage sites included in the PrPSc N-terminus, making difficult the interpretation of the results obtained [25].
To explore the issue of concurrent PrPSc types in sCJD in depth, we carried out a systematic regional study in a large series of cases. To this aim, we applied a refined western blot assay with increased sensitivity for the detection of PrPSc types coexistence [25] and correlated the molecular data with those obtained by clinical and pathological examinations.
Materials and methods
Case and tissue selection
This study was based on 225 cases with an established diagnosis of sCJD obtained after clinical, neuropathological, and molecular examination. Cases of genetic and acquired forms of CJD were excluded. The study was restricted to cases from which large amounts of both frozen and fixed brain tissue were available. 200 patients were obtained from a group of ~240 consecutive cases referred for diagnosis (~40 cases lacked sufficient frozen tissue), whereas the remaining 25 were specifically chosen to increase the number of cases with mixed phenotypes. More precisely, the 25 additional cases were selected based on the demonstration of a mixed synaptic and perivacuolar pattern of PrP deposition by PrP immunohistochemistry (see “Results”). 61 patients died in the USA between 1990 and 2001, and 164 in Europe (124 in Italy, 34 in Germany, and 6 in Belgium) between 1993 and 2007.
Brains were removed at autopsy and either one half or selected coronal sections of tissue, including all major brain structures and nuclei, were immediately frozen and stored at −80°C. The remaining tissue was fixed in formalin and was used for neuropathological examination and PrP immunohistochemistry. Samples of frozen gray matter (between 50 and 100 mg) for protein analysis were obtained from the following regions: frontal (superior and middle frontal gyri), temporal (superior and middle temporal gyri), parietal (inferior parietal lobule), and occipital (calcarine cortex and lateral occipital gyrus) cortices, hippocampus (Ammon’s horn), limbic cortices (entorhinal cortex anterior, insular cortex, and cingulate gyrus), striatum (caudate and putamen nuclei), thalamus (medial and lateral nuclei), hypothalamus, brainstem (midbrain periaqueductal gray, pontine periaqueductal gray including locus coeruleus, medullar periventricular gray), cerebellum (hemisphere and vermis). The parietal cortex and the 3 samples from the brainstem were not available in 30 cases from Germany. Furthermore, 1 or 2 samples were occasionally lacking in some other cases [altogether 34 samples, mostly from the hypothalamus (n = 15) and amygdala (n = 11)]. Sampling of frozen tissues was performed by the same investigator (PP) according to a defined protocol across the whole series of cases. Blocks of fixed tissue were taken from the opposite half of the brain and were used for histopathologic examination. Sampling of fixed tissues was performed by the same investigator (PP) according to a defined protocol in 160 cases. The remaining 65 cases were sampled in Munich (Germany) and Indianapolis (USA). In these Centers, which are both involved in CJD National surveillance and Brain banking, CJD brains are sampled extensively according to protocols, which included all the areas that were of interest for this study. Sampling of both fixed and frozen tissue was performed twice in a subgroup of 10 codon 129 MM subjects.
Protein analysis
Preparation of samples, Western blotting (WB), and PrPSc typing were performed according to established methods [25, 26, 33]. In particular, for the sensitive detection of PrPSc types 1 and 2 concurrence, we followed the method of Notari et al. [25]. All samples were homogenized in lysis buffer plus at pH 6.9, digested with proteinase K (PK) (Roche Diagnostics, specific activity by certificate of analysis: 47.9 U/mg) at a final concentration of 10 U/mL and run in a 6.5 cm long separating gel. In addition, selected samples from MV2 cases with kuru plaques (MV 2K) and MM or MV subjects showing mixed pathological features but only PrPSc type 1 after the run in the 6.5 cm long gel were also run in a 15 cm long gel. To this aim, at least one sample from each cortical lobe was re-analyzed. All immunoblots were probed with the monoclonal antibody 3F4 with epitope at PrP residues 108-111 at 0.1 μg/ml concentration (Signet Labs, MA, USA). In addition, immunoblots were probed with the monoclonal antibody 1E4 with epitope mapped at PrP residues 98–100 at 2 μg/ml concentration (Sanquin Reagents, Amsterdam, The Netherlands) in the following cases: (a) all samples from VV1 cases; (b) 20 neocortical samples from 10 MM1 cases with pure synaptic PrP deposition, and (c) all samples from 30 MM or MV cases, which showed a mixed pattern of PrP deposition by immunohistochemistry and either no evidence or a very scanty and focal (1 or 2 areas) type 2 accumulation after immunoblotting with the 3F4 antibody. Finally, selected blots from MV 2K samples were also probed with SAF60 against PrP residues 157–161, at 0.4 μg/ml.
We quantitatively calculated the ratio between type 1 and type 2 in the non-glycosylated PrPSc after densitometric analyses of immunoblots probed with 3F4 using the AIDA Image Analyzer software (RayTest). This choice was in part driven by our experience indicating that deglycosylation is associated to a loss of definition in the type 1 (21 kDa) and type 2 (19 kDa) bands which makes their distinction and relative quantitation less reliable. Furthermore, given that it has been previously shown [28, 30] that the relative abundance of glycosylated versus non-glycosylated PrPSc shows only relatively minor differences among the sCJD subtypes associated to either PrPSc type 1 or PrPSc type 2, we assumed that the analyses of the unglycosylated form would be representative of the whole protein. We tested this hypothesis by comparing densitometric analyses of non-glycosylated glycoforms versus deglycosylated preparations from representative regions of MM cases with either a dominant type 1 or a dominant type 2. Overall, we found that, compared to the latter, the first approach is associated to a relatively minor overestimation of type 2 protein. In particular, the difference was almost negligible for the group of samples with dominant type 1 (69/31% on non-glycosylated isoforms vs. 72/28% after deglycosylation, n = 20), and slightly more evident in the group of samples with dominant type 2 (29/71% on non-glycosylated isoforms vs. 37/63% after deglycosylation, n = 30).
To express quantitatively with a single number the relative amount of type 1 and type 2 in a given case (total type 1 or type 2 “load”) we calculated the mean of the relative amounts of either type 1 or type 2, expressed in % of total PrPSc signal, in the different brain regions examined.
Molecular genetic analysis
Genomic DNA from all subjects was used to amplify the coding region of PRNP in the polymerase chain reaction (PCR). PRNP open reading frame was amplified as previously described [11]. The PCR product was visualized on a 1% agarose gel to detect potential insertion mutations or deletions. Potential point mutations were revealed by dHPLC analyses. Mutations were also ruled out by direct sequencing of PRNP open reading frame in about 50% of cases. Finally, the codon 129 genotype was examined by digestion with the restriction endonuclease Nsp 1.
Neuropathology
Semiquantitative evaluation of spongiosis and gliosis was carried out in all cases by comparing hematoxylin and eosin stained sections obtained from the same 21 brain regions evaluated biochemically. Spongiosis was rated on a 0–4 scale (not detectable, mild, moderate, severe, and status spongiosis), whereas gliosis was scored on a 0–3 scale (not detectable, mild, moderate, and severe). A lesion profile for each patient was obtained by averaging the two scores.
For PrP immunohistochemistry, paraffin sections from formalin-fixed and formic acid treated blocks of the areas selected for neuropathologic examination were processed using the monoclonal antibodies 3F4, according to published protocols [30]. PrP deposits were described as diffuse or synaptic, perivacuolar, coarse or patchy and plaque-like.
Clinical analysis
Clinical data were available in most cases, and medical records always included at least one neurological examination. Duration of symptoms was calculated from the time of presentation of neurological signs suggesting an organic cause. Prodromal symptoms were not taken into account. Clinical signs were classified “at onset” when observed within the first quarter of the mean duration of symptoms of the group to which the patient belonged (i.e. 1 month if the mean duration was 4).
Results
PrPSc detection and typing
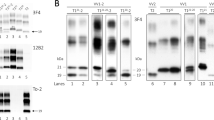
In the present study, we tried to maximize the detection sensitivity of PrPSc type co-occurrence in sCJD-affected brains. At variance with previous studies, we typed PrPSc after a detailed topographic analysis of 21 brain regions and using the method recently described by Notari et al. [25], which has shown increased sensitivity for the detection of bona-fide concurrence of PrPSc types 1 and 2. Furthermore, we probed selected immunoblots with 1E4, a recently characterized monoclonal antibody with a higher affinity for type 2 than for type 1 [44]. The latter approach allowed us an increased protein loading in the gel and a better identification of PrPSc type 2 when present in relatively low amounts (Fig. 1a) in the protein types mixture.

a mAb 1E4 shows a relative higher affinity for PrPSc type 2 than for PrPSc type 1. Western blot profiles of PrPSc from 3 sCJD MM subjects showing various relative proportions of PrPSc type 1 and type 2. Each sample has been loaded twice at the same concentration and probed either with 3F4 (left) and 1E4 (right). Lane 1 sample with dominant type 1 and only 3–5% of type 2; Lane 2 sample with 60/40% type1/type2 ratio; Lane 3 sample with pure type 2. b Western blot profiles of PrPSc (3F4 antibody) from sCJD subjects with concurrent PrPSc types 1 and 2 carrying different codon 129 PRNP genotypes (lanes 2–4). PrPSc type 1 from a MM1 case (lane 1), and PrPSc type 2 from a VV2 case (lane 6) are included as controls. The PrPSc profile associated with the sCJD MV2 phenotype with kuru plaques (sCJD MV2K) is shown in lane 5. The upper band of the PrPSc doublet in lane 5 migrates faster than the type 1 band. Frontal cortex homogenates were treated with PK and probed with 3F4. Approximate molecular masses are in kilodaltons
The results obtained in the largely unselected series of 200 cases after using both 3F4 and 1E4 antibodies are shown in Table 1. The overall prevalence of PrPSc types 1 and 2 concurrence was 35% (32% when only the results with 3F4 were considered). The same incidence of mixed cases was also found in the two largest groups of subjects from Italy and USA when considered individually. Although the protein mixture was detected in each codon 129 genotype (Fig. 1b), PrPSc types 1 and 2 co-occurred more frequently in MM than in MV or VV subjects. A PrPSc profile comprising two distinct bands also characterized the MV2 subtype with kuru plaques (MV 2K) in many brain regions, but the relative size of the associated band (i.e. 20 kDa) distinguished these profiles from bona-fide types 1 and 2 concurrence (Fig. 1b), as previously shown [24, 25].
Regional variation and relative “load” of PrPSc Types 1 and 2 concurrence
MM subjects
The regional distribution of co-existing PrPSc types 1 and 2 in MM subjects reproduced features previously observed in the pure MM1 and MM2-cortical (MM 2C) sCJD subtypes [30], although quantitatively the relative proportion of type 1 and type 2 accumulation in these cases varied extensively. Most cases had PrPSc type 1 in all areas and PrPSc type 2 detectable only focally in the cerebral neocortex or thalamus (Fig. 2a), whereas only a small group of subjects showed a predominant PrPSc type 2 “load” (Fig. 2b). The two groups will be indicated as MM 1+2C and MM 2C+1 throughout the manuscript, MM 2C+1 being defined as a case in which the mean total amount of type 2 (total type 2 “load”) expressed in % of total PrPSc signal and calculated by averaging the results obtained from all brain regions examined, is higher than that of type 1 (i.e. >51%).

Western blot profiles of PrPSc (3F4 antibody) from different brain regions of a one sCJD MM 1+2C subject showing the type 2 protein only focally in the cerebral cortex (arrowheads) and b one sCJD MM 2C+1 subject showing a widespread PrPSc type 2 accumulation, a focal deposition of the type 1 protein (arrowheads) in the cerebral cortex, putamen, and thalamus, and a dominant type 1 in the cerebellum. Areas shown include the middle frontal gyrus (MFG), middle temporal gyrus (MTG), parietal cortex (PC), lateral occipital cortex (OCC2), insular cortex (INS), putamen nucleus (PUT), medial thalamic nuclei (TH1), and cerebellum (CE). Approximate molecular masses are in kilodaltons
The detailed regional PrPSc typing in the MM 1+2C group provided the following results. When 3F4 was used, 74% of cases showed type 2 in 7 or fewer of the 21 regions analyzed, and 42% of them in only 1 or 2 (Fig. 3). In the latter group, 3F4 detected the PrPSc types mixture only in the cerebral cortex or thalamus in 37 of the 504 samples analyzed (Fig. 3). However, when 1E4 was used, the number of mixed samples in the cerebral cortex or thalamus raised about 2.6 times to 98 (data not shown). Noteworthy, the number of cases showing type 2 in only one or two areas dropped from 24 to 8. Furthermore, 1E4 detected PrPSc type 2 in 6 out of 7 subjects in which 3F4 failed to demonstrate the type 2 protein despite its presence was strongly suggested by the results of PrP immunohistochemistry (see below). Finally, in the remaining samples from subcortical areas, 1E4 confirmed the results obtained with 3F4 and failed to reveal a significant type 2 accumulation.

Prevalence and regional distribution of PrPSc type 2 in a series of 57 MM 1+2C subjects. PrPSc type 1 was detected in all areas. Subjects are divided in 3 groups based on the number of areas with a detectable PrPSc type 2 signal on WB. For each group, the % of type 2 positive samples in each brain region is shown. The mean total type 2 “load” for each group, as defined in “Materials and methods”, is given in brackets. FC frontal cortex, TC temporal cortex, PC parietal cortex, OCC occipital cortex, Limbic CTX limbic cortices, Hipp + AMG hippocampus+amygdala, STR striatum, HYP hypothalamus, TH thalamus, BRST brainstem, CE cerebellum
In the group of MM 1+2C with type 2 in up to 7 brain regions (i.e. 74% of cases), the amount of PrPSc type 1 was higher than that of type 2 in 84% of the mixed samples, while type 2 had a stronger signal than type 1 in the remaining 16%. The mean total type 2 “load” in this group, calculated on immunoblots probed with 3F4, was 3.4% (range 0.6–14%).
The remaining 26% of MM 1+2C cases showed a more widespread PrPSc type 2 accumulation involving from 10 to 16 different brain regions. In this group, PrPSc type 1 was predominant over type 2 in 43% of the mixed samples, while type 2 had a stronger signal than type 1 in the remaining 57%. The mean total type 2 “load”, again calculated on immunoblots probed with 3F4 raised to 30% (range 18–38%).
The study of the regional protein distribution in MM 1+2C subjects showed that PrPSc type 2 was overall more frequently seen in the cerebral cortex than in subcortical areas (Fig. 3). The temporal, parietal, and occipital neocortices were the cortical areas which preferentially accumulated the type 2 protein (Fig. 3). Among subcortical areas, the thalamus was by far the most frequent site of type 2 accumulation, whereas the brainstem and, to a greater extent, the cerebellum (Fig. 3) were the regions with the lowest frequency (1 and 0 cases, respectively) of type 2 detection.
At variance with MM 1+2C cases, in MM 2C+1 subjects type 2 was the only detectable protein species in some areas (Fig. 2b, Table 2) and its amount was higher than that of type 1 also in most areas where the two proteins coexisted (Table 2).
MV subjects
PrPSc types 1 and 2 concurrence in MV cases was detected in two phenotypically distinct groups of subjects. The absence or presence of kuru plaques best distinguished the two groups. In subjects lacking kuru plaques, the concurrence of PrPSc types reproduced the features seen in MM 1+2C subjects. At variance with the MM group, however, the relative proportion of MV 1+2C and MV 2C+1 was significantly different. Indeed, only one case had type 1 in all brain regions and the concurrent type 2 in 5, two had 12 and 14 areas with a mixed molecular phenotype, respectively (but a predominant type 1 load), while the remaining 3 showed a predominant type 2 accumulation (Table 3).
PrPSc types 1 and 2 concurrence was more difficult to assess in MV subjects with kuru plaques (i.e. MV 2K subtype) because of the presence of the 20 kDa band (Fig. 1), which we detected in virtually all samples from subcortical structures and in 75% of those taken from the neocortex (data not shown). Thus, a relatively weak 21 kDa signal cannot be considered proof of bona-fide PrPSc type 1 concurrence, since it would also be compatible with a partially cleaved fragment of the 20 kDa PrPSc core [25]. As a consequence, we considered the 21 kDa band a bona-fide type 1 only when its intensity was similar or higher than that of the 20 kDa fragment. Using these stringent criteria, we detected an undisputable type 1 signal in a least two cortical samples in only 2 of the 24 MV 2K cases analyzed (data not shown). Interestingly, at variance with the other MV 2K cases, the PrPSc profile in these two cases included the so called 13 kDa CTF (C-terminal truncated fragment) (data not shown), which at variance with MV 2K is consistently found in both MM1 and VV1 cases [26].
VV subjects
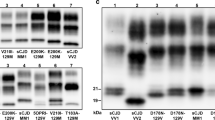
Six VV cases with concurrent PrPSc types 1 and 2 were found. PrPSc type 2 was always dominant and detectable in almost all samples. By contrast, type 1 was only present focally in the cerebral cortex, in the striatum or in both. In two cases, however, PrPSc type 1 was seen in most samples from cerebral cortex and striatum and in a single case, it showed an even more widespread accumulation involving all the cerebral cortical samples, the striatum, amygdala, hippocampus, hypothalamus and thalamus (Fig. 4a). When concurring, type 2 was predominant over type 1 in 76% of samples, while type 1 had a stronger signal than type 2 in the remaining 24%. The mean total type 1 “load” in VV 2+1 was 9.4% (range 1.5–38%).

a Western blot profiles of PrPSc (3F4 antibody) from different brain regions of the sCJD VV 2+1 subject with the more significant type 1 deposition. Areas shown include the middle frontal gyrus (MFG), middle temporal gyrus (MTG), parietal cortex (PC), lateral occipital cortex (OCC2), insular cortex (INS), putamen nucleus (PUT), medial thalamic nuclei (TH1), and cerebellum (CE). Approximate molecular masses are in kilodaltons. b Western blot profiles of PrPSc doublets in sCJD VV2, sCJD MM 1+2 and sCJD MV 2K. Lanes 1 and 2 show the association of a ~18.5 kDa fragment with the typical 19 kDa type 2 band in sCJD VV2 cerebellum (CE, V = vermis, H = hemisphere). Approximate molecular masses are in kilodaltons
Although clearly distinguishable from a type 1 and 2 concurrence, also the PrPSc type 2 profile in VV subjects was sometimes characterized by a doublet comprising a ~18.5 kDa fragment in addition to the typical 19 kDa band (Fig. 4b). These two bands concurred in the cerebellum in all cases, in the thalamus in 50% of them and in the midbrain in 18%. Since epitope mapping indicated that the 18.5 peptide, like the 19 kDa fragment, has an intact C-terminus (data not shown), the two fragments likely differ in their N-terminal PK cleavage site (i.e. the 18.5 fragment is further cleaved beyond residues 97–99).
Molecular-phenotypic correlations: the pathological phenotype
The pathological phenotype in MM/MV 1+2C and MM/MV 2+1C subjects displayed varying relative amounts of features as previously described in the pure MM/MV 1 and MM/MV 2C subtypes [30, 33]. The lesion profile in MM/MV 1+2C largely overlapped with that of pure MM/MV 1 cases, with the exception of the cerebellum, which was less affected relatively to the cerebral cortex (Fig. 5, and data not shown). Histopathologic hallmarks in these cases were (a) “morula-like”, sometimes referred to as “grape-like”, confluent foci of spongiform degeneration with large vacuoles in addition to the classic microvacuolation, and (b) a mixed synaptic/perivacuolar or coarse pattern of PrP deposition (Fig. 6a, b). With the exception of one case, in which, only the type 1 protein could be demonstrated by immunoblotting even after the 1E4 staining, in this group we found a 100% correlation between the large vacuoles and the perivacuolar staining on one hand and the PrPSc type 2 detection by immunoblotting on the other. The extension of the perivacuolar or coarse PrP staining, co-localizing with the confluent vacuoles varied significantly from case to case and was overall more common in the cerebral cortex than in subcortical areas (Fig. 7). The temporal, parietal and occipital cortices were the most affected cortical areas (Fig. 7). Among subcortical structures, the thalamus most often showed the perivacuolar staining, whereas the brainstem and to a greater extent the cerebellum (Fig. 7) were the areas with the lowest frequency.

Relative severity of lesions (i.e. spongiform degeneration and astrogliosis) in the cerebral cortex (mean of scores in neocortical and limbic cortical areas) and cerebellum: comparison between MM/MV 1 (n = 65), MM/MV 1+2C (n = 63), MM/MV 2C+1 (n = 9), and MM/MV 2C (n = 4) sCJD groups. The line slope expresses the ratio between the degree of cortical and cerebellar pathology. Group MM/MV 1+2C* includes only the cases within the MM/MV 1+2C group showing type 2 in at least 5 areas (n = 17)

a A “grape-like” focus of spongiform degeneration with large confluent vacuoles surrounded by typical spongiform degeneration with fine, relatively small vacuoles in sCJD MM 1+2C (hematoxylin and eosin ×200, frontal cortex); b focal perivacuolar staining surrounded by a synaptic pattern of PrP immunoreactivity (×200) in the cerebral cortex of a sCJD MM 1+2C; c spongiform degeneration with large vacuoles (hematoxylin and eosin ×200) and d perivacuolar PrP deposition (×200) in the cerebral cortex of a sCJD MV 2K

Regional distribution of the coarse/perivacuolar pattern of PrPSc deposition in 40 MM/MV 1+2C cases divided in 3 groups according to the number of regions showing the perivacuolar pattern in individual cases. For each group, the % of cases revealing the perivacuolar pattern in each brain region is shown. FC frontal cortex, TC temporal cortex, PC parietal cortex, OCC occipital cortex, Limbic CTX limbic cortices, Hipp + AMG hippocampus+amygdala, STR striatum, HYP hypothalamus, TH thalamus, BRST brainstem, CE cerebellum
The pathological phenotype of MM/MV 2C+1 largely overlapped to that of MM/MV 2C subjects. However, as a distinctive feature from the pure MM/MV 2C phenotype, all these cases had a synaptic pattern of PrP staining in the cerebellum, which well correlated with the presence of PrPSc type 1 on western blots.
The lesion profile and the pattern of PrP staining in all 6 VV subjects with concurrent PrPSc types (VV2+1) were very similar to those of typical VV2 sCJD cases. As the only distinctive feature, the two cases with the most significant type 1 accumulation showed a less consistent laminar pattern of spongiform degeneration in the cerebral cortex and a milder cerebellar pathology when compared to VV2 cases with similar disease duration.
Lastly, the two MV 2K+1 cases had no clear distinguishing pathological features although they both showed a relatively mild pathology and the lack of a laminar pattern of spongiform degeneration in the cerebral cortex. Of notice, however, we observed a perivacuolar pattern of PrPSc deposition co-localizing with the presence of larger vacuoles in 6 out of 25 of the sCJD MV 2K subjects (Fig. 6c, d) as well as in one MM2-thalamic (MM 2T) case. The regional distribution of the perivacuolar staining was not distinguishable from that observed in MM/MV 1+2C and mainly involved samples from the cerebral cortex (data not shown).
Molecular-phenotypic correlations: clinical features
MM/MV 1+2C and MM/MV 2C+1 subjects showed a significant difference in both disease duration and frequency of symptoms at onset according to the relative load of the two protein types (Tables 4, 5). Clinical signs in MM/MV 1+2C with a limited PrPSc type 2 deposition (up to 5 brain regions involved) did not significantly differ from those of MM/MV 1, but the disease duration became significantly longer and cerebellar signs less frequent at onset with increasing type 2 load. Thus, MM/MV 2C+1 subjects showed both a disease duration and relative frequency of symptoms at onset consistent with those of sCJD MM/MV 2C. Age at onset and disease duration in VV 2+1 subjects were consistent with those of sCJD VV2, but the relative frequency of cognitive decline at onset was higher than in VV2 cases, although the difference was not statistically significant, probably because of the low number of VV 2+1 cases. Last, clinical features in the 2 MV 2K+1 subjects were within the spectrum of the MV 2K group.
Discussion
Previous studies have addressed the issue of PrPSc types 1 and 2 co-occurrence in sCJD. Most of them raised the question of the influence of the number of cases and brain areas analyzed and emphasized the possibility that the co-occurrence of PrPSc types 1 and 2 is underestimated [13, 18, 20, 30, 37, 40, 43]. On the other hand, the use of a novel, potentially very sensitive approach, later shown to have pitfalls related to the detection of unspecific bands generated by partially digested PrPSc fragments [25], likely led other investigators to overestimate the incidence of the concurrent PrPSc types [35, 45]. Thus, the overall results on the phenomenon of the coexistence of molecular and clinico-pathological sCJD subtypes are at present inconclusive with respect to incidence, effect on disease phenotype and criteria for disease classification. To contribute to the full understanding of these issues, in the present study, we combined a systematic analysis of several brain regions in a large series of case including all codon 129 genotypes and the rarest phenotypes with the use of a refined methodology for the detection of the PrPSc type concurrence, which provides good sensitivity combined with high specificity [25].
After screening about 4,200 samples from a largely consecutive series of 200 cases, we estimated that PrPSc types 1 and 2 coexist in about 35% of sCJD cases, which is overall consistent with figures from some of the previous studies [13, 37, 43] in which the number of cases and areas analyzed were significantly lower. This finding supports the idea that PrPSc types co-occurrence involves a relevant but limited group of sCJD subjects and indicates that the incidence of the phenomenon had not been significantly underestimated.
As far as the characteristics of the CJD population with mixed phenotypes are concerned, our data show that the PrPSc types 1 and 2 co-occur more frequently in the MM than in the MV and VV genotypes. More specifically, the large majority of sCJD cases with concurrent PrPSc types combines features of the MM and MM 2C sCJD subtypes, in variable proportions. Most commonly, in such cases, the MM1 phenotype is predominant over the MM 2C phenotype, but the opposite situation also rarely occurs. The latter results significantly differ from those obtained in most previous studies. Indeed, Head et al. [13] mainly found a focal type 1 co-occurrence in MM and MV subjects with dominant type 2, Schoch et al. [40] detected the mixed protein types mostly in MV2 cases showing the type 1 only focally in subcortical areas, and Uro-Coste et al. [43] mainly detected a random co-occurrence of type 1 in MV or VV cases with dominant type 2. Given that only our study was based on a large series of consecutive cases, we attribute such heterogeneity of previous results to case selection biases, although methodological differences may also have contributed [43].
Since subjects with mixed PrPSc types represent a significant proportion of the sCJD population, show distinctive phenotypic features, and potentially represent a distinct subtype in terms of biological relevance, it is important that they are properly identified and are added as new subtypes in the current sCJD classification (Table 6).
Despite the emphasis on molecular features of current sCJD classification, it has become increasingly clear that PrPSc typing alone, when limited to a single or even a few brain samples, fails to provide an accurate classification in a significant proportion of cases. This is mainly related to the focal nature of the “mixed features” in many sCJD cases with PrPSc types 1 and 2 concurrence. Indeed, we would have misclassified the disease subtype in about 27.5% (using 3F4) of cases with MM or MV genotype, if we had analyzed PrPSc only in the frontal cortex, the area more commonly used for typing worldwide. For the same reason, discrepancies may arise when PrPSc-typing and PrP immunohistochemistry are performed from individual samples taken from opposite hemispheres or even adjacent cortical gyri. However, our study shows that the regional deposition of either type 1 or type 2 when concurrent is not random and that a relatively limited number of critical brain structures must be assessed to reach an accurate classification. Furthermore, our results further underline the importance of applying both molecular and neuropathological assessment for sCJD subtype classification. In this regard, the lack of detection of PrPSc type 2 in a minority of MM subjects, despite the presence of a mixed synaptic and perivacuolar pattern of PrP deposition, indicates that when type 2 is very focal or limited in amount, histopathologic examination is more sensitive in identifying such cases than PrPSc typing, at least when only the 3F4 antibody is used. Given the very strong correlation in MM subjects between PrPSc type 2 detection and the large “grape-like” vacuoles and the perivacuolar pattern of PrP deposition on histopathologic examination, which is in line with results previously obtained in other studies [18, 37], we propose that these cases are classified as MM 1+2C or MV 1+2C even without the final proof of type 2 detection by western blot. Alternatively, PrPSc typing using the antibody 1E4 was in our hand as sensitive as the histopathologic examination in the detection of cases with very focal type 2.
In the light of the present results, the most important regions to be assessed pathologically include the cerebral cortex from each of the 4 lobes, the striatum, hippocampus, thalamus and cerebellum. The cerebellum, in particular, is critical for the recognition of the synaptic pattern of PrP deposition as marker of PrPSc type 1 concurrence in the cases with dominant type 2.
Taken together, our data indicate that a protocol including the neuropathologic assessment of the eight brain regions mentioned above and PrPSc typing in four critical regions such as the temporal, parietal and occipital neocortices, and medial thalamus is strongly recommended for a reliable sCJD group classification addressing the issue of mixed phenotypes. Indeed, by applying this protocol instead of examining all 21 brain regions, we would have reached the same classification of cases in the present series.
We also wish to underline the importance of identifying correctly the sCJD cases with mixed features for transmission purposes. Indeed, the question of whether the concurrence of PrPSc types 1 and 2 in CJD reflects a co-infection by two prion strains related to specific undiscovered human genotypes, or determined by epigenetic factors remains unanswered and will largely rely on transmission studies in which the careful selection of samples will be of critical importance. Concerning this critical question, we find intriguing that the large, confluent vacuoles and the perivacuolar pattern of PrPSc deposition, we originally linked to sCJD MM 2C are also found in a subgroup of MV 2K subjects in addition to MM/MV 1+2C. In addition, we have described here the same morphological features in one case of fatal insomnia (i.e. the MM2-thalamic subtype or MM 2T) which adds to two previously reported cases [19, 30, 31]. Thus, it seems that large confluent vacuoles and the perivacuolar pattern of PrPSc deposition may be found in sCJD associated with all phenotypes linked to MM or MV at codon 129. Although this observation remains difficult to interpret at present, it appears relevant for our future understanding of the molecular basis and the extent of strain variation in sCJD. In any case, our observation strongly suggests that the phenomenon of mixed phenotypes in sCJD goes beyond PrPSc types 1 and 2 coexistence and also involves subtypes which shares the same PrPSc type. This, in turn, further underlines the importance of combining histopathological assessment and biochemical PrPSc typing for sCJD subtype characterization.
The present data also show that the association of two PrP27-30 fragments, which does not represent a bona-fide type 1 and 2 concurrence, may also be a feature of some sCJD cases. Thus, the PrP27-30 profile in VV2 cases in the cerebellum, thalamus and midbrain is sometime characterized by a doublet comprising a 18.5 kDa in addition to the typical 19 kDa band, while the western blot profile of PrP27-30 in the MV 2K cases appears almost invariably characterized by the association of two PrPSc core fragments including a classic 19 kDa type 2 band and a slower migrating band of about 20 kDa. Although these profiles truly represent concurrent PrPSc fragments, and the 20 and 18.5 kDa fragments likely reflect specific PK cleavage sites, the 20 and 18.5 kDa bands are distinguished from the type 1 and type 2 fragments because, at least to date, they were never detected independently from types 1 and 2, and are not markers of specific clinico-pathological phenotypes. Knowledge of these regional variations is nonetheless important to avoid misinterpreting a PrPSc profile as novel when only one brain region is analyzed [21].
Finally, the results obtained from the analyses of lesion profiles and clinical features in the subgroups of sCJD cases with mixed features deserve further comment. By showing that the relative “load” of each of the two PrPSc types significantly correlates with disease duration, the relative frequency of certain symptoms, and the ratio between cortical and cerebellar pathology, our study provides further strong evidence for the PrPSc type being a major biological determinant in human prion disease.
In conclusion, the present data add to our knowledge of the prevalence and phenotypic spectrum of the sCJD variants with mixed molecular and pathological features, provide an updated molecular classification of the disease subtypes and will serve for future epidemiologic and transmission studies aimed at disclosing the etiology and extent of strain variation in sCJD.
References
Baron T, Biacabe AG, Arsac JN, Benestad S, Groschup MH (2007) Atypical transmissible spongiform encephalopathies (TSEs) in ruminants. Vaccine 25:5625–5630
Barron RM, Thomson V, Jamieson E et al (2001) Changing a single amino acid in the N-terminus of murine PrP alters TSE incubation time across three species barriers. EMBO J 20:5070–5078
Bessen RA, Marsh RF (1994) Distinct PrP properties suggest the molecular basis of strain variation in transmissible mink encephalopathy. J Virol 68:7859–7868
Bolton DC, McKinley MP, Prusiner SB (1982) Identification of a protein that purifies with the scrapie prion. Science 218:1309–1311
Brown P, Coker-Vann M, Pomeroy K et al (1986) Diagnosis of Creutzfeldt–Jakob disease by Western blot identification of marker protein in human brain tissue. N Engl J Med 314:547–551
Bruce ME, McConnell I, Fraser H, Dickinson AG (1991) The disease characteristics of different strains of scrapie in Sinc congenic mouse lines: implications for the nature of the agent and host control of pathogenesis. J Gen Virol 72:595–603
Cali I, Castellani R, Yuan J et al (2006) Classification of sporadic Creutzfeldt–Jakob disease revisited. Brain 129:2266–2277
Caughey BW, Dong A, Bhat KS, Ernst D, Hayes SF, Caughey WS (1991) Secondary structure analysis of the scrapie-associated protein PrP27–30 in water by infrared spectroscopy. Biochemistry 30:7672–7680
Caughey B, Raymond GJ, Bessen RA (1998) Strain-dependent differences in beta-sheet conformations of abnormal prion protein. J Biol Chem 273:32230–32235
Collinge J, Sidle KC, Meads J et al (1996) Molecular analysis of prion strain variation and the aetiology of ‘new variant’ CJD. Nature 383:685–690
Ferrer I, Armstrong J, Capellari S et al (2007) Effects of formalin fixation, paraffin embedding, and time of storage on DNA preservation in brain tissue: a BrainNet Europe study. Brain Pathol 17:297–303
Goldmann W, Hunter N, Smith G, Foster J, Hope J (1994) PrP genotype and agent effects in scrapie: change in allelic interaction with different isolates of agent in sheep, a natural host of scrapie. J Gen Virol 75:989–995
Head MW, Bunn TJ, Bishop MT et al (2004) Prion protein heterogeneity in sporadic but not variant Creutzfeldt–Jakob Disease: U.K. cases. 1991–2002. Ann Neurol 55:851–859
Kascsak RJ, Rubenstein R, Merz PA et al (1986) Immunological comparison of scrapie-associated fibrils isolated from animals infected with four different scrapie strains. J Virol 59:676–683
Kimberlin RH, Cole S, Walker CA (1987) Temporary and permanent modifications to a single strain of mouse scrapie on transmissions to rats and hamsters. J Gen Virol 68:1875–1881
Kobayashi A, Asano M, Mohri S, Kitamoto T (2004) Cross-sequence transmission of sporadic Creutzfeldt–Jakob disease creates a new prion strain. J Biol Chem 282:30022–30028
Korth C, Kaneko K, Groth D et al (2003) Abbreviated incubation times for human prions in mice expressing a chimeric mouse-human prion protein transgene. Proc Natl Acad Sci USA 100:4784–4789
Kovács GG, Head MW, Hegyi I et al (2002) Immunohistochemistry for the prion protein: comparison of different monoclonal antibodies in human prion disease subtypes. Brain Pathol 12:1–11
Kretzschmar H, Giese A, Zerr I et al (1998) The German FFI cases. Brain Pathol 8:559–561
Lewis V, Hill AF, Klug GM, Boyd A, Masters CL, Collins SJ (2005) Australian sporadic CJD analysis supports endogenous determinants of molecular-clinical profiles. Neurology 65:113–118
Mead S, Joiner S, Desbruslais M et al (2007) Creutzfeldt–Jakob disease, prion protein gene codon 129VV, and a novel PrPSc type in a young British woman. Arch Neurol 64:1780–1784
Monari L, Chen SG, Brown P et al (1994) Fatal Familial Insomnia and familial Creutzfeldt–Jakob disease: Different prion proteins determined by a DNA polymorphism. Proc Natl Acad Sci USA 91:2839–2842
Nonno R, Di Bari MA, Cardone F et al (2005) Efficient transmission and characterization of Creutzfeldt–Jakob disease strains in bank voles. PLoS Pathog 2:e12
Notari S, Capellari S, Giese A et al (2004) Effects of different experimental conditions on the PrPSc core generated by protease digestion. J Biol Chem 279:16797–16804
Notari S, Capellari S, Langeveld J et al (2007) A refined method for molecular typing reveals that co-occurence of PrPSc types in Creutzfeldt–Jakob disease is not the rule. Lab Invest 87:1103–1112
Notari S, Strammiello R, Capellari S et al (2008) Characterization of truncated forms of abnormal prion protein in Creutzfeldt–Jakob disease. J Biol Chem 283:30557–30565
Pan KM, Baldwin M, Nguyen J et al (1993) Conversion of alpha-helices into beta-sheets features in the formation of the scrapie prion proteins. Proc Natl Acad Sci USA 90:10962–10966
Parchi P, Castellani R, Capellari S et al (1996) Molecular basis of phenotypic variability in sporadic Creutzfeldt–Jakob disease. Ann Neurol 39:767–778
Parchi P, Capellari S, Chen SG et al (1997) Typing prion isoforms. Nature 386:232–233
Parchi P, Giese A, Capellari S et al (1999) Classification of sporadic Creutzfeldt–Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol 46:224–233
Parchi P, Capellari S, Chin S et al (1999) A subtype of sporadic prion disease mimicking fatal familial insomnia. Neurology 52:1757–1763
Parchi P, Zou W, Wang W et al (2000) Genetic influence on the structural variations of the abnormal prion protein. Proc Natl Acad Sci USA 97:10168–10172
Parchi P, Notari S, Weber P et al (2009) Inter-laboratory assessment of PrPSc typing in Creutzfeldt–Jakob disease: a western blot study within the NeuroPrion Consortium. Brain Pathol 19:384–391
Pattison IH, Millson GC (1961) Scrapie produced experimentally in goats with special reference to the clinical syndrome. J Comp Pathol 71:101–108
Polymenidou M, Stoeck K, Glatzel M, Vey M, Bellon A, Aguzzi A (2005) Coexistence of multiple PrPSc types in individuals with Creutzfeldt–Jakob disease. Lancet Neurol 4:805–814
Prusiner SB (1998) Prions. Proc Natl Acad Sci USA 95:13363–13383
Puoti G, Giaccone G, Rossi G, Canciani B, Bugiani O, Tagliavini F (1999) Sporadic Creutzfeldt–Jakob disease: co-occurrence of different types of PrPSc in the same brain. Neurology 53:2173–2176
Roberts GW, Lofthouse R, Brown R, Crow TJ, Barry RA, Prusiner SB (1986) Prion protein immunoreactivity in human transmissible dementias. N Engl J Med 315:1231–1233
Safar J, Wille H, Itri V et al (1998) Eight prion strains have PrPSc molecules with different conformations. Nat Med 4:1157–1165
Schoch G, Seeger H, Bogousslavsky J et al (2006) Analysis of prion strains by PrPSc profiling in sporadic Creutzfeldt–Jakob disease. PLoS Med 3:e14
Somerville RA, Chong A, Mulqueen OU, Birkett CR, Wood SC, Hope J (1997) Biochemical typing of scrapie strains. Nature 386:564
Telling GC, Parchi P, DeArmond SJ et al (1996) Evidence for the conformation of the pathologic isoform of the prion protein enciphering and propagating prion diversity. Science 274:2079–2082
Uro-Coste E, Cassard H, Simon S et al (2008) Beyond PrPres type 1/type 2 dichotomy in Creutzfeldt–Jakob disease. PLoS Pathog 4:e1000029
Yuan J, Dong Z, Guo JP et al (2008) Accessibility of a critical prion protein region involved in strain recognition and its implications for the early detection of prions. Cell Mol Life Sci 65:631–643
Yull HM, Ritchie DL, Langeveld JP et al (2006) Detection of type 1 prion protein in variant Creutzfeldt–Jakob disease. Am J Pathol 168:151–157
Acknowledgments
We wish to thank Barbara Polischi and Sabrina Boninsegna for her technical assistance. We also thank all the physicians who provided clinical data and helped in the collection of tissues and all family members who consented to the use of tissue for research. This study was funded in the frame of the bilateral Italy (ISS)–USA (NIH, Office for Rare Diseases) agreement on joint research on rare diseases, by the European Commission (FOOD-CT-2004-506579), the Italian Ministry of University, Research and Technology (FIRB-2003-RBNE03FMCJ_006), the Federal Ministry of Health (ZV2-1369-340): grant PHS P30 AG010133, and the Gino Galletti Foundation.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Parchi, P., Strammiello, R., Notari, S. et al. Incidence and spectrum of sporadic Creutzfeldt–Jakob disease variants with mixed phenotype and co-occurrence of PrPSc types: an updated classification. Acta Neuropathol 118, 659–671 (2009). https://doi.org/10.1007/s00401-009-0585-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-009-0585-1