
Abstract
Abeta accumulation has an important function in the etiology of Alzheimer’s disease (AD) with its typical clinical symptoms, like memory impairment and changes in personality. However, the mode of this toxic activity is still a matter of scientific debate. We used the APP/PS1KI mouse model for AD, because it is the only model so far which develops 50% hippocampal CA1 neuron loss at the age of 1 year. Previously, we have shown that this model develops severe learning deficits occurring much earlier at the age of 6 months. This observation prompted us to study the anatomical and cellular basis at this time point in more detail. In the current report, we observed that at 6 months of age there is already a 33% CA1 neuron loss and an 18% atrophy of the hippocampus, together with a drastic reduction of long-term potentiation and disrupted paired pulse facilitation. Interestingly, at 4 months of age, there was no long-term potentiation deficit in CA1. This was accompanied by reduced levels of pre- and post-synaptic markers. We also observed that intraneuronal and total amount of different Abeta peptides including N-modified, fibrillar and oligomeric Abeta species increased and coincided well with CA1 neuron loss. Overall, these data provide the basis for the observed robust working memory deficits in this mouse model for AD at 6 months of age.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Transgenic mouse models are valuable tools to study the etiology of human disorders, like Alzheimer’s disease (AD). Many AD mouse models have been described so far, however, none of them exhibits all AD-typical hallmarks. Early pathological changes, such as deficits in synaptic transmission [16], changes in behavior, differential glutamate responses and deficits in long-term potentiation were reported in mouse models for AD overexpressing amyloid precursor protein (APP), the precursor of Aβ peptides [26]. Learning deficits [1, 14, 29, 33] were evident in different APP models, however, Aβ-amyloid deposition did not correlate with the behavioral phenotype [15]. The molecular mechanism of Aβ initiating the neurodegenerative process remains so far unknown. In the past, toxic Aβ has been regarded as acting extracellulary, while recent evidence points to toxic effects of Aβ in intracellular compartments (see [43, 47] for review). Moreover, it is now generally accepted that the conformation of the most toxic forms of Aβ are oligomers, in addition to aggregated β-sheet containing amyloid fibrils [19, 40]. It has been demonstrated that soluble oligomeric Aβ42, and not plaque-associated Aβ correlates best with cognitive dysfunction in AD [25, 27]. Moreover, oligomeres are formed preferentially within neuronal processes and synapses rather than extracellularly [41, 44]. Previously, we have reported that transient intraneuronal Abeta rather than extracellular plaque pathology correlates with neuron loss in the frontal cortex of APP/PS1KI mice [5].
In the present work, we used APP/PS1KI mice, expressing transgenic human mutant APP751 including the Swedish and London mutations on a murine knock-in (KI) Presenilin 1 (PS1) background with two FAD-linked mutations (PS1M233T and PS1L235P). Aβ42 comprises 85% of total Aβ in this model, consisting of full-length and heterogeneous N-terminal modified Aβ variants. This is the only mouse model so far, developing abundant hippocampal neuron loss at the age of 1 year [2]. We have shown previously that these mice exhibit severe and early learning deficits at the age of 6 months [46]. This observation prompted us to study the basis of these early learning deficits using anatomical and physiological techniques in more detail in the present report. We observed that intraneuronal accumulation of different Aβ peptides, together with overall accumulation of fibrillar and oligomeric species, coincided with 30% CA1 neuron loss, 18% hippocampus atrophy, and drastic reduction of synaptic plasticity.
Materials and methods
Transgenic mice
The generation of APP/PS1KI mice has been described previously [2]. All mice named as PS1KI were homozygous for the mutant PS1 knock-in transgene (KI). The APP/PS1KI mice harbored one hemizygous APP751SL transgene in addition to homozygous PS1KI. The mice were backcrossed for more than ten generations on a C57BL/6J genetic background. APP/PS1KI were crossed with PS1KI mice generating again APP/PS1KI and PS1KI mice, therefore all offspring carried homozygous PS1KI alleles. Mice carrying the additional APP transgene were identified by tail biopsy followed by PCR specific for the APP transgene sequence (forward primer: 5′-GTAGCAGAGGAGGAAGAAGTG-3′; reverse primer: 5′-CATGACCTGGGACATTCTC-3′). Only littermates were used for the described experiments. Hemizygous APP/PS1KI mice (hemizygous for PS1KI and for APP) were generated by backcrossing APP/PS1KI mice with wildtype C57Bl6/J6 mice. All animals were handled according to German guidelines for animal care.
Immunohistochemistry and histology
Mice were anaesthetized and transcardially perfused with ice-cold phosphate-buffered saline (PBS) followed by 4% paraformaldehyde. Brain samples were carefully dissected and post-fixed in 4% phosphate-buffered formalin at 4°C. Immunohistochemistry was performed on 4 μm paraffin sections as described previously [48]. We used antibodies against N-unmodified Aβ at position 1 [AβN1(D)], racemized aspartate at position 1 [AβN1(rD)], against pyroglutamate at position 3 [AβN3(pE)] (generous gift of Dr. T. Saido, RIKEN Institute, Japan) [36], and OC antibody detecting fibrillar oligomers (generous gift of Drs. C. Glabe and R. Kayed) [18]. Biotinylated secondary anti-rabbit and anti-mouse antibodies (1:200) were purchased from DAKO. Staining was visualized using the ABC method, with a Vectastain kit (Vector Laboratories) and diaminobenzidine as chromogen. Counterstaining was carried out with hematoxylin.
Immunohistochemical quantification of Aβ peptides
Three hippocampal sections from each APP/PS1KI mouse (n = 5), which were at least 50 μm far from each other, were randomly chosen and stained with antibodies against N-modified and fibrillar Aβ. Quantification of the relative density of intraneuronal immunoreactivity was done using the software program Cell F (Olympus) in single optical images. For this purpose, high-magnification images (400×) of amyloid plaque-free CA1 regions were taken. After defining the color threshold, which remains unchanged during measurement of the whole series, the CA1 granule cell layer was manually delineated as the region of interest. The image was then binarized and a relative staining intensity of the surface area in the defined region of interest was calculated. For quantification of the total hippocampal Aβ load, lower magnification (100×) images were used and the whole hippocampus was manually delineated as the region of interest. All images were taken using an Olympus BX-51 microscope equipped with an Olympus DP-50 camera. All data are given as mean values ± SEM.
Stereologic quantification of total numbers of neurons
Mice were anaesthetized and transcardially perfused as previously described [38]. The brains were carefully removed from the skull, post-fixed for 2 h and dissected. The left brain halves were cryoprotected in 30% sucrose, quickly frozen and cut frontally into series of 30 μm thick sections. Every tenth section was systematically sampled, stained with cresyl violet and used for stereological analysis of the hippocampal volume and the number of CA1 neurons. The hippocampal cell layer CA1 was delineated on cresyl violet-stained sections. Using a stereology workstation (StereoInvestigator; MicroBrightField, Williston, VT, USA) and a 100× oil lens, all neurons whose nucleus top came into focus were counted using the Optical Fractionator. In addition, volumes of the investigated brain regions were calculated from the delineated areas with Cavalieri’s principle.
Protein extraction
For dot blot assays and western blotting, proteins were homogenized in a Dounce-homogenizer in 2.5 ml 2% SDS, containing complete protease inhibitor (Roche). Homogenates were sonified for 30 s and subsequently centrifuged at 80,000g for 1 h at 4°C. The supernatants were immediately frozen and stored at −80°C until further analysis. For synaptosome enrichment, brain hemispheres were homogenized in 10× volume of 0.32 M sucrose, 5 mM HEPES, pH 7.5 including complete protease inhibitor (Roche) using a glass-teflon homogenizer. Homogenates were centrifuged at 10,000g for 10 min and the supernatant was centrifuged at 12,000g for additional 20 min. The resulting pellet contains the crude synaptosomal fraction.
Dot blot
For the dot blot, protein extracts were applied to nitrocellulose membranes and air dried. Membranes were blocked in 10% non-fat dry milk in TBS-T (10 mM Tris pH 8.0, 150 mM NaCl, 0.05% Tween 20) and incubated with the primary antibody (N1D, generous gift of Dr. T. Saido; OC and I-11 were a generous gift of Drs. C. Glabe and R. Kayed) for 1 h at room temperature. After washing in TBS-T, the membranes were incubated with horseradish-peroxidase conjugated secondary antibodies and developed using enhanced chemiluminescence. Quantification was done by densitometry of Dot blot signals using ImageJ software (NIH, http://rsb.info.nih.gov/ij).
Western blot
Electrophoresis was performed using 4–12% sodium dodecylsulfate–polyacrylamide gels (Vario-Gel, Anamed, Germany). Proteins were transferred to nitrocellulose membranes by semi-dry blotting and membranes were probed with antibodies against PSD-95, SNAP25 and neuron-specific clathrin light chain (generous gift from Synaptic Systems, Göttingen, Germany). Quantification was done by densitometry of Western blot bands using ImageJ software (NIH, http://rsb.info.nih.gov/ij). Normalization was carried out using an antibody detecting Tubulin (Chemicon).
Slice preparation for electrophysiology
Hippocampal slices were prepared using standard methods. The brain was rapidly removed and cooled in an ice cold solution containing 87 mM NaCl, 25 mM NaHCO3, 2.5 mM KCl, 1.25 mM NaH2PO4·H2O, 7 mM MgCl2, 0.5 mM CaCl2, 10 mM glucose, 75 mM sucrose. Brain slices (400 μm) were cut with a vibratome. Slices were stored submerged at room temperature (20°C) in an artificial cerebrospinal fluid (ACSF) solution for at least 1.5 h before recording. The composition of the ACSF solution was: 136 mM NaCl, 25 mM NaHCO3, 2.5 mM KCl, 1.25 mM NaH2PO4·H2O, 1 mM MgCl2, 2 mM CaCl2, 10 mM glucose. All solutions were bubbled with 95%O2/5%CO2.
Electrophysiological recording and data analysis
A glass stimulating electrode filled with ACSF solution (1–5 MΩ resistance) was placed on the surface of the stratum radiatum of the hippocampus CA3 area for activation. The extracellular field potentials were recorded in the stratum radiatum of area CA1 with another glass electrode filled with ACSF solution (1–5 MΩ resistance). Stimuli were applied by an optically isolated stimulator (model 2100, A-M Systems Inc., USA). Signals were low-pass filtered at 3 kHz and recorded by a low noise high gain amplifier. Data were collected at 5 kHz using NeuroMatic (Version 1.86) in Igor Pro (WaveMetrics, Inc, USA) via a PCI 6024 (National Instruments Inc., USA). An input–output curve was generated to determine the stimulus strength needed to elicit a half-maximal field excitatory post-synaptic potential (fEPSP). Baseline fEPSP was determined using the half-maximal stimulus intensity, and a low stimulation frequency (0.033 Hz) by a dual-pulse protocol (interpulse interval was 40 ms). After establishment of the baseline fEPSP, we determined whether long-term potentiation (LTP) could be elicited in the slice. The LTP protocol consisted of three trains of 100 Hz for 1 s, with an interval of 10 s. After the third high frequency stimulus (HFS), the baseline stimulus was applied again, and the post-HFS fEPSP was determined. The amplitude of the first negative peak of the fEPSP was measured as an index of synaptic efficacy. Paired pulse facilitation (PPF) was calculated as the ratio between the second and first negative peak amplitude of the fEPSP (40 ms interpulse interval) before (before high frequency stimulation, HFS) and after LTP (between 15 and 25 min after the last HFS). LTP was calculated by averaging the normalized fEPSP amplitude collected between 15 and 25 min after the last HFS.
Statistical analysis
Differences between groups were tested with either one-way analysis of variance (ANOVA) or unpaired t tests. All data were given as mean values ± SEM. Significance levels were given as follows: ***P < 0.001; **P < 0.01; *P < 0.05. All calculations were performed using GraphPad Prism version 4.03 for Windows (GraphPad Software, San Diego, CA, USA).
Results
Immunohistochemical analysis of Aβ peptides
Strong immunoreactivity with antibodies AβN1(D) starting at position 1 with aspartate, AβN1(rD) starting at position 1 with racemized aspartate, AβN3(pE) starting at position 3 with cyclized glutamate and OC recognizing fibrillar oligomers was detected already at 2 months of age within CA1 neurons. Their staining intensity increased significantly at the age of 6 months (Fig. 1). PS1KI control animals were constantly negative (not shown). Quantification of the different Aβ peptides in CA1 neurons revealed that all four antibodies showed a strong increased intraneuronal staining between 2 and 6 months of age, however, with varying degrees. The amount increased as follows: AβN1(D) by 301%, AβN1(rD) by 297%, AβN3(pE) by 435%, and OC by 51%. In addition, the total Aβ load of Aβ peptides was assessed in the entire hippocampal area and showed an age-dependent increase for AβN1(D) by 529%, for AβN1(rD) by 486%, for AβN3(pE) by 1,076%, and for OC by 178%. Interestingly, the spatial pattern of plaque deposition did not correlate with the observed CA1 neuron loss since extracellular plaques were not found in a significant amount decorating the somata of CA1 pyramidal neurons (Fig. 1).

Representative hippocampus sections and quantification of 6-month-old APP/PS1KI mice stained with antibodies against Aβ forms including peptides starting with aspartate AβN1(D) (a, b), racemized aspartate AβN1(rD) (c, d), AβN3(pE) against pyroglutamate at position 3 (e, f), and OC against fibrillar oligomers and fibrils (g, h). Quantitative analysis revealed a significant increase of all Aβ peptides in total hippocampus, as well as in CA1 pyramidal neurons (i–t) at 6 months compared with 2-month-old APP/PS1KI mice (n = 5 per group). Values are given as mean values ± SEM, ***P < 0.001; **P < 0.01; *P < 0.05. Scale bars 200 μm (hippocampus: a, c, e, g) and 50 μm (CA1: i, j, l, m, o, p, r, s)
Dot blot analysis of Aβ peptides
The antibodies OC and I-11 have been previously shown in dot blot assays to detect oligomeric and fibrillar Aβ species depending on their conformation. OC recognizes fibrillar oligomers and mature fibrils, whereas I-11 recognizes only Aβ oligomers [18]. Therefore, we used this established method to study the accumulation profile of the respective Aβ peptides in dot blots of whole brain protein lysates. Between 2 and 6 months of age, all antibodies detected significantly elevated peptide levels (N1D with 227%, I-11 with 50% and OC with 157%) (Fig. 2). Antibody I-11 did not work in immunohistochemistry.

Dot blot analysis of APP/PS1KI brain extracts at 2 and 6 months of age. Values are given as mean values ± SEM ***P < 0.001; **P < 0.01; *P < 0.05
CA1 neuron loss and hippocampus atrophy
Neuronal loss was quantitatively assessed by design-based stereology. In 6-month-old APP/PS1KI mice, a 33% loss of pyramidal cells within the pyramidal layer of CA1 in the hippocampus was detected, compared to 6-month-old PS1KI littermate control mice (Fig. 3a). Previously we have demonstrated that 2-month-old APP/PS1KI mice showed a normal number of CA1 neurons and a 50% reduction at the age of 1 year [2]. In addition, 6-month-old APP/PS1KI mice exhibited a decreased volume of the CA1 pyramidal cell layer of 30% (Fig. 3b), and a total hippocampal atrophy of 18% (Fig. 3c).

a Stereological cell counting of CA1 neurons of 6-month-old APP/PS1KI (n = 7) and PS1KI (n = 6) mice revealed a 33% reduction. b A significant difference in the volume of the CA1 pyramidal layer was detected at 6 months, corresponding to a reduction of 30% (n = 6–7 per group). c This results in an overall hippocampus volume reduction of 18%. d Representative sections of cresyl-violett stained PS1KI and APP/PS1KI mice e at 6 months of age. Values are given as mean values ± SEM, **P < 0.01; *P < 0.05
Western blot analysis of pre- and postsynaptic markers
Synaptosome-associated protein of 25 kDa (SNAP25) is a highly conserved protein anchored to the cytosolic face of membranes and is required for exocytosis of presynaptic vesicles. Clathrin consists of heavy chains and light chains. The neuron-specific light chains are enriched in synaptic nerve terminals participating in presynaptic vesicle endocytosis. PSD-95 is a major protein at the postsynaptic density and has an important function in regulating the surface expression of glutamate receptors, thereby representing a critical factor regulating synaptic plasticity. We quantified the levels by Western blot in whole brain extracts and synaptosome enriched fractions of 2 and 6 month-old mice. At 2 months, there was no difference between APP/PS1KI and PS1KI control mice. In whole brain Western blots only PSD-95 levels were significantly reduced at the age of 6 months in APP/PS1KI mice (Fig. 4a). Of interest, the levels of all three markers were significantly decreased in synaptosome-enriched fractions at that time point (Fig. 4b).

Western blot analysis of pre- (clathrin light chain; SNAP25) and post-synaptic markers (PSD-95). a While in whole brain lysates, only the levels of PSD-95 declined significantly, b the levels of all synaptic markers were decreased in synaptosome enriched fractions in 6-month-old APP/PS1KI mice. At 2 months, there was no difference between APP/PS1KI and PS1KI control mice. Values are given as mean values ± SEM, ***P < 0.001; **P < 0.01, *P < 0.05
Synaptic transmission
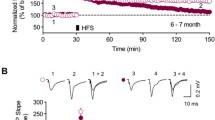
In order to study synaptic plasticity, we recorded field excitatory postsynaptic potentials (fEPSP) evoked by stimulation of the Schaffer collaterals in CA1 stratum radiatum in slices taken from mice at 2, 4 and 6 months of age. We found no significant differences of fEPSPs between APP/PS1KI (0.51 ± 0.08 mV) and PS1KI (0.8 ± 0.17 mV) mice at the age of 2 months. At 6 months of age, we observed a significant reduction of fEPSPs in APP/PS1KI (0.26 ± 0.05 mV) compared to PS1KI mice (0.80 ± 0.17 mV) (Fig. 5a). We next examined long-lasting synaptic plasticity in hippocampal slices from mice at 2, 4 and 6 months of age by inducing long-term potentiation (LTP) by tetanic stimulation. While no difference in LTP was observed at 2 months between APP/PS1KI (184.0 ± 13.2%) and PS1KI (162.9 ± 13.3%) and at 4 months of age between APP/PS1KI (155.4 ± 8.5%) and PS1KI (161.2 ± 8.4%), at 6 months of age, APP/PS1KI mice (106.3 ± 9.3%) showed a dramatic reduction. APP/PS1KI hemizygous (with only 1 knock-in PS1 allele; 175.2 ± 16.3%), PS1KI (167.2 ± 8.2%) and wildtype controls (169.6 ± 7.0%) showed the normal LTP pattern (Fig. 5b–e). To analyze short-term synaptic plasticity, we calculated PPF before and after LTP induction. At the age of 6 months, WT and PS1KI mice exhibited a robust PPF, which was significantly lower after induction of LTP. APP/PS1KI mice failed to show this pattern and elicited no significant difference at both time points. This lack of difference was attributable to a reduced PPF ratio before LTP rather than after LTP (Fig. 5f).

Electrophysiological recordings of hippocampus slices. a Half-maximal field excitatory post-synaptic potentiation (fEPSP) of APP/PS1KI and PS1KI demonstrate an age-dependent significant decrease of fEPSP in APP/PS1KI mice. b Quantitative analysis of LTP at 2, 4 and 6 months of age. c The pattern of LTP was similar in 2-month-old APP/PS1KI (6 slices/5 mice) and PS1KI mice (n = 5/4). d The pattern of LTP was also similar in 4-month-old APP/PS1KI (9 slices/3 mice) and PS1KI mice (n = 7 slices/3 mice). e LTP was almost abolished in APP/PS1KI (n = 4 slices/3 mice), compared to WT (n = 8 slices/5 mice) and PS1KI control mice (n = 10 slices/4 mice) at the age of 6 months. f As expected the difference between PPF before and after high frequency stimulation was significantly reduced from that induced prior to HFS in PS1KI and WT control mice. This normal PPF pattern was clearly absent in 6-month-old APP/PS1KI mice. All data are given as mean values ± SEM ***P < 0.001; **P < 0.01; *P < 0.05
Discussion
The data presented herein provide evidence that intraneuronal accumulation of different Aβ peptides correlates well with a robust deficiency in synaptic transmission, neuron loss, and hippocampus atrophy in the APP/PS1KI mouse model for AD. Despite the fact that plaque-associated Aβ also increases with age, the spatial distribution did not match with the pattern of neuron loss. In addition, a possible toxic effect of soluble Aβ species is likely to be negligible, because other groups of neurons like dentate gyrus granule cells, which do not accumulate intraneuronal Aβ are unaffected. Previously, we also have shown that (a) 85% of total Aβ in APP/PS1KI mice consist of Aβ42, (b) that 1-year-old mice exhibit a 50% CA1 neuron loss [2], and (c) that robust learning deficits occur already at 6 months of age [46].
An emerging role of intracellular Aβ accumulation has been previously shown in human AD [7, 12]. It has been observed that Aβ localizes predominantly to abnormal endosomes [3], multivesicular bodies and within pre- and postsynaptic compartments [22, 42]. Takahashi et al. [41] demonstrated that Aβ42 aggregates into oligomers within endosomal vesicles and along microtubules of neuronal processes, both in Tg2576 neurons with time in culture, as well as in Tg2576 and human AD brain. In good agreement with these reports, we observed that different Aβ peptides strongly accumulate within CA1 neurons in the critical period between 2 and 6 months of age. Pyroglutamate AβN3(pE) species have been reported to be the dominant Aβ species in AD brain [36]. AβN3(pE)-42 has a higher aggregation rate and shows an increased toxicity compared to full-length Aβ [13, 35] and in addition, N3(pE)-modified peptides display an up to 250-fold increased acceleration of the initial formation of Aβ aggregates [37]. In the present report, AβN3(pE) showed the strongest relative increase in the accumulation profile, compared to the other investigated Aβ peptides. As expected, we also observed an age-dependent increase of fibrillar and oligomeric Aβ species as shown by immunohistochemical and dot blot assays, as it has been previously shown in the 3xTg-AD mouse model [31]. Soluble oligomeric Aβ42, and not plaque-associated Aβ correlates best with cognitive dysfunction in AD [25, 27]. Oligomers are formed preferentially intracellulary within neuronal processes and synapses rather than extracellularly [41, 44], and cause synaptic alterations [20, 21]. On the other side, previous studies using post-mortem AD brain sections have shown that intraneuronal Aβ immunoreactivity is not a predictor of brain amyloidosis, neurofibrillary degeneration or neuronal loss [45].
Impairment of basal synaptic transmission has been shown in a variety of other APP transgenic mouse models [9, 16, 26]. In addition, deficits in induction of long-term synaptic changes have also been observed in several APP transgenic lines, like PDAPP mice [10, 23], APP London [8, 26], Tg2576 [4] and the 3xTg-AD line [30].
In good agreement with these studies, we have shown that the APP/PS1KI mouse line also exhibits strong deficits in synaptic plasticity expressed LTP. Schaeffer collateral LTP is considered a post-synaptic form of LTP in which activation of NMDA receptors leads to calcium entry, which then induces LTP [6, 24, 28]. We observed that APP/PS1KI mice with only one mutant PS1 allele, wildtype and PS1KI mice showed a normal LTP pattern at 6 months. Previously, synaptic transmission and hippocampal LTP has been shown to be already altered in transgenic mice overexpressing only FAD linked PS1 mutations [32, 39]. Therefore, the normal LTP pattern in PS1KI control mice could be due to low expression levels of endogenous murine PS1. We speculate that there is a narrow period for the onset of the synaptotoxic effect as (1) 4-month-old APP/PS1KI mice, and (2) 6-month-old APP/PS1KI hemizygous mice with only one PS1 knock-in allele show no synaptic deficits. The LTP normal pattern in 6-month-old APP/PS1KI hemizygous mice also demonstrate that mutant APP751 Swedish/London expression alone is not sufficient to elicit an effect.
The reduced LTP in 6-month-old APP/PS1KI mice is corroborated by strongly reduced levels of the pre- and postsynaptic markers in the synaptosome fraction. Interestingly, post-synaptic density protein PSD-95 level was already reduced in whole brain fractions, which might indicate a stronger pathological effect at the post-synaptic site.
Volumetric measures of the hippocampus correlate well with neuropathological markers to identify non-demented individuals at risk to develop AD at a later age [11]. However, APP transgenic mouse models have not been reported to develop considerable age-dependent hippocampal atrophy. PDAPP mice revealed a 12.3% reduction of hippocampus volume in transgenic mice compared with wildtype controls. This reduction persisted without progression to an age of 21 months [34], thereby representing a pattern more compatible with a developmental process. We have shown in the present report that APP/PS1KI mice develop hippocampal atrophy at the age of 6 months, thereby reflecting an additional hallmark of human AD pathology. The observed volume reduction is in the range of patients with mild cognitive impairment (for example [17]).
In conclusion, we provide evidence that a variety of different Aβ peptides accumulate intra- and extracellularly in the critical period between 2 and 6 months. Some of these peptides, like AβN3pE and oligomeric species have been reported to possess highly toxic properties. This coincided with hippocampus atrophy, beginning CA1 neuron loss and synaptic failure, thereby linking Aβ accumulation with neurodegenerative mechanisms. These observations also provide additional evidence that Aβ is able to trigger typical neurodegenerative mechanisms in AD independent of neurofibrillary tangle formation.
References
Billings LM, Oddo S, Green KN, McGaugh JL, Laferla FM (2005) Intraneuronal Abeta causes the onset of early Alzheimer’s disease-related cognitive deficits in transgenic mice. Neuron 45:675–688
Casas C, Sergeant N, Itier JM, Blanchard V, Wirths O, van der Kolk N, Vingtdeux V, van de Steeg E, Ret G, Canton T, Drobecq H, Clark A, Bonici B, Delacourte A, Benavides J, Schmitz C, Tremp G, Bayer TA, Benoit P, Pradier L (2004) Massive CA1/2 Neuronal Loss with Intraneuronal and N-Terminal Truncated Abeta42 Accumulation in a Novel Alzheimer Transgenic Model. Am J Pathol 165:1289–1300
Cataldo AM, Petanceska S, Terio NB, Peterhoff CM, Durham R, Mercken M, Mehta PD, Buxbaum J, Haroutunian V, Nixon RA (2004) Abeta localization in abnormal endosomes: association with earliest Abeta elevations in AD and Down syndrome. Neurobiol Aging 25:1263–1272
Chapman PF, White GL, Jones MW, Cooper-Blacketer D, Marshall VJ, Irizarry M, Younkin L, Good MA, Bliss TV, Hyman BT, Younkin SG, Hsiao KK (1999) Impaired synaptic plasticity and learning in aged amyloid precursor protein transgenic mice. Nat Neurosci 2:271–276
Christensen DZ, Kraus SL, Flohr A, Cotel MC, Wirths O, Bayer TA (2008) Transient intraneuronal Abeta rather than extracellular plaque pathology correlates with neuron loss in the frontal cortex of APP/PS1KI mice. Acta Neuropathol 116:647–655
Collingridge GL, Kehl SJ, McLennan H (1983) The antagonism of amino acid-induced excitations of rat hippocampal CA1 neurones in vitro. J Physiol 334:19–31
D’Andrea MR, Nagele RG, Wang HY, Lee DH (2002) Consistent immunohistochemical detection of intracellular beta-amyloid42 in pyramidal neurons of Alzheimer’s disease entorhinal cortex. Neurosci Lett 333:163–166
Dewachter I, Reverse D, Caluwaerts N, Ris L, Kuiperi C, Van den Haute C, Spittaels K, Umans L, Serneels L, Thiry E, Moechars D, Mercken M, Godaux E, Van Leuven F (2002) Neuronal deficiency of presenilin 1 inhibits amyloid plaque formation and corrects hippocampal long-term potentiation but not a cognitive defect of amyloid precursor protein [V717I] transgenic mice. J Neurosci 22:3445–3453
Fitzjohn SM, Morton RA, Kuenzi F, Rosahl TW, Shearman M, Lewis H, Smith D, Reynolds DS, Davies CH, Collingridge GL, Seabrook GR (2001) Age-related impairment of synaptic transmission but normal long-term potentiation in transgenic mice that overexpress the human APP695SWE mutant form of amyloid precursor protein. J Neurosci 21:4691–4698
Giacchino J, Criado JR, Games D, Henriksen S (2000) In vivo synaptic transmission in young and aged amyloid precursor protein transgenic mice. Brain Res 876:185–190
Gosche KM, Mortimer JA, Smith CD, Markesbery WR, Snowdon DA (2002) Hippocampal volume as an index of Alzheimer neuropathology: findings from the Nun Study. Neurology 58:1476–1482
Gouras GK, Tsai J, Naslund J, Vincent B, Edgar M, Checler F, Greenfield JP, Haroutunian V, Buxbaum JD, Xu H, Greengard P, Relkin NR (2000) Intraneuronal Abeta42 accumulation in human brain. Am J Pathol 156:15–20
He W, Barrow CJ (1999) The A beta 3-pyroglutamyl and 11-pyroglutamyl peptides found in senile plaque have greater beta-sheet forming and aggregation propensities in vitro than full-length A beta. Biochemistry 38:10871–10877
Holcomb L, Gordon MN, McGowan E, Yu X, Benkovic S, Jantzen P, Wright K, Saad I, Mueller R, Morgan D, Sanders S, Zehr C, O’Campo K, Hardy J, Prada CM, Eckman C, Younkin S, Hsiao K, Duff K (1998) Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat Med 4:97–100
Holcomb LA, Gordon MN, Jantzen P, Hsiao K, Duff K, Morgan D (1999) Behavioral changes in transgenic mice expressing both amyloid precursor protein and presenilin-1 mutations: lack of association with amyloid deposits. Behav Genet 29:177–185
Hsia AY, Masliah E, McConlogue L, Yu GQ, Tatsuno G, Hu K, Kholodenko D, Malenka RC, Nicoll RA, Mucke L (1999) Plaque-independent disruption of neural circuits in Alzheimer’s disease mouse models. Proc Natl Acad Sci USA 96:3228–3233
Jack CR Jr, Petersen RC, Xu YC, O’Brien PC, Smith GE, Ivnik RJ, Boeve BF, Waring SC, Tangalos EG, Kokmen E (1999) Prediction of AD with MRI-based hippocampal volume in mild cognitive impairment. Neurology 52:1397–1403
Kayed R, Head E, Sarsoza F, Saing T, Cotman CW, Necula M, Margol L, Wu J, Breydo L, Thompson JL, Rasool S, Gurlo T, Butler P, Glabe CG (2007) Fibril specific, conformation dependent antibodies recognize a generic epitope common to amyloid fibrils and fibrillar oligomers that is absent in prefibrillar oligomers. Mol Neurodegener 2:18
Klein WL (2002) Abeta toxicity in Alzheimer’s disease: globular oligomers (ADDLs) as new vaccine and drug targets. Neurochem Int 41:345–352
Lacor PN, Buniel MC, Chang L, Fernandez SJ, Gong Y, Viola KL, Lambert MP, Velasco PT, Bigio EH, Finch CE, Krafft GA, Klein WL (2004) Synaptic targeting by Alzheimer’s-related amyloid beta oligomers. J Neurosci 24:10191–10200
Lacor PN, Buniel MC, Furlow PW, Sanz Clemente A, Velasco PT, Wood M, Viola KL, Klein WL (2007) Abeta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer’s Disease. J Neurosci 27:796–807
Langui D, Girardot N, El Hachimi KH, Allinquant B, Blanchard V, Pradier L, Duyckaerts C (2004) Subcellular topography of neuronal Abeta peptide in APPxPS1 transgenic mice. Am J Pathol 165:1465–1477
Larson J, Lynch G, Games D, Seubert P (1999) Alterations in synaptic transmission and long-term potentiation in hippocampal slices from young and aged PDAPP mice. Brain Res 840:23–35
MacDermott AB, Mayer ML, Westbrook GL, Smith SJ, Barker JL (1986) NMDA-receptor activation increases cytoplasmic calcium concentration in cultured spinal cord neurones. Nature 321:519–522
McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, Bush AI, Masters CL (1999) Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer’s disease. Ann Neurol 46:860–866
Moechars D, Dewachter I, Lorent K, Reverse D, Baekelandt V, Naidu A, Tesseur I, Spittaels K, Haute CV, Checler F, Godaux E, Cordell B, Van Leuven F (1999) Early phenotypic changes in transgenic mice that overexpress different mutants of amyloid precursor protein in brain. J Biol Chem 274:6483–6492
Naslund J, Haroutunian V, Mohs R, Davis KL, Davies P, Greengard P, Buxbaum JD (2000) Correlation between elevated levels of amyloid beta-peptide in the brain and cognitive decline. Jama 283:1571–1577
Nowak L, Bregestovski P, Ascher P, Herbet A, Prochiantz A (1984) Magnesium gates glutamate-activated channels in mouse central neurones. Nature 307:462–465
Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, Van Eldik L, Berry R, Vassar R (2006) Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s Disease mutations: potential factors in amyloid plaque formation. J Neurosci 26:10129–10140
Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM (2003) Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron 39:409–421
Oddo S, Caccamo A, Tran L, Lambert MP, Glabe CG, Klein WL, LaFerla FM (2006) Temporal profile of amyloid-beta (Abeta) oligomerization in an in vivo model of Alzheimer disease. A link between Abeta and tau pathology. J Biol Chem 281:1599–1604
Parent A, Linden DJ, Sisodia SS, Borchelt DR (1999) Synaptic transmission and hippocampal long-term potentiation in transgenic mice expressing FAD-linked presenilin 1. Neurobiol Dis 6:56–62
Puolivali J, Wang J, Heikkinen T, Heikkila M, Tapiola T, van Groen T, Tanila H (2002) Hippocampal A beta 42 levels correlate with spatial memory deficit in APP and PS1 double transgenic mice. Neurobiol Dis 9:339–347
Redwine JM, Kosofsky B, Jacobs RE, Games D, Reilly JF, Morrison JH, Young WG, Bloom FE (2003) Dentate gyrus volume is reduced before onset of plaque formation in PDAPP mice: A magnetic resonance microscopy and stereologic analysis. Proc Natl Acad Sci USA 100:1381–1386
Russo C, Violani E, Salis S, Venezia V, Dolcini V, Damonte G, Benatti U, D’Arrigo C, Patrone E, Carlo P, Schettini G (2002) Pyroglutamate-modified amyloid bete-peptides—AbetaN3(pE)—strongly affect cultured neuron and astrocyte survival. J Neurochem 82:1480–1489
Saido TC, Iwatsubo T, Mann DM, Shimada H, Ihara Y, Kawashima S (1995) Dominant and differential deposition of distinct beta-amyloid peptide species, Abeta N3(pE), in senile plaques. Neuron 14:457–466
Schilling S, Lauber T, Schaupp M, Manhart S, Scheel E, Bohm G, Demuth HU (2006) On the seeding and oligomerization of pGlu-amyloid peptides (in vitro). Biochemistry 45:12393–12399
Schmitz C, Rutten BP, Pielen A, Schafer S, Wirths O, Tremp G, Czech C, Blanchard V, Multhaup G, Rezaie P, Korr H, Steinbusch HW, Pradier L, Bayer TA (2004) Hippocampal neuron loss exceeds amyloid plaque load in a transgenic mouse model of Alzheimer’s disease. Am J Pathol 164:1495–1502
Schneider I, Reverse D, Dewachter I, Ris L, Caluwaerts N, Kuiperi C, Gilis M, Geerts H, Kretzschmar H, Godaux E, Moechars D, Van Leuven F, Herms J (2001) Mutant presenilins disturb neuronal calcium homeostasis in the brain of transgenic mice, decreasing the threshold for excitotoxicity and facilitating long-term potentiation. J Biol Chem 276:11539–11544
Selkoe DJ (2001) Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev 81:741–766
Takahashi RH, Almeida CG, Kearney PF, Yu F, Lin MT, Milner TA, Gouras GK (2004) Oligomerization of Alzheimer’s beta-amyloid within processes and synapses of cultured neurons and brain. J Neurosci 24:3592–3599
Takahashi RH, Milner TA, Li F, Nam EE, Edgar MA, Yamaguchi H, Beal MF, Xu H, Greengard P, Gouras GK (2002) Intraneuronal Alzheimer abeta42 accumulates in multivesicular bodies and is associated with synaptic pathology. Am J Pathol 161:1869–1879
Tseng BP, Kitazawa M, LaFerla FM (2004) Amyloid beta-peptide: the inside story. Curr Alzheimer Res 1:231–239
Walsh DM, Tseng BP, Rydel RE, Podlisny MB, Selkoe DJ (2000) The oligomerization of amyloid beta-protein begins intracellularly in cells derived from human brain. Biochemistry 39:10831–10839
Wegiel J, Kuchna I, Nowicki K, Frackowiak J, Mazur-Kolecka B, Imaki H, Wegiel J, Mehta PD, Silverman WP, Reisberg B, Deleon M, Wisniewski T, Pirttilla T, Frey H, Lehtimaki T, Kivimaki T, Visser FE, Kamphorst W, Potempska A, Bolton D, Currie JR, Miller DL (2007) Intraneuronal Abeta immunoreactivity is not a predictor of brain amyloidosis-beta or neurofibrillary degeneration. Acta Neuropathol 113:389–402
Wirths O, Breyhan H, Schäfer S, Roth C, Bayer TA (2008) Deficits in working memory and motor performance in the APP/PS1ki mouse model for Alzheimer’s disease. Neurobiol Aging 29:891–901
Wirths O, Multhaup G, Bayer TA (2004) A modified beta-amyloid hypothesis: intraneuronal accumulation of the beta-amyloid peptide—the first step of a fatal cascade. J Neurochem 91:513–520
Wirths O, Multhaup G, Czech C, Blanchard V, Moussaoui S, Tremp G, Pradier L, Beyreuther K, Bayer TA (2001) Intraneuronal Abeta accumulation precedes plaque formation in beta-amyloid precursor protein and presenilin-1 double-transgenic mice. Neurosci Lett 306:116–120
Acknowledgments
The excellent technical support by David Stevens, Katrin Rubly, Ulrike Schuler and Stephanie Schäfer is gratefully acknowledged. Financial support was provided by the European Commission, Marie Curie Early Stage Training, MEST-CT-2005-020013 (NEURAD), Alzheimer Ph.D. Graduate School, the German Research Council, the Research program, Faculty of Medicine, Georg-August-University Göttingen and Alzheimer Forschung Initiative e.V. (to O.W.) and a Klaus Murmann-Ph.D.-scholarship from the Foundation of German Businesses (to H.B.). Disclosure statement: There are no further actual or potential conflicts of interest to disclose.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
H. Breyhan, O. Wirths and K. Duan contributed equally to this work.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Breyhan, H., Wirths, O., Duan, K. et al. APP/PS1KI bigenic mice develop early synaptic deficits and hippocampus atrophy. Acta Neuropathol 117, 677–685 (2009). https://doi.org/10.1007/s00401-009-0539-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-009-0539-7