
Abstract
Heart failure continues to be a significant global health concern, causing substantial morbidity and mortality. The limited ability of the adult heart to regenerate has posed challenges in finding effective treatments for cardiac pathologies. While various medications and surgical interventions have been used to improve cardiac function, they are not able to address the extensive loss of functioning cardiomyocytes that occurs during cardiac injury. As a result, there is growing interest in understanding how the cell cycle is regulated and exploring the potential for stimulating cardiomyocyte proliferation as a means of promoting heart regeneration. This review aims to provide an overview of current knowledge on cell cycle regulation and mechanisms underlying cardiomyocyte proliferation in cases of heart failure, while also highlighting established and novel therapeutic strategies targeting this area for treatment purposes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Heart failure is becoming a growing concern globally in industrialized societies. The prevalence of heart failure has resulted in significant mortality rates and poor prognoses for individuals affected by these conditions. Loss of cardiomyocytes, or CMs, is observed in various human heart failure including hypertensive cardiomyopathy, myocarditis, myocardial infarction (MI), and aortic stenosis. This loss ultimately leads to decreased cardiac function and potential progression to heart failure [58, 72, 93, 158]. Unlike certain amphibians and fish that can regenerate their hearts following injury [54, 111, 140], the regenerative capacity of the adult mammalian heart is limited [17, 154]. Less than 1% of adult mammalian cardiomyocytes have the ability to proliferate within one year, with this rate declining as individuals age [103]. Regrettably, lost cardiomyocytes are replaced by non-functional scar tissue that cannot be effectively restored after injury occurs [66, 141]. One of the primary reasons for the limited regenerative capacity in adult mammalian cardiomyocytes is their gradual exit from the cell cycle during maturation [68, 94]. As a result, they lose their ability to proliferate and have very restricted potential to re-enter the cell cycle.
Promoting cardiomyocytes to re-enter the cell cycle and complete cytokinesis after cardiac injury is a crucial approach to addressing heart failure. In the past four decades, numerous studies have investigated various methods of inducing cardiomyocyte cell cycle activity to enhance proliferation, including regulating cell cycle regulators, signaling pathways, non-coding RNAs (ncRNAs), and metabolism [16, 29, 45, 124]. So far, we have reached a stage of consolidation with new tools at hand, such as gene therapy and delivery methods. This review will focus on the current understanding of how cardiomyocyte cell cycle activity is regulated as a potential mechanism for cardiac repair. Based on current literature, we will also discuss targeting the cell cycle as a strategy for cardiac regeneration therapy in treating heart failure.
The mammalian cell cycle
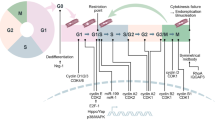
The cell cycle is the process in which a cell duplicates and divides into two daughter cells. In mammals, the cell cycle consists of G1 phase, synthesis (S) phase, G2 phase, and M phase (mitosis and cytokinesis) [65, 96]. During G1 and G2 phases, the cell grows and prepares for division [48, 143]. The S phase is when DNA synthesis occurs [22]. After interphase, the cell enters a checkpoint before progressing to mitosis [163]. Mitosis encompasses several phases including prophase, prometaphase, metaphase, anaphase telophase, culminating in cytokinesis [185]. If the cell exits the cell cycle in response to molecular cues during any part of the cell cycle, it enters the arrested quiescent G0 state.
Cellular processes involved in the cell cycle are tightly regulated. In G1 phase, cyclin-dependent kinase 4 (CDK4) and CDK6 complexes phosphorylate retinoblastoma (RB) proteins, leading to the inhibition of E2F repressor RB and increased activity of the E2F family (Fig. 1). This enhances expression of gene driving DNA synthesis and promotes cell cycle progression [145, 162]. As the cell cycle advances towards the G1 restriction point, formation of the cyclin E-CDK2 complex occurs. This complex facilitates phosphorylation of RB proteins, stimulating G1/S transition and initiating S phase [188]. Following this transition, progression through S phase is controlled by cyclin A-CDK2 complex which regulates chromosome duplication and early mitotic events [153]. While during the G2/M transition, cyclin A forms complex with CDK1 which facilitates entry into the M phase along with cyclin B-CDK1 complex [153]. In the context of myocardial proliferation, it is suggested that targeting the regulation of the cell cycle could be a potential therapeutic approach for cardiac repair. However, due to the intricate and diverse nature of myocardial cell cycle regulation, there is still a need for more comprehensive strategies to control these processes effectively.

Schematic diagram of physiological and pathological processes during cardiomyocyte cell cycle progression
In mature mammalian cells, negative regulators known as cyclin-dependent kinase inhibitors (CKIs) have an important role in regulating cell cycle activity by binding to cyclin-CDK complexes and inhibiting their functions [20]. The CKI complex consists of two distinct protein groups: INK4 family members (p14, p15, p16, p18 and p19) inhibit cyclin D function on CDK4 and CDK6 leading to cell cycle arrest in G1 phase [27, 87]; while CIP/KIP family members (p21, p27 and p57) act as broad blockers preventing progression through different phases by inhibiting various cyclin-CDK complexes [40, 170]. Thus, both activating and inhibiting factors collaborate to ensure precise regulation of the cell cycle.
Manipulation of the cell cycle to induce cardiomyocyte proliferation
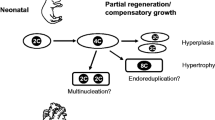
In adult mammals, cardiomyocytes are specialized cells with the majority having lost their ability to divide. After birth, most of these cardiomyocytes undergo a process where they cease cell division and become polyploid and multinucleated [7, 86, 100, 161]. Polyploid and multinucleated cardiomyocytes lose the ability to proliferate and regenerate [88]. The exit from the cell cycle can occur at various checkpoints either prior to or after mitosis [8, 23, 100]. Shortly after birth, the majority of postnatal cardiomyocytes pause at the G1/S transition phase in order to prepare for their designated functions. During this transition phase, genes related to promoting cell division such as cyclins are downregulated while levels of cell cycle inhibitors like p21 and p57 increase [4, 83, 89, 137]. Although some cardiomyocytes manage to pass through the G1/S checkpoint and proceed into mitosis, most are unable to complete chromosome separation or cytokinesis resulting in polyploidy and cell cycle exit [17, 88, 94]. While the majority of adult mammalian cardiomyocytes are unable to re-enter the cell cycle, a small subset has demonstrated proliferative potential within the adult heart [6, 17], particularly in response to injury or disease [126, 154].
In recent years, numerous studies have investigated different strategies aimed at modulating the cardiomyocyte cell cycle for enhanced proliferation. A deeper understanding of the molecular mechanisms governing this process may hold promise for developing therapies that can replenish cardiomyocyte populations and restore cardiac function following cardiac injury. In this review, we will focus on the effects on cardiomyocyte cell cycle from the following perspectives: cell cycle regulators, signaling pathways, ncRNAs, metabolic pathways, epigenetic and extrinsic factors.
In the adult mammalian heart, cardiomyocytes have the ability to re-enter the cell cycle. However, this does not result in functional regeneration after injury due to the limited number of cardiomyocytes that successfully complete the cell cycle and undergo proliferation. Therefore, most research efforts focus on identifying paracrine cues that can stimulate cardiac muscle cell growth. This is particularly challenging because the majority of cardiomyocytes in adult mammals are polyploid or multi-nucleated, unlike naturally regenerative non-mammalian vertebrates. Cell cycle regulation in mammalian cardiomyocytes involves the repression of cell cycle activators, such as cyclins, CDKs, and CDK-activating kinases (CAKs), along with CKIs [23]. This leads to cardiomyocyte cell cycle arrest during cardiac development [78]. The activity of cyclin-CDK complexes is tightly controlled and plays a crucial role in facilitating cell cycle progression through phosphorylation of downstream proteins [78].
Numerous studies have been conducted to investigate the potential reactivation of cell cycle activators in cardiomyocytes. For instance, enhancing the expression of cyclin D1 has been shown to stimulate DNA synthesis in adult mouse cardiomyocytes, resulting in more than 40% of these cells re-entering the cell cycle [161, 171]. However, it should be noted that many of these stimulated cardiomyocytes experienced M-phase arrest or endoreplication. On the other hand, constitutive cardiac expression of cyclin A2 (CCNA2) in adult mice or rats leads to increased cardiomyocyte mitosis and improved cardiac function after heart injury [32, 37, 189]. Additionally, delivering the CCNA2 gene via adenovirus into peri-infarct myocardium in pig hearts promotes cytokinesis and an approximately 18% increase in ejection fraction [157]. Similarly, researchers have explored the regulation of other cyclins or CDKs, such as exogenously increasing the expression of CDK2, cyclin D2, or cyclin B1 with constitutively activated cell division cycle 2 kinase (CDC2), to enhance adult cardiomyocyte proliferation [21, 110, 133]. Additionally, specific overexpression of cyclin D2 has been found to promote cardiomyocyte proliferation, improve cardiac function, and decrease infarct size after MI in both mice and pigs in vivo [165]. Recognizing that a single cyclin or CDK may have limited impact, Mohamed et al. recently conducted combinatorial screening and reported that ectopic overexpression of CDK1-CDK4-CCNB-CCND (4F) can increase cardiomyocyte proliferation by approximately 15% to 20% in cultured mouse and human cardiomyocytes [1, 124]. Furthermore, overexpressing 4F has shown promising results by significantly increasing cardiomyocyte proliferation and enhancing cardiac function following MI in mouse models as well as rat models of subacute heart failure [1, 124].
In contrast, several studies have aimed to promote cardiomyocyte re-entry into the cell cycle by inhibiting CKIs. One study found that p16 knockdown extends the regeneration period in neonatal mice through CDK4/6 and reactive oxygen species (ROS)-related autophagy [167]. Another study demonstrated that specifically inactivating p16 in cardiomyocytes improves heart function and reduces scar size after a myocardial infarction [148]. Additionally, triple knockdown of CKIs (p21, p27 and p57) promotes cell cycle progression into the S phase and cytokinesis in adult rat cardiomyocytes, leading to an increase in their number [40]. Recently, researchers reported that silencing cyclin L1 (CCNL1) via AAV9-cTnT-CCNL1 shRNA enhances the percentage of Ki67-positive and phospho-histone H3 (pHH3)-positive cardiomyocytes in MI mice, thereby promoting cardiac repair within the infarct zone and improving cardiac function [56]. Thus, CCNL1 appears to inhibit cardiomyocyte proliferation and indicates that cyclins can have diverse, and sometimes conflicting, functions in cardiomyocyte cell cycle regulation. A summary of these findings is depicted in Table 1.
These studies collectively indicate that promoting cardiomyocyte proliferation can be achieved by reactivating cell cycle activators or inhibiting CKIs. However, it should be noted that certain findings demonstrate that increased DNA synthesis in cardiomyocytes may lead to multinucleation or polyploidy without complete cytokinesis. Hence, additional research is required to not only understand how to facilitate cardiomyocyte re-entry into the cell cycle but also ensure successful completion of cytokinesis.
Manipulation of signaling pathways to induce cardiomyocyte proliferation
Studies have demonstrated that signaling pathways play a crucial role in inducing cardiomyocyte proliferation by influencing cell cycle regulators. These intricate molecular networks interact with each other to ensure precise control of the cell cycle in response to internal or environmental cues. Signaling pathways coordinate cellular responses and collaborate with transcription factors to regulate various cellular processes. Numerous pathways and proteins have been identified as key players in regulating cardiomyocyte proliferation following cardiac injury.
Hippo signaling pathway is evolutionarily conserved to control organ size and development by controlling proliferation, apoptosis, viability and differentiation [62, 130, 200]. The Hippo pathway in mammals comprises core components sterile 20-like protein kinases 1 and 2 (MST1/2), Salvador homolog 1 (Sav1), large tumor suppressors 1 and 2 (LATS1/2), MOB kinase activator 1A and 1B (MOB1A/1B), the two Yorkie homologs Yes-associated protein (YAP) and transcriptional coactivator with PDZ-binding motif (TAZ), and TEA domain family member (TEAD) [199]. Recent findings have revealed that Hippo pathway is essential for mammalian cardiomyocyte regeneration, along with its downstream effector Yes-associated protein (YAP)/ transcriptional co-activator with PDZ-binding motif (TAZ) [70, 112, 193]. Overexpression of YAP extends neonatal cardiomyocyte cell cycle activity and promotes proliferation [179], whereas cardiomyocyte-specific deletion of Yap impairs regenerative responses and the hearts display extensive cardiac fibrosis [193]. Based on the importance of YAP in regeneration, pharmaceutical interventions targeting YAP activity have significant potential for the treatment of heart failure. Nathaniel Kastan et al. have screened a small molecule inhibitor, TRULI, which can reversibly activate YAP and promote proliferation in postnatal day 0 (P0) murine cardiomyocyte in vitro [84]. Consistent with these results, cardiomyocyte-specific knockout of Sav, MST1/2 or LATS2 in mouse hearts also increases cardiomyocyte proliferation in the neonatal heart after adult myocardial infarction as well as after P8 myocardial infarction [70, 71]. Furthermore, knockdown of the Sav in border zone cardiomyocytes in pigs 2 weeks after I/R can significantly promote cardiomyocyte proliferation, reduce scar size and improve cardiac function without any tumor formation [114]. Taken together, Hippo pathway has great potential to be the clinical target for heart failure.
Phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT) signaling pathway serves as one of the key mechanisms in cell cycle progression through regulating p21 and p27, cyclin D1 and CDK2 [118, 122, 132]. Studies in cardiomyocytes have also shown that PI3K/AKT pathway plays an important role in cardiomyocyte proliferation [121, 159]. Specific nuclear overexpression of AKT in mouse heart results in an increase in smaller cardiomyocytes, prolonged cell cycling and enhanced cardiac contractility [60, 151]. Mechanistically, PI3K/AKT pathway induces cardiomyocyte proliferation by coordinating with other signaling pathways to regulate downstream molecules. For example, acting as a downstream effector, PI3K subunit beta (PIK3CB) has been identified to mediate the effect of YAP on the activation of the PI3K-AKT pathway and thus the induction of CM proliferation [113]. As an upstream regulator, AKT phosphorylates and inhibits glycogen synthase kinase-3β (GSK-3β), which also plays central role in regulating Wnt signaling pathway. Deletion of GSK-3β in mice causes upregulated expression of GATA4, cyclin D1 and c-Myc, leading to cardiomyocyte hyperproliferation [85]. Cardiomyocyte-specific knockout of GSK-3β significantly promotes cardiomyocyte proliferation and preserves the cardiac function in mice after MI [202].
In addition, Jiang et al., have also shown that the Wnt/β-catenin signaling pathway can stimulate cardiomyocyte proliferation and cardiac development [81, 175]. In their study, activation of β-catenin decreases the ploidy and nuclear number of cardiomyocytes both in vitro and in vivo in mice, suggesting that β-catenin could promote cell division completion of polyploid and multinucleated cardiomyocytes. Similarly, overexpression of serine/threonine‐protein kinase 3 (SGK3) or its direct activator CDK9 can promote cardiomyocyte proliferation mainly through inactivating GSK-3β and upregulating β-catenin expression. As new potential therapeutic targets, overexpression of SGK3 or CDK9 after myocardial injury in mouse in vivo promotes cardiac repair and aims to partially restore the cardiac function [107, 166]. In addition, low-density lipoprotein receptor-related protein 5 (LRP5) and low-density lipoprotein receptor-related protein 6 (LRP6), co-receptors of Wnt signaling, are key regulators of cardiomyocyte cell cycle activity. Cardiac-specific knockout of LRP5 inhibits myocardial regeneration in the mouse after injury, whereas specific deletion of LRP6 in cardiomyocytes induces robust regeneration in mouse heart after MI and promoted recovery of cardiac function [191, 206].
The neuregulin-1 (NRG1)/ErbB signaling pathway, consisting mainly of growth factor NRG1 and its tyrosine kinase receptors ErbB2 and ErbB4, has been demonstrated to regulate cardiomyocyte proliferation [205]. Injection of recombinant NRG1, cardiac-specific overexpression of ErbB4 or transient overexpression of activated ErbB2 can all promote adult cardiomyocyte proliferation in mouse in vivo. Further studies in mouse disease models show that exogenous NRG1 or activated ErbB2 can induce cardiac regeneration, reduce the scar size and improve cardiac function by activating downstream PI3K or YAP after cardiac injury [3, 19].
The Notch signaling pathway regulates the activity of bone morphogenetic protein 10 (BMP10) and expression of NRG1, therefore it is essential for cardiomyocyte proliferation [55, 59]. Inhibition of Notch pathway impairs cardiac regeneration in zebrafish [204]; loss of BMP10 in mouse also leads to elevated expression of p57 and inhibited cardiomyocyte proliferation [33]. Consistently, activation of Notch pathway induces expression of cyclin D1 and cell cycle re-entry in quiescent neonatal murine cardiomyocytes [26]. However, hyperactivation of Notch signaling also blocks cardiomyocyte proliferation in zebrafish [204]. These findings indicate that the activity of targeted signaling pathway should be precisely controlled if it is to be used as therapeutic targets for cardiac regeneration. Summary of the information is shown in Table 2.
Apart from these canonical signaling pathways, many genes have been shown to promote cardiomyocyte re-entry into cell cycle, leading to cardiomyocyte proliferation. For example, overexpression of E2F1 or E2F2 in mouse heart induces cardiomyocyte cell cycle re-entry through regulating the expression of CDK4 or cyclin A and E respectively. However, overexpression of E2F1 leads to apoptosis [2], whereas E2F2 induces hypertrophic cell growth in cardiomyocytes [43]. Another important family of transcription factors in cardiomyocyte proliferation is T-box (TBX) gene family. Overexpression of TBX20 or TBX6 in adult mouse heart induces cardiomyocyte proliferation by regulating the expression of multiple cell cycle regulators, TBX20 overexpression also promotes cardiac repair and improves heart function in mouse after MI [61, 192]. A recent study reports that double knockdown of RB1 and Meis homeobox 2 (MEIS2) increased proliferation rate of human induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs) in vitro and in rat in vivo [5]. Similarly, double knockout of Meis homeobox 1 (MEIS1) and HOXB13 in mouse in vivo can promote cardiomyocyte proliferation and improve cardiac function after MI [127]. As a deacetylase of p21, sirtuin1 (SIRT1) has been shown to positively regulate cardiomyocyte proliferation and protect cardiac function in mouse in vivo after MI via regulating p21 [99]. In addition, the anti-inflammatory cytokine interleukin 13 (IL-13) and the critical subunit of its type II receptor interleukin4Rα (IL4Rα) are also involved in regulating cardiomyocyte cell cycle and heart regeneration in neonatal mice [129, 187]. Another cytokine oncostatin M (OSM) and its receptor heterodimers oncostatin M receptor (OSMR)/glycoprotein 130 (gp130) are essential for cardiomyocyte proliferation, when activated can improve heart regeneration and function in mouse in vivo after MI [106]. Moreover, nuclear lamina filament Lamin B2 can promote cardiomyocyte M-phase progression and cytokinesis in neonatal mice [63]. Furthermore, knockout of the RNA binding protein muscleblind-like 1 (MBNL1) can promote cardiomyocyte proliferation in mouse heart after MI [14], and osteopontin (OPN) was found to enable cardiomyocyte re-entry into cell cycle in mouse heart, but also to stimulate other cells to improve scar formation and left ventricular remodeling [152].
Manipulation of non-coding RNAs to induce cardiomyocyte proliferation
The ncRNAs are the functional RNAs without protein coding ability, including microRNA (miRNA), long non-coding RNA (lncRNA) and circular RNA (circRNA). These ncRNAs act as regulators to coordinate gene expression at the epigenetic, post-transcriptional, and translational levels [31]. A large number of ncRNAs have been reported to be associated with cardiomyocyte cell cycle regulation and cardiomyocyte proliferation [201].
miRNAs are small RNAs that bind to 3'-untranslated regions (3'-UTR) of mRNAs, leading to the degradation or repression of targeted mRNAs. Recent studies have shown that a range of miRNAs can induce cardiomyocyte proliferation by targeting the cell cycle regulators. A high-throughput functional phenotypic screen by using a whole-genome miRNA library has identified miR-199a, miR-302b, miR-518, miR-590 and miR-1825 among 204 potential human miRNAs that can increase 5-ethynyl-2'-deoxyuridine (EdU)-incorporation, Ki67, and pHH3 in mouse or rat cardiomyocytes [45]. Moreover, miR-199a and miR-590 have been linked to the activation of cell cycle induction and progression, resulting in the enhancement of cardiomyocyte proliferation in vitro and in vivo [45]. Further studies show that miR-199a directly targets two mRNAs, the upstream YAP inhibitory kinase TAOK1 and the E3 ubiquitin ligase β-TrCP, leading to YAP degradation [174]. Also, expression human microRNA-199a AAV serotype 6 (AAV6) in infarcted pig hearts can rescue and repair cardiac function via stimulating endogenous cardiomyocyte proliferation [53]. Another independent proliferation screen has been performed in hiPSC-CMs, and 96 miRNAs have been identified as drivers of DNA synthesis and cell division in cardiomyocytes, and 67 of the 96 miRNAs stimulate cardiomyocyte proliferation in a YAP-dependent manner [41]. Overexpression of miR-25 can increase the percentage of EdU and Ki67 positive cardiomyocytes by targeting FBXW7 and regulating cell cycle genes in human embryonic stem cell derived cardiomyocytes, as well as in zebrafish [180]. miR-106b ~ 25 cluster stimulates cardiomyocyte regeneration by targeting negative cell cycle regulators including E2F5, CDKN1C, CCNE1 and WEE1. Exogenous viral delivery of miR-106b ~ 25 induces cardiac regeneration and restores heart function after MI [146]. In addition, overexpression of miR-204 promotes cardiomyocyte proliferation in rat in vitro and mouse in vivo by targeting Jarid2 thus leading to the upregulation of cell cycle regulators Cyclin A, Cyclin B, Cyclin D2, Cyclin E, CDC2 and PCNA [109]. miR-499, a myocyte-specific miRNA (myomiR) expressed within one of the introns of β-myosin heavy chain (Myh7b) gene, can promote neonatal cardiomyocyte proliferation through regulating cyclin D1 and SRY-Box Transcription Factor 6 (Sox6) [104]. Similarly, miR-1825 is able to induce both DNA synthesis and cytokinesis in adult rat cardiomyocytes and improve cardiac function in vivo following MI, possibly by regulating the expression levels of miR-199a and its downstream targets RB1 and MEIS2 [131]. The studies provide the potential targets for gene therapy for cardiac regeneration in human patients.
In contrast, a large number of miRNAs have recently been identified that suppress the cell cycle in cardiomyocytes, such as miRNA let-7i-5p, miR-1/133a, miR-15 family, miR-26a, miR-29a/b, miR-34a and miR-128. miRNA let-7i-5p inhibits cardiomyocyte proliferation via targeting E2F2 and CCND2, while inhibition of let-7i-5p promotes mouse cardiomyocytes proliferation and enhance cardiac function after MI [74]. Similarly, miR-1/133a can prevent re-entry of adult rat cardiomyocytes into the cell cycle by suppressing FGFR1 and OSMR, while its inhibition promotes cardiomyocytes proliferation in mouse in vivo [176]. The miR-15 family (including miR-15a, miR-15b, miR-16-1, miR-16-2, miR-195 and miR-497), modulates cardiac regenerative capacity in neonatal mice and adult myocyte proliferation by regulating cell cycle genes and mitochondrial genes [139]. Overexpression of miR-195 impairs the cardiomyocyte proliferation and regenerative capability of P1 mouse heart after MI by repressing a number of cell cycle genes, including checkpoint kinase 1 (Chek1) in vivo [138, 139]. Inhibition of miR-26a enhances neonatal mouse cardiomyocyte proliferation through the regulation of cell cycle inhibitors in vitro and in vivo [39]. Overexpression of miR-29a suppresses cardiomyocyte proliferation, while its inhibition promotes cell division via upregulating the expression level of cyclin D2 in neonatal rat cardiomyocytes [28]. Similarly, inhibition of miR-29b promotes cardiomyocyte proliferation by targeting notch receptor 2 (NOTCH2) in mouse in vitro and zebrafish in vivo [195]. In addition, inhibition of miR-34a also results in enhanced cardiomyocyte proliferation and improved cardiac function after MI by regulating silent information regulator factor 2 related enzyme 1 (Sirt1), B-cell lymphoma 2 (Bcl2) and Cyclin D1 [196]. Likewise, inhibition of miR-128 promotes cardiomyocyte proliferation and improves cardiac function in response to MI through activating cyclin E and CDK2 [76].
lncRNAs are noncoding RNA molecules with nucleotides longer than 200, functioning through interactions with DNA, RNA or proteins. Similar to miRNAs, lncRNAs also engaged in the process of cardiomyocyte proliferation. As evidence, endogenous cardiac regeneration-associated regulator (ECRAR) was discovered to provoke myocardial regeneration and heart repair in rat in vivo after MI by activating cyclin D1 and cyclin E1 through E2F1-ECRAR-ERK1/2 signaling [35]. In addition, the downstream target of RNA-binding protein LIN28a, long noncoding RNA-H19, mediated the reprogramming of cardiomyocyte metabolism and enhancing of cell cycle activity, thereby protecting mouse heart from MI [150]. Conversely, cardiomyocyte proliferation regulator (CPR), LncDACH1 and natriuretic peptide A antisense RNA 1 (NPPA‑AS1) are recently reported to negatively regulate cardiomyocyte proliferation through binding and interacting with DNMT3A, PP1A/YAP1, splicing factor proline and glutamine rich (SFPQ), respectively. Deletion of CPR, LncDACH1 or NPPA-AS1 can restore cardiac function by promoting cardiomyocyte regeneration in mouse after MI [25, 52, 136]. These studies suggested lncRNA-based approaches as potential therapeutic treatments for heart failure.
circRNAs are stable RNA molecules formed closed loop structure by back-splicing. Although circRNAs are reported to be involved in regulation of many biological processes, the current scientific evidence supporting the role of circRNAs in regulating the cardiomyocyte cell cycle remains scarce. In the past years, only a few studies linked cicrRNAs to cardiomyocyte proliferation: overexpression of circNfix repressed cyclin A2 and cyclin B1 expression by inducing Y-box binding protein 1 (YBX1) ubiquitin-dependent degradation; it also increased miR-214 activity to promote GSK3β expression thus inhibiting cardiomyocyte proliferation. In contrast, downregulation of circNfix facilitated mouse heart regeneration and repair after MI [75]. Similar to circNfix, circMdc1 plays negative role in cardiomyocyte proliferation by blocking translation of MDC1 and when silenced can improve cardiomyocyte regeneration and heart function in vivo after injury [117]. The exploration of circRNAs in heart regeneration is still in the early stage, and deeper understanding of the molecular mechanisms underlying is required. Summary of the information is shown in Table 3.
Although initially considered as junk RNAs, more and more evidence highlights the indispensable role of ncRNAs in cellular processes including cardiomyocyte proliferation. Discovering potential ncRNAs targets in cardiac regeneration and understanding the mechanisms will help developing innovative strategies for heart regeneration.
Manipulation of metabolism to induce cardiomyocyte cell cycle
During maturation, mammalian cardiomyocytes gradually exit the cell cycle and lose their regenerative potential, accompanied by a metabolic shift from glycolysis to fatty acid oxidation to meet the energy requirements of adult cardiomyocytes [115]. This highlights the critical role of cardiac metabolism in cardiac regeneration and further studies have identified potential metabolic targets that can be regulated to promote cardiomyocyte proliferation.
The metabolic shift during maturation suggests a negative correlation between cardiac regeneration and fatty acid oxidation, and a positive correlation with glycolysis. Despite the energetic advantage for adult cardiomyocytes, the increased production of ROS resulting from fatty acid oxidation can induce DNA damage and activate DNA damage response pathway, thus contributing to cardiomyocyte cell cycle arrest [123]. As reported by Cardoso et al., conditional knockout of pyruvate dehydrogenase kinase 4 (PDK4) in cardiomyocytes, which results in increased glucose relative to fatty acid oxidation, is able to reduce DNA damage and promote cardiomyocyte proliferation as well as cardiac function in response to MI in mouse in vivo [29]. In addition, inhibition of succinate dehydrogenase (SDH) by malonate promoted mouse cardiac regeneration after MI through stimulating metabolic switch to glycolysis [12]; knockdown of acyl CoA synthase long chain family member 1 (ACSL1), the key enzyme regulating lipid metabolism, can effectively induce myocardial regeneration after MI [108]. Recent study shows that blocking fatty acid oxidation in mouse cardiomyocytes by genetically deletion of carnitine palmitoyltransferase 1b (Cpt1b) reshapes epigenetic landscape thereby promoting cardiomyocyte proliferation and enabling cardiac regeneration after injury [105]. These studies further demonstrate that inhibition of the fatty acid utilization or acceleration of glycolysis can promote cardiomyocyte proliferation and provide potential therapeutic targets for heart failure.
Further, it has been reported that overexpression of pyruvate kinase muscle isozyme M2 (PKM2), an enzyme involved in the final step of glycolysis, using a novel cardiomyocyte-targeting strategy with modified RNA in mouse in vivo after MI can successfully promote cardiomyocyte cell division thereby improving cardiac function and long term survival [119]. Controversially, another study has shown that PKM2 inactivation in mouse heart promotes cardiomyocyte proliferation in the infarct zone after MI, thus indicating the opposite antiproliferative function of PKM2 [69]. Although more experiments are needed to clarify the function of PKM2, both of the studies show that PKM2 interacts with β-catenin to regulate cardiomyocyte proliferation, highlighting the crosstalk between metabolism and signaling pathway.
Epigenetic regulation of cardiomyocyte cell cycle
In addition to the metabolic shift, an increase in global methylation was observed during rat heart development, suggesting a potential role of epigenetic modification in cardiomyocyte differentiation and proliferation [90]. As DNA methylome and histone modifications can regulate the chromatin condensation, resulting in differential chromatin accessibility and ultimately affecting the gene expression, epigenetics play an essential role in molecular networks and biological processes. In recent decades, increasing evidence has linked epigenetics to cardiomyocyte cell cycle re-entry.
N6-methyladenosine (m6A) is one of the most common modifications of messenger RNAs (mRNAs), and recent studies have shown that targeting m6A methylation can efficiently regulate cardiomyocyte re-entry into the cell cycle [64, 102, 207]. The knockdown of methyltransferase-like 3 (METTL3) has also been shown to reduce m6A-mediated pri-miR-143 maturation into miR-143-3p, thus promotes cardiac regeneration after MI through miR-143-YAP/CTNND1 (catenin delta-1) axis [57]; whereas forced expression of m6A demethylase ALKBH5 also promotes cardiac regeneration after MI in mice and CM proliferation in hiPSC-CM by improving the mRNA stability of YTH N6-methyladenosine RNA-binding protein 1 (YTHDF1) and consequently promoting the translation of YAP [64]. In addition, deletion of abraxas brother 1 (ABRO1) improved cardiac regeneration and mouse heart function after MI by targeting METTL3-mediated m6A methylation of PSPH mRNA and downstream CDK2 [182].
In contrast, mitochondrial transmembrane protein 11 (TMEM11)-mediated N7-methylguanosine (m7G) methylation negatively regulates cardiomyocyte proliferation and cardiac function in mouse in vivo after MI via targeting TMEM11-methyltransferase 1, tRNA methylguanosine (METTL1)-activating transcription factor 5 (ATF5)-inhibitor of CDK, cyclin A1 interacting protein 1 (INCA1) axis[34]. Several potential epigenetic targets for cardiomyocyte cell cycle regulation have also been recently reported. As shown by Paola Cattaneo et al., DOT1L was involved in cardiomyocyte cell cycle exit by catalyzing H3K79me2 and subsequently regulating transcriptional networks [30]. Moreover, knockdown of dual-specificity tyrosine regulated kinase 1A (DYRK1A) can enhance cardiomyocyte cell cycle activity via epigenetic modification of H3K4me3 and H3K27ac [95]. Furthermore, ablation of chromobox 7 (CBX7), a polycomb group (PcG) protein, can promote cardiac regeneration in both neonatal and adult injured hearts by targeting TAR DNA-binding protein 43 (TARDBP) and RNA binding motif protein 38 (RBM38) [38].
Taken together, the evidence to date highlights the importance of chromatin structure in cardiac cell cycle and identifies key factors with potential therapeutic value.
Extrinsic factors driving the cardiomyocyte cell cycle
In the cellular context, cardiomyocytes reside in a complex and dynamic microenvironment with multiple cell types. Although the cardiomyocytes are the primary population and are responsible for the contractile function in the heart, proper cardiac function requires the cooperation of various cell populations. Recently, a number of extrinsic factors affecting cardiomyocyte cell-cycle arrest has recently been identified, including the oxygen concentration, extracellular matrix and the immune system.
The postnatal high oxygen level has been reported to be an important driver of mammalian cardiomyocyte cell cycle arrest after birth, mainly through the increase of reactive oxygen species (ROS) generated by the increased mitochondrial oxidative metabolism accompanied by a temporal shift from glycolytic to oxidative metabolism and the subsequent DNA damage response (DDR) [144]. On top of which, hypoxia or ROS scavenging can delay the cell cycle exit, thus prolonging the postnatal proliferative window of cardiomyocytes [144]. In adult mouse, gradual exposure to severe systemic hypoxemia (O2 is gradually decreased by 1% and maintained at 7% for 2 weeks) induces cardiomyocyte hypertrophy and promotes right ventricular cardiomyocyte proliferation [82], while in MI mouse, it promotes cardiac regeneration and thus improves cardiac function [126]. However, in the fetal rat heart, hypoxia (1% O2) inhibits cardiomyocyte proliferation [172]. In addition to this, maternal hypoxia (10.5% O2) decreases cardiomyocyte proliferation in both fetal and neonatal rat hearts [173]. The role of hypoxia in cardiomyocyte cell cycle regulation remains controversial. As shown by Ye et al., only the moderate range of hypoxia can promote cardiomyocyte cell cycle activities in both hiPSC-CMs and human heart samples [197]. They also showed that hypoxic treatment (10% O2) promotes the proliferation of neonatal rat cardiomyocytes, but inhibits cell cycle activities of fetal cardiomyocytes, suggesting that the role of hypoxia is related to development stage [168]. Taken together, the level of hypoxia and the phase or duration of cardiomyocytes experiencing hypoxia are key factors in regulating cardiomyocyte cell cycle.
The extracellular matrix (ECM), including structural proteins, matricellular proteins and carbohydrates, provides structural support to cells in the heart and mediates the cellular interactions, thus playing a vital role in regulating cell events [18, 49, 50]. A growing body of evidence suggests that the composition and rigidity of ECM are critical for the regulation of cardiomyocyte proliferation and cardiac regeneration [128, 164, 181, 183, 190]. It has been reported that the ECM proteins secreted by mouse embryonic cardiac fibroblasts including fibronectin, collagen and heparin-binding EGF-like growth factor, promote cardiomyocyte proliferation through β1 integrin and cell cycle-related genes [77]. Another separate study comparing the rat embryonic and postnatal cardiac fibroblast-derived ECM found that two embryonic ECM proteins SLIT2 and nephronectin (NPNT) can promote cardiomyocyte cytokinesis both in vitro and in vivo [190]. Individual ECM protein is also able to induce cardiac regeneration. For example, periostin has been shown to facilitate cardiomyocyte cell cycle re-entry and mitosis, thereby improving cardiac function after MI, in rat via activation of integrins and PI3K pathway [92]. However, the function of periostin in mouse remains controversial. In neonatal mice, knockout of periostin inhibits post-MI cardiac regeneration [36]; whereas in adult mice both overexpression and knockout periostin show no change in cardiomyocyte proliferation in the peri-infarct area [116]. Agrin, found in neonatal ECM, is able to drive hiPSC-CM proliferation and promote post-MI cardiac regeneration, thereby improving cardiac function via Yap signaling in both adult mice and pigs [13, 15]. Recently, the versican, a cardiac fibroblast–derived ECM protein, has been reported to promote hiPSC-CM proliferation. Intramyocardial injection of versican after MI results in reduced fibrosis and increased cardiomyocyte proliferation along with improved cardiac function in adult mice through activation of β1 integrin and downstream ERK1/2 and Akt [47]. These studies highlight the clinical potential of targeting ECM components for the treatment of heart failure. Meanwhile, ECM stiffness is also involved in the regulation of cardiomyocyte proliferation. As reported, compliant elastic matrices promote mouse cardiomyocyte dedifferentiation and cytokinesis in mice via regulating organization of the myoskeleton [194]. Correspondingly, decreased stiffness of ECM achieved by pharmacological inhibition of cross-linker enzyme lysyl oxidase (LOX) preserves the cardiac regenerative capacity in 3-day-old mice and enhances post-injury cardiac regeneration which induced by exogenous fetal ECM through increasing YAP localization to nuclei [128, 183].
In addition to fibroblast and ECM, the cardiac microenvironment consists of various cell types such as immune cells, adipocytes, vascular cells and neurons. Immune cells play an essential role in remodeling and inflammation after cardiac injury, and recent studies suggest that they also play an important role in cardiomyocyte proliferation. Specific ablation of CD4+ T-cells after MI by monoclonal antibody promotes cardiomyocyte proliferation in juvenile mice but not in adult mice [101]. Consistent with this, infarcted 1-day-old mice reconstituted with adult T cells have more monocyte-derived macrophage recruitment and show irreversible impairment of cardiac function along with increased fibrosis[42]. Furthermore, deletion of macrophages in neonatal mice blocks cardiac regeneration after MI [11]. Although immune cells show significant impact in regulating cardiomyocyte proliferation, the mechanisms involved remain unclear. Therefore, further efforts are needed to link immune cells to cardiomyocyte cell cycle. In addition, human adipose-derived stromal cells primed under hypoxic and pro-inflammatory conditions can facilitate the proliferation of rat neonatal cardiomyocytes via secreted interleukin-6 (IL-6) and activation of downstream Janus kinase-signal transducer and activator of transcription (JAK/STAT) and mitogen-activated protein kinases (MAPK) mitogenic pathways [142].
To date, studies of extrinsic factors show great clinical promise for the treatment of heart failure and highlight the importance of cardiac microenvironment which must be carefully considered when trying to develop interventional therapeutic strategies of cardiac regeneration.
Different approaches of monitoring cardiomyocyte proliferation rate
Adult mammalian cardiomyocytes are terminally differentiated cells with limited mitotic activity. Similar to many terminally differentiated cell types, most of adult mammalian cardiomyocytes are polyploid or multinucleation [88, 97, 120]. Multiple genome copies resulted by polyploidization may enable cardiomyocytes to acquire elevated energy level thus increasing contractile force and enhance the tolerance to stress [9, 24, 97]. The polyploidization is induced by two different issues, karyokinesis failure and cytokinesis failure, during cell cycle [88]. Karyokinesis failure during which cell exit cell cycle without nuclear division results in polyploidy in single nuclear. While cytokinesis failure after successful karyokinesis generates multinucleated cell [24, 98].
Although a small proportion of the cardiomyocytes are able to enter cell cycle, the majority fail to complete karyokinesis or cytokinesis leading to polyploidization instead of authentic division into two new daughter cells [134]. Thereby, cardiac regeneration requires precise control of cardiomyocyte cell division and polyploidization. Even though numerous studies in the adult mammalian context have claimed successful regulation of cardiomyocyte re-entry into the cell cycle, the proliferation markers and indicators used have obvious limitations in identifying authentic cell division from polyploidization.
A commonly used approach is to measure the DNA synthesis activity by using thymidine analogs, such as bromodeoxyuridine (BrdU) or EdU, which is incorporated into the replicating DNA during S phase. Other common markers such as the nuclear protein Ki67 which present from G1 to M phase, and pHH3, present from G2 to M phase and responsible for chromatin condensation, are all limited in that they do not indicate the completion of cytokinesis [8]. Thus, these assays could detect the DNA synthesis, but could not distinguish between the multinucleation, polyploidization, or de novo generation of cardiomyocytes. However, cardiomyocyte DNA replication often results in a polyploid cardiomyocyte and only rarely leads to a new cardiomyocyte by cell division. Besides, Aurora B is a quantifiable marker for cardiomyocytes that is ultimately localized to the cleavage furrow during cytokinesis [10]. However, it also has significant shortcomings. On the one hand, it is a very rare event with a short window of opportunity, only about 0–0.04% of adult mammalian cardiomyocytes are Aurora B positive as shown by in vivo studies [184, 192]. The extremely low proportion of Aurora B-positive cardiomyocytes makes it extremely difficult to quantify the proliferation rate. The complexity of cardiac tissue also makes it difficult to identify the cytokinetic furrow and associated cells in vivo. On the other hand, Aurora B-positive cardiomyocytes may also fail to complete cytokinesis, leading to multinucleation, as indicated by the position of midbody and the distance between the daughter nuclei [44, 73]. An additional marker, anillin, is necessary in this setting to assess if cardiomyocytes form multi-nucleated cells or complete the cell cycle through formation of daughter cells. Multinucleation is characterized by the asymmetric constriction of the cleavage furrow and defective midbody formation, as indicated by Aurora B and Anillin colocalization [44, 98]. In conclusion, each of the markers used so far has served as an indicator of cell cycle activity, but none of them alone can be considered as definitive indicators of authentic cell division but not polyploidization in cardiomyocytes.
To date, many transgenic reporter mice have been generated based on tracking these proliferative indicators for real-time cell cycle monitoring. Kretzschmar et al. generate a mouse model to label Ki67 in cycling cells with red fluorescent tdTomato [91]. In addition, Raulf et al. generate the double transgenic Myh6-H2B-mCherry/CAG-eGFP-Anillin transgenic mouse. By labeling cardiomyocyte nuclei with red fluorescence and Anillin with green fluorescence, the mouse model is able to show the kinetic localization of Anillin and thus indicate the proliferation status of cardiomyocytes [147]. Moreover, an Aurora B reporter–based mouse system with a tdTomato fluorescence labeling to monitor the proliferating cardiomyocytes has also been generated [51, 80]. However, these models using a single proliferative marker is not sufficient to quantify the authentic cytokinesis in cardiomyocytes.
In addition to these mouse systems, fluorescent ubiquitination-based cell cycle indicator (FUCCI) system has been specifically developed to study cell cycle dynamics and the cardiomyocyte proliferation. Cardiomyocyte-specific FUCCI system consists of two different fluorescent reporters driven by the α-myosin heavy chain (αMHC) promoter and relies on the ubiquitination and degradation of DNA replication factor Cdt1 and Geminin [8]. The FUCCI system allows the assessment of cycling and non-cycling cells, but the approach is limited by the rare activation of cycling cardiomyocytes in adult hearts after cardiac injury and the inability to label daughter cells to identify endoreplicative and authentic cell division in vivo. In addition, another mouse model, mosaic analysis with double markers (MADM) system, has been developed to label cardiomyocytes that have completed cytokinesis. MADM system requires introduction of two reciprocal chimeric marker genes, each consisting of part of green and red fluorescent protein coding sequence separated by loxP site, to identical loci on homologous chromosomes. Based on Cre-mediated recombination between loxP sites, 4 types of daughter cells are possible after mitosis including single-colored green, single-colored red, double colored yellow and colorless cells [208]. If mitotic recombination occurs during G0 or G1 phase, a double-labeled cell is produced without changing the genotype of the cells, only completed cytokinesis is capable of producing single-colored daughter cells, making MADM a gold standard for lineage tracing in cardiomyocyte proliferation. Despite potential limitations of this model, such as Cre combination efficiency and the individual variability, MADM remains a powerful tool for investigating cardiomyocyte proliferation.
In conclusion, these newly developed systems allow distinguishing between authentic cell division and polyploidization thus offering an opportunity to validate previous research on the proliferation of cardiomyocytes. However, it is important to acknowledge the limitations of using only proliferative markers and address potential challenges by utilizing a combination of different approaches.
Challenges in cardiac regeneration research
The ultimate goal of improving cardiac regeneration is to repair damaged heart and improve cardiac function by increasing the number of cardiomyocytes. Although much effort has been put into understanding cell cycle regulation and gene or cell therapy to induce cardiomyocyte proliferation, therapeutic cardiac regeneration has not yet been achieved. Before translation, there are still several conceptual and technical challenges needed to be addressed to ensure the safety and efficacy of the gene therapy for the treatment of heart disease.
Most studies to date have only utilized in vitro or small rodent models to study cardiomyocyte cell cycle regulation. However, despite their homology, there are significant differences between human and rodent genomes. The mouse genome is approximately 14% smaller than the human genome with approximately 20% of genes having no identified orthologue in human genome, especially non-coding genes [125]. To bridge the gap between rodents and humans and to advance preclinical translational studies for cardiac regeneration, large animal models, such as dogs, pigs, sheep and non-human primates are an indispensable step in translating basic research into clinical applications. So far, only the CCNA2, CCND2, YAP, Sav and miR-199a have been reached in pig studies [13, 53, 114, 157, 165]. Therefore, further investigations are needed to validate the potential therapeutic effects of the targets in large animal models before clinical translation. However, also need to be noted that, large animals such as pigs also have obvious limitations as preclinical research models for human translational medicine. Despite the similarities in terms of organ size and physiology of pig and human, they are different in heart development, anatomy and genome, especially ncRNAs [135, 156, 178]. Thus, the impact of regenerative strategies being tested in the pigs needs to be considered, but the use of the pig system for studies of cardiac regeneration still offers inspiring and exciting possibilities to improve clinical translation to humans.
In addition, the molecular cell cycle control system of cardiomyocytes involves proto-oncogenes and tumor suppressors, and regulation of these genes to induce cell cycle re-entry may result in uncontrolled cardiomyocyte proliferation and even tumorigenesis, and also may lead to cardiac dysfunction in the long term [67, 177]. For example, transient overexpression of Oct4, Sox2, Klf4, and c-Myc (OSKM) in adult mouse cardiomyocytes induces cell cycle re-entry and promotes non-tumorigenic cardiac regeneration, whereas prolonged expression of OSKM leads to cardiac tumor formation [36]. Despite promising improvement of gene therapy in cardiac regeneration, these studies have been limited by tumor formation, strong immunological response, and high mortality rate. Thus, the precise control of cardiomyocyte proliferation with respect to reversibility is required to prevent tumor formation and maintain cardiac function, and is critical for the advancement of gene therapy in the clinical setting.
In addition, the adult cardiomyocytes tend to fail karyokinesis or cytokinesis resulting in polyploidy or multinucleation during the cell cycle. Studies have shown that the transcription levels are different between the diploid and polyploid cardiomyocytes [186, 198]. Furthermore, recent studies have shown that polyploid and multi-nucleated cardiomyocytes are also able to re-enter into cell cycle and complete cell division [53, 169]. Therefore, it is important to investigate the molecular mechanisms regulating cardiomyocyte cell division and polyploidization in cardiac regeneration prior to clinical translation studies. Currently the single-cell RNA sequencing (scRNA-seq) can potentially characterize the individual cells and elucidate the biological mechanism at the cellular level, but it still does not have the sufficient depth to reveal all differences at the single cell level. Thus, the further studies including electromechanical coupling and contractile function are needed, and new tools also need to be developed.
Moreover, the efficiency of RNA delivery directly related to the efficacy of gene therapy. Therefore, the specific while efficient gene delivery to the heart is fundamental for the clinical application of cardiomyocyte regeneration. Most of the studies discussed above have relied on adeno-associated virus (AAV), ultrasound-targeted microbubble destruction (UTMD) which combines ultrasound-mediated delivery with microbubbles, exosome-mediated delivery or nanoparticle delivery [46, 149, 155, 203]. However, RNA delivery has the limitation of low gene transduction, and have resulted in orthotropic delivery of therapeutics into the myocardium via catheter or intracardiac injection, limiting the potential accessibility of such therapeutics [79]. Therefore, optimization of the delivery method and reduction of the off-target effects for gene therapy need to be addressed in the further investigations.
Finally, most studies are performed in young and healthy animals. However, heart failure in humans mostly affects older adults. Aging is also associated with an increase in deaths from heart disease [160]. Therefore, it is really important to know if the regenerative therapy can induce cardiomyocyte proliferation in aged animals as it does in young adults. Besides, experiments in chronic heart failure models are also required. Although in vivo studies show cardiomyocyte regeneration, almost all in vivo studies have been performed in the acute stage of MI. It remains unknown whether the therapy could be applied to chronic heart failure models. Therefore, the current regenerative therapy in the aged animals and chronic heart failure models need to be addressed before clinical setting.
In summary, although many challenges remain, the clinical application of cardiomyocyte proliferation induction by regulating cell cycle related targets may offer a bright future for the treatment of heart diseases.
Conclusion
The massive loss of cardiomyocytes after cardiac injury and their subsequent replacement by fibrotic tissue is the major cause of heart failure, while the cardiomyocytes have limited proliferative capacity. Therefore, inducing resident cardiomyocytes to re-enter the cell cycle and progress through mitosis and cytokinesis is the most physiological approach to achieve cardiomyocyte regeneration with improved cardiac function in the diseased heart. In this review, we have discussed several aspects targeting cardiomyocyte regeneration via cell cycle regulation. While research in these areas is promising, challenges remain in controlling authentic cardiomyocyte cell division and translating these approaches into clinical practice. In addition, a combinatorial approach to induce cardiomyocyte regeneration may need to be developed to overcome the current limitations and challenges for therapeutic cardiac regeneration in patients in the future.
References
Abouleisa RRE, Salama ABM, Ou Q, Tang X-L, Solanki M, Guo Y, Nong Y, McNally L, Lorkiewicz PK, Kassem KM, Ahern BM, Choudhary K, Thomas R, Huang Y, Juhardeen HR, Siddique A, Ifthikar Z, Hammad SK, Elbaz AS, Ivey KN, Conklin DJ, Satin J, Hill BG, Srivastava D, Bolli R, Mohamed TMA (2022) Transient cell cycle induction in cardiomyocytes to treat subacute ischemic heart failure. Circulation 145:1339–1355. https://doi.org/10.1161/CIRCULATIONAHA.121.057641
Agah R, Kirshenbaum LA, Abdellatif M, Truong LD, Chakraborty S, Michael LH, Schneider MD (1997) Adenoviral delivery of E2F–1 directs cell cycle reentry and p53-independent apoptosis in postmitotic adult myocardium in vivo. J Clin Invest 100:2722–2728. https://doi.org/10.1172/JCI119817
Aharonov A, Shakked A, Umansky KB, Savidor A, Genzelinakh A, Kain D, Lendengolts D, Revach O-Y, Morikawa Y, Dong J, Levin Y, Geiger B, Martin JF, Tzahor E (2020) ERBB2 drives YAP activation and EMT-like processes during cardiac regeneration. Nat Cell Biol 22:1346–1356. https://doi.org/10.1038/s41556-020-00588-4
Ahuja P, Sdek P, MacLellan WR (2007) Cardiac myocyte cell cycle control in development, disease, and regeneration. Physiol Rev 87:521–544. https://doi.org/10.1152/physrev.00032.2006
Alam P, Haile B, Arif M, Pandey R, Rokvic M, Nieman M, Maliken BD, Paul A, Wang Y, Sadayappan S, Ahmed RPH, Kanisicak O (2019) Inhibition of senescence-associated genes Rb1 and Meis2 in adult cardiomyocytes results in cell cycle reentry and cardiac repair post-myocardial infarction. JAHA 8:e012089. https://doi.org/10.1161/JAHA.119.012089
Ali SR, Hippenmeyer S, Saadat LV, Luo L, Weissman IL, Ardehali R (2014) Existing cardiomyocytes generate cardiomyocytes at a low rate after birth in mice. Proc Natl Acad Sci USA 111:8850–8855. https://doi.org/10.1073/pnas.1408233111
Alkass K, Panula J, Westman M, Wu T-D, Guerquin-Kern J-L, Bergmann O (2015) No evidence for cardiomyocyte number expansion in preadolescent mice. Cell 163:1026–1036. https://doi.org/10.1016/j.cell.2015.10.035
Alvarez R, Wang BJ, Quijada PJ, Avitabile D, Ho T, Shaitrit M, Chavarria M, Firouzi F, Ebeid D, Monsanto MM, Navarrete N, Moshref M, Siddiqi S, Broughton KM, Bailey BA, Gude NA, Sussman MA (2019) Cardiomyocyte cell cycle dynamics and proliferation revealed through cardiac-specific transgenesis of fluorescent ubiquitinated cell cycle indicator (FUCCI). J Mol Cell Cardiol 127:154–164. https://doi.org/10.1016/j.yjmcc.2018.12.007
Anatskaya OV, Vinogradov AE (2007) Genome multiplication as adaptation to tissue survival: evidence from gene expression in mammalian heart and liver. Genomics 89:70–80. https://doi.org/10.1016/j.ygeno.2006.08.014
Auchampach J, Han L, Huang GN, Kühn B, Lough JW, O’Meara CC, Payumo AY, Rosenthal NA, Sucov HM, Yutzey KE, Patterson M (2022) Measuring cardiomyocyte cell-cycle activity and proliferation in the age of heart regeneration. Am J Physiol-Heart Circ Physiol 322:H579–H596. https://doi.org/10.1152/ajpheart.00666.2021
Aurora AB, Porrello ER, Tan W, Mahmoud AI, Hill JA, Bassel-Duby R, Sadek HA, Olson EN (2014) Macrophages are required for neonatal heart regeneration. J Clin Invest 124:1382–1392. https://doi.org/10.1172/JCI72181
Bae J, Salamon RJ, Brandt EB, Paltzer WG, Zhang Z, Britt EC, Hacker TA, Fan J, Mahmoud AI (2021) Malonate promotes adult cardiomyocyte proliferation and heart regeneration. Circulation 143:1973–1986. https://doi.org/10.1161/CIRCULATIONAHA.120.049952
Baehr A, Umansky KB, Bassat E, Jurisch V, Klett K, Bozoglu T, Hornaschewitz N, Solyanik O, Kain D, Ferraro B, Cohen-Rabi R, Krane M, Cyran C, Soehnlein O, Laugwitz KL, Hinkel R, Kupatt C, Tzahor E (2020) Agrin promotes coordinated therapeutic processes leading to improved cardiac repair in pigs. Circulation 142:868–881. https://doi.org/10.1161/CIRCULATIONAHA.119.045116
Bailey LRJ, Bugg D, Reichardt IM, Ortaç CD, Gunaje J, Johnson R, MacCoss MJ, Sakamoto T, Kelly DP, Regnier M, Davis JM (2023) MBNL1 regulates programmed postnatal switching between regenerative and differentiated cardiac states. Cell Biol. https://doi.org/10.1161/CIRCULATIONAHA.123.066860
Bassat E, Mutlak YE, Genzelinakh A, Shadrin IY, Baruch Umansky K, Yifa O, Kain D, Rajchman D, Leach J, Riabov Bassat D, Udi Y, Sarig R, Sagi I, Martin JF, Bursac N, Cohen S, Tzahor E (2017) The extracellular matrix protein agrin promotes heart regeneration in mice. Nature 547:179–184. https://doi.org/10.1038/nature22978
Beigi F, Schmeckpeper J, Pow-anpongkul P, Payne JA, Zhang L, Zhang Z, Huang J, Mirotsou M, Dzau VJ (2013) C3orf58, a novel paracrine protein, stimulates cardiomyocyte cell-cycle progression through the PI3K–AKT–CDK7 pathway. Circ Res 113:372–380. https://doi.org/10.1161/CIRCRESAHA.113.301075
Bergmann O, Bhardwaj RD, Bernard S, Zdunek S, Barnabé-Heider F, Walsh S, Zupicich J, Alkass K, Buchholz BA, Druid H, Jovinge S, Frisén J (2009) Evidence for cardiomyocyte renewal in humans. Science 324:98–102. https://doi.org/10.1126/science.1164680
Berk BC, Fujiwara K, Lehoux S (2007) ECM remodeling in hypertensive heart disease. J Clin Invest 117:568–575. https://doi.org/10.1172/JCI31044
Bersell K, Arab S, Haring B, Kühn B (2009) Neuregulin1/ErbB4 signaling induces cardiomyocyte proliferation and repair of heart injury. Cell 138:257–270. https://doi.org/10.1016/j.cell.2009.04.060
Besson A, Dowdy SF, Roberts JM (2008) CDK inhibitors: cell cycle regulators and beyond. Dev Cell 14:159–169. https://doi.org/10.1016/j.devcel.2008.01.013
Bicknell KA, Coxon CH, Brooks G (2004) Forced expression of the cyclin B1–CDC2 complex induces proliferation in adult rat cardiomyocytes. Biochem J 382:411–416. https://doi.org/10.1042/BJ20031481
Blow JJ, Tanaka TU (2005) The chromosome cycle: coordinating replication and segregation: second in the cycles review series. EMBO Rep 6:1028–1034. https://doi.org/10.1038/sj.embor.7400557
Brooks G (1998) Arresting developments in the cardiac myocyte cell cycle: role of cyclin-dependent kinase inhibitors. Cardiovasc Res 39:301–311. https://doi.org/10.1016/S0008-6363(98)00125-4
Broughton KM, Sussman MA (2019) Adult cardiomyocyte cell cycle detour: off-ramp to quiescent destinations. Trends Endocrinol Metab 30:557–567. https://doi.org/10.1016/j.tem.2019.05.006
Cai B, Ma W, Wang X, Sukhareva N, Hua B, Zhang L, Xu J, Li X, Li S, Liu S, Yu M, Xu Y, Song R, Xu B, Yang F, Han Z, Ding F, Huang Q, Yu Y, Zhao Y, Wang J, Bamba D, Zagidullin N, Li F, Tian Y, Pan Z, Yang B (2020) Targeting LncDACH1 promotes cardiac repair and regeneration after myocardium infarction. Cell Death Differ 27:2158–2175. https://doi.org/10.1038/s41418-020-0492-5
Campa VM, Gutiérrez-Lanza R, Cerignoli F, Díaz-Trelles R, Nelson B, Tsuji T, Barcova M, Jiang W, Mercola M (2008) Notch activates cell cycle reentry and progression in quiescent cardiomyocytes. J Cell Biol 183:129–141. https://doi.org/10.1083/jcb.200806104
Cánepa ET, Scassa ME, Ceruti JM, Marazita MC, Carcagno AL, Sirkin PF, Ogara MF (2007) INK4 proteins, a family of mammalian CDK inhibitors with novel biological functions. IUBMB Life 59:419–426. https://doi.org/10.1080/15216540701488358
Cao X, Wang J, Wang Z, Du J, Yuan X, Huang W, Meng J, Gu H, Nie Y, Ji B, Hu S, Zheng Z (2013) MicroRNA profiling during rat ventricular maturation: a role for miR-29a in regulating cardiomyocyte cell cycle re-entry. FEBS Lett 587:1548–1555. https://doi.org/10.1016/j.febslet.2013.01.075
Cardoso AC, Lam NT, Savla JJ, Nakada Y, Pereira AHM, Elnwasany A, Menendez-Montes I, Ensley EL, Bezan Petric U, Sharma G, Sherry AD, Malloy CR, Khemtong C, Kinter MT, Tan WLW, Anene-Nzelu CG, Foo RS-Y, Nguyen NUN, Li S, Ahmed MS, Elhelaly WM, Abdisalaam S, Asaithamby A, Xing C, Kanchwala M, Vale G, Eckert KM, Mitsche MA, McDonald JG, Hill JA, Huang L, Shaul PW, Szweda LI, Sadek HA (2020) Mitochondrial substrate utilization regulates cardiomyocyte cell-cycle progression. Nat Metab 2:167–178. https://doi.org/10.1038/s42255-020-0169-x
Cattaneo P, Hayes MGB, Baumgarten N, Hecker D, Peruzzo S, Aslan GS, Kunderfranco P, Larcher V, Zhang L, Contu R, Fonseca G, Spinozzi S, Chen J, Condorelli G, Dimmeler S, Schulz MH, Heinz S, Guimarães-Camboa N, Evans SM (2022) DOT1L regulates chamber-specific transcriptional networks during cardiogenesis and mediates postnatal cell cycle withdrawal. Nat Commun 13:7444. https://doi.org/10.1038/s41467-022-35070-2
Cech TR, Steitz JA (2014) The noncoding RNA revolution—trashing old rules to forge new ones. Cell 157:77–94. https://doi.org/10.1016/j.cell.2014.03.008
Chaudhry HW, Dashoush NH, Tang H, Zhang L, Wang X, Wu EX, Wolgemuth DJ (2004) Cyclin A2 mediates cardiomyocyte mitosis in the postmitotic myocardium. J Biol Chem 279:35858–35866. https://doi.org/10.1074/jbc.M404975200
Chen H, Shi S, Acosta L, Li W, Lu J, Bao S, Chen Z, Yang Z, Schneider MD, Chien KR, Conway SJ, Yoder MC, Haneline LS, Franco D, Shou W (2004) BMP10 is essential for maintaining cardiac growth during murine cardiogenesis. Development 131:2219–2231. https://doi.org/10.1242/dev.01094
Chen X-Z, Li X-M, Xu S-J, Hu S, Wang T, Li R-F, Liu C-Y, Xue J-Q, Zhou L-Y, Wang Y-H, Li P-F, Wang K (2023) TMEM11 regulates cardiomyocyte proliferation and cardiac repair via METTL1-mediated m7G methylation of ATF5 mRNA. Cell Death Differ 30:1786–1798. https://doi.org/10.1038/s41418-023-01179-0
Chen Y, Li X, Li B, Wang H, Li M, Huang S, Sun Y, Chen G, Si X, Huang C, Liao W, Liao Y, Bin J (2019) Long non-coding RNA ECRAR triggers post-natal myocardial regeneration by activating ERK1/2 signaling. Mol Ther 27:29–45. https://doi.org/10.1016/j.ymthe.2018.10.021
Chen Z, Xie J, Hao H, Lin H, Wang L, Zhang Y, Chen L, Cao S, Huang X, Liao W, Bin J, Liao Y (2017) Ablation of periostin inhibits post-infarction myocardial regeneration in neonatal mice mediated by the phosphatidylinositol 3 kinase/glycogen synthase kinase 3β/cyclin D1 signalling pathway. Cardiovasc Res 113:620–632. https://doi.org/10.1093/cvr/cvx001
Cheng RK, Asai T, Tang H, Dashoush NH, Kara RJ, Costa KD, Naka Y, Wu EX, Wolgemuth DJ, Chaudhry HW (2007) Cyclin A2 induces cardiac regeneration after myocardial infarction and prevents heart failure. Circ Res 100:1741–1748. https://doi.org/10.1161/CIRCRESAHA.107.153544
Cho K-W, Andrade M, Bae S, Kim S, Eyun Kim J, Jang EY, Lee S, Husain A, Sutliff RL, Calvert JW, Park C, Yoon Y (2023) Polycomb group protein CBX7 represses cardiomyocyte proliferation through modulation of the TARDBP/RBM38 axis. Circulation 147:1823–1842. https://doi.org/10.1161/CIRCULATIONAHA.122.061131
Crippa S, Nemir M, Ounzain S, Ibberson M, Berthonneche C, Sarre A, Boisset G, Maison D, Harshman K, Xenarios I, Diviani D, Schorderet D, Pedrazzini T (2016) Comparative transcriptome profiling of the injured zebrafish and mouse hearts identifies miRNA-dependent repair pathways. Cardiovasc Res 110:73–84. https://doi.org/10.1093/cvr/cvw031
Di Stefano V, Giacca M, Capogrossi MC, Crescenzi M, Martelli F (2011) Knockdown of cyclin-dependent kinase inhibitors induces cardiomyocyte re-entry in the cell cycle. J Biol Chem 286:8644–8654. https://doi.org/10.1074/jbc.M110.184549
Diez-Cuñado M, Wei K, Bushway PJ, Maurya MR, Perera R, Subramaniam S, Ruiz-Lozano P, Mercola M (2018) miRNAs that induce human cardiomyocyte proliferation converge on the Hippo pathway. Cell Rep 23:2168–2174. https://doi.org/10.1016/j.celrep.2018.04.049
Dolejsi T, Delgobo M, Schuetz T, Tortola L, Heinze KG, Hofmann U, Frantz S, Bauer A, Ruschitzka F, Penninger JM, Campos Ramos G, Haubner BJ (2022) Adult T-cells impair neonatal cardiac regeneration. Eur Heart J 43:2698–2709. https://doi.org/10.1093/eurheartj/ehac153
Ebelt H, Zhang Y, Kampke A, Xu J, Schlitt A, Buerke M, Muller-Werdan U, Werdan K, Braun T (2008) E2F2 expression induces proliferation of terminally differentiated cardiomyocytes in vivo. Cardiovasc Res 80:219–226. https://doi.org/10.1093/cvr/cvn194
Engel FB, Schebesta M, Keating MT (2006) Anillin localization defect in cardiomyocyte binucleation. J Mol Cell Cardiol 41:601–612. https://doi.org/10.1016/j.yjmcc.2006.06.012
Eulalio A, Mano M, Ferro MD, Zentilin L, Sinagra G, Zacchigna S, Giacca M (2012) Functional screening identifies miRNAs inducing cardiac regeneration. Nature 492:376–381. https://doi.org/10.1038/nature11739
Fan C, Joshi J, Li F, Xu B, Khan M, Yang J, Zhu W (2020) Nanoparticle-Mediated Drug Delivery for Treatment of Ischemic Heart Disease. Front Bioeng Biotechnol 8:687. https://doi.org/10.3389/fbioe.2020.00687
Feng J, Li Y, Li Y, Yin Q, Li H, Li J, Zhou B, Meng J, Lian H, Wu M, Li Y, Dou K, Song W, Lu B, Liu L, Hu S, Nie Y (2023) Versican promotes cardiomyocyte proliferation and cardiac repair. Circulation. https://doi.org/10.1161/CIRCULATIONAHA.123.066298
Foster DA, Yellen P, Xu L, Saqcena M (2010) Regulation of G1 cell cycle progression: distinguishing the restriction point from a nutrient-sensing cell growth checkpoint(s). Genes Cancer 1:1124–1131. https://doi.org/10.1177/1947601910392989
Frangogiannis NG (2019) The extracellular matrix in ischemic and nonischemic heart failure. Circ Res 125:117–146. https://doi.org/10.1161/CIRCRESAHA.119.311148
French KM, Boopathy AV, DeQuach JA, Chingozha L, Lu H, Christman KL, Davis ME (2012) A naturally derived cardiac extracellular matrix enhances cardiac progenitor cell behavior in vitro. Acta Biomater 8:4357–4364. https://doi.org/10.1016/j.actbio.2012.07.033
Fu W, Liao Q, Li L, Shi Y, Zeng A, Zeng C, Wang WE (2020) An aurora kinase B-based mouse system to efficiently identify and analyze proliferating cardiomyocytes. Front Cell Dev Biol 8:570252. https://doi.org/10.3389/fcell.2020.570252
Fu W, Ren H, Shou J, Liao Q, Li L, Shi Y, Jose PA, Zeng C, Wang WE (2022) Loss of NPPA-AS1 promotes heart regeneration by stabilizing SFPQ–NONO heteromer-induced DNA repair. Basic Res Cardiol 117:10. https://doi.org/10.1007/s00395-022-00921-y
Gabisonia K, Prosdocimo G, Aquaro GD, Carlucci L, Zentilin L, Secco I, Ali H, Braga L, Gorgodze N, Bernini F, Burchielli S, Collesi C, Zandonà L, Sinagra G, Piacenti M, Zacchigna S, Bussani R, Recchia FA, Giacca M (2019) MicroRNA therapy stimulates uncontrolled cardiac repair after myocardial infarction in pigs. Nature 569:418–422. https://doi.org/10.1038/s41586-019-1191-6
Gamba L, Harrison M, Lien C-L (2014) Cardiac regeneration in model organisms. Curr Treat Options Cardio Med 16:288. https://doi.org/10.1007/s11936-013-0288-8
Gomez AH, Joshi S, Yang Y, Tune JD, Zhao M-T, Yang H (2021) Bioengineering systems for modulating notch signaling in cardiovascular development, disease, and regeneration. JCDD 8:125. https://doi.org/10.3390/jcdd8100125
Gong R, Gao X, Liu Y, Shen Y, Jiang Z, Wang X, Zagidullin N, Ma W, Wang N, Cai B (2023) Cyclin L1 controls cardiomyocyte proliferation and heart repair after injury. Sig Transduct Target Ther 8:243. https://doi.org/10.1038/s41392-023-01444-1
Gong R, Wang X, Li H, Liu S, Jiang Z, Zhao Y, Yu Y, Han Z, Yu Y, Dong C, Li S, Xu B, Zhang W, Wang N, Li X, Gao X, Yang F, Bamba D, Ma W, Liu Y, Cai B (2021) Loss of m6A methyltransferase METTL3 promotes heart regeneration and repair after myocardial injury. Pharmacol Res 174:105845. https://doi.org/10.1016/j.phrs.2021.105845
Gonzalez A (2003) Cardiomyocyte apoptosis in hypertensive cardiomyopathy. Cardiovasc Res 59:549–562. https://doi.org/10.1016/S0008-6363(03)00498-X
Grego-Bessa J, Luna-Zurita L, Del Monte G, Bolós V, Melgar P, Arandilla A, Garratt AN, Zang H, Mukouyama Y, Chen H, Shou W, Ballestar E, Esteller M, Rojas A, Pérez-Pomares JM, De La Pompa JL (2007) Notch signaling is essential for ventricular chamber development. Dev Cell 12:415–429. https://doi.org/10.1016/j.devcel.2006.12.011
Gude N, Muraski J, Rubio M, Kajstura J, Schaefer E, Anversa P, Sussman MA (2006) Akt promotes increased cardiomyocyte cycling and expansion of the cardiac progenitor cell population. Circ Res 99:381–388. https://doi.org/10.1161/01.RES.0000236754.21499.1c
Haginiwa S, Sadahiro T, Kojima H, Isomi M, Tamura F, Kurotsu S, Tani H, Muraoka N, Miyake N, Miyake K, Fukuda K, Ieda M (2019) Tbx6 induces cardiomyocyte proliferation in postnatal and adult mouse hearts. Biochem Biophys Res Commun 513:1041–1047. https://doi.org/10.1016/j.bbrc.2019.04.087
Halder G, Johnson RL (2011) Hippo signaling: growth control and beyond. Development 138:9–22. https://doi.org/10.1242/dev.045500
Han L, Choudhury S, Mich-Basso JD, Ammanamanchi N, Ganapathy B, Suresh S, Khaladkar M, Singh J, Maehr R, Zuppo DA, Kim J, Eberwine JH, Wyman SK, Wu YL, Kühn B (2020) Lamin B2 levels regulate polyploidization of cardiomyocyte nuclei and myocardial regeneration. Dev Cell 53:42-59.e11. https://doi.org/10.1016/j.devcel.2020.01.030
Han Z, Wang X, Xu Z, Cao Y, Gong R, Yu Y, Yu Y, Guo X, Liu S, Yu M, Ma W, Zhao Y, Xu J, Li X, Li S, Xu Y, Song R, Xu B, Yang F, Bamba D, Sukhareva N, Lei H, Gao M, Zhang W, Zagidullin N, Zhang Y, Yang B, Pan Z, Cai B (2021) ALKBH5 regulates cardiomyocyte proliferation and heart regeneration by demethylating the mRNA of YTHDF1. Theranostics 11:3000–3016. https://doi.org/10.7150/thno.47354
Harper JV, Brooks G (2004) The mammalian cell cycle: an overview. Cell cycle control. Humana Press, New Jersey, pp 113–154
Hashimoto H, Olson EN, Bassel-Duby R (2018) Therapeutic approaches for cardiac regeneration and repair. Nat Rev Cardiol 15:585–600. https://doi.org/10.1038/s41569-018-0036-6
Hatzistergos KE, Williams AR, Dykxhoorn D, Bellio MA, Yu W, Hare JM (2019) Tumor suppressors RB1 and CDKN2a cooperatively regulate cell-cycle progression and differentiation during cardiomyocyte development and repair: implications for stimulating neomyogenesis with cell-based therapy. Circ Res 124:1184–1197. https://doi.org/10.1161/CIRCRESAHA.118.314063
Haubner BJ, Adamowicz-Brice M, Khadayate S, Tiefenthaler V, Metzler B, Aitman T, Penninger JM (2012) Complete cardiac regeneration in a mouse model of myocardial infarction. Aging 4:966–977. https://doi.org/10.18632/aging.100526
Hauck L, Dadson K, Chauhan S, Grothe D, Billia F (2021) Inhibiting the Pkm2/b-catenin axis drives in vivo replication of adult cardiomyocytes following experimental MI. Cell Death Differ 28:1398–1417. https://doi.org/10.1038/s41418-020-00669-9
Heallen T, Morikawa Y, Leach J, Tao G, Willerson JT, Johnson RL, Martin JF (2013) Hippo signaling impedes adult heart regeneration. Development 140:4683–4690. https://doi.org/10.1242/dev.102798
Heallen T, Zhang M, Wang J, Bonilla-Claudio M, Klysik E, Johnson RL, Martin JF (2011) Hippo pathway inhibits Wnt signaling to restrain cardiomyocyte proliferation and heart size. Science 332:458–461. https://doi.org/10.1126/science.1199010
Hein S, Arnon E, Kostin S, Schönburg M, Elsässer A, Polyakova V, Bauer EP, Klövekorn W-P, Schaper J (2003) Progression from compensated hypertrophy to failure in the pressure-overloaded human heart: structural deterioration and compensatory mechanisms. Circulation 107:984–991. https://doi.org/10.1161/01.CIR.0000051865.66123.B7
Hesse M, Doengi M, Becker A, Kimura K, Voeltz N, Stein V, Fleischmann BK (2018) Midbody positioning and distance between daughter nuclei enable unequivocal identification of cardiomyocyte cell division in mice. Circ Res 123:1039–1052. https://doi.org/10.1161/CIRCRESAHA.118.312792
Hu Y, Jin G, Li B, Chen Y, Zhong L, Chen G, Chen X, Zhong J, Liao W, Liao Y, Wang Y, Bin J (2019) Suppression of miRNA let-7i-5p promotes cardiomyocyte proliferation and repairs heart function post injury by targeting CCND2 and E2F2. Clin Sci 133:425–441. https://doi.org/10.1042/CS20181002
Huang S, Li X, Zheng H, Si X, Li B, Wei G, Li C, Chen Y, Chen Y, Liao W, Liao Y, Bin J (2019) Loss of super-enhancer-regulated circRNA Nfix induces cardiac regeneration after myocardial infarction in adult mice. Circulation 139:2857–2876. https://doi.org/10.1161/CIRCULATIONAHA.118.038361
Huang W, Feng Y, Liang J, Yu H, Wang C, Wang B, Wang M, Jiang L, Meng W, Cai W, Medvedovic M, Chen J, Paul C, Davidson WS, Sadayappan S, Stambrook PJ, Yu X-Y, Wang Y (2018) Loss of microRNA-128 promotes cardiomyocyte proliferation and heart regeneration. Nat Commun 9:700. https://doi.org/10.1038/s41467-018-03019-z
Ieda M, Tsuchihashi T, Ivey KN, Ross RS, Hong T-T, Shaw RM, Srivastava D (2009) Cardiac fibroblasts regulate myocardial proliferation through β1 integrin signaling. Dev Cell 16:233–244. https://doi.org/10.1016/j.devcel.2008.12.007
Ikenishi A, Okayama H, Iwamoto N, Yoshitome S, Tane S, Nakamura K, Obayashi T, Hayashi T, Takeuchi T (2012) Cell cycle regulation in mouse heart during embryonic and postnatal stages. Dev Growth Differ 54:731–738. https://doi.org/10.1111/j.1440-169X.2012.01373.x
Ishikawa K, Weber T, Hajjar RJ (2018) Human cardiac gene therapy. Circ Res 123:601–613. https://doi.org/10.1161/CIRCRESAHA.118.311587
Jang J, Engleka KA, Liu F, Li L, Song G, Epstein JA, Li D (2020) An engineered mouse to identify proliferating cells and their derivatives. Front Cell Dev Biol 8:388. https://doi.org/10.3389/fcell.2020.00388
Jiang Y-H, Zhu Y, Chen S, Wang H-L, Zhou Y, Tang F-Q, Jian Z, Xiao Y-B (2019) Re-enforcing hypoxia-induced polyploid cardiomyocytes enter cytokinesis through activation of β-catenin. Sci Rep 9:17865. https://doi.org/10.1038/s41598-019-54334-4
Johnson J, Yang Y, Bian Z, Schena G, Li Y, Zhang X, Eaton DM, Gross P, Angheloiu A, Shaik A, Foster M, Berretta R, Kubo H, Mohsin S, Tian Y, Houser SR (2023) Systemic hypoxemia induces cardiomyocyte hypertrophy and right ventricular specific induction of proliferation. Circ Res 132:723–740. https://doi.org/10.1161/CIRCRESAHA.122.321604
Kang MJ, Koh GY (1997) Differential and dramatic changes of cyclin-dependent kinase activities in cardiomyocytes during the neonatal period. J Mol Cell Cardiol 29:1767–1777. https://doi.org/10.1006/jmcc.1997.0450
Kastan N, Gnedeva K, Alisch T, Petelski AA, Huggins DJ, Chiaravalli J, Aharanov A, Shakked A, Tzahor E, Nagiel A, Segil N, Hudspeth AJ (2021) Small-molecule inhibition of Lats kinases may promote Yap-dependent proliferation in postmitotic mammalian tissues. Nat Commun 12:3100. https://doi.org/10.1038/s41467-021-23395-3
Kerkela R, Kockeritz L, MacAulay K, Zhou J, Doble BW, Beahm C, Greytak S, Woulfe K, Trivedi CM, Woodgett JR, Epstein JA, Force T, Huggins GS (2008) Deletion of GSK-3β in mice leads to hypertrophic cardiomyopathy secondary to cardiomyoblast hyperproliferation. J Clin Invest 118:3609–3618. https://doi.org/10.1172/JCI36245
Kim H (1992) Human fetal heart development after mid-term: morphometry and ultrastructural study. J Mol Cell Cardiol 24:949–965. https://doi.org/10.1016/0022-2828(92)91862-Y
Kim WY, Sharpless NE (2006) The regulation of INK4/ARF in cancer and aging. Cell 127:265–275. https://doi.org/10.1016/j.cell.2006.10.003
Kirillova A, Han L, Liu H, Kühn B (2021) Polyploid cardiomyocytes: implications for heart regeneration. Development 148:dev199401. https://doi.org/10.1242/dev.199401
Koh KN, Kang MJ, Frith-Terhune A, Park SK, Kim I, Lee CO, Koh GY (1998) Persistent and heterogenous expression of the cyclin-dependent kinase inhibitor, p27KIP1, in rat hearts during development. J Mol Cell Cardiol 30:463–474. https://doi.org/10.1006/jmcc.1997.0611
Kou CY-C, Lau SL-Y, Au K-W, Leung P-Y, Chim SS-C, Fung K-P, Waye MM-Y, Tsui SK-W (2010) Epigenetic regulation of neonatal cardiomyocytes differentiation. Biochem Biophys Res Commun 400:278–283. https://doi.org/10.1016/j.bbrc.2010.08.064
Kretzschmar K, Post Y, Bannier-Hélaouët M, Mattiotti A, Drost J, Basak O, Li VSW, Van Den Born M, Gunst QD, Versteeg D, Kooijman L, Van Der Elst S, Van Es JH, Van Rooij E, Van Den Hoff MJB, Clevers H (2018) Profiling proliferative cells and their progeny in damaged murine hearts. Proc Natl Acad Sci USA. https://doi.org/10.1073/pnas.1805829115
Kühn B, Del Monte F, Hajjar RJ, Chang Y-S, Lebeche D, Arab S, Keating MT (2007) Periostin induces proliferation of differentiated cardiomyocytes and promotes cardiac repair. Nat Med 13:962–969. https://doi.org/10.1038/nm1619
Kytö V, Saraste A, Saukko P, Véronique H, Pulkki K, Vuorinen T, Voipio-Pulkki L-M (2004) Apoptotic cardiomyocyte death in fatal myocarditis. Am J Cardiol 94:746–750. https://doi.org/10.1016/j.amjcard.2004.05.056
Laflamme MA, Murry CE (2011) Heart regeneration. Nature 473:326–335. https://doi.org/10.1038/nature10147
Lan C, Chen C, Qu S, Cao N, Luo H, Yu C, Wang N, Xue Y, Xia X, Fan C, Ren H, Yang Y, Jose PA, Xu Z, Wu G, Zeng C (2022) Inhibition of DYRK1A, via histone modification, promotes cardiomyocyte cell cycle activation and cardiac repair after myocardial infarction. EBioMedicine 82:104139. https://doi.org/10.1016/j.ebiom.2022.104139
Leblond CP, El-Alfy M (1998) The eleven stages of the cell cycle, with emphasis on the changes in chromosomes and nucleoli during interphase and mitosis. Anat Rec 252:426–443. https://doi.org/10.1002/(SICI)1097-0185(199811)252:3%3c426::AID-AR11%3e3.0.CO;2-3
Leone M, Engel FB (2019) Advances in heart regeneration based on cardiomyocyte proliferation and regenerative potential of binucleated cardiomyocytes and polyploidization. Clin Sci 133:1229–1253. https://doi.org/10.1042/CS20180560
Leone M, Magadum A, Engel FB (2015) Cardiomyocyte proliferation in cardiac development and regeneration: a guide to methodologies and interpretations. Am J Physiol-Heart Circ Physiol 309:H1237–H1250. https://doi.org/10.1152/ajpheart.00559.2015
Li B, Li M, Li X, Li H, Lai Y, Huang S, He X, Si X, Zheng H, Liao W, Liao Y, Bin J (2019) Sirt1-inducible deacetylation of p21 promotes cardiomyocyte proliferation. Aging 11:12546–12567. https://doi.org/10.18632/aging.102587
Li F (1996) Rapid transition of cardiac myocytes from hyperplasia to hypertrophy during postnatal development. J Mol Cell Cardiol 28:1737–1746. https://doi.org/10.1006/jmcc.1996.0163
Li J, Liang C, Yang KY, Huang X, Han MY, Li X, Chan VW, Chan KS, Liu D, Huang Z-P, Zhou B, Lui KO (2020) Specific ablation of CD4 + T-cells promotes heart regeneration in juvenile mice. Theranostics 10:8018–8035. https://doi.org/10.7150/thno.42943
Li L, Xu N, Liu J, Chen Z, Liu X, Wang J (2022) m6A methylation in cardiovascular diseases: from mechanisms to therapeutic potential. Front Genet 13:908976. https://doi.org/10.3389/fgene.2022.908976
Li M, Izpisua Belmonte JC (2016) Mending a faltering heart. Circ Res 118:344–351. https://doi.org/10.1161/CIRCRESAHA.115.306820
Li X, Wang J, Jia Z, Cui Q, Zhang C, Wang W, Chen P, Ma K, Zhou C (2013) MiR-499 regulates cell proliferation and apoptosis during late-stage cardiac differentiation via Sox6 and cyclin D1. PLoS ONE 8:e74504. https://doi.org/10.1371/journal.pone.0074504
Li X, Wu F, Günther S, Looso M, Kuenne C, Zhang T, Wiesnet M, Klatt S, Zukunft S, Fleming I, Poschet G, Wietelmann A, Atzberger A, Potente M, Yuan X, Braun T (2023) Inhibition of fatty acid oxidation enables heart regeneration in adult mice. Nature 622:619–626. https://doi.org/10.1038/s41586-023-06585-5
Li Y, Feng J, Song S, Li H, Yang H, Zhou B, Li Y, Yue Z, Lian H, Liu L, Hu S, Nie Y (2020) gp130 controls cardiomyocyte proliferation and heart regeneration. Circulation 142:967–982. https://doi.org/10.1161/CIRCULATIONAHA.119.044484
Li Y, Wei T, Fan Y, Shan T, Sun J, Chen B, Wang Z, Gu L, Yang T, Liu L, Du C, Ma Y, Wang H, Sun R, Wei Y, Chen F, Guo X, Kong X, Wang L (2021) Serine/threonine-protein kinase 3 facilitates myocardial repair after cardiac injury possibly through the glycogen synthase kinase-3β/β-catenin pathway. JAHA 10:e022802. https://doi.org/10.1161/JAHA.121.022802
Li Y, Yang M, Tan J, Shen C, Deng S, Fu X, Gao S, Li H, Zhang X, Cai W (2022) Targeting ACSL1 promotes cardiomyocyte proliferation and cardiac regeneration. Life Sci 294:120371. https://doi.org/10.1016/j.lfs.2022.120371
Liang D, Li J, Wu Y, Zhen L, Li C, Qi M, Wang L, Deng F, Huang J, Lv F, Liu Y, Ma X, Yu Z, Zhang Y, Chen Y-H (2015) miRNA-204 drives cardiomyocyte proliferation via targeting Jarid2. Int J Cardiol 201:38–48. https://doi.org/10.1016/j.ijcard.2015.06.163
Liao H-S, Kang PM, Nagashima H, Yamasaki N, Usheva A, Ding B, Lorell BH, Izumo S (2001) Cardiac-specific overexpression of cyclin-dependent kinase 2 increases smaller mononuclear cardiomyocytes. Circ Res 88:443–450. https://doi.org/10.1161/01.RES.88.4.443
Liao S, Dong W, Lv L, Guo H, Yang J, Zhao H, Huang R, Yuan Z, Chen Y, Feng S, Zheng X, Huang J, Huang W, Qi X, Cai D (2017) Heart regeneration in adult Xenopus tropicalis after apical resection. Cell Biosci 7:70. https://doi.org/10.1186/s13578-017-0199-6
Lin Z, Von Gise A, Zhou P, Gu F, Ma Q, Jiang J, Yau AL, Buck JN, Gouin KA, Van Gorp PRR, Zhou B, Chen J, Seidman JG, Wang D-Z, Pu WT (2014) Cardiac-specific YAP activation improves cardiac function and survival in an experimental murine mi model. Circ Res 115:354–363. https://doi.org/10.1161/CIRCRESAHA.115.303632
Lin Z, Zhou P, Von Gise A, Gu F, Ma Q, Chen J, Guo H, Van Gorp PRR, Wang D-Z, Pu WT (2015) Pi3kcb links Hippo-YAP and PI3K-AKT signaling pathways to promote cardiomyocyte proliferation and survival. Circ Res 116:35–45. https://doi.org/10.1161/CIRCRESAHA.115.304457
Liu S, Li K, Wagner Florencio L, Tang L, Heallen TR, Leach JP, Wang Y, Grisanti F, Willerson JT, Perin EC, Zhang S, Martin JF (2021) Gene therapy knockdown of Hippo signaling induces cardiomyocyte renewal in pigs after myocardial infarction. Sci Transl Med 13:eabd6892. https://doi.org/10.1126/scitranslmed.abd6892
Lopaschuk GD, Collins-Nakai RL, Itoi T (1992) Developmental changes in energy substrate use by the heart. Cardiovasc Res 26:1172–1180. https://doi.org/10.1093/cvr/26.12.1172
Lorts A, Schwanekamp JA, Elrod JW, Sargent MA, Molkentin JD (2009) Genetic manipulation of periostin expression in the heart does not affect myocyte content, cell cycle activity, or cardiac repair. Circ Res. https://doi.org/10.1161/CIRCRESAHA.108.188649
Ma W, Wang X, Sun H, Xu B, Song R, Tian Y, Zhao L, Xu Y, Zhao Y, Yang F, Chen H, Gong R, Yu Y, Li X, Li S, Zhang W, Zhang T, Ne J, Cai B (2022) Oxidant stress-sensitive circRNA Mdc1 controls cardiomyocyte chromosome stability and cell cycle re-entry during heart regeneration. Pharmacol Res 184:106422. https://doi.org/10.1016/j.phrs.2022.106422
Maddika S, Ande SR, Wiechec E, Hansen LL, Wesselborg S, Los M (2008) Akt-mediated phosphorylation of CDK2 regulates its dual role in cell cycle progression and apoptosis. J Cell Sci 121:979–988. https://doi.org/10.1242/jcs.009530
Magadum A, Singh N, Kurian AA, Munir I, Mehmood T, Brown K, Sharkar MTK, Chepurko E, Sassi Y, Oh JG, Lee P, Santos CXC, Gaziel-Sovran A, Zhang G, Cai C-L, Kho C, Mayr M, Shah AM, Hajjar RJ, Zangi L (2020) Pkm2 regulates cardiomyocyte cell cycle and promotes cardiac regeneration. Circulation 141:1249–1265. https://doi.org/10.1161/CIRCULATIONAHA.119.043067
Mahiny-Shahmohammady D, Hauck L, Billia F (2022) Defining the molecular underpinnings controlling cardiomyocyte proliferation. Clin Sci 136:911–934. https://doi.org/10.1042/CS20211180
McDevitt TC, Laflamme MA, Murry CE (2005) Proliferation of cardiomyocytes derived from human embryonic stem cells is mediated via the IGF/PI 3-kinase/Akt signaling pathway. J Mol Cell Cardiol 39:865–873. https://doi.org/10.1016/j.yjmcc.2005.09.007
Medema RH, Kops GJPL, Bos JL, Burgering BMT (2000) AFX-like Forkhead transcription factors mediate cell-cycle regulation by Ras and PKB through p27kip1. Nature 404:782–787. https://doi.org/10.1038/35008115
Menendez-Montes I, Abdisalaam S, Xiao F, Lam NT, Mukherjee S, Szweda LI, Asaithamby A, Sadek HA (2021) Mitochondrial fatty acid utilization increases chromatin oxidative stress in cardiomyocytes. Proc Natl Acad Sci USA 118:e2101674118. https://doi.org/10.1073/pnas.2101674118
Mohamed TMA, Ang Y-S, Radzinsky E, Zhou P, Huang Y, Elfenbein A, Foley A, Magnitsky S, Srivastava D (2018) Regulation of cell cycle to stimulate adult cardiomyocyte proliferation and cardiac regeneration. Cell 173:104-116.e12. https://doi.org/10.1016/j.cell.2018.02.014
Mouse Genome Sequencing Consortium (2002) Initial sequencing and comparative analysis of the mouse genome. Nature 420:520–562. https://doi.org/10.1038/nature01262
Nakada Y, Canseco DC, Thet S, Abdisalaam S, Asaithamby A, Santos CX, Shah AM, Zhang H, Faber JE, Kinter MT, Szweda LI, Xing C, Hu Z, Deberardinis RJ, Schiattarella G, Hill JA, Oz O, Lu Z, Zhang CC, Kimura W, Sadek HA (2017) Hypoxia induces heart regeneration in adult mice. Nature 541:222–227. https://doi.org/10.1038/nature20173
Nguyen NUN, Canseco DC, Xiao F, Nakada Y, Li S, Lam NT, Muralidhar SA, Savla JJ, Hill JA, Le V, Zidan KA, El-Feky HW, Wang Z, Ahmed MS, Hubbi ME, Menendez-Montes I, Moon J, Ali SR, Le V, Villalobos E, Mohamed MS, Elhelaly WM, Thet S, Anene-Nzelu CG, Tan WLW, Foo RS, Meng X, Kanchwala M, Xing C, Roy J, Cyert MS, Rothermel BA, Sadek HA (2020) A calcineurin–Hoxb13 axis regulates growth mode of mammalian cardiomyocytes. Nature 582:271–276. https://doi.org/10.1038/s41586-020-2228-6
Notari M, Ventura-Rubio A, Bedford-Guaus SJ, Jorba I, Mulero L, Navajas D, Martí M, Raya Á (2018) The local microenvironment limits the regenerative potential of the mouse neonatal heart. Sci Adv 4:eaao5553. https://doi.org/10.1126/sciadv.aao5553
Paddock SJ, Swift SK, Alencar-Almeida V, Kenarsary A, Alvarez-Argote S, Flinn MA, Patterson M, O’Meara CC (2021) IL4Rα signaling promotes neonatal cardiac regeneration and cardiomyocyte cell cycle activity. J Mol Cell Cardiol 161:62–74. https://doi.org/10.1016/j.yjmcc.2021.07.012
Pan D (2010) The Hippo signaling pathway in development and cancer. Dev Cell 19:491–505. https://doi.org/10.1016/j.devcel.2010.09.011
Pandey R, Velasquez S, Durrani S, Jiang M, Neiman M, Crocker JS, Benoit JB, Rubinstein J, Paul A, Ahmed RP (2017) MicroRNA-1825 induces proliferation of adult cardiomyocytes and promotes cardiac regeneration post ischemic injury. Am J Transl Res 9:3120–3137
Parekh P, Motiwale L, Naik N, Rao KVK (2011) Downregulation of cyclin D1 is associated with decreased levels of p38 MAP kinases, Akt/PKB and Pak1 during chemopreventive effects of resveratrol in liver cancer cells. Exp Toxicol Pathol 63:167–173. https://doi.org/10.1016/j.etp.2009.11.005
Pasumarthi KBS, Nakajima H, Nakajima HO, Soonpaa MH, Field LJ (2005) Targeted expression of cyclin D2 results in cardiomyocyte DNA synthesis and infarct regression in transgenic mice. Circ Res 96:110–118. https://doi.org/10.1161/01.RES.0000152326.91223.4F
Patterson M, Swift SK (2019) Residual diploidy in polyploid tissues: a cellular state with enhanced proliferative capacity for tissue regeneration? Stem Cells Dev 28:1527–1539. https://doi.org/10.1089/scd.2019.0193
Piórkowska K, Ropka-Molik K (2021) Pig genomics and genetics. Genes 12:1692. https://doi.org/10.3390/genes12111692
Ponnusamy M, Liu F, Zhang Y-H, Li R-B, Zhai M, Liu F, Zhou L-Y, Liu C-Y, Yan K-W, Dong Y-H, Wang M, Qian L-L, Shan C, Xu S, Wang Q, Zhang Y-H, Li P-F, Zhang J, Wang K (2019) Long noncoding RNA CPR (cardiomyocyte proliferation regulator) regulates cardiomyocyte proliferation and cardiac repair. Circulation 139:2668–2684. https://doi.org/10.1161/CIRCULATIONAHA.118.035832
Poolman RA, Gilchrist R, Brooks G (1998) Cell cycle profiles and expressions of p21CIP1 and p27KIP1 during myocyte development. Int J Cardiol 67:133–142. https://doi.org/10.1016/S0167-5273(98)00320-9
Porrello ER, Johnson BA, Aurora AB, Simpson E, Nam Y-J, Matkovich SJ, Dorn GW, Van Rooij E, Olson EN (2011) miR-15 family regulates postnatal mitotic arrest of cardiomyocytes. Circ Res 109:670–679. https://doi.org/10.1161/CIRCRESAHA.111.248880
Porrello ER, Mahmoud AI, Simpson E, Johnson BA, Grinsfelder D, Canseco D, Mammen PP, Rothermel BA, Olson EN, Sadek HA (2013) Regulation of neonatal and adult mammalian heart regeneration by the miR-15 family. Proc Natl Acad Sci USA 110:187–192. https://doi.org/10.1073/pnas.1208863110
Poss KD, Wilson LG, Keating MT (2002) Heart regeneration in zebrafish. Science 298:2188–2190. https://doi.org/10.1126/science.1077857
Prabhu SD, Frangogiannis NG (2016) The biological basis for cardiac repair after myocardial infarction: from inflammation to fibrosis. Circ Res 119:91–112. https://doi.org/10.1161/CIRCRESAHA.116.303577
Przybyt E, Krenning G, Brinker MG, Harmsen MC (2013) Adipose stromal cells primed with hypoxia and inflammation enhance cardiomyocyte proliferation rate in vitro through STAT3 and Erk1/2. J Transl Med 11:39. https://doi.org/10.1186/1479-5876-11-39
Puck TT, Steffen J (1963) Life cycle analysis of mammalian cells. Biophys J 3:379–397. https://doi.org/10.1016/S0006-3495(63)86828-9
Puente BN, Kimura W, Muralidhar SA, Moon J, Amatruda JF, Phelps KL, Grinsfelder D, Rothermel BA, Chen R, Garcia JA, Santos CX, Thet S, Mori E, Kinter MT, Rindler PM, Zacchigna S, Mukherjee S, Chen DJ, Mahmoud AI, Giacca M, Rabinovitch PS, Asaithamby A, Shah AM, Szweda LI, Sadek HA (2014) The oxygen-rich postnatal environment induces cardiomyocyte cell-cycle arrest through DNA damage response. Cell 157:565–579. https://doi.org/10.1016/j.cell.2014.03.032
Quelle DE, Ashmun RA, Shurtleff SA, Kato JY, Bar-Sagi D, Roussel MF, Sherr CJ (1993) Overexpression of mouse D-type cyclins accelerates G1 phase in rodent fibroblasts. Genes Dev 7:1559–1571. https://doi.org/10.1101/gad.7.8.1559
Raso A, Dirkx E, Sampaio-Pinto V, El Azzouzi H, Cubero RJ, Sorensen DW, Ottaviani L, Olieslagers S, Huibers MM, De Weger R, Siddiqi S, Moimas S, Torrini C, Zentillin L, Braga L, Nascimento DS, Da Costa Martins PA, Van Berlo JH, Zacchigna S, Giacca M, De Windt LJ (2021) A microRNA program regulates the balance between cardiomyocyte hyperplasia and hypertrophy and stimulates cardiac regeneration. Nat Commun 12:4808. https://doi.org/10.1038/s41467-021-25211-4
Raulf A, Horder H, Tarnawski L, Geisen C, Ottersbach A, Röll W, Jovinge S, Fleischmann BK, Hesse M (2015) Transgenic systems for unequivocal identification of cardiac myocyte nuclei and analysis of cardiomyocyte cell cycle status. Basic Res Cardiol 110:33. https://doi.org/10.1007/s00395-015-0489-2
Redgrave RE, Dookun E, Booth LK, Camacho Encina M, Folaranmi O, Tual-Chalot S, Gill JH, Owens WA, Spyridopoulos I, Passos JF, Richardson GD (2023) Senescent cardiomyocytes contribute to cardiac dysfunction following myocardial infarction. NPJ Aging 9:15. https://doi.org/10.1038/s41514-023-00113-5
Reichart D, Newby GA, Wakimoto H, Lun M, Gorham JM, Curran JJ, Raguram A, DeLaughter DM, Conner DA, Marsiglia JDC, Kohli S, Chmatal L, Page DC, Zabaleta N, Vandenberghe L, Liu DR, Seidman JG, Seidman C (2023) Efficient in vivo genome editing prevents hypertrophic cardiomyopathy in mice. Nat Med 29:412–421. https://doi.org/10.1038/s41591-022-02190-7
Rigaud VOC, Hoy RC, Kurian J, Zarka C, Behanan M, Brosious I, Pennise J, Patel T, Wang T, Johnson J, Kraus LM, Mohsin S, Houser SR, Khan M (2023) RNA-binding protein LIN28a regulates new myocyte formation in the heart through long noncoding RNA-H19. Circulation 147:324–337. https://doi.org/10.1161/CIRCULATIONAHA.122.059346
Rota M, Boni A, Urbanek K, Padin-Iruegas ME, Kajstura TJ, Fiore G, Kubo H, Sonnenblick EH, Musso E, Houser SR, Leri A, Sussman MA, Anversa P (2005) Nuclear targeting of Akt enhances ventricular function and myocyte contractility. Circ Res 97:1332–1341. https://doi.org/10.1161/01.RES.0000196568.11624.ae
Rotem I, Konfino T, Caller T, Schary Y, Shaihov-Teper O, Palevski D, Lewis N, Lendengolts D, Naftali-Shani N, Leor J (2022) Osteopontin promotes infarct repair. Basic Res Cardiol 117:51. https://doi.org/10.1007/s00395-022-00957-0
Secco I, Giacca M (2023) Regulation of endogenous cardiomyocyte proliferation: the known unknowns. J Mol Cell Cardiol 179:80–89. https://doi.org/10.1016/j.yjmcc.2023.04.001
Senyo SE, Steinhauser ML, Pizzimenti CL, Yang VK, Cai L, Wang M, Wu T-D, Guerquin-Kern J-L, Lechene CP, Lee RT (2013) Mammalian heart renewal by pre-existing cardiomyocytes. Nature 493:433–436. https://doi.org/10.1038/nature11682
Shafei S, Khanmohammadi M, Ghanbari H, Nooshabadi VT, Tafti SHA, Rabbani S, Kasaiyan M, Basiri M, Tavoosidana G (2022) Effectiveness of exosome mediated miR-126 and miR-146a delivery on cardiac tissue regeneration. Cell Tissue Res 390:71–92. https://doi.org/10.1007/s00441-022-03663-4
Shah A, Goerlich CE, Pasrija C, Hirsch J, Fisher S, Odonkor P, Strauss E, Ayares D, Mohiuddin MM, Griffith BP (2022) Anatomical differences between human and pig hearts and their relevance for cardiac xenotransplantation surgical technique. JACC Case Rep 4:1049–1052. https://doi.org/10.1016/j.jaccas.2022.06.011
Shapiro SD, Ranjan AK, Kawase Y, Cheng RK, Kara RJ, Bhattacharya R, Guzman-Martinez G, Sanz J, Garcia MJ, Chaudhry HW (2014) Cyclin A2 induces cardiac regeneration after myocardial infarction through cytokinesis of adult cardiomyocytes. Sci Transl Med. https://doi.org/10.1126/scitranslmed.3007668
Shernan SK (2003) Perioperative myocardial ischemia reperfusion injury. Anesthesiol Clin North Am 21:465–485. https://doi.org/10.1016/S0889-8537(03)00038-5
Shiojima I, Walsh K (2006) Regulation of cardiac growth and coronary angiogenesis by the Akt/PKB signaling pathway. Genes Dev 20:3347–3365. https://doi.org/10.1101/gad.1492806
Sidney S, Sorel ME, Quesenberry CP, Jaffe MG, Solomon MD, Nguyen-Huynh MN, Go AS, Rana JS (2018) Comparative trends in heart disease, stroke, and all-cause mortality in the United States and a large integrated healthcare delivery system. Am J Med 131:829-836.e1. https://doi.org/10.1016/j.amjmed.2018.02.014
Soonpaa MH, Kim KK, Pajak L, Franklin M, Field LJ (1996) Cardiomyocyte DNA synthesis and binucleation during murine development. Am J Physio-Heart Circ Physiol 271:H2183–H2189. https://doi.org/10.1152/ajpheart.1996.271.5.H2183
Soonpaa MH, Koh GY, Pajak L, Jing S, Wang H, Franklin MT, Kim KK, Field LJ (1997) Cyclin D1 overexpression promotes cardiomyocyte DNA synthesis and multinucleation in transgenic mice. J Clin Invest 99:2644–2654. https://doi.org/10.1172/JCI119453
Stark GR, Taylor WR (2004) Analyzing the G2/M checkpoint. Checkpoint controls and cancer. Humana Press, New Jersey, pp 051–082
Sun J, Peterson EA, Wang AZ, Ou J, Smith KE, Poss KD, Wang J (2022) hapln1 defines an epicardial cell subpopulation required for cardiomyocyte expansion during heart morphogenesis and regeneration. Circulation 146:48–63. https://doi.org/10.1161/CIRCULATIONAHA.121.055468
Sun J, Wang L, Matthews RC, Walcott GP, Lu Y-A, Wei Y, Zhou Y, Zangi L, Zhang J (2023) CCND2 modified mRNA activates cell cycle of cardiomyocytes in hearts with myocardial infarction in mice and pigs. Circ Res 133:484–504. https://doi.org/10.1161/CIRCRESAHA.123.322929
Sun J, Yang T, Wei T, Zhou L, Shan T, Chen J, Gu L, Chen B, Liu L, Jiang Q, Du C, Ma Y, Wang H, Chen F, Guo X, Ji Y, Wang L (2022) CDK9 binds and activates SGK3 to promote cardiac repair after injury via the GSK-3β/β-catenin pathway. Front Cardiovasc Med 9:970745. https://doi.org/10.3389/fcvm.2022.970745
Sun J, Zhou L, Yang T, Deng B, Bao Y, Gu L, Wang H, Wang L (2023) P16INK4a regulates ros-related autophagy and CDK4/6-mediated proliferation: a new target of myocardial regeneration therapy. Oxid Med Cell Longev 2023:1–23. https://doi.org/10.1155/2023/1696190
Sun Y, Jiang C, Hong H, Liu J, Qiu L, Huang Y, Ye L (2019) Effects of hypoxia on cardiomyocyte proliferation and association with stage of development. Biomed Pharmacother 118:109391. https://doi.org/10.1016/j.biopha.2019.109391
Swift SK, Purdy AL, Kolell ME, Andresen KG, Lahue C, Buddell T, Akins KA, Rau CD, O’Meara CC, Patterson M (2023) Cardiomyocyte ploidy is dynamic during postnatal development and varies across genetic backgrounds. Development 150:dev201318. https://doi.org/10.1242/dev.201318
Tane S, Ikenishi A, Okayama H, Iwamoto N, Nakayama KI, Takeuchi T (2014) CDK inhibitors, p21Cip1 and p27Kip1, participate in cell cycle exit of mammalian cardiomyocytes. Biochem Biophys Res Commun 443:1105–1109. https://doi.org/10.1016/j.bbrc.2013.12.109
Tane S, Kubota M, Okayama H, Ikenishi A, Yoshitome S, Iwamoto N, Satoh Y, Kusakabe A, Ogawa S, Kanai A, Molkentin JD, Nakamura K, Ohbayashi T, Takeuchi T (2014) Repression of cyclin D1 expression is necessary for the maintenance of cell cycle exit in adult mammalian cardiomyocytes. J Biol Chem 289:18033–18044. https://doi.org/10.1074/jbc.M113.541953
Tong W, Xiong F, Li Y, Zhang L (2013) Hypoxia inhibits cardiomyocyte proliferation in fetal rat hearts via upregulating TIMP-4. Am J Physiol-Regul Integr Comp Physiol 304:R613–R620. https://doi.org/10.1152/ajpregu.00515.2012
Tong W, Xue Q, Li Y, Zhang L (2011) Maternal hypoxia alters matrix metalloproteinase expression patterns and causes cardiac remodeling in fetal and neonatal rats. Am J Physiol-Heart Circ Physiol 301:H2113–H2121. https://doi.org/10.1152/ajpheart.00356.2011
Torrini C, Cubero RJ, Dirkx E, Braga L, Ali H, Prosdocimo G, Gutierrez MI, Collesi C, Licastro D, Zentilin L, Mano M, Zacchigna S, Vendruscolo M, Marsili M, Samal A, Giacca M (2019) Common regulatory pathways mediate activity of MicroRNAs inducing cardiomyocyte proliferation. Cell Rep 27:2759-2771.e5. https://doi.org/10.1016/j.celrep.2019.05.005
Tzahor E (2007) Wnt/β-catenin signaling and cardiogenesis: timing does matter. Dev Cell 13:10–13. https://doi.org/10.1016/j.devcel.2007.06.006
Valussi M, Besser J, Wystub-Lis K, Zukunft S, Richter M, Kubin T, Boettger T, Braun T (2021) Repression of Osmr and Fgfr1 by miR-1/133a prevents cardiomyocyte dedifferentiation and cell cycle entry in the adult heart. Sci Adv 7:eabi6648. https://doi.org/10.1126/sciadv.abi6648
Velayutham N, Calderon MU, Alfieri CM, Padula SL, van Leeuwen FN, Scheijen B, Yutzey KE (2023) Btg1 and Btg2 regulate neonatal cardiomyocyte cell cycle arrest. J Mol Cell Cardiol 179:30–41. https://doi.org/10.1016/j.yjmcc.2023.03.016
Velayutham N, Yutzey KE (2022) Porcine models of heart regeneration. JCDD 9:93. https://doi.org/10.3390/jcdd9040093
Von Gise A, Lin Z, Schlegelmilch K, Honor LB, Pan GM, Buck JN, Ma Q, Ishiwata T, Zhou B, Camargo FD, Pu WT (2012) YAP1, the nuclear target of Hippo signaling, stimulates heart growth through cardiomyocyte proliferation but not hypertrophy. Proc Natl Acad Sci USA 109:2394–2399. https://doi.org/10.1073/pnas.1116136109
Wang B, Xu M, Li M, Wu F, Hu S, Chen X, Zhao L, Huang Z, Lan F, Liu D, Wang Y (2020) miR-25 promotes cardiomyocyte proliferation by targeting FBXW7. Mol Ther Nucleic Acids 19:1299–1308. https://doi.org/10.1016/j.omtn.2020.01.013
Wang RM, Mesfin JM, Hunter J, Cattaneo P, Guimarães-Camboa N, Braden RL, Luo C, Hill RC, Dzieciatkowska M, Hansen KC, Evans S, Christman KL (2022) Myocardial matrix hydrogel acts as a reactive oxygen species scavenger and supports a proliferative microenvironment for cardiomyocytes. Acta Biomater 152:47–59. https://doi.org/10.1016/j.actbio.2022.08.050
Wang T, Zhou L-Y, Li X-M, Liu F, Liang L, Chen X-Z, Ju J, Ponnusamy M, Wang K, Liu C-Y, Yan K-W, Wang K (2023) ABRO1 arrests cardiomyocyte proliferation and myocardial repair by suppressing PSPH. Mol Ther 31:847–865. https://doi.org/10.1016/j.ymthe.2023.01.011
Wang X, Senapati S, Akinbote A, Gnanasambandam B, Park PS-H, Senyo SE (2020) Microenvironment stiffness requires decellularized cardiac extracellular matrix to promote heart regeneration in the neonatal mouse heart. Acta Biomater 113:380–392. https://doi.org/10.1016/j.actbio.2020.06.032
Wang X, Wan TC, Lauth A, Purdy AL, Kulik KR, Patterson M, Lough JW, Auchampach JA (2022) Conditional depletion of the acetyltransferase Tip60 protects against the damaging effects of myocardial infarction. J Mol Cell Cardiol 163:9–19. https://doi.org/10.1016/j.yjmcc.2021.09.012
Wang Z (2022) Cell cycle progression and synchronization: an overview. In: Wang Z (ed) Cell-cycle synchronization. Springer, US, New York, NY, pp 3–23
Windmueller R, Leach JP, Babu A, Zhou S, Morley MP, Wakabayashi A, Petrenko NB, Viatour P, Morrisey EE (2020) Direct comparison of mononucleated and binucleated cardiomyocytes reveals molecular mechanisms underlying distinct proliferative competencies. Cell Rep 30:3105-3116.e4. https://doi.org/10.1016/j.celrep.2020.02.034
Wodsedalek DJ, Paddock SJ, Wan TC, Auchampach JA, Kenarsary A, Tsaih S-W, Flister MJ, O’Meara CC (2019) IL-13 promotes in vivo neonatal cardiomyocyte cell cycle activity and heart regeneration. Am J Physiol-Heart Circ Physiol 316:H24–H34. https://doi.org/10.1152/ajpheart.00521.2018
Woo RA, Poon RYC (2003) Cyclin-dependent kinases and S phase control in mammalian cells. Cell Cycle 2:316–324
Woo YJ, Panlilio CM, Cheng RK, Liao GP, Atluri P, Hsu VM, Cohen JE, Chaudhry HW (2006) Therapeutic delivery of cyclin A2 induces myocardial regeneration and enhances cardiac function in ischemic heart failure. Circulation. https://doi.org/10.1161/CIRCULATIONAHA.105.000455
Wu C-C, Jeratsch S, Graumann J, Stainier DYR (2020) Modulation of mammalian cardiomyocyte cytokinesis by the extracellular matrix. Circ Res 127:896–907. https://doi.org/10.1161/CIRCRESAHA.119.316303
Wu Y, Zhou L, Liu H, Duan R, Zhou H, Zhang F, He X, Lu D, Xiong K, Xiong M, Zhuang J, Liu Y, Li L, Liang D, Chen Y-H (2021) LRP6 downregulation promotes cardiomyocyte proliferation and heart regeneration. Cell Res 31:450–462. https://doi.org/10.1038/s41422-020-00411-7
Xiang F, Guo M, Yutzey KE (2016) Overexpression of Tbx20 in adult cardiomyocytes promotes proliferation and improves cardiac function after myocardial infarction. Circulation 133:1081–1092. https://doi.org/10.1161/CIRCULATIONAHA.115.019357
Xin M, Kim Y, Sutherland LB, Murakami M, Qi X, McAnally J, Porrello ER, Mahmoud AI, Tan W, Shelton JM, Richardson JA, Sadek HA, Bassel-Duby R, Olson EN (2013) Hippo pathway effector yap promotes cardiac regeneration. Proc Natl Acad Sci USA 110:13839–13844. https://doi.org/10.1073/pnas.1313192110
Yahalom-Ronen Y, Rajchman D, Sarig R, Geiger B, Tzahor E (2015) Reduced matrix rigidity promotes neonatal cardiomyocyte dedifferentiation, proliferation and clonal expansion. Elife 4:e07455. https://doi.org/10.7554/eLife.07455
Yang Q, Wu F, Mi Y, Wang F, Cai K, Yang X, Zhang R, Liu L, Zhang Y, Wang Y, Wang X, Xu M, Gui Y, Li Q (2020) Aberrant expression of miR-29b-3p influences heart development and cardiomyocyte proliferation by targeting NOTCH2. Cell Prolif 53:e12764. https://doi.org/10.1111/cpr.12764
Yang Y, Cheng H-W, Qiu Y, Dupee D, Noonan M, Lin Y-D, Fisch S, Unno K, Sereti K-I, Liao R (2015) MicroRNA-34a plays a key role in cardiac repair and regeneration following myocardial infarction. Circ Res 117:450–459. https://doi.org/10.1161/CIRCRESAHA.117.305962
Ye L, Qiu L, Feng B, Jiang C, Huang Y, Zhang H, Zhang H, Hong H, Liu J (2020) Role of blood oxygen saturation during post-natal human cardiomyocyte cell cycle activities. JACC Basic Transl Sci 5:447–460. https://doi.org/10.1016/j.jacbts.2020.02.008
Yekelchyk M, Guenther S, Preussner J, Braun T (2019) Mono- and multi-nucleated ventricular cardiomyocytes constitute a transcriptionally homogenous cell population. Basic Res Cardiol 114:36. https://doi.org/10.1007/s00395-019-0744-z
Yu F-X, Zhao B, Guan K-L (2015) Hippo pathway in organ size control, tissue homeostasis, and cancer. Cell 163:811–828. https://doi.org/10.1016/j.cell.2015.10.044
Yuan T, Annamalai K, Naik S, Lupse B, Geravandi S, Pal A, Dobrowolski A, Ghawali J, Ruhlandt M, Gorrepati KDD, Azizi Z, Lim D-S, Maedler K, Ardestani A (2021) The Hippo kinase LATS2 impairs pancreatic β-cell survival in diabetes through the mTORC1-autophagy axis. Nat Commun 12:4928. https://doi.org/10.1038/s41467-021-25145-x
Yuan T, Krishnan J (2021) Non-coding RNAs in cardiac regeneration. Front Physiol 12:650566. https://doi.org/10.3389/fphys.2021.650566
Yusuf AM, Qaisar R, Al-Tamimi AO, Jayakumar MN, Woodgett JR, Koch WJ, Ahmad F (2022) Cardiomyocyte-GSK-3β deficiency induces cardiac progenitor cell proliferation in the ischemic heart through paracrine mechanisms. J Cell Physiol 237:1804–1817. https://doi.org/10.1002/jcp.30644
Zhang L, Sun Z, Ren P, You M, Zhang J, Fang L, Wang J, Chen Y, Yan F, Zheng H, Xie M (2017) Localized Delivery of shRNA against PHD2 Protects the Heart from Acute Myocardial Infarction through Ultrasound-Targeted Cationic Microbubble Destruction. Theranostics 7:51–66. https://doi.org/10.7150/thno.16074
Zhao L, Borikova AL, Ben-Yair R, Guner-Ataman B, MacRae CA, Lee RT, Burns CG, Burns CE (2014) Notch signaling regulates cardiomyocyte proliferation during zebrafish heart regeneration. Proc Natl Acad Sci USA 111:1403–1408. https://doi.org/10.1073/pnas.1311705111
Zhao Y, Sawyer DR, Baliga RR, Opel DJ, Han X, Marchionni MA, Kelly RA (1998) Neuregulins promote survival and growth of cardiac myocytes. J Biol Chem 273:10261–10269. https://doi.org/10.1074/jbc.273.17.10261
Zhou H, Zhang F, Wu Y, Liu H, Duan R, Liu Y, Wang Y, He X, Zhang Y, Ma X, Guan Y, Liu Y, Liang D, Zhou L, Chen Y (2022) LRP5 regulates cardiomyocyte proliferation and neonatal heart regeneration by the AKT/P21 pathway. J Cell Mol Medi 26:2981–2994. https://doi.org/10.1111/jcmm.17311
Zhu B, Gong Y, Shen L, Li J, Han J, Song B, Hu L, Wang Q, Wang Z (2020) Total panax notoginseng saponin inhibits vascular smooth muscle cell proliferation and migration and intimal hyperplasia by regulating WTAP/p16 signals via m6A modulation. Biomed Pharmacother 124:109935. https://doi.org/10.1016/j.biopha.2020.109935
Zong H, Espinosa JS, Su HH, Muzumdar MD, Luo L (2005) Mosaic analysis with double markers in mice. Cell 121:479–492. https://doi.org/10.1016/j.cell.2005.02.012
Funding
Open Access funding enabled and organized by Projekt DEAL. This work was supported by the European Innovation Council (GA: 822455) and the German Research Foundation (SFB/TR267), the LOEWE Center for Cell and Gene Therapy and the Messer Foundation grant.
Author information
Authors and Affiliations
Contributions
CZ wrote the manuscript and put the Figure and tables together. TY and JK edited the manuscript, the figure and tables, and helped to structure and revise the manuscript. All authors contributed to the article and approved the submitted version.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhu, C., Yuan, T. & Krishnan, J. Targeting cardiomyocyte cell cycle regulation in heart failure. Basic Res Cardiol 119, 349–369 (2024). https://doi.org/10.1007/s00395-024-01049-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00395-024-01049-x