
Abstract
There remains an unmet need to identify novel therapeutic strategies capable of protecting the myocardium against the detrimental effects of acute ischemia–reperfusion injury (IRI), to reduce myocardial infarct (MI) size and prevent the onset of heart failure (HF) following acute myocardial infarction (AMI). In this regard, perturbations in mitochondrial morphology with an imbalance in mitochondrial fusion and fission can disrupt mitochondrial metabolism, calcium homeostasis, and reactive oxygen species production, factors which are all known to be critical determinants of cardiomyocyte death following acute myocardial IRI. As such, therapeutic approaches directed at preserving the morphology and functionality of mitochondria may provide an important strategy for cardioprotection. In this article, we provide an overview of the alterations in mitochondrial morphology which occur in response to acute myocardial IRI, and highlight the emerging therapeutic strategies for targeting mitochondrial shape to preserve mitochondrial function which have the future therapeutic potential to improve health outcomes in patients presenting with AMI.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cardiovascular diseases (CVD) remain the leading causes of death and disability worldwide [216], with acute myocardial infarction (AMI) and heart failure (HF) that often follow being the main contributors to this healthcare burden [250]. Therefore, novel therapies capable of protecting the myocardium from the detrimental effects of acute ischemia–reperfusion injury (IRI) are needed to reduce myocardial infarct (MI) size and preserve cardiac function to prevent the onset of HF following AMI [103].
Morphological and metabolic alterations in mitochondria are known to be associated with the onset and progression of cardiac diseases including AMI and HF [145, 204]. An imbalance in mitochondrial morphology is known to disturb energy production, mitochondrial reactive oxygen species (ROS) generation, and calcium homeostasis, factors which act in concert to contribute to cardiomyocyte death following acute IRI in the setting of AMI [102, 145, 204]. The complex signaling pathways underlying mitochondrial morphology may offer potential therapeutic targets for preventing mitochondrial dysfunction following AMI, so much so that finely tuning the balance between fission and fusion to preserve mitochondrial shape, may lie at the heart of cardioprotection.
In this article, we review how changes in the balance between mitochondrial fission and fusion affect susceptibility to acute myocardial IRI and highlight mitochondrial morphology as a therapeutic target for cardioprotection and for potentially improving health outcomes in patients with AMI. Although the focus of this article is on the role of cardiomyocyte mitochondria in IRI and cardioprotection, it must be appreciated that those studies investigating the role of mitochondria in the heart at the tissue level may not necessarily be restricting their findings to cardiomyocyte mitochondria given the presence of non-cardiomyocyte cells such as immune cells, endothelial cells, and fibroblasts.
Mitochondrial morphology in the healthy heart
Normal mitochondrial homeostasis and function is determined by a number of different factors including mitochondrial structure, location, morphology, biogenesis, and mitophagy.
Mitochondrial structure: membrane and lipid composition
Mitochondria are organelles of endosymbiotic origin that harbor two membranes and two aqueous compartments, the outer mitochondrial membrane (OMM) and the inner mitochondrial membrane (IMM), which divide the organelle into an inner boundary membrane and a cristae membrane [114]. Advances in electron microscopy and computer reconstruction algorithms have revealed the cristae to exhibit both tubular and lamellar forms, reflecting their functional specialization in different metabolic microcompartments [114, 199]. The OMM and IMM show significant differences in lipid composition and permeability. The lipid-rich OMM is generally permeable to ions and small uncharged molecules through pore-forming membrane proteins. Among its constituents, the OMM has a voltage-dependent anion channel that provides a route for metabolic substrates (e.g., pyruvate, glutamate, and malate) and nucleotides (e.g., ADP and ATP) to gain access to the intermembranous space (IMS) [147, 205]. There is no membrane potential across the OMM because of its porosity. Furthermore, the OMM provides a dynamic platform for cell signaling and tethers subcellular compartments to form membrane contact sites, including the endoplasmic reticulum (ER), plasma membrane, lysosomes, peroxisomes, endosomes, and lipid droplets [177, 208]. In contrast, the IMM has restricted permeability, with an electrochemical membrane potential (120–180 mV, negative inside) needed to drive oxidative phosphorylation. The IMM contains specific transporters, and translocases that facilitate the passage of substrates into the matrix, where they are metabolized by enzymes, including those of the tricarboxylic acid cycle and fatty acid oxidation, as well as antioxidant enzymes [117]. In addition, the IMM contains cardiolipin (CL), the signature phospholipid of energy-transducing membranes, which has been reported in rat heart mitochondria to become oxidized during ischemia and reduced upon reperfusion [112].
Mitochondrial distribution in cardiomyocytes
Advancements in live-cell imaging techniques, including 3D reconstruction and electron tomography, have revolutionized our appreciation of the spatial distribution of three distinct subpopulations of mitochondria within cardiomyocytes [186, 194, 207, 221]: interfibrillar mitochondria (IFM), subsarcolemmal mitochondria (SSM), and perinuclear mitochondria (PNM) [221]. Despite their shared cellular environment, these mitochondrial compartments exhibit remarkable heterogeneity in their morphological attributes and biochemical functionalities. This divergence is particularly noticeable in their responses to metabolic and physiological processes.
For instance, these populations demonstrate differences in protein content [70], redox potentials, signifying variations in their oxidative metabolic activity [186]. IFM in adult ventricular cardiomyocytes are typically oval in shape and organized in longitudinal rows alongside the myofibrils and possess a higher rate of substrate oxidation (approximately 1.5 times) than the other two mitochondrial subpopulations [104]. The close proximity of the IFM to the intense energy-requiring demands of the myofilaments results in higher levels of substrate oxidation and increased activity of key oxidative phosphorylation enzymes, including succinate dehydrogenase and citrate synthase. The SSM located directly beneath the sarcolemma may provide the energy supply for the active sarcolemmal transport of electrolytes and metabolites [23, 25]. PNM typically appear more spherical in shape and are distributed around the nucleus in the cardiomyocyte and provide ATP for nuclear transcription. PNM also regulates various nuclear functions, including modifications of promoters to alter transcriptional complex assembly and mRNA expression [166, 207]. Proteomics studies have demonstrated that IFM and SSM possess variations in protein content and synthesis rates [122, 127]. Isotopic tracer methods and peptide analysis by liquid chromatography–mass spectrometry (LC–MS/MS) allow the measurement of mitochondrial protein synthesis in vivo [33, 70, 203]. The heavy water (2H2O) method with LC–MS/MS analysis has determined that the turnover rates of SSM proteins are faster in mice (average half-life 17 days) [127] than in rats. In contrast, the mitochondrial protein half-life in rats was significantly lower (average half-life 30 days) [122]—the faster turnover of SMM protein in mice correlated with its higher metabolic rate. Interestingly, ischemic damage appears to be progress more rapidly in the SSM subpopulation when compared to IFM.
Additionally, it has been reported that mitochondrial protein synthesis in SSM subpopulations is 15% faster than IFM [122]. Interestingly myocardial protective effects have been shown mainly in SSM subpopulations [139]. Forty-five minutes of ischemia decreased oxidative phosphorylation through cytochrome oxidase in SSM [140]. Most research on cardioprotective therapies that protect the heart against a greater subsequent ischemic insult suggests that the SSM subpopulation is predominantly impacted due to heightened sensitivity to ischemic conditions and Ca2 + overload [57]. This is attributed to the environment associated with the subsarcolemmal and extracellular spaces. Furthermore, connexin 43, associated with cardioprotection, is reported to exist solely in the SSM of cardiomyocytes, further hinting at the role of SSM in protection [25].
Tracking mitochondrial dynamics by photoactivation of mtPA-GFP has revealed marked differences in mitochondrial fusion and fission between the PNM and IFM populations. The PNM population displays significantly heightened fusion and fission activity compared to the IFM population. Intriguingly, sensitivity to mdivi-1, an inhibitor of mitochondrial fission was more pronounced in PNM. This is an intriguing phenomenon, especially considering the similarity in the distribution of fusion–fission proteins between the IFM and PNM. These findings suggest that despite the shared presence of fusion–fission proteins, PNM demonstrates a more dynamic state of fusion and fission [148]. This implies that other regulatory mechanisms or local conditions within the PNM may promote these dynamic activities, shedding light on nuanced differences in the behavior of distinct mitochondrial populations. An improved understanding of these distinct mitochondrial populations may provide a more informed view of cardiac mitochondrial function and regulation and facilitate the development of targeted therapeutic strategies to address mitochondrial dysfunction in cardiac disorders.
The mitochondrial shaping proteins
Mitochondria are dynamic organelles that constantly change their shape between a fragmented disconnected phenotype by undergoing fission and an elongated interconnected morphology by undergoing fusion, processes that are coordinated by specific proteins. Mitochondrial fusion plays a vital role in the exchange of genetic material between the mitochondria, enhancing their functionality and resilience, especially under metabolic and environmental stressors (Fig. 1A). Mitochondrial fission is a fundamental process required for dividing organelles and maintaining their quality through mitophagy, ensuring that they function optimally within the cell [98, 178, 180]. In the following section, we provide a detailed description of the cellular machinery involved in orchestrating mitochondrial fusion and fission.

Mitochondrial morphology dynamically adapts to diverse environmental stimuli, resulting in morphological modifications that have implications for cell survival. A In healthy hearts, mitochondrial quality control is managed by the dynamic balance between fragmented and elongated phenotypes, enhancing both functionality and metabolism. Mitochondrial fusion, a characteristic process, commences with the interlinking of two proximate mitochondria facilitated by the OMM fusion GTPase proteins, Mfn1 and Mfn2. This action mediates the fusion of the OMM. Subsequently, OPA1 directs the fusion of the IMM and matrix material, culminating in a single elongated mitochondrion. B Mitochondrial fission is over-stimulated under IRI conditions, predominantly driven by Drp1 phosphorylation at Ser-616. Adaptor proteins such as Fis1, Mid49, Mid51, and MFF mediate initial mitochondrial constriction preceding Drp1 recruitment. The recruited Drp1 helps form helical ring oligomers, which in turn stimulate the constriction and scission of the outer mitochondrial membrane through GTP hydrolysis. A multitude of upstream signals, some shared and some distinct across cell types, such as cardiomyocytes, endothelial cells, fibroblasts, and vascular smooth muscle cells, incite this excessive mitochondrial fission. This heightened fission often culminates in cellular dysfunction and death, particularly in cardiac tissues subjected to IRI. This figure was created using the Adobe Illustrator 2023. IRI ischemia–reperfusion injury, IFM interfibrillar mitochondria, SSM subsarcolemmal mitochondria, PNM perinuclear mitochondria, OMM outer mitochondrial membrane, IMM inner mitochondrial membrane, CM cristae membrane, Mfn1 mitofusin 1, Mfn2 mitofusin 2, OPA1 optic atrophy 1, Drp1 dynamin-related protein 1, Mid49 mitochondrial dynamics 49, Mid51 mitochondrial dynamics 51, MFF fission factor, ER endoplasmic reticulum, mPTP mitochondrial permeability transition pore, ATP adenosine triphosphate; PPCI: primary percutaneous coronary intervention
Mitochondrial fusion
A series of highly conserved GTPase proteins play vital roles in the dynamics of mitochondrial morphology. Among these, mitofusin 1 (Mfn1) and 2 (Mfn2) are transmembrane GTPases that mediate the fusion process of the OMM [43, 46]. Mfn2 plays an additional role as a tether between mitochondria and endoplasmic reticulum through the interaction of two newly discovered Mfn2 variants ERMIN2 and ERMIT2 [167]. Both Mfn1 and Mfn2 facilitate the docking of two juxtaposed mitochondria through the oligomerization of their GTPase domains, a process which requires guanosine triphosphate hydrolysis [227]. Recent insights into the topology of mitofusins suggest the existence of only one transmembrane domain in human Mfns, suggesting an alternative mechanism for oligomerization of Mfn molecules, which is essential for OMM fusion (Fig. 1) [142, 201]. Mattie et al. showed that two cysteine residues located within the HR2 domains (situated in the IMS) could undergo oxidation when exposed to elevated levels of oxidized glutathione. This oxidation leads to the formation of disulphide bonds between Mfn molecules, facilitating oligomerization. This represents a crucial step in the understanding of the mitochondrial fusion process [157]. Researchers have also demonstrated that introducing glutathione (GSH) to previously formed glutathione disulphide (GSSG)-induced Mfn2 oligomers reversed oligomerization. This novel mechanism underscores the crucial role of redox signaling in OMM fusion [225]. Further investigations are warranted to fully elucidate the mechanistic underpinnings of mitochondrial fusion and understand the impact of aberrant redox signaling on this process.
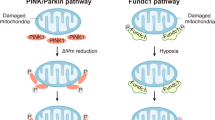
Post-translational modifications, including phosphorylation, ubiquitination, and deacetylation, modulate Mfns activity. For instance, phosphorylation of Mfn1 in the HR1 domain by extracellular signal-regulated kinase (ERK) inhibits mitochondrial fusion, thereby favoring apoptosis. Conversely, the deacetylation of Mfn2 by histone deacetylase 6 activates it, promoting fusion under conditions of glucose deprivation. Moreover, cellular stress induces the phosphorylation of Mfn2 by JNK, which activates E3 ubiquitin ligase. This ligase ubiquitinates Mfn2, prompting its proteasomal degradation [72]. This intricate interplay of regulatory modifications underscores the complexity of the mitochondrial fusion control mechanisms. The degradation of Mfn2 following its ubiquitination triggers mitochondrial fragmentation and increases the risk of apoptotic cell death. Furthermore, Mfn2 can be phosphorylated by PINK1, a modification that paves the way for its ubiquitination by parkin [86, 98]. This sequence of events culminates in mitophagy, demonstrating the vital role of these processes in regulating mitochondrial dynamics and cellular health.
Another important member of this conserved GTPase family, which belongs to the dynamin class, is optic atrophy 1 (OPA1) [6]. This protein resides in the IMM facing the intermembrane space and regulates mitochondrial fusion of the latter [98] and encompasses eight isoforms in human. These isoforms are generated by alternative splicing [174, 243] of three small exons, namely 4, 4b, and 5b, which are located in the N-terminal region of the gene. Each isoform of OPA1 can feature between one and three proteolytic cleavage sites conventionally labeled S1, S2, and S3 [6]. The S1 site is a common feature of all the eight OPA1 isoforms. In contrast, the S2 and S3 sites each appear in only four isoforms, underscoring the variable proteolytic susceptibility of different OPA1 isoforms. Proteolytic cleavage of the S1 is regulated by the metalloprotease, OMA1. Proteolytic cleavage at sites S2 and S3 is constitutive and is mediated by YME1L [229, 243]. However, cleavage at the S1 site in the OPA1 isoforms is condition specific and occurs exclusively under certain stress-related conditions.
Currently, the precise mechanism of IMM fusion is not fully understood. Ban et al. demonstrated that when recombinant L-OPA1 was incubated with liposomes containing reconstituted CL, a heterotypic interaction was occurring and between L-OPA1 and CL culminating in the fusion of IMM, highlighting the essential role of CL in this process. Further investigation demonstrated that CL is essential for membrane fusion, even when L-OPA1 was present on both sides of the membrane [14, 15]. Ban et al. extended their findings to confirm that the GTPase of OPA1 was necessary to maintain the fusion activity. This implies the pivotal roles of CL-OPA1 binding and OPA1 GTP hydrolysis in IMM fusion [13]. To expand this understanding, further research is necessary to elucidate the exact roles of CL in IMM fusion and to uncover the underlying molecular mechanisms involved. In addition to the proteolytic modifications of OPA1 orchestrated by YME1L and OMA1, there is an additional layer of regulation. Sirtuin-3 (Sirt3), a NAD-dependent deacetylase, targets the GTPase effector domain of OPA1 at lysine residues 926 and 931. This molecular modification increases the GTPase activity of OPA1, thereby promoting an environment conducive to mitochondrial fusion [262]. Other than mitochondrial fusion, OPA1 has a central role in controlling cristae shape in the IMM impinging on mitochondrial metabolism by respiratory chain supercomplexes assembly [54, 239] and on apoptosis, blunting cytochrome c release [79]. We recently found that a redox-insensitive mutant of OPA1 dissociates the mitochondrial fusion and the cristae shape activity of OPA1 [217].
Mitochondrial fission
Mitochondrial fission is a critical cellular process in which a single mitochondrion segregates into two distinct entities. This mechanism has several essential functions, including appropriate apportioning and inheritance of organelles during cellular division, ensuring an even distribution of mitochondria within the cell, and facilitating mitophagy and release of cytochrome c (cytc), a step integral to apoptosis. If fission is inhibited, the balance between fusion and fission results in the accumulation of elongated damaged mitochondria owing to unopposed fusion activity [98, 202, 262]. Conversely, the disruption of fusion mechanisms results in an overabundance of fragmented mitochondria. The precise mechanisms underlying this phenomenon remain uncertain; however, one plausible explanation suggests that this fragmentation may be a compensatory measure to maintain a consistent ATP supply within the cells.
In mammals, fission is coordinated by dynamin-related protein 1 (Drp1), fission protein 1 (Fis1), mitochondrial fission factor (MFF), and mitochondrial dynamic proteins of 49 kDa and 51 kDa (Mid49 and Mid51) [32]. The preliminary phase of mitochondrial division is facilitated by the ER. In this process, ER tubules establish contact with mitochondria, mediating constriction at these sites prior to the recruitment of Drp1. Once Drp1 is recruited to the outer mitochondrial membrane by the adaptors proteins MFF, Mid49, Mid51, and Fis1, where it forms a ring-like structure around the mitochondrion, amplifying the existing constriction [132, 218]. Subsequently, Drp1 undergoes GTP hydrolysis, leading to the recruitment of dynamin 2 (DNM2) to the site of mitochondrial constriction where it assembles to complete the division process. However, another perspective indicates that DNM2 may not be required for mitochondrial fission and that Drp1 alone, with its constricting and severing capabilities, might suffice to complete the fission process. Whether complete mitochondrial fission occurs in the absence of DNM2 remains to be elucidated [120]. Constriction of the inner IMM is a calcium-dependent process that takes place at the point of contact between the mitochondria and ER. This process is initiated by calcium release from the ER into the mitochondria, leading to IMM constriction and division before the recruitment of Drp1. Notably, CL, in addition to its role in mitochondrial fusion, interacts with Drp1. This interaction promotes the oligomerization of Drp1 and stimulates its GTPase activity, thereby increasing the constriction of liposome membranes. Further research is required to understand how CL modulates the balance between mitochondrial fusion and fission, and the triggers for its diverse roles (Fig. 2) [119].

Exercise training is the most accessible and effective intervention for many cardiovascular diseases, primarily because it amplifies the intracellular production of ROS and energy-regulating molecules, such as ATP and AMP. These molecules serve as potent signaling transducers capable of activating a range of protein kinases, including AMPK, an important mediator of glucose and fatty acid oxidation. Moderate interval training has been correlated with cardioprotection against IRI, often attributed to the role of the HSP72, AMPK and PGC-1α. Strikingly, consecutive days of endurance exercise can enhance mitochondrial dynamics in male rats with myocardial infarction by increasing the expression of Mfn2 and PGC-1α, while reducing the levels of Drp1. This figure was created using the Adobe Illustrator 2023. AMPK AMP-activated protein kinase, ROS reactive oxygen species, HSP72 heat shock protein 72, PGC-1α peroxisome proliferator-activated receptor-γ coactivator-1 alpha, Drp1 dynamin-related protein 1, Mfn2 mitofusin 2
Drp1 is subjected to an extensive array of post-translational modifications. These include phosphorylation [40], SUMOylation [4], palmitoylation [168], ubiquitination [254], S-nitrosylation [28], and O-GlcNAcylation [51]. These modifications occur predominantly in the B-insert region of the protein and collectively orchestrate the functional regulation of Drp1. Phosphorylation is one of the most extensively studied post-translational modifications of Drp1. Depending on the specific phosphorylation site, it can boost or dampen Drp1 activity. Several sites have been identified, including Ser-579, Ser-40, Ser-585, Ser-44, Ser-592, Ser-656, Ser-616, Ser-637, and Ser-693. Among these, Ser-616 and Ser-637 have garnered scientific attention because of their significant roles in the regulation of Drp1 [200]. Phosphorylation of Ser-616 is an activating event that contributes to the OMM localization of Drp1 and subsequent mitochondrial fission. The most recent kinases known to regulate Drp1 phosphorylation include CDK1/CYCLINB [155], FAK-Erk1/2 [44], AMPK (adenosine monophosphate-activated protein kinase) [69], PINK1 (PTEN-induced putative kinase 1) [224], CAMKII (Ca2 + /calmodulin-dependent kinase II) [245, 255], GSK3ß [141], ROCK1 and DDAH2 (dimethylarginine dimethylaminohydrolase 2)-PKA-NO (nitric oxide) [108].
Focal adhesion kinase (FAK) regulates the phosphorylation of Drp1 via extracellular signal-regulated kinases 1 and 2 (Erk 1/2) in cardiomyocytes. FAK–Erk1/2–Drp1 pathway mediates metabolic adaptation in response to extracellular environment change, and inhibiting this pathway reduces 50% of ATP levels [44]. Chang et al. [44] and Ikeda et al. [111] have demonstrated that FAK–Erk1/2–Drp1 Ser-616 is essential for maintaining the basal energy supply of cardiomyocytes. Fibronectin-activated FAK is associated with mitochondrial fission and respiration via Drp1 Ser1-616 in CMs. However, it has been reported that increased fibronectin expression is associated with cardiac hypertrophy via impaired adrenergic receptors (ARs) [150]. Erk1/2–Drp1 616 activation has been associated with cardiotoxicity in vitro and in vivo rat models. Transient receptor potential cation channel subfamily C member 6 (TRPC6) has been correlated to cardiac pathologies, including MI [149], cardiac hypertrophy [172] and fibrosis [175]. TRPC6–Erk1/2–Drp1 activation induces mitochondrial fission and cell death in a model of (AIC) anthracycline in rat cardiomyocytes [253]. It has been demonstrated that ARs stimulation induces mPTP opening through activating CaMKII via phosphorylation of Drp1 at Ser-616. Inhibiting CAMKII activity or mutating the phosphorylation site Ser-616 rescues cardiomyocytes death by mPTP opening [255].
PINK1 is another kinase that phosphorylates Drp1 at the Ser-616 site, thereby regulating mitochondrial fission [91]. Studies have shown that PINK1 overexpression boosts mitochondrial fission via Drp1 Ser-616, which slows the progression of HFpEF (heart failure with preserved ejection fraction). The same research team found that without PINK1, there is a decrease in genes related to mitochondrial function, membrane potential, and ATP production, pointing to mitochondrial dysfunction. Interestingly, in cells lacking PINK1, the restoration of mitochondrial function was observed with Drp1 overexpression but not with Drp1 Ser-616 [224]. The phosphorylation of the Ser-616 site is vital for Drp1's role in regulating mitochondrial fission and overall function. Consequently, PINK1 acts to phosphorylate Drp1 at this specific site, enhancing mitochondrial performance.
The phosphorylation of Ser-637 prevents the translocation of Drp1 to mitochondria and keeps it inactive in the cytosol thereby preventing mitochondrial fission [261]. The balance of Ser-616 and Ser-637 phosphorylation in Drp1 is not only integral to the function of the protein but also linked to the onset of various diseases [200]. Interestingly, phosphorylation of Ser-616 alone did not induce mitochondrial fission [4] [260]. Given the spatial proximity of Ser-616 and Ser-637 in the three-dimensional structure of Drp1, research has demonstrated that the level of phosphorylation at the Ser-637 site can affect the phosphorylation of Ser-616 [231]. However, the phosphorylation levels at the Ser-637 site were not influenced by the phosphorylation state of Ser-616 [31, 200]. This leads to the intriguing possibility that the basal phosphorylation level of Ser-637 could be instrumental in maintaining the basal phosphorylation state of Ser-616, suggesting a priming role for Ser-637 phosphorylation of Ser-616 [115, 264]. This hypothesis needs to be further tested.
AMPK is an upstream kinase that regulates Drp1 phosphorylation. Intravenous pre-administration with AICAR, an activator of AMPK, improves mitochondrial membrane potential, reduces reactive oxygen species production, and inhibits mitochondrial damage by enhancing phosphorylation of Drp1 at Ser-637 and inhibiting the phosphorylation of Drp1 at Ser-616 [69]. Recently, studies have indicated that DDAH2 modulates Drp1 activity through the nitric oxide synthase (NOS) and subsequent NO generation, leading to Drp1 phosphorylation and mitochondrial fission [108].
Studying mitochondrial morphology
Our understanding of mitochondrial dynamics has been mainly derived from in vitro studies and non-mammalian models, leaving certain aspects of mammalian mitochondrial function that have not been entirely explored. Factors such as tissue type, specific cell population, and even mitochondrial subpopulations can influence mitochondrial structure and behavior. Yet, the ramifications of this diversity are, in many cases, largely undefined. Consequently, the demand for innovative genetic tools capable of meticulously monitoring dynamic fission–fusion events across diverse tissues, developmental stages, and disease conditions such as cardiovascular diseases is increasingly critical. In this section, we will discuss various strategies for studying mitochondrial dynamics.
Over the last decade, research has recognized the potential of using mitochondrial-targeted fluorescent proteins (FPs) [80, 156, 190], photoactivation [121, 184], and photo-switching [195] to assess mitochondrial dynamics. Genetic tools have also been developed to monitor the dynamic fusion and fission events in different tissues. Several transgenic mouse strains express mitochondria-targeted fluorescent reporters in green- and red-light spectra. These included Mito-EGFP, mtGFP-tg, mtDsRed2-Tg, Acr3–EGFP–CAG–su9-DsRed2, PhAM-excised, Mito-QC, and Mt-Keimareporters. The limitation of green/red spectral emission applies similarly to tissue-specific and inducible reporter strains, including, Mito-Timer, PhAMfloxed, Thy1–mitoDsRed, Thy1–mito-TagRFP, Nse–mitoYFP, CaMKIIa–mitoYFP, Hb9–MitoEGFP, and Endo–mitoEGFP and Thy1.2–mitoDendra [1, 93, 158, 195, 223, 257].
Photoactivation offers external control over the intensity or color of fluorescent emission. This process enables a distinct group of proteins to be marked and tracked, thereby revealing their subsequent dynamics and interactions within individual cells, tissues, and even whole organisms. This precise level of control and visibility presents new opportunities for the in-depth exploration and understanding of biological processes. Two distinct forms of photoactivation have been observed [53, 189, 235]. The first involves reversible photo-switching between the fluorescent and non-fluorescent states, which is brought about by the isomerization of the chromophore. The second is irreversible photoconversion, which occurs due to light-induced covalent modification [7, 184, 195, 248].
Dendra2 (D2) is a monomeric photoconvertible fluorescent protein originally cloned from the soft coral Dendronephthya sp., with a structure similar to that of the green fluorescent protein from the jellyfish Aequorea Victoria (avGFP). Similar to avGFP, the unconverted form of D2 showed a peak excitation at 490 nm and a peak emission at 507 nm [84, 185]. However, in D2, short-wavelength light-induced structural photoconversion that shifted the spectral properties to longer wavelengths, with a peak excitation at 553 nm and a peak emission at 573 nm. In contrast to numerous other photo-switchable proteins, the transition from the green (gD2) to the red (rD2) state is irreversible, with the red signal fading solely because of protein degradation. Therefore, D2 fluorescence serves as a robust and enduring marker that enables cells to be tagged and monitored noninvasively across both space and time. The mito-Dendra2 mouse model enables the study of mitochondrial dynamics across a broad range of primary cells and tissues, including disease conditions. Specifically, studies using cardiomyocytes isolated from mito-Dendra2 heart mice demonstrated that mitochondria undergo fission in response to simulated ischemia–reperfusion injury (SIRI). Moreover, it has been observed that hydralazine, a drug commonly prescribed to manage hypertension and heart failure, can prevent mitochondrial fission and reduce MI size. This study underscores the potential of the mito-Dendra2 mouse as a powerful tool for understanding and treating conditions associated with mitochondrial dynamics [195, 197].
Moreover, a diverse range of mitochondrial biosensors have been developed to monitor various processes. These processes include energy production, generation of ROS, redox state, secondary messenger activities (such as those involving cAMP or Ca2+), and Zn2+ homeostasis. Most of these markers emit fluorescence in the blue and green spectral range. Therefore, the scarcity of ubiquitous mitochondrial reporters that function outside the blue/green/red color spectrum restricts the ability of researchers to track mitochondrial dynamics and other processes visualized using different biosensors [131].
Mito::mKate2, a far-red FPs, has the unique capability to be observed simultaneously with traditional fluorescent markers (including GFP, YFP, CFP, and DsRed), as well as mitochondria-specific biosensors. mito::mKate2 is an effective tool for tracking mitochondrial behavior and cell cycle changes during embryonic development and in adult tissues in mice. The superior brightness and photostability of far-red FPs, such as mito::mKate2, permit a deeper imaging scope than traditional green and red markers. Consequently, mito::mKate2 is better suited for in vivo and ex vivo imaging of mitochondrial activity in living tissues [16].
In parallel with other scientific fields, cutting-edge technologies such as genomics, proteomics, transcriptomics, metabolomics, and epigenomics have spearheaded revolutionary discoveries in mitochondrial biology. The deployment of compartment-specific sensors and techniques for assessing mitochondrial respiration in intact cells has greatly augmented our understanding of mitochondrial physiology. However, despite these knowledge gains, we still face the challenge of comprehensively characterizing and understanding the role of the mitochondria in cardiac diseases. Consequently, it is imperative to expedite the development and application of enabling tools and technologies to bridge the gap between the basic discoveries and their translation into clinical practice.
Mitochondrial dynamics in cardiac diseases
An imbalance in mitochondrial morphology can impact on energy and mitochondrial ROS production, Ca2+ homeostasis, and protein stability, potentially inducing cardiomyocyte death within the heart in a variety of cardiac diseases. In this section, we discuss alterations in mitochondrial morphology linked to cardiac diseases and explore prospective therapeutic strategies aimed at counteracting mitochondrial dysfunction. Such strategies show substantial promise for the prevention and treatment of a range of cardiac conditions.
Changes in mitochondrial morphology in acute myocardial ischemia–reperfusion injury (IRI)
Currently, the most effective therapeutic intervention for reducing acute myocardial IRI and limiting MI size in AMI patients is timely and effective myocardial reperfusion using either thrombolytic therapy or primary percutaneous coronary intervention (PPCI). However, myocardial reperfusion itself can induce further cardiomyocyte death, a phenomenon known as acute myocardial IRI [101]. The sequence of events that occur during IRI has been extensively explored and has been detailed in several recent reviews [96, 179, 181, 204].
Mitochondria can trigger cell death in cardiomyocytes via two main pathways. The first involves excessive permeability of the OMM, leading to cytc leakage into the cytoplasm. Cytochrome c activates caspase-9, which initiates the cleavage of caspase-3. It is characterized by a reduction in mitochondrial membrane potential, increased levels of ROS, increased BAX expression, and decreased Blc-2 expression, a classical route to mitochondria-induced apoptosis [59, 87]. It has been reported that Drp1 acts with Bcl-2 family proteins to accelerate mitochondrial fragmentation and apoptosis. During IRI, Drp1 is recruited to the OMM, instigating the division of these organelles. Several post-translational modifications can affect Drp1 fission activity. In particular, Drp1 phosphorylation at Ser-616 increases its translocation toward the OMM increasing mitochondrial fragmentation and mitochondrial ROS generation. Concurrently, cyt c is discharged into the cytoplasm, which triggers an inflammatory response and initiates cell apoptosis [126, 220, 221]. The second pathway is triggered by the sustained opening of the mitochondrial permeability transition pore (mPTP) due to the formation of a non-selective pore in the IMM whose molecular composition is still debated. Prolonged opening of the mPTP induces mitochondrial swelling, collapse of the mitochondrial member potential, and impairment of oxidative phosphorylation, leading ultimately to cell death by necrosis (Fig. 1B) [204].
Mitochondria have been demonstrated to undergo fragmentation during acute myocardial IRI. A study conducted by Ong S. and colleagues revealed that overexpression of Mfn1, Mfn2, or the dominant-negative mutant of Drp1 (Drp1 K38A) to induce mitochondrial elongation delayed the opening of the mPTP and significantly reduce cell death after SIRI in HL-1 cells [182]. Drp1 activation and excessive mitochondrial fission have been observed in peri-infarcted regions of mouse hearts during the initial phase of ischemia [173] and continue to be sustained throughout the reperfusion process [67]. Pharmacological Drp1 inhibition protected adult CMs against simulated IRI, inhibited mPTP opening, and reduced MI size in an in vivo murine model [173].
Mfn2 plays a pivotal role in IRI and HF, given its ability to regulate mitochondrial fusion, ER–ER–mitochondria interaction, cellular metabolism, and cell death. Some studies have suggested that Mfn2 overexpression in heart diseases, such as HF and myocardial ischemia, can mitigate cardiac hypertrophy and dysfunction under various stressors. Conversely, other studies have indicated that deletion of Mfn2 in cardiomyocytes could confer protection against IRI. Thus, there is a pressing need for further research to delve deeper into the detailed molecular mechanisms of Mfn2 in cardiovascular diseases, as it may reveal a potential therapeutic target for patients [50]. Interestingly, acute genetic ablation of both Mfn1 and Mfn2 in murine cardiomyocytes paradoxically reduced MI size following IRI, although one may have expected MI size to be increased due to unopposed mitochondrial fission [89]. The apparent explanation was that the non-fusion pleiotropic effect of Mfn2 as a tethering protein between sarcoplasmic reticulum (SR) and mitochondria had a more dominant effect than the role of Mfn2 on fusion. Therefore, the genetic ablation of Mfn2 protected the mitochondria against mPTP opening and mitochondrial dysfunction by disrupting the association between mitochondria and SR and reducing mitochondrial calcium overload. However, it is worth noting that while acute ablation of Mfn1 and Mfn2 offers protection against acute IRI, long-term ablation of these proteins could be detrimental, leading to cardiomyopathy and sudden cardiac death [89].
Also, OPA1 plays a central role in IRI. A mouse model expressing an increased level of OPA1 displayed protection against cardiac ischemia–reperfusion injury by blunting cristae remodeling and preventing cell death [242]. During ischemia–reperfusion, OPA1 undergoes proteolytic cleavage that is due to the loss of activity of the protein. OPA1 deficiency has been associated with increased sensitivity to IRI with an imbalance in mitochondrial Ca2+ uptake [135]. Moreover, increased ROS production occurring during ischemia–reperfusion injury leads to cysteine oxidation of OPA1 contributing to mitochondrial damage and cell death [217].
Mitochondrial dynamics in heart failure
Heart failure has emerged as a significant health crisis worldwide, particularly in the elderly population. It represents the final stage of a range of cardiovascular diseases and is distinguished by its high incidence rate, frequent hospitalization, and elevated mortality [212, 233]. Heart failure can primarily be categorized into ischemic and non-ischemic types [143]. Ischemic HF is closely linked to coronary artery disease, particularly myocardial infarction, and constitutes approximately 50% of all cases of HF [164].
During the pathological development of HF, cardiomyocytes undergo alterations in their energy metabolism. This transition manifests as an increased dependence on glucose while simultaneously experiencing diminished utilization of fatty acids through beta-oxidation. This metabolic reconfiguration of substrate utilization prompts a shift in cardiac metabolism, reverting it back to a state reminiscent of fetal energy metabolism. When glucose serves as the substrate for energy production, the associated oxygen consumption is reduced compared to when fatty acids are utilized [17]. Severe hypoxia profoundly impairs oxidative phosphorylation. The shift to anaerobic glycolysis fails to produce sufficient ATP to satisfy the energy demands of the heart and leads to lactate accumulation. The depletion of ATP and resultant acidosis contribute to reduced myocardial contractility and damage to membrane pumps and ion channels. These alterations trigger mitochondrial swelling, accumulation of Ca2+, and opening of the mPTP. These changes are particularly noticeable in cardiomyocytes affected by myocardial ischemic injury. The role of mPTP has been highlighted as being critical in various forms of cell death associated with myocardial IRI [22, 55, 258]. Indeed, endocardial biopsies from 48 patients, comprising a total of 66 samples, were examined. These patients were diagnosed with cardiomyopathy, a specific form of HF. The analyses revealed diverse cellular architectures, with a notable frequency of changes involving mitochondria. Certain cells exhibit notable mitochondrial changes, including a significant increase in their number, thinning of their matrices, and the occasional emergence of unusually large mitochondria. Initial biochemical investigations were conducted using tissue homogenates from explanted hearts, bypassing the use of isolated mitochondria. These homogenate studies revealed a considerable decrease in both creatine and creatine phosphate levels, implying the depletion of the energy reserves of the cells [10].
Downregulation of Mfn2, a key regulator of mitochondrial dynamics, causes mitochondrial fragmentation, which contributes to the onset of HF. This has been noted both in rat models and patients with pulmonary arterial hypertension (PAH) [215]. In a study conducted by Chen L and colleagues, significant mitochondrial fragmentation was observed in adult Sprague–Dawley (SD) rats with HF post-myocardial infarction. Despite steady levels of OPA1 mRNA, there was a noticeable decline in the protein content of OPA1. In contrast, the protein contents of Mfn1 and Mfn2 remained unaltered [49]. Other studies have shown that in the HF dog model, mitochondrial fission and fusion proteins in the left ventricular myocardium are dysregulated. The expression levels of Drp1 and Fis1 were significantly upregulated. Research has indicated that mice with mutations in the Mff gene experienced mortality at 13 weeks, which is attributable to HF induced by severe dilated cardiomyopathy. The mutant tissues presented a reduction in mitochondrial density and respiratory chain activity while exhibiting an increase in mitochondrial size. These findings suggest that Mff-mediated mitochondrial fission could potentially contribute to the progression of HF [47]. Similarly, a study confirmed that homozygous Mff-deficient (Mffgt) mice exhibited a smaller MI size, restored cardiac function, improved blood flow, and reduced microcirculatory perfusion defects [273].
The inclination toward mitochondrial fission in HF presents several challenges. Primarily, it reduces the number of mitochondria within the cell. This reduction may impair cardiac function owing to diminished ATP production. Second, escalated fission may result in the generation of small, dysfunctional mitochondria. These underperforming mitochondria are not only inefficient in ATP production, but also have a greater likelihood of releasing ROS, which can impact on oxidation of DNA, proteins, and lipids. Along with increased fission, HF induces a decrease in mitochondrial fusion. This decrease in fusion can result in a reduced size and number of mitochondria within the cell, and foster an increase in the aggregation of mitochondria into large clumps. These sizeable mitochondrial aggregations are less efficient in ATP production and more prone to ROS release.
Research into mitochondrial dynamics in HF is currently in its nascent stage, and findings have been mixed, primarily because of two main challenges. First, the complex dynamic system encompassing mitochondrial dynamics and metabolism is involved in the progression of HF, leading to comprehensive studies of the function of mitochondria in HF. Second, mitochondrial dynamics play varying roles in different stages of HF and are influenced by a plethora of pathological conditions. Therefore, more in-depth research is necessary to elucidate the mechanisms underlying mitochondrial dynamics in HF. Such insights could potentially identify critical timing and novel molecular targets, paving the way for the development of innovative therapies for HF. Finally, targeting mitochondrial morphology as a therapeutic strategy for HF will be challenging as it will require chronic treatment which in itself can induce adverse effects such as cardiomyopathy with prolonged inhibition of mitochondrial fission.
Changes in mitochondrial morphology in other cardiac diseases
Diabetic cardiomyopathy (DMCM)
Despite growing evidence highlighting the functional and structural alterations in the myocardium as a consequence of diabetes, the underlying pathological mechanisms, particularly in type 2 diabetic cardiomyopathy (DMCM), remain unclear [249, 256]. DMCM is characterized by abnormal myocardial structure and function in individuals with diabetes, occurring independently of other cardiac risk factors, such as coronary artery disease, hypertension, or significant valvular disease [63, 193, 214]. Recent studies have shown that mitochondrial oxidative damage, mitochondrial dysfunction [76], and diminished cardiomyocyte function [116, 188] are observed in diabetic hearts and contribute to DMCM development. Reduced Mfn2 expression and excessive mitochondrial fission have been demonstrated in diabetic hearts, which results in mitochondrial dysfunction and DMCM. db/db mouse hearts showed reduced Mfn2 expression and impaired cardiac function at 12 weeks of age compared with db/ + mice [76]. Mitochondrial morphological abnormalities, mitochondrial dysfunction, and disrupted Ca2+ handling contribute to the development of DMCM [50, 66]. In patients with DMCM, cardiomyocytes exhibit a range of detrimental changes, including fragmented mitochondria and decreased expression of Mfn1. Interestingly, Mfn1 expression was inversely correlated with HbA1c levels, a critical marker of long-term blood glucose control [163, 246]. Bach D et al. showed that mitochondrial fission activity was higher in the hearts of db/db mice with type 2 diabetes mellitus, possibly due to diminished Mfn2 expression. The protective effects of Mfn2 in high-glucose and high-fat medium (HG/HF)-treated cardiomyocytes were blunted by fission activation FFCP, while a Mfn2 activator restored mitochondrial fusion and exerted the protective effects in Mfn2-knockdown CMs, suggesting that imbalanced mitochondrial dynamics induced by down-regulated Mfn2 could be the main cause of cardiac dysfunction in diabetic hearts [106]. This was likely linked to a reduction in the expression of peroxisome proliferator-activated receptor α (PPARα) and a subsequent decrease in PPARα binding to the Mfn2 promoter. Given that mitochondrial dynamics serve as the foundation of mitochondrial function, more in-depth investigations are warranted to devise effective interventions targeting mitochondrial fusion and fission in diabetes to retard DMCM progression [12, 106].
Hypertension
Hypertension is closely linked to endothelial dysfunction and structural remodeling. Oxidative stress, which is considered a key player in both disease progression and aging, emanates primarily from mitochondria, which are also major targets of ROS [134, 165]. Impairments in mitochondrial biogenesis and dynamics can significantly undermine bioenergetic supply, thereby contributing to endothelial dysfunction and the development of cardiovascular diseases [142]. Activation of the sympathetic nervous system has been recognized as a pivotal factor in the development of hypertension among obese individuals. It also plays a critical role in driving cardiac remodeling processes that occur in association with hypertension. Norepinephrine initiates cardiomyocyte hypertrophy by activating specific signaling cascades, particularly calcium-activated protein phosphatase calcineurin. In hypertensive rats, there was a notable decrease in the mRNA levels of fusion proteins Mfn1, Mfn2, and OPA1 [230]. This suggests a tendency toward increased mitochondrial fragmentation during hypertension. In relation to this, studies on cultured neonatal rat cardiomyocytes treated with norepinephrine have shown it stimulates mitochondrial fission. This event is associated with a decline in mean mitochondrial volume and an increase in the relative number of mitochondria per cell [192]. This change is driven by the norepinephrine-mediated elevation of cytoplasmic Ca2+, which in turn activates calcineurin, promoting the relocation of the fission protein Drp1 to the mitochondria. A mutation in Drp1 has been linked to cardiomyopathy, highlighting the essential role of Drp1-mediated processes in preserving normal cardiac function [230, 237]. These findings have led to the speculation that norepinephrine might stimulate mitochondrial fission as a compensatory mechanism to uphold heart contractility under hypertensive conditions, potentially leading to thickening of the ventricular wall. Therefore, it has been proposed that curbing Drp1-mediated mitochondrial fission may help prevent the progression of cardiac pathologies. However, experimental observations also suggest that total loss of Drp1 function may have adverse effects.
Another critical aspect of norepinephrine-induced mitochondrial fission is its connection to both ROS production and cellular apoptosis. It is widely accepted that cyt c is released through Bax-lined pores at sites of Drp1-mediated mitochondrial fission, triggering cellular apoptosis [220]. Notably, in the context of hypertension-related left ventricular hypertrophy, both ROS production and myocardial cellular apoptosis are commonly theorized as mechanisms implicated in the onset and progression of the disease. Furthermore, as previously mentioned, hypertension-induced mitochondrial alterations are also associated with changes in mitochondrial energy metabolism, including diminished respiration and ATP production. It has been proposed that, while fusion enhances respiratory efficiency, mitochondrial fission is linked to a decline in oxidative metabolism.
The process of cytosolic Drp1 recruitment to mitochondria during fission is complex and is regulated by post-translational modifications of Drp1. One such regulatory modification is phosphorylation by cyclic AMP-dependent protein kinase A (PKA) at Ser-637 in the GTPase effector domain of Drp1. This action mitigates Drp1 GTPase activity, inhibiting mitochondrial fission. In cardiomyocytes, after a 48-h incubation period with norepinephrine, a reduction in the phosphorylation of Drp1 at Ser-637 was observed. This finding supports the notion that norepinephrine induces mitochondrial fission in cardiomyocytes. Mitochondria predominantly produce ROS, notably superoxide and hydrogen peroxide, which are critical contributors to cellular damage, functional impairment, tissue enlargement, and inflammation in various organs. Hypertension is intricately linked to the reduction and inactivation of the crucial mitochondrial enzyme, sirtuin-3. They play a pivotal role in the management of essential metabolic processes. The absence of sirtuin-3 can precipitate the onset of hypertension and stimulate the progression of cardiac fibrosis, a condition characterized by excess fibrous connective tissue in the heart [64].
Ang II treatment has been observed to significantly enhance the protein expression of Drp1 while inhibiting OPA1 expression in HUVECs. This disruption in mitochondrial dynamics results in cell apoptosis, a process by which acacetin can counteract apoptosis by readjusting the protein expression of Drp1 and OPA1. Other studies have reported similar findings where Ang II provokes the phosphorylation of Drp1 and induces mitochondrial fission in abdominal aortic VSMCs and adventitial fibroblasts, conditions that can be thwarted through Drp1 silencing [62, 107, 209].
Obesity
Metabolically unhealthy obesity is linked to an increased risk of obesity-related cardiovascular conditions and overall mortality [19]. The primary cause of obesity is typically identified as an energy imbalance that occurs when calorie intake exceeds the number of calories burned [20, 61, 144]. Evidence suggests that the overconsumption of nutrients can have a detrimental impact on mitochondrial function. Studies have shown that obesity is associated with mitochondrial dysfunction. Introducing chemical uncouplers, such as FCCP or CCCP, triggers complete fragmentation of the mitochondrial network, recruitment of Drp1 to the outer membrane, and degradation of OPA1 [241, 243]. Furthermore, recent research has demonstrated that CCCP-induced depolarization triggers proteasome-dependent degradation of other mitochondrial fusion proteins, including Mfn1 and Mfn2, as well as other outer membrane proteins. Notably, proteasome-dependent degradation of mitofusins necessitates overexpression of the E3–ubiquitin–ligase Parkin [42, 137, 240]. Consistent with this, uncouplers can simulate conditions of excessive nutrient availability, thereby augmenting nutrient oxidation and electron transport chain activity, as observed in activated brown fat or beta cells. Based on this concept, studies involving beta cells subjected to excess nutrients or conditions that decouple the mitochondria under physiological stimuli have demonstrated an upsurge in respiration and pronounced fragmentation within the mitochondrial network [162].
Abundant evidence from both clinical and experimental environments has substantiated the role of obesity in the development of cardiovascular diseases, including HF. Obesity also influences the structure and pumping efficiency of the myocardium, which are notable characteristics of obesity-induced cardiomyopathy [9, 36, 88, 183]. In recent decades, substantial efforts have been made to decipher the intricacies of mitochondrial biogenesis, dynamics, quality control, and their roles in advancing obesity-associated cardiomyocyte dysfunction. Mitochondrial proliferation was increased in db/db hearts [30]. A notable morphological shift from a mitochondrial network to fragmented mitochondria has been observed in cardiomyocytes that are affected by obesity. In neonatal rat cardiomyocytes, the initial exposure to palmitate triggers an increase in mitochondrial respiration and heightened mitochondrial polarization and ATP generation. However, prolonged exposure to palmitate (beyond 8 h) produced ROS and induced mitochondrial fission [241]. The occurrence of cardiomyocyte apoptosis and cardiac dysfunction caused by lipid overload may be attributed to changes in post-translational modifications of proteins involved in mitochondrial fission and fusion. This includes an increase in ubiquitination of A-Kinase Anchor Protein 121 (AKAP121), Drp1, and OPA1. The mitochondria and ER are interlinked organelles. Many proteins have been proposed to bind to these two structures at specific locations, known as mitochondria-associated ER membranes. Interestingly, although the disruption of MAMs leads to irregular calcium signaling and cardiac anomalies, a recent study suggested that excessive glucose triggers FUNDC1-mediated mitochondria-associated membrane formation and mitochondrial calcium overload in cardiomyocytes, resulting in functional cardiac abnormalities [251, 252].
Research has confirmed cardiac structure and function alterations in cases of both genetically predisposed and diet-induced obesity [129]. Current understanding of the mechanisms underlying obesity-induced cardiomyopathy includes metabolic disruptions (such as insulin resistance, abnormal glucose transport, increased fatty acids, lipotoxicity, and amino acid imbalance), changes in intracellular calcium homeostasis, oxidative stress, impaired autophagy regulation, myocardial fibrosis, and cardiac autonomic neuropathy (manifesting as either denervation or overflow of adrenergic and renin–angiotensin–aldosterone). Furthermore, factors such as inflammation, small coronary vessel disease (microangiopathy), impaired coronary flow reserve, coronary artery endothelial dysfunction, and epigenetic modifications contribute to the pathogenesis of obesity-induced cardiomyopathy. Although practical targeted medications and procedures are still lacking, a substantial body of research has been devoted to managing obesity-induced cardiomyopathy. Non-pharmacological interventions, such as lifestyle modifications including regular exercise and dietary regulation, could also prove beneficial for cardiac health in individuals with obesity.
Aging
Although aging itself is not classified as a disease, it notably affects the functionality of cardiac mitochondria. The research presents differing views, with some studies suggesting a reduction in the number of mitochondria present within the cytoplasm of aged cardiac muscle cells, while others propose that the fraction of cellular volume occupied by mitochondria remains stable throughout the aging process. As we age, the form of the mitochondria changes, becoming less elongated and more spherical. In addition, the surface area of the IMM in the aging heart muscle decreases, although the structure of the cristae, remains unaffected [24, 56, 73, 196]. Generally, older hearts demonstrate less responsiveness to cardioprotective treatments than younger hearts, with all other factors being constant. Aging, a primary risk factor for HF, is linked to the deterioration of nuclear and mitochondrial genetic integrity due to telomere shortening [226]. This process is counteracted by the enzyme telomerase reverse transcriptase. SSM isolated from the heart muscles of aged rodents predominantly preserves their respiration ability. However, IFM exhibited reduced oxygen consumption with age. This decrease in oxygen consumption aligns with the observed decline in the activity of electron transport chain complexes in IFM. Respiratory complex III and IV activities in the IFM of aging heart muscles were diminished. Remarkably, the function of mitochondria remains intact, mainly in aged cardiomyocytes, with their outer membranes disrupted [74, 138]. This age-related decrease in mitochondrial function could influence cellular energy generation, consequently affecting cardiac function. While ATP levels may remain steady in the resting state, evidence from various studies indicates a possible reduction in either ATP content or production [171].
A recently engineered Mito-Timer mouse model revealed a heterogeneous distribution of newly synthesized and aged mitochondria within the heart [234]. Upon examining the expression of proteins integral to mitochondrial fusion and fission, a decrease in the levels of Mfn1 and Mfn2 was observed with age. However, this study found that aging did not affect the OPA1 and Drp1 protein levels [271]. In contrast, a study by Ljubicic et al. showed increased expression of OPA1 and Drp1 with age [146]. These mice demonstrated a build-up of impaired mitochondria, eventually leading to HF. However, moderate catalase expression, explicitly targeted to the mitochondria, normalized ROS production and mitigated structural alterations in hearts deficient in Mfn2. Interestingly, high levels of mitochondrial catalase did not improve mitochondrial function or HF. These data imply that no dose–effect relationship exists between local ROS formation and cardiac degeneration [228].
Progress in mitigating aging-induced health complications will likely hinge on a deeper understanding of the mechanisms that drive aging. In particular, focusing on systems that maintain mitochondrial homeostasis could offer strategies to address mitochondrial damage with aging.
Targeting mitochondrial morphology in acute myocardial ischemia–reperfusion injury
Currently, therapeutic strategies against acute myocardial IRI that target the mitochondria are mainly focused on the prevention of mitochondrial ROS production and Ca2+ overload [98]. In this section, we review cardioprotective interventions aimed at preserving mitochondrial morphology and functionality which may provide new treatment strategies for reduce MI size and preventing HF post-AMI.
Exercise
Exercise is a nonpharmacological strategy that promotes health and serves as a key strategy for preventing age-related diseases [78, 198]. Notably, exercise induces temporary modifications in the functionality and metabolism of the mitochondria [124]. The influence of exercise training on energy production and its subsequent effects on mitochondrial and metabolic processes have been comprehensively studied. As these adaptations provide insights into the pivotal role of mitochondria in exercise-induced cardioprotection, we will highlight the protective effects of exercise training on cardiac mitochondria in the following section [81, 109].
The metabolic profile of the heart is altered with moderated exercise when compared to the sedentary state. An exercise-trained heart is distinguished by its heightened capacity for fatty acid and glucose oxidation paired with a reduced rate of glycolysis, and the heart boasts a superior capacity to adjust its metabolism in response to acute stress. This adaptability stems from the elevated expression of AMPK, peroxisome proliferator-activated receptor-γ coactivator-1 alpha (PGC-1α), and phosphoinositide 3-kinase (PI3K), all of which enhance fatty acid and glucose oxidation, glucose uptake, and the formation of new mitochondria. Engaging in immediate bouts of exercise has been linked to increased production of ROS, primarily as by-products of the electron transport chain [35]. Alleman et al. discovered that energetic mitochondrial recovery, characterized by the oxygen consumption rates following hypoxia–reoxygenation, was enhanced compared to sedentary counterparts in animals subjected to exercise. They also found that the ratio of mitochondrial hydrogen peroxide (H2O2) production to oxygen consumption was twice as high in mitochondria sourced from sedentary animals than in those from exercised animals. This implies that exercise training may reduce ROS production relative to oxygen consumption. This finding is consistent with other studies, which indicate that exercise training can curb the disruption of the respiratory control ratio in mitochondria exposed to hypoxia–reoxygenation in vitro [5, 8]. Exercise training influences the redox state of cardiac cells and the regulation of Ca2+ homeostasis, which could indirectly reduce the susceptibility of mitochondria to IRI. Repeated bouts of endurance exercise protect against IRI arrhythmias, myocardial stunning, and myocardial infarction. Interestingly, only 3–5 consecutive days of endurance exercise is required to achieve a significant level of cardioprotection against IRI [90, 110]. In this line, it has been demonstrated that moderated interval training resulted in improved mitochondrial fusion and fission in male rats with myocardial infarction increasing Mfn2 and PGC-1α and reducing Drp1 (Fig. 2) [11]. However, prolonged exercise resulted in a significant reduction in the gene expression of Mfn1 and Mfn2 and it was an increase in the expression of Fis1 in skeletal muscle of male rats. The magnitude of these alterations was exercise duration dependent. These findings suggest that mitochondrial fusion and fission protein expression are rapidly altered in response to changing energy demand [65]. The direct effect of exercise on mitochondrial dynamics in the heart remains controversial. Future research should aim to discern how exercise training can influence 1) the regulation of mitochondrial dynamics, 2) the control of Ca2+ handling and mPTP opening, 3) the intricate interplay between inflammation and mitochondria, and 4) the interaction between mitochondria and redox signaling induced by exercise. This could enhance our understanding of cardioprotective mechanisms and pave the way for the discovery of novel cardioprotective pathways.
Caloric restriction
Another nonpharmacological strategy that promotes health is the decrease in caloric intake without compromising nutritional needs, also known as caloric restriction (CR) [219]. CR represents the most potent and thoroughly researched dietary intervention across a multitude of non-human species [247]. Furthermore, CR has also been demonstrated to confer broad health benefits in humans, whether dietary restriction is adopted by choice or through unavoidable circumstances. A recent meta-analysis of randomized human trials showed that caloric restriction was associated with a reduction in cardiovascular risk. This was associated with a significant decrease in both blood pressure and heart rate [128]. Mild-to-moderate CR has been found to alleviate cardiac dysfunction in various experimental scenarios including, cardiomyocyte hypertrophy, cardiac fibrosis, inflammation, and mitochondrial damage in middle-aged and aged mice [21, 169, 170].
Short- and long-term caloric restriction also offered protective benefits against acute myocardial IRI [71, 123, 161, 211, 222], and ischemic conditioning mitigates post-ischemic dysfunction in isolated perfused hearts from food-restricted aging rats [2]. However, this effect was not observed in the hearts of aging rats fed ad libitum [2, 3]. Recent studies have indicated that caloric restriction does not alter the susceptibility to mPTP opening in mitochondria isolated from cardiac muscle [219]. In this line, it is important to recognize that the process of isolating mitochondria from tissues can alter their morphology and distribution within cells [133]. This underscores the necessity for more sophisticated tools and standardized experimental models, specifically tailored for studying mitochondria in the context of cardiovascular diseases. Conversely, research has indicated that caloric restriction enhances the expression of Mfn2 in various organs [41]. During nutrient deprivation, protein kinase A (PKA) is activated and phosphorylates Drp1 keeping the latter within the cytoplasm, thereby maintaining mitochondrial fusion [85].
The widespread adoption of caloric restriction appears improbable given the challenge of sustaining long-term CR in contemporary society. Therefore, initiatives are underway to devise pharmacological alternatives that replicate the effects of CR including, metformin [18], resveratrol [45], and rapamycin [34, 125]. These substances, known as caloric restriction mimetics, can confer the advantageous metabolic, hormonal, and physiological effects of CR without necessitating a change in dietary intake.
Pharmacological modulators—fission
Altered cardiac mitochondrial dynamics with excessive fission are the predominant cause of cardiac dysfunction during IRI. Therefore, several studies have explored the pharmaceutical means for modulating mitochondrial fusion and fission, specifically by manipulating Mfn1, Mnf2, and Drp1 (Table 1, Fig. 3) [98, 154, 180, 181]. Among the available inhibitors, mdivi-1, a quinazoline derivative, is the most extensively studied reversible allosteric inhibitor of Drp1. Mdivi-1 has been demonstrated to effectively inhibit the GTPase activity of Dnm1, a yeast counterpart of Drp1. Its inhibitory activity was observed at a half-maximal inhibitory concentration (IC50) ranging between 1 and 10 μM, indicating its potent inhibitory effect [38]. Ong et al. were one of the first to demonstrate cardioprotection with pharmacological Drp1 inhibition with mdivi-1. Forty minutes of pre-treatment with 50 μmol/L of mdivi-1 decreased mPTP sensitivity and decreased cell death after SIRI in murine cardiomyocytes. A single intravenous bolus of mdivi-1 (1.2 mg/Kg) administered 10 min before acute coronary occlusion significantly reduced myocardial infarct size [182]. In another study by Maneechote et al. investigated the effects of inhibiting mitochondrial fission using mdivi-1. This was performed at three distinct time frames: prior to ischemia, throughout the ischemic phase, and at the initiation of reperfusion, all within the rat cardiac IRI model. The results indicated the most pronounced improvement in cardiac performance when mdivi-1 treatment was implemented before ischemia, which was accompanied by a marked decrease in mitochondrial fragmentation and a notable increase in mitochondrial functionality. Although the administration of mdivi-1 during ischemia and at the onset of reperfusion also resulted in cardiac function enhancement, the level of improvement was comparatively lower than that achieved with the pre-ischemia treatment strategy. Maneechote et al. proposed that the protective effect exerted by mdivi-1 on the left ventricle during IRI incidents might be attributed to its ability to enhance mitochondrial function. They argued that this enhancement was achieved by attenuating excessive mitochondrial fission, which in turn mitigates the incidence of cell death in heart muscle cells or cardiomyocyte death [152]. These preclinical studies indicate the considerable therapeutic potential of Drp1 inhibition. However, the specificity of mdivi-1 has been questioned, highlighting the need for further investigation to validate its selective inhibitory effects [26, 27, 267].

Pharmacological manipulation of mitochondrial fission and fusion processes. Pharmaceutical strategies aimed at modulating mitochondrial dynamics are depicted; those focusing on the regulation of the mitochondrial fission process via strategic manipulation of Drp1 are highlighted in green. In contrast, agents that activate Mfn1 and Mfn2, thereby influencing the fusion process, are shown in purple. This figure was created using the Adobe Illustrator 2023. Drp1 dynamin-related protein 1, Mfn2 mitofusin 2, Mfn1 mitofusin 1
DRP1i27, a novel small molecule that interacts directly with human isoform 3 of Drp1. Rosdah et al. have demonstrated the protective capabilities of this new molecule. Remarkably, it was shown to shield cells from IRI and toxic conditions, acting in a way which is consistent with the modulatory role of Drp1. The treatment with 50μM of DRP1i27 increased fused mitochondrial networks of mouse fibroblasts in a Drp1-dependent manner. DRP1i27 induced cardioprotection against SIRI in murine atrial HL-1 cells. Additionally, DRP1i27 showed cytoprotective effects against doxorubicin-induced toxicity in human iPSC-derived cardiomyocytes. Insights from molecular docking suggest that DRP1i27 attached to the GTPase site of Drp1, establishing hydrogen bonds with Gln34 and Asp218. The successful identification of DRP1i27 as a binding participant underscores the potential of this compound as a novel small-molecule inhibitor of Drp1 [213].
Another attractive strategy to inhibit fission that has attracted scientific interest is Isosteviol sodium or STVNa [159, 266]. STVNa, a sodium derivative of isosteviol, protects H9c2 cardiomyocytes from IRI by inhibiting the mitochondrial fission pathway [269]. Several studies have examined its diverse therapeutic properties, including the anti-hyperglycaemic, anti-hypertensive, anti-inflammatory, and anti-tumor effects. STVNa effectively maintained mitochondrial membrane potential (Δψ) and notably reduced the overproduction of ROS during reperfusion in a dose-dependent manner. Moreover, compared with diazoxide, a known selective opener of the mitochondrial ATP-sensitive potassium channel reported to safeguard cardiac mitochondria, STVNa presented compelling results [160].
We recently demonstrated the cardioprotective effect of hydralazine [118], a Food and Drug Administration (FDA)-approved therapy for treating essential hypertension, severe hypertension in pregnancy [232], and chronic HF when used in combination with isosorbide-dinitrate [176]. Using photo-switched mitochondrial Dendra2 mice, we demonstrated that pre-treatment with hydralazine inhibited mitochondrial fission, preserved mitochondrial fusion events, and prevented cell death in adult cardiomyocytes following SIRI.
These findings provide new insights into future innovative therapeutic strategies for patients with MI. Future treatments could focus on targeting surplus mitochondrial fission observed during cardiac ischemia or at the initiation of reperfusion, thus providing a potentially effective approach to alleviate the damage caused by such cardiac events.
An imbalance in inositol levels has been reported to affect mitochondrial dynamics and provide valuable insights into the pathogenesis of mitochondrial fission and fusion-related human diseases. Hsu et al. demonstrate that inositol serves as a key metabolite, which directly limits AMPK-dependent mitochondrial fission, independent of its conventional role as a precursor for phosphoinositide creation. A reduction in inositol due to inositol monophosphatase 1 and 2 (IMPA1/2) deficiency triggers AMPK activation and mitochondrial fission, irrespective of ATP levels, whereas inositol accumulation prevents AMPK-dependent mitochondrial fission [105]. Both metabolic stress and mitochondrial damage can lead to decreased inositol levels in cells and mice, thereby inducing AMPK-dependent mitochondrial fission. Inositol directly interacts with AMPK and competes with AMP for this binding, resulting in limited AMPK activation and mitochondrial fission. This research suggests that the AMP/inositol ratio is a pivotal factor in AMPK activation, and proposes a model in which inositol decline is necessary to free AMPK for AMP binding. Therefore, AMPK is an inositol sensor and its deactivation by inositol acts as a mechanism to limit mitochondrial fission. Interventions such as inositol treatment, activation of IMPA1/2, or targeting CDIPT (CDP-diacylglycerol-inositol 3-phosphotidyltransferase)could potentially be effective strategies for addressing a range of human diseases linked to aberrant AMPK-dependent mitochondrial dynamics [105].
The derivative of AS-IV, LS-102, has shown significant efficacy in protecting against IRI damage. LS-102 demonstrated considerable efficacy in reducing apoptosis; reducing the levels of ROS, creatine kinase (CK), lactate dehydrogenase (LDH), and calcium; enhancing the mitochondrial membrane potential; and regulating the Bax/Bcl-2 ratio in cardiomyocytes during IRI. Notably, LS-102 induced IRI-induced mitochondrial fission by reducing the mitochondrial localization of Drp1 via the downregulation of Drp1 phosphorylation at Ser-616 and upregulation of its phosphorylation at Ser-637 in H9c2 cells. LS-102 provides cardioprotection against IRI by inhibiting mitochondrial fission, primarily by blocking GSK-3β-mediated and Drp1 phosphorylation at Ser-616 [48, 206].
Pharmacological modulators—fusion
Franco et al. first identified mini-peptides derived from Mfn2 that could either specifically activate or inhibit Mfn1 and Mfn2, thereby allowing the manipulation of mitochondrial fusion with the use of mitofusin agonists or antagonists [77]. They proposed that the mechanism of action for the fusion-promoting (agonist) peptide was its ability to compete with intramolecular (between HR1 and HR2 domain) interactions, which normally maintain a closed, non-fusion-permitting conformation. Intramolecular peptide binding results in a more open fusion-friendly conformation. In contrast, the antagonist peptide was thought to function by encouraging the opposite conformational shift, that is, pushing toward a more closed, non-fusion-permitting state. The conformational changes observed in this study were monitored using Mfn2 FRET probes labeled with fluorophores at the amino and carboxyl termini. However, the precise structure of fully intact mitofusin proteins in either a “closed” inactive or “open” active conformations has not been definitively determined and remains a topic of ongoing debate in the field, as discussed earlier [37, 83, 109, 157].
In a subsequent study, Rocha et al. designed a strategy to generate small-molecule peptidomimetics, which demonstrated enhanced in vitro effectiveness over the original mitofusin-activating peptide [210]. They began by identifying a mitofusin-activating peptide with just 11 amino acids. Subsequently, the team used alanine scanning to highlight the amino acids pivotal to the function of the peptide. Finally, they employed a pharmacophore model to facilitate an in silico screening process. This study aimed to identify commercially available compounds that shared structural features with amino acids instrumental in the function of the agonist peptide. Biological screening of 55 potential matches led to the identification of two compounds with observable agonist activity. From these, the novel synthesis of what can be termed as "Franken-molecules,” possessing varying chemical segments from these fusogenic compounds, culminated in the creation of the first-of-its-kind small-molecule mitofusin agonist, Chimera B-A/l.
Chimera B-A/l showed the ability to bind to Mfn2 HR2 and replace the original agonist peptide, from which it was designed. Similar to the mitofusin agonist peptide, Chimera B-A/l effectively reversed mitochondrial fragmentation and depolarization in cultured mouse neurons expressing the human Charcot–Marie–Tooth Disease type 2A (CMT2A) mutant Mfn2 protein. Furthermore, this principle mitofusin agonist quickly restored normal axonal mitochondrial trafficking both in vitro, using cultured CMT2A neurons, and ex vivo, using CMT2A mouse sciatic nerves. Collectively, these investigations present an opportunity for future experimental endeavors and potential clinical treatments, leveraging either cell-permeable mini-peptides or small-molecule peptidomimetics that allosterically activate Mfn1 and Mfn2 [210].
M1 is another agent that induces cardioprotection by activating fusion in preclinical models of IRI. M1 has demonstrated significant cardioprotective properties in normal [151] and prediabetic rats [154]. M1 has been shown to restore the expression of mitochondrial fusion proteins, thereby ameliorating mitochondrial function [244]. Another class of small molecule activator of mitofusins with better pharmacokinetics properties has recently been described. This investigation has led to the development of a series of 6-phenylhexanamide derivatives. Through pharmacokinetic optimization, a 4-hydroxy cyclohexyl analog, compound 13, was synthesized. This compound demonstrated potency, selectivity, and oral bioavailability as a preclinical candidate. Intriguingly, further studies of the cis- and trans- 4-hydroxy cyclohexyl isostereomers of compound 13 revealed that the functional activity and protein interaction were exclusive to the trans-form, referred to as 13 B [58].
Finally, using structural and biochemical insights into the direct modulation of Mfn1 and Mfn2 conformations, Zacharioudakis et al. developed rational pharmacophore methodologies to perform computational screening of small molecules [263]. These strategic screenings yielded small molecules that could either activate or inhibit the fusion activity of mitofusins by modulating their tethering-permissive structure. Their results demonstrated that the mitofusin activator MASM7 and the mitofusin inhibitor MFI8 directly interacted with the recombinant HR2 domain of Mfn2. Moreover, these compounds can also interact with the intact Mfn2 protein found in the mitochondria within cells. MASM7 fosters the pro-tethering structure of Mfn1 and Mfn2, thereby facilitating mitochondrial fusion. In contrast, MFI8 disrupts mitochondrial fusion by actively obstructing the tethering-favorable conformation of mitofusins. The small molecules MASM7 and MFI8 were found to elevate or reduce the levels of GTP-dependent Mfn2 higher-order oligomers. This study establishes a novel strategic avenue for pharmacological intervention with mitofusins using small molecules, thereby enriching the domain of molecular therapeutics. Thus, a deeper exploration of MASM7 and MFI8 in relation to IRI is required [263].
Despite the prevailing challenges, the prospect of applying pharmacological methodologies, either supplementary to or in place of genetic manipulation, opens a new avenue to further understand fusion and fission processes by manipulating their components, which not only enriches the current research, but also potentially holds promise for clinical applications.
Ischemic conditioning
Ischemic conditioning strategies, including local preconditioning (IPC), postconditioning (IPost), and remote ischemic conditioning (RIC), are potentially promising avenues for therapy, although their mechanisms are not entirely understood and likely involve multiple pathways. IPC, for instance, delays the recovery of intracellular pH, prevents NOS uncoupling, and the subsequent production of reactive oxygen and nitrogen species while amplifying the signaling of protein kinase G (PKG), reperfusion injury salvage kinase (RISK), and survivor activating factor enhancement (SAFE) in reperfused cardiomyocytes. Interestingly, RIC appears to be similar to IPC in that it affects nitrosylation and conserves PKG activity [100] However, RIC also influences mitochondrial function and activates the RISK and SAFE pathways, further expanding its cardioprotective potential [94, 96, 104, 130, 261]
Within the scope of IRI, conditioning strategies aimed at reducing mitochondrial fission or augmenting mitochondrial fusion have been shown to be correlated with diminished IRI. Particularly, remote ischemic conditioning has emerged as a prominent strategy. Heusch [99] accentuates its potential in reducing infarct size among patients with acute myocardial infarction undergoing percutaneous coronary intervention. Meanwhile, Kleinbongard et al. [130] further investigated RIC, examining the various levels of this approach and its associated signal transduction pathways, drawing attention to its successful clinical applications. Chong et al. [52] offered a comprehensive review of the signaling mechanisms associated with RIC, pointing out the divergent outcomes observed in various clinical trials. He further highlighted inconsistent findings in clinical trials, advocating for enhanced research efforts to optimize RIC’s application in cardiac surgery.
The cardioprotective effects of remote RIC are linked to an upregulation in the expression of the mitochondrial fusion protein OPA1 coupled with a reduction in the mitochondrial fission protein Drp1 in the heart. Cellier et al. observed a decrease in Drp1 levels in the mitochondrial fraction following RIC. This implies that RIC can potentially disrupt Drp1 translocation to the mitochondria, thereby obstructing the fission process initiated by ischemia–reperfusion. This suggests that the safeguarding mechanisms of RIC significantly influence the mitochondrial activity dynamics [39]. More recently, we have shown that IPC and IPost preserved the mitochondrial network by inhibiting fission and promoting fusion in H9c2 and adult murine cardiomyocytes subjected to IRI [113].
Mitochondria-targeted antioxidants
Excessive fragmentation of mitochondria following IRI is a key determinant of mitochondrial damage and cardiomyocyte death [182]. Additionally, mitochondrial dysfunction induced by IRI can result in increased ROS production, which in turn causes more mitochondrial damage and further ROS release, a phenomenon referred to as “ROS-induced ROS release” [97]. Given that mitochondria are the primary source of ROS, scavenging mitochondrial ROS in reperfused cardiomyocytes has long been suggested as a potential therapeutic target for myocardial IRI. Suppressing mitochondrial fission reduces mitochondrial ROS, mitigates mitochondrial dysfunction, and decreases cell apoptosis. Consequently, these effects collectively lead to an improvement in cardiac function.
Melatonin, chemically known as N-acetyl-5-methoxytryptamine, is primarily produced by the pineal gland. It plays a multifaceted role in several bodily systems, including immune regulation, the prevention of cancer metastasis, sleep regulation, and circadian rhythms, beyond its basic hormonal functions [92]. Given its antioxidant, anti-inflammatory, and apoptotic properties, melatonin is believed to play a crucial role in mitigating the myocardial damage caused by reperfusion [75, 265]. Preclinical studies have demonstrated that melatonin can inhibit mitochondrial fission under certain pathological conditions [66, 259]. From a mechanistic standpoint, melatonin impedes the mitochondrial translocation of fission proteins, such as the mitochondrial Fis1 and Drp1, as well as the pro-apoptotic protein Bax. Concurrently, it upregulates the expression of mitochondrial fusion proteins, namely Mfn1, Mfn2, and OPA1 [187, 191]. Melatonin inhibits the translocation of Fis1 and Drp1 to the OMM, thereby reducing fission. The mechanisms by which melatonin regulates mitochondrial fusion proteins are intricate and multifaceted. Melatonin may elevate the expression of Mfn1 through Notch1 signaling or, alternatively, it can downregulate both Mfn1 and OPA1 [191, 238]. It has been reported that, melatonin influences the stabilization of Opa1 through the AMPK signaling pathway, and inhibiting AMPK results in decreased OPA1 expression, compromising the cardioprotective benefits of melatonin. In essence, these results validate that OPA1-associated mitochondrial fusion is indeed modulated by melatonin in the context of IRI. Furthermore, orchestrating the AMPK-OPA1-mitochondrial fusion-mitophagy axis via melatonin may represent a novel therapeutic strategy to mitigate myocardial IRI [270].
The clinical intrigue surrounding melatonin as a cardioprotective agent was highlighted in the MARIA trial, which assessed its efficacy in STEMI patients undergoing PPCI [68]. However, it is imperative to approach these results with caution. Notably, the experimental data advocating melatonin’s beneficial impact on MI size primarily originated from small animal MI models devoid of comorbidities and comedications. Much of this research primarily determines melatonin’s long-term effects on post-MI adverse remodeling. More critically, in a pertinent large animal closed-chest reperfused porcine MI model, both intravenous and intracoronary melatonin, when administered pre-reperfusion, did not exhibit a reduction in MI size. This indicates a possible inconsistency in melatonin’s cardioprotective efficacy, even in controlled experimental conditions. Such revelations underscore the need for rigorous vetting of emerging cardioprotective treatments in laboratory settings before transitioning into clinical trials [95].
Resveratrol, a polyphenolic phytoalexin primarily found in grapes, berries, peanuts, and wines, exhibits a wide array of beneficial properties. Chemically identified as 3,4',5-trihydroxystilbene (C14H12O3), it is known for its antioxidant, anti-inflammatory, anti-apoptotic, and anticancer potential. In animal research, resveratrol has been demonstrated to protect cardiomyocytes against oxidative stress, mitigating autophagy, cardiac fibrosis, and apoptosis [268]. Recently, it has been shown that resveratrol promotes the protection of mitochondria from damage caused by hypoxia–reoxygenation events by activating the Sirt1–Sirt3–Mfn2–Parkin–PGC1α pathway [272]. It re-establishes mitochondrial dynamics by accelerating the partitioning of damaged mitochondria and facilitating the interchange of components within the mitochondrial network through the stimulation of fission and fusion processes via the Sirt1–Sirt3 pathway, thereby effectively controlling mitochondrial quantity. Besides, it has been also reported that resveratrol regulates Mfn1 expression via the AMPK pathway and that the inhibition of the MPKA pathway also neutralized the anti-apoptotic effect of resveratrol on re-oxygenated cells [82].
Despite extensive research conducted using animal models, which suggests that antioxidants could potentially be a significant strategy for treating cardiovascular diseases, the therapeutic efficacy of these compounds in humans still requires confirmation. Thus, the field of clinical medicine faces the ongoing task of enhancing our understanding of antioxidants and its viability as a commercially viable therapy, making this an ongoing challenge.
Summary and conclusions
Changes in mitochondrial morphology in response to acute myocardial IRI are known to mediate mitochondrial dysfunction and cardiomyocyte death providing a therapeutic target for cardioprotection in terms of reducing MI size and preventing HF following AMI. These therapeutic strategies may help restore the balance in mitochondrial fission and fusion perturbed by IRI and could be implemented by acute transient inhibition of mitochondrial fission to preserve mitochondrial function which could potentially be applied at the time of reperfusion following AMI. However, although this treatment approach may be applied to other cardiac diseases characterized by disturbances in the balance of mitochondrial fusion and fission such as diabetic cardiomyopathy and post-AMI HF, the chronic manipulation of mitochondrial morphology such as by sustained inhibition of fission may inadvertently result in cardiomyopathy by allowing the accumulation of damaged mitochondria due to disruption of mitophagy. This may restrict pharmacological manipulation of mitochondrial morphology to acute settings such as for limiting MI size following AMI.
Risk factors, underlying comorbidities, and concurrent medications can significantly impact mitochondrial function, often through multifaceted mechanisms and, in some instances, irreversibly. Consequently, a singular approach targeting only one mechanism for mitoprotection may need to be more holistic [29]. Although the molecular structure of the mPTP remains elusive, and specific inhibitors to deter its activation are yet to be identified, it stands as a promising avenue to counteract reperfusion injury. When it comes to modulating mitochondrial fusion at the onset of reperfusion, a different approach may be needed based on inhibiting Mfn2 to dissociate mitochondria from SR and preventing mitochondrial calcium overload and subsequent mPTP opening and cardiomyocyte death. This supports the notion that targeting singular intracellular components like mPTP or Mfn2 may fall short in cardioprotection. The emphasis should be on a broader understanding of the underlying cardioprotective mechanisms.
Our understanding of how targeting mitochondrial dynamics offers cardioprotection is still evolving. Since the (EU)-CARDIOPROTECTION Cooperation in Science and Technology (COST) discussion in 2019, major advances have been made, and significant molecular information has rapidly accumulated, bringing this classic discipline back to renewed attention. Mitochondrial morphology has been classified under the second category of targets, which includes mechanisms activated endogenously for cardioprotection [60]. Additionally, the introduction of a detailed set of criteria titled 'IMproving Preclinical Assessment of Cardioprotective Therapies (IMPACT) has been stablished to enhance the successful translation of cardioprotective therapies to benefit patients in clinical settings [136].
In conclusion, targeting mitochondrial morphology has the therapeutic potential to reduce MI size and prevent HF following AMI and may also be beneficial in other chronic cardiac conditions characterized by disturbed mitochondrial morphology. However, further studies are needed to investigate the optimal therapeutic approach to manipulating mitochondrial morphology that confers cardioprotection in order that health outcomes following AMI can be improved.
References
Abe T, Kiyonari H, Shioi G, Inoue K, Nakao K, Aizawa S, Fujimori T (2011) Establishment of conditional reporter mouse lines at ROSA26 locus for live cell imaging. Genesis 49:579–590. https://doi.org/10.1002/dvg.20753
Abete P, Testa G, Ferrara N, De Santis D, Capaccio P, Viati L, Calabrese C, Cacciatore F, Longobardi G, Condorelli M, Napoli C, Rengo F (2002) Cardioprotective effect of ischemic preconditioning is preserved in food-restricted senescent rats. Am J Physiol Heart Circ Physiol 282:H1978-1987. https://doi.org/10.1152/ajpheart.00929.2001
Abete P, Testa G, Galizia G, Mazzella F, Della Morte D, de Santis D, Calabrese C, Cacciatore F, Gargiulo G, Ferrara N, Rengo G, Sica V, Napoli C, Rengo F (2005) Tandem action of exercise training and food restriction completely preserves ischemic preconditioning in the aging heart. Exp Gerontol 40:43–50. https://doi.org/10.1016/j.exger.2004.10.005
Adaniya SM, OU J, Cypress MW, Kusakari Y, Jhun BS (2019) Posttranslational modifications of mitochondrial fission and fusion proteins in cardiac physiology and pathophysiology. Am J Physiol Cell Physiol 316:C583–C604. https://doi.org/10.1152/ajpcell.00523.2018
Alleman RJ, Tsang AM, Ryan TE, Patteson DJ, McClung JM, Spangenburg EE, Shaikh SR, Neufer PD, Brown DA (2016) Exercise-induced protection against reperfusion arrhythmia involves stabilization of mitochondrial energetics. Am J Physiol Heart Circ Physiol 310:H1360-1370. https://doi.org/10.1152/ajpheart.00858.2015
Anand R, Wai T, Baker MJ, Kladt N, Schauss AC, Rugarli E, Langer T (2014) The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission. J Cell Biol 204:919–929. https://doi.org/10.1083/jcb.201308006
Ando R, Hama H, Yamamoto-Hino M, Mizuno H, Miyawaki A (2002) An optical marker based on the UV-induced green-to-red photoconversion of a fluorescent protein. Proc Natl Acad Sci USA 99:12651–12656. https://doi.org/10.1073/pnas.202320599
Ascensao A, Ferreira R, Oliveira PJ, Magalhaes J (2006) Effects of endurance training and acute Doxorubicin treatment on rat heart mitochondrial alterations induced by in vitro anoxia-reoxygenation. Cardiovasc Toxicol 6:159–172. https://doi.org/10.1385/ct:6:3:159
Aurigemma GP, de Simone G, Fitzgibbons TP (2013) Cardiac remodeling in obesity. Circ Cardiovasc Imaging 6:142–152. https://doi.org/10.1161/CIRCIMAGING.111.964627
Baandrup U, Florio RA, Roters F, Olsen EG (1981) Electron microscopic investigation of endomyocardial biopsy samples in hypertrophy and cardiomyopathy. A semiquantitative study in 48 patients. Circulation 63:1289–1298. https://doi.org/10.1161/01.cir.63.6.1289
Babak Ebadi AD (2018) Effect of exercise training intensity on mitochondrial dynamics and mitophagy in post myocardial infarction rats. Int J Appl Exerc Physiol 7(2):46–53. https://doi.org/10.22631/ijaep.v7i2.278
Bach D, Naon D, Pich S, Soriano FX, Vega N, Rieusset J, Laville M, Guillet C, Boirie Y, Wallberg-Henriksson H, Manco M, Calvani M, Castagneto M, Palacin M, Mingrone G, Zierath JR, Vidal H, Zorzano A (2005) Expression of Mfn2, the Charcot-Marie-Tooth neuropathy type 2A gene, in human skeletal muscle: effects of type 2 diabetes, obesity, weight loss, and the regulatory role of tumor necrosis factor alpha and interleukin-6. Diabetes 54:2685–2693. https://doi.org/10.2337/diabetes.54.9.2685
Ban T, Ishihara N (2020) Analysis of mitochondrial membrane fusion GTPase OPA1 expressed by the silkworm expression system. Methods Mol Biol 2159:115–127. https://doi.org/10.1007/978-1-0716-0676-6_9
Ban T, Ishihara T, Kohno H, Saita S, Ichimura A, Maenaka K, Oka T, Mihara K, Ishihara N (2017) Molecular basis of selective mitochondrial fusion by heterotypic action between OPA1 and cardiolipin. Nat Cell Biol 19:856–863. https://doi.org/10.1038/ncb3560
Ban T, Kohno H, Ishihara T, Ishihara N (2018) Relationship between OPA1 and cardiolipin in mitochondrial inner-membrane fusion. Biochim Biophys Acta Bioenerg 1859:951–957. https://doi.org/10.1016/j.bbabio.2018.05.016
Barrasso AP, Tong X, Poche RA (2018) The mito::mKate2 mouse: a far-red fluorescent reporter mouse line for tracking mitochondrial dynamics in vivo. Genesis. https://doi.org/10.1002/dvg.23087
Benjamin EJ, Blaha MJ, Chiuve SE, Cushman M, Das SR, Deo R, de Ferranti SD, Floyd J, Fornage M, Gillespie C, Isasi CR, Jimenez MC, Jordan LC, Judd SE, Lackland D, Lichtman JH, Lisabeth L, Liu S, Longenecker CT, Mackey RH, Matsushita K, Mozaffarian D, Mussolino ME, Nasir K, Neumar RW, Palaniappan L, Pandey DK, Thiagarajan RR, Reeves MJ, Ritchey M, Rodriguez CJ, Roth GA, Rosamond WD, Sasson C, Towfighi A, Tsao CW, Turner MB, Virani SS, Voeks JH, Willey JZ, Wilkins JT, Wu JH, Alger HM, Wong SS, Muntner P (2017) Heart disease and stroke statistics-2017 update: a report from the American Heart Association. Circulation 135:e146–e603. https://doi.org/10.1161/CIR.0000000000000485
Bhamra GS, Hausenloy DJ, Davidson SM, Carr RD, Paiva M, Wynne AM, Mocanu MM, Yellon DM (2008) Metformin protects the ischemic heart by the Akt-mediated inhibition of mitochondrial permeability transition pore opening. Basic Res Cardiol 103:274–284. https://doi.org/10.1007/s00395-007-0691-y
Blaak EE, Goossens GH (2023) Metabolic phenotyping in people living with obesity: implications for dietary prevention. Rev Endocr Metab Disord 24:825–838. https://doi.org/10.1007/s11154-023-09830-4
Bluher M (2019) Obesity: global epidemiology and pathogenesis. Nat Rev Endocrinol 15:288–298. https://doi.org/10.1038/s41574-019-0176-8
Boengler K, Buechert A, Heinen Y, Roeskes C, Hilfiker-Kleiner D, Heusch G, Schulz R (2008) Cardioprotection by ischemic postconditioning is lost in aged and STAT3-deficient mice. Circ Res 102:131–135. https://doi.org/10.1161/CIRCRESAHA.107.164699
Boengler K, Hilfiker-Kleiner D, Heusch G, Schulz R (2010) Inhibition of permeability transition pore opening by mitochondrial STAT3 and its role in myocardial ischemia/reperfusion. Basic Res Cardiol 105:771–785. https://doi.org/10.1007/s00395-010-0124-1
Boengler K, Ruiz-Meana M, Gent S, Ungefug E, Soetkamp D, Miro-Casas E, Cabestrero A, Fernandez-Sanz C, Semenzato M, Di Lisa F, Rohrbach S, Garcia-Dorado D, Heusch G, Schulz R (2012) Mitochondrial connexin 43 impacts on respiratory complex I activity and mitochondrial oxygen consumption. J Cell Mol Med 16:1649–1655. https://doi.org/10.1111/j.1582-4934.2011.01516.x
Boengler K, Schulz R, Heusch G (2009) Loss of cardioprotection with ageing. Cardiovasc Res 83:247–261. https://doi.org/10.1093/cvr/cvp033
Boengler K, Stahlhofen S, van de Sand A, Gres P, Ruiz-Meana M, Garcia-Dorado D, Heusch G, Schulz R (2009) Presence of connexin 43 in subsarcolemmal, but not in interfibrillar cardiomyocyte mitochondria. Basic Res Cardiol 104:141–147. https://doi.org/10.1007/s00395-009-0007-5
Bordt EA, Clerc P, Roelofs BA, Saladino AJ, Tretter L, Adam-Vizi V, Cherok E, Khalil A, Yadava N, Ge SX, Francis TC, Kennedy NW, Picton LK, Kumar T, Uppuluri S, Miller AM, Itoh K, Karbowski M, Sesaki H, Hill RB, Polster BM (2017) The putative Drp1 inhibitor mdivi-1 Is a reversible mitochondrial complex I inhibitor that modulates reactive oxygen species. Dev Cell 40(583–594):e586. https://doi.org/10.1016/j.devcel.2017.02.020
Bordt EA, Zhang N, Waddell J, Polster BM (2022) The non-specific Drp1 inhibitor Mdivi-1 has modest biochemical antioxidant activity. Antioxidants (Basel). https://doi.org/10.3390/antiox11030450
Bossy B, Petrilli A, Klinglmayr E, Chen J, Lutz-Meindl U, Knott AB, Masliah E, Schwarzenbacher R, Bossy-Wetzel E (2010) S-Nitrosylation of DRP1 does not affect enzymatic activity and is not specific to Alzheimer’s disease. J Alzheimers Dis 20(Suppl 2):S513-526. https://doi.org/10.3233/JAD-2010-100552
Botker HE, Cabrera-Fuentes HA, Ruiz-Meana M, Heusch G, Ovize M (2020) Translational issues for mitoprotective agents as adjunct to reperfusion therapy in patients with ST-segment elevation myocardial infarction. J Cell Mol Med 24:2717–2729. https://doi.org/10.1111/jcmm.14953
Boudina S, Sena S, Theobald H, Sheng X, Wright JJ, Hu XX, Aziz S, Johnson JI, Bugger H, Zaha VG, Abel ED (2007) Mitochondrial energetics in the heart in obesity-related diabetes: direct evidence for increased uncoupled respiration and activation of uncoupling proteins. Diabetes 56:2457–2466. https://doi.org/10.2337/db07-0481
Brand CS, Tan VP, Brown JH, Miyamoto S (2018) RhoA regulates Drp1 mediated mitochondrial fission through ROCK to protect cardiomyocytes. Cell Signal 50:48–57. https://doi.org/10.1016/j.cellsig.2018.06.012
Breitzig MT, Alleyn MD, Lockey RF, Kolliputi N (2018) A mitochondrial delicacy: dynamin-related protein 1 and mitochondrial dynamics. Am J Physiol Cell Physiol 315:C80–C90. https://doi.org/10.1152/ajpcell.00042.2018
Busch R, Kim YK, Neese RA, Schade-Serin V, Collins M, Awada M, Gardner JL, Beysen C, Marino ME, Misell LM, Hellerstein MK (2006) Measurement of protein turnover rates by heavy water labeling of nonessential amino acids. Biochim Biophys Acta 1760:730–744. https://doi.org/10.1016/j.bbagen.2005.12.023
Buss SJ, Muenz S, Riffel JH, Malekar P, Hagenmueller M, Weiss CS, Bea F, Bekeredjian R, Schinke-Braun M, Izumo S, Katus HA, Hardt SE (2009) Beneficial effects of Mammalian target of rapamycin inhibition on left ventricular remodeling after myocardial infarction. J Am Coll Cardiol 54:2435–2446. https://doi.org/10.1016/j.jacc.2009.08.031
Campos JC, Marchesi Bozi LH, Krum B, Grassmann Bechara LR, Ferreira ND, Arini GS, Albuquerque RP, Traa A, Ogawa T, van der Bliek AM, Beheshti A, Chouchani ET, Van Raamsdonk JM, Blackwell TK, Ferreira JCB (2023) Exercise preserves physical fitness during aging through AMPK and mitochondrial dynamics. Proc Natl Acad Sci USA 120:e2204750120. https://doi.org/10.1073/pnas.2204750120
Cao L, Qin X, Peterson MR, Haller SE, Wilson KA, Hu N, Lin X, Nair S, Ren J, He G (2016) CARD9 knockout ameliorates myocardial dysfunction associated with high fat diet-induced obesity. J Mol Cell Cardiol 92:185–195. https://doi.org/10.1016/j.yjmcc.2016.02.014
Cao YL, Meng S, Chen Y, Feng JX, Gu DD, Yu B, Li YJ, Yang JY, Liao S, Chan DC, Gao S (2017) MFN1 structures reveal nucleotide-triggered dimerization critical for mitochondrial fusion. Nature 542:372–376. https://doi.org/10.1038/nature21077
Cassidy-Stone A, Chipuk JE, Ingerman E, Song C, Yoo C, Kuwana T, Kurth MJ, Shaw JT, Hinshaw JE, Green DR, Nunnari J (2008) Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev Cell 14:193–204. https://doi.org/10.1016/j.devcel.2007.11.019
Cellier L, Tamareille S, Kalakech H, Guillou S, Lenaers G, Prunier F, Mirebeau-Prunier D (2016) Remote ischemic conditioning influences mitochondrial dynamics. Shock 45:192–197. https://doi.org/10.1097/SHK.0000000000000500
Cereghetti GM, Stangherlin A, Martins de Brito O, Chang CR, Blackstone C, Bernardi P, Scorrano L (2008) Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc Natl Acad Sci USA 105:15803–15808. https://doi.org/10.1073/pnas.0808249105
Cerqueira FM, Laurindo FR, Kowaltowski AJ (2011) Mild mitochondrial uncoupling and calorie restriction increase fasting eNOS, akt and mitochondrial biogenesis. PLoS One 6:e18433. https://doi.org/10.1371/journal.pone.0018433
Chan NC, Salazar AM, Pham AH, Sweredoski MJ, Kolawa NJ, Graham RL, Hess S, Chan DC (2011) Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum Mol Genet 20:1726–1737. https://doi.org/10.1093/hmg/ddr048
Chandhok G, Lazarou M, Neumann B (2018) Structure, function, and regulation of mitofusin-2 in health and disease. Biol Rev Camb Philos Soc 93:933–949. https://doi.org/10.1111/brv.12378
Chang YW, Song ZH, Chen CC (2022) FAK regulates cardiomyocyte mitochondrial fission and function through Drp1. FEBS J 289:1897–1910. https://doi.org/10.1111/febs.16263
Chen CJ, Yu W, Fu YC, Wang X, Li JL, Wang W (2009) Resveratrol protects cardiomyocytes from hypoxia-induced apoptosis through the SIRT1-FoxO1 pathway. Biochem Biophys Res Commun 378:389–393. https://doi.org/10.1016/j.bbrc.2008.11.110
Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC (2003) Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol 160:189–200. https://doi.org/10.1083/jcb.200211046
Chen H, Ren S, Clish C, Jain M, Mootha V, McCaffery JM, Chan DC (2015) Titration of mitochondrial fusion rescues Mff-deficient cardiomyopathy. J Cell Biol 211:795–805. https://doi.org/10.1083/jcb.201507035
Chen L, Chen XY, Wang QL, Yang SJ, Zhou H, Ding LS, Qing LS, Luo P (2020) Astragaloside IV derivative (LS-102) alleviated myocardial ischemia reperfusion injury by inhibiting Drp1(Ser616) phosphorylation-mediated mitochondrial fission. Front Pharmacol 11:1083. https://doi.org/10.3389/fphar.2020.01083
Chen L, Gong Q, Stice JP, Knowlton AA (2009) Mitochondrial OPA1, apoptosis, and heart failure. Cardiovasc Res 84:91–99. https://doi.org/10.1093/cvr/cvp181
Chen Y, Liu Y, Dorn GW 2nd (2011) Mitochondrial fusion is essential for organelle function and cardiac homeostasis. Circ Res 109:1327–1331. https://doi.org/10.1161/CIRCRESAHA.111.258723
Cheng X, Hart GW (2001) Alternative O-glycosylation/O-phosphorylation of serine-16 in murine estrogen receptor beta: post-translational regulation of turnover and transactivation activity. J Biol Chem 276:10570–10575. https://doi.org/10.1074/jbc.M010411200
Chong J, Bulluck H, Fw Ho A, Boisvert WA, Hausenloy DJ (2019) Chronic remote ischemic conditioning for cardiovascular protection. Cond Med 2:164–169
Chudakov DM, Belousov VV, Zaraisky AG, Novoselov VV, Staroverov DB, Zorov DB, Lukyanov S, Lukyanov KA (2003) Kindling fluorescent proteins for precise in vivo photolabeling. Nat Biotechnol 21:191–194. https://doi.org/10.1038/nbt778
Cogliati S, Frezza C, Soriano ME, Varanita T, Quintana-Cabrera R, Corrado M, Cipolat S, Costa V, Casarin A, Gomes LC, Perales-Clemente E, Salviati L, Fernandez-Silva P, Enriquez JA, Scorrano L (2013) Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell 155:160–171. https://doi.org/10.1016/j.cell.2013.08.032
Cohen MV, Yang XM, Downey JM (2008) Acidosis, oxygen, and interference with mitochondrial permeability transition pore formation in the early minutes of reperfusion are critical to postconditioning’s success. Basic Res Cardiol 103:464–471. https://doi.org/10.1007/s00395-008-0737-9
Corsetti G, Pasini E, D’Antona G, Nisoli E, Flati V, Assanelli D, Dioguardi FS, Bianchi R (2008) Morphometric changes induced by amino acid supplementation in skeletal and cardiac muscles of old mice. Am J Cardiol 101:26E-34E. https://doi.org/10.1016/j.amjcard.2008.02.078
Crost S, Martin MK, Palmer JW (1987) Hormonal effects on mitochondrial respiration: potential role of endogenous lipolytic activities. Arch Biochem Biophys 256:421–429. https://doi.org/10.1016/0003-9861(87)90598-4
Dang X, Zhang L, Franco A, Li J, Rocha AG, Devanathan S, Dolle RE, Bernstein PR, Dorn GW 2nd (2020) Discovery of 6-phenylhexanamide derivatives as potent stereoselective mitofusin activators for the treatment of mitochondrial diseases. J Med Chem 63:7033–7051. https://doi.org/10.1021/acs.jmedchem.0c00366
Davidson SM, Adameova A, Barile L, Cabrera-Fuentes HA, Lazou A, Pagliaro P, Stenslokken KO, Garcia-Dorado D, Action E-CC (2020) Mitochondrial and mitochondrial-independent pathways of myocardial cell death during ischaemia and reperfusion injury. J Cell Mol Med 24:3795–3806. https://doi.org/10.1111/jcmm.15127
Davidson SM, Ferdinandy P, Andreadou I, Botker HE, Heusch G, Ibanez B, Ovize M, Schulz R, Yellon DM, Hausenloy DJ, Garcia-Dorado D, Action CC (2019) Multitarget strategies to reduce myocardial ischemia/reperfusion injury: JACC review topic of the week. J Am Coll Cardiol 73:89–99. https://doi.org/10.1016/j.jacc.2018.09.086
Dehlin M, Jacobsson L, Roddy E (2020) Global epidemiology of gout: prevalence, incidence, treatment patterns and risk factors. Nat Rev Rheumatol 16:380–390. https://doi.org/10.1038/s41584-020-0441-1
Deng Y, Li S, Chen Z, Wang W, Geng B, Cai J (2021) Mdivi-1, a mitochondrial fission inhibitor, reduces angiotensin-II-induced hypertension by mediating VSMC phenotypic switch. Biomed Pharmacother 140:111689. https://doi.org/10.1016/j.biopha.2021.111689
Dia M, Gomez L, Thibault H, Tessier N, Leon C, Chouabe C, Ducreux S, Gallo-Bona N, Tubbs E, Bendridi N, Chanon S, Leray A, Belmudes L, Coute Y, Kurdi M, Ovize M, Rieusset J, Paillard M (2020) Reduced reticulum-mitochondria Ca(2+) transfer is an early and reversible trigger of mitochondrial dysfunctions in diabetic cardiomyopathy. Basic Res Cardiol 115:74. https://doi.org/10.1007/s00395-020-00835-7
Dikalov S, Dikalova A (2022) Mitochondrial deacetylase Sirt3 in vascular dysfunction and hypertension. Curr Opin Nephrol Hypertens 31:151–156. https://doi.org/10.1097/MNH.0000000000000771
Ding H, Jiang N, Liu H, Liu X, Liu D, Zhao F, Wen L, Liu S, Ji LL, Zhang Y (2010) Response of mitochondrial fusion and fission protein gene expression to exercise in rat skeletal muscle. Biochim Biophys Acta 1800:250–256. https://doi.org/10.1016/j.bbagen.2009.08.007
Ding M, Feng N, Tang D, Feng J, Li Z, Jia M, Liu Z, Gu X, Wang Y, Fu F, Pei J (2018) Melatonin prevents Drp1-mediated mitochondrial fission in diabetic hearts through SIRT1-PGC1alpha pathway. J Pineal Res 65:e12491. https://doi.org/10.1111/jpi.12491
Disatnik MH, Ferreira JC, Campos JC, Gomes KS, Dourado PM, Qi X, Mochly-Rosen D (2013) Acute inhibition of excessive mitochondrial fission after myocardial infarction prevents long-term cardiac dysfunction. J Am Heart Assoc 2:e000461. https://doi.org/10.1161/JAHA.113.000461
Dominguez-Rodriguez A, Abreu-Gonzalez P, de la Torre-Hernandez JM, Gonzalez-Gonzalez J, Garcia-Camarero T, Consuegra-Sanchez L, Garcia-Saiz MD, Aldea-Perona A, Virgos-Aller T, Azpeitia A, Reiter RJ, Investigators M (2017) Effect of intravenous and intracoronary melatonin as an adjunct to primary percutaneous coronary intervention for acute ST-elevation myocardial infarction: results of the melatonin adjunct in the acute myocardial infarction treated with angioplasty trial. J Pineal Res. https://doi.org/10.1111/jpi.12374
Du J, Li H, Song J, Wang T, Dong Y, Zhan A, Li Y, Liang G (2022) AMPK activation alleviates myocardial ischemia-reperfusion injury by regulating Drp1-mediated mitochondrial dynamics. Front Pharmacol 13:862204. https://doi.org/10.3389/fphar.2022.862204
Dufner DA, Bederman IR, Brunengraber DZ, Rachdaoui N, Ismail-Beigi F, Siegfried BA, Kimball SR, Previs SF (2005) Using 2H2O to study the influence of feeding on protein synthesis: effect of isotope equilibration in vivo vs. in cell culture. Am J Physiol Endocrinol Metab 288:E1277-1283. https://doi.org/10.1152/ajpendo.00580.2004
Edwards AG, Donato AJ, Lesniewski LA, Gioscia RA, Seals DR, Moore RL (2010) Life-long caloric restriction elicits pronounced protection of the aged myocardium: a role for AMPK. Mech Ageing Dev 131:739–742. https://doi.org/10.1016/j.mad.2010.09.007
El-Hattab AW, Suleiman J, Almannai M, Scaglia F (2018) Mitochondrial dynamics: biological roles, molecular machinery, and related diseases. Mol Genet Metab 125:315–321. https://doi.org/10.1016/j.ymgme.2018.10.003
El’darov ChM, Vays VB, Vangeli IM, Kolosova NG, Bakeeva LE (2015) Morphometric examination of mitochondrial ultrastructure in aging cardiomyocytes. Biochemistry (Mosc) 80:604–609. https://doi.org/10.1134/S0006297915050132
Fannin SW, Lesnefsky EJ, Slabe TJ, Hassan MO, Hoppel CL (1999) Aging selectively decreases oxidative capacity in rat heart interfibrillar mitochondria. Arch Biochem Biophys 372:399–407. https://doi.org/10.1006/abbi.1999.1508
Feng D, Wang B, Wang L, Abraham N, Tao K, Huang L, Shi W, Dong Y, Qu Y (2017) Pre-ischemia melatonin treatment alleviated acute neuronal injury after ischemic stroke by inhibiting endoplasmic reticulum stress-dependent autophagy via PERK and IRE1 signalings. J Pineal Res. https://doi.org/10.1111/jpi.12395
Fillmore N, Mori J, Lopaschuk GD (2014) Mitochondrial fatty acid oxidation alterations in heart failure, ischaemic heart disease and diabetic cardiomyopathy. Br J Pharmacol 171:2080–2090. https://doi.org/10.1111/bph.12475
Franco A, Kitsis RN, Fleischer JA, Gavathiotis E, Kornfeld OS, Gong G, Biris N, Benz A, Qvit N, Donnelly SK, Chen Y, Mennerick S, Hodgson L, Mochly-Rosen D, Dorn GW II (2016) Correcting mitochondrial fusion by manipulating mitofusin conformations. Nature 540:74–79. https://doi.org/10.1038/nature20156
Frasier CR, Moore RL, Brown DA (2011) Exercise-induced cardiac preconditioning: how exercise protects your achy-breaky heart. J Appl Physiol (1985) 111:905–915. https://doi.org/10.1152/japplphysiol.00004.2011
Frezza C, Cipolat S, Martins de Brito O, Micaroni M, Beznoussenko GV, Rudka T, Bartoli D, Polishuck RS, Danial NN, De Strooper B, Scorrano L (2006) OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell 126:177–189. https://doi.org/10.1016/j.cell.2006.06.025
Fukasawa Y, Tsuji J, Fu SC, Tomii K, Horton P, Imai K (2015) MitoFates: improved prediction of mitochondrial targeting sequences and their cleavage sites. Mol Cell Proteomics 14:1113–1126. https://doi.org/10.1074/mcp.M114.043083
Fulghum K, Hill BG (2018) Metabolic mechanisms of exercise-induced cardiac remodeling. Front Cardiovasc Med 5:127. https://doi.org/10.3389/fcvm.2018.00127
Gao J, Wang H, Li Y, Li W (2019) Resveratrol attenuates cerebral ischaemia reperfusion injury via modulating mitochondrial dynamics homeostasis and activating AMPK-Mfn1 pathway. Int J Exp Pathol 100:337–349. https://doi.org/10.1111/iep.12336
Giacomello M, Scorrano L (2018) The INs and OUTs of mitofusins. J Cell Biol 217:439–440. https://doi.org/10.1083/jcb.201801042
Gligorijevic B, Kedrin D, Segall JE, Condeelis J, van Rheenen J (2009) Dendra2 photoswitching through the Mammary Imaging Window. J Vis Exp. https://doi.org/10.3791/1278
Gomes LC, Di Benedetto G, Scorrano L (2011) During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol 13:589–598. https://doi.org/10.1038/ncb2220
Gong G, Song M, Csordas G, Kelly DP, Matkovich SJ, Dorn GW 2nd (2015) Parkin-mediated mitophagy directs perinatal cardiac metabolic maturation in mice. Science 350:aad2459. https://doi.org/10.1126/science.aad2459
Grosse L, Wurm CA, Bruser C, Neumann D, Jans DC, Jakobs S (2016) Bax assembles into large ring-like structures remodeling the mitochondrial outer membrane in apoptosis. EMBO J 35:402–413. https://doi.org/10.15252/embj.201592789
Gupta PP, Fonarow GC, Horwich TB (2015) Obesity and the obesity paradox in heart failure. Can J Cardiol 31:195–202. https://doi.org/10.1016/j.cjca.2014.08.004
Hall AR, Burke N, Dongworth RK, Kalkhoran SB, Dyson A, Vicencio JM, Dorn GW II, Yellon DM, Hausenloy DJ (2016) Hearts deficient in both Mfn1 and Mfn2 are protected against acute myocardial infarction. Cell Death Dis 7:e2238. https://doi.org/10.1038/cddis.2016.139
Hamilton KL, Staib JL, Phillips T, Hess A, Lennon SL, Powers SK (2003) Exercise, antioxidants, and HSP72: protection against myocardial ischemia/reperfusion. Free Radic Biol Med 34:800–809. https://doi.org/10.1016/s0891-5849(02)01431-4
Han H, Tan J, Wang R, Wan H, He Y, Yan X, Guo J, Gao Q, Li J, Shang S, Chen F, Tian R, Liu W, Liao L, Tang B, Zhang Z (2020) PINK1 phosphorylates Drp 1(S616) to regulate mitophagy-independent mitochondrial dynamics. EMBO Rep 21:e48686. https://doi.org/10.15252/embr.201948686
Han L, Wang H, Li L, Li X, Ge J, Reiter RJ, Wang Q (2017) Melatonin protects against maternal obesity-associated oxidative stress and meiotic defects in oocytes via the SIRT3-SOD2-dependent pathway. J Pineal Res. https://doi.org/10.1111/jpi.12431
Hasuwa H, Muro Y, Ikawa M, Kato N, Tsujimoto Y, Okabe M (2010) Transgenic mouse sperm that have green acrosome and red mitochondria allow visualization of sperm and their acrosome reaction in vivo. Exp Anim 59:105–107. https://doi.org/10.1538/expanim.59.105
Hausenloy DJ, Botker HE, Engstrom T, Erlinge D, Heusch G, Ibanez B, Kloner RA, Ovize M, Yellon DM, Garcia-Dorado D (2017) Targeting reperfusion injury in patients with ST-segment elevation myocardial infarction: trials and tribulations. Eur Heart J 38:935–941. https://doi.org/10.1093/eurheartj/ehw145
Hausenloy DJ, Garcia-Dorado D, Erik Botker H, Davidson SM, Downey J, Engel FB, Jennings R, Lecour S, Leor J, Madonna R, Ovize M, Perrino C, Prunier F, Schulz R, Sluijter JPG, Van Laake LW, Vinten-Johansen J, Yellon DM, Ytrehus K, Heusch G, Ferdinandy P (2017) Melatonin as a cardioprotective therapy following ST-segment elevation myocardial infarction: is it really promising? Reply. Cardiovasc Res 113:1418–1419. https://doi.org/10.1093/cvr/cvx137
Hausenloy DJ, Yellon DM (2013) Myocardial ischemia-reperfusion injury: a neglected therapeutic target. J Clin Invest 123:92–100. https://doi.org/10.1172/JCI62874
Hernandez-Resendiz S, Chinda K, Ong SB, Cabrera-Fuentes H, Zazueta C, Hausenloy DJ (2018) The role of redox dysregulation in the inflammatory response to acute myocardial ischaemia-reperfusion injury—adding fuel to the fire. Curr Med Chem 25:1275–1293. https://doi.org/10.2174/0929867324666170329100619
Hernandez-Resendiz S, Prunier F, Girao H, Dorn G, Hausenloy DJ, Action E-CC (2020) Targeting mitochondrial fusion and fission proteins for cardioprotection. J Cell Mol Med 24:6571–6585. https://doi.org/10.1111/jcmm.15384
Heusch G (2018) EURO-CRIPS: another piece of evidence for systemic cardiovascular protection by remote ischemic conditioning. Int J Cardiol 257:30–31. https://doi.org/10.1016/j.ijcard.2017.12.043
Heusch G (2015) Molecular basis of cardioprotection: signal transduction in ischemic pre-, post-, and remote conditioning. Circ Res 116:674–699. https://doi.org/10.1161/CIRCRESAHA.116.305348
Heusch G (2020) Myocardial ischaemia-reperfusion injury and cardioprotection in perspective. Nat Rev Cardiol 17:773–789. https://doi.org/10.1038/s41569-020-0403-y
Heusch G, Andreadou I, Bell R, Bertero E, Botker HE, Davidson SM, Downey J, Eaton P, Ferdinandy P, Gersh BJ, Giacca M, Hausenloy DJ, Ibanez B, Krieg T, Maack C, Schulz R, Sellke F, Shah AM, Thiele H, Yellon DM, Di Lisa F (2023) Health position paper and redox perspectives on reactive oxygen species as signals and targets of cardioprotection. Redox Biol 67:102894. https://doi.org/10.1016/j.redox.2023.102894
Heusch G, Libby P, Gersh B, Yellon D, Bohm M, Lopaschuk G, Opie L (2014) Cardiovascular remodelling in coronary artery disease and heart failure. Lancet 383:1933–1943. https://doi.org/10.1016/S0140-6736(14)60107-0
Hollander JM, Thapa D, Shepherd DL (2014) Physiological and structural differences in spatially distinct subpopulations of cardiac mitochondria: influence of cardiac pathologies. Am J Physiol Heart Circ Physiol 307:H1-14. https://doi.org/10.1152/ajpheart.00747.2013
Hsu CC, Zhang X, Wang G, Zhang W, Cai Z, Pan BS, Gu H, Xu C, Jin G, Xu X, Manne RK, Jin Y, Yan W, Shao J, Chen T, Lin E, Ketkar A, Eoff R, Xu ZG, Chen ZZ, Li HY, Lin HK (2021) Inositol serves as a natural inhibitor of mitochondrial fission by directly targeting AMPK. Mol Cell 81(3803–3819):e3807. https://doi.org/10.1016/j.molcel.2021.08.025
Hu L, Guo Y, Song L, Wen H, Sun N, Wang Y, Qi B, Liang Q, Geng J, Liu X, Fu F, Li Y (2022) Nicotinamide riboside promotes Mfn2-mediated mitochondrial fusion in diabetic hearts through the SIRT1-PGC1α-PPAR α pathway. Free Radic Biol Med 183:75–88. https://doi.org/10.1016/j.freeradbiomed.2022.03.012
Huang G, Cong Z, Wang X, Yuan Y, Xu R, Lu Z, Wang X, Qi J (2020) Targeting HSP90 attenuates angiotensin II-induced adventitial remodelling via suppression of mitochondrial fission. Cardiovasc Res 116:1071–1084. https://doi.org/10.1093/cvr/cvz194
Huang S, Li Z, Wu Z, Liu C, Yu M, Wen M, Zhang L, Wang X (2021) DDAH2 suppresses RLR-MAVS-mediated innate antiviral immunity by stimulating nitric oxide-activated, Drp1-induced mitochondrial fission. Sci Signal. https://doi.org/10.1126/scisignal.abc7931
Huertas JR, Casuso RA, Agustin PH, Cogliati S (2019) Stay fit, stay young: mitochondria in movement: the role of exercise in the new mitochondrial paradigm. Oxid Med Cell Longev 2019:7058350. https://doi.org/10.1155/2019/7058350
Hutter JJ, Mestril R, Tam EK, Sievers RE, Dillmann WH, Wolfe CL (1996) Overexpression of heat shock protein 72 in transgenic mice decreases infarct size in vivo. Circulation 94:1408–1411. https://doi.org/10.1161/01.cir.94.6.1408
Ikeda Y, Shirakabe A, Maejima Y, Zhai P, Sciarretta S, Toli J, Nomura M, Mihara K, Egashira K, Ohishi M, Abdellatif M, Sadoshima J (2015) Endogenous Drp1 mediates mitochondrial autophagy and protects the heart against energy stress. Circ Res 116:264–278. https://doi.org/10.1161/CIRCRESAHA.116.303356
Ikon N, Ryan RO (2017) Cardiolipin and mitochondrial cristae organization. Biochim Biophys Acta Biomembr 1859:1156–1163. https://doi.org/10.1016/j.bbamem.2017.03.013
Ismail NI, Michel NA, Katwadi K, Lim MM, Chan TK, Rahman A, Xu D, Ong SG, Hausenloy DJ, Ong SB (2022) Ischemic preconditioning and postconditioning protect the heart by preserving the mitochondrial network. Biomed Res Int 2022:6889278. https://doi.org/10.1155/2022/6889278
Jezek P, Jaburek M, Holendova B, Engstova H, Dlaskova A (2023) Mitochondrial cristae morphology reflecting metabolism, superoxide formation, redox homeostasis, and pathology. Antioxid Redox Signal. https://doi.org/10.1089/ars.2022.0173
Jhun BS, Uchi JO, Adaniya SM, Mancini TJ, Cao JL, King ME, Landi AK, Ma H, Shin M, Yang D, Xu X, Yoon Y, Choudhary G, Clements RT, Mende U, Sheu SS (2018) Protein kinase D activation induces mitochondrial fragmentation and dysfunction in cardiomyocytes. J Physiol 596:827–855. https://doi.org/10.1113/JP275418
Jia G, Hill MA, Sowers JR (2018) Diabetic cardiomyopathy: an update of mechanisms contributing to this clinical entity. Circ Res 122:624–638. https://doi.org/10.1161/CIRCRESAHA.117.311586
Joubert F, Puff N (2021) Mitochondrial cristae architecture and functions: lessons from minimal model systems. Membranes (Basel). https://doi.org/10.3390/membranes11070465
Kalkhoran SB, Kriston-Vizi J, Hernandez-Resendiz S, Crespo-Avilan GE, Rosdah AA, Lees JG, Costa J, Ling NXY, Holien JK, Samangouei P, Chinda K, Yap EP, Riquelme JA, Ketteler R, Yellon DM, Lim SY, Hausenloy DJ (2022) Hydralazine protects the heart against acute ischaemia/reperfusion injury by inhibiting Drp1-mediated mitochondrial fission. Cardiovasc Res 118:282–294. https://doi.org/10.1093/cvr/cvaa343
Kameoka S, Adachi Y, Okamoto K, Iijima M, Sesaki H (2018) Phosphatidic acid and cardiolipin coordinate mitochondrial dynamics. Trends Cell Biol 28:67–76. https://doi.org/10.1016/j.tcb.2017.08.011
Kamerkar SC, Kraus F, Sharpe AJ, Pucadyil TJ, Ryan MT (2018) Dynamin-related protein 1 has membrane constricting and severing abilities sufficient for mitochondrial and peroxisomal fission. Nat Commun 9:5239. https://doi.org/10.1038/s41467-018-07543-w
Karbowski M, Arnoult D, Chen H, Chan DC, Smith CL, Youle RJ (2004) Quantitation of mitochondrial dynamics by photolabeling of individual organelles shows that mitochondrial fusion is blocked during the Bax activation phase of apoptosis. J Cell Biol 164:493–499. https://doi.org/10.1083/jcb.200309082
Kasumov T, Dabkowski ER, Shekar KC, Li L, Ribeiro RF Jr, Walsh K, Previs SF, Sadygov RG, Willard B, Stanley WC (2013) Assessment of cardiac proteome dynamics with heavy water: slower protein synthesis rates in interfibrillar than subsarcolemmal mitochondria. Am J Physiol Heart Circ Physiol 304:H1201-1214. https://doi.org/10.1152/ajpheart.00933.2012
Katare RG, Kakinuma Y, Arikawa M, Yamasaki F, Sato T (2009) Chronic intermittent fasting improves the survival following large myocardial ischemia by activation of BDNF/VEGF/PI3K signaling pathway. J Mol Cell Cardiol 46:405–412. https://doi.org/10.1016/j.yjmcc.2008.10.027
Kavazis AN (2009) Exercise preconditioning of the myocardium. Sports Med 39:923–935. https://doi.org/10.2165/11317870-000000000-00000
Khan S, Salloum F, Das A, Xi L, Vetrovec GW, Kukreja RC (2006) Rapamycin confers preconditioning-like protection against ischemia-reperfusion injury in isolated mouse heart and cardiomyocytes. J Mol Cell Cardiol 41:256–264. https://doi.org/10.1016/j.yjmcc.2006.04.014
Kim H, Scimia MC, Wilkinson D, Trelles RD, Wood MR, Bowtell D, Dillin A, Mercola M, Ronai ZA (2011) Fine-tuning of Drp1/Fis1 availability by AKAP121/Siah2 regulates mitochondrial adaptation to hypoxia. Mol Cell 44:532–544. https://doi.org/10.1016/j.molcel.2011.08.045
Kim TY, Wang D, Kim AK, Lau E, Lin AJ, Liem DA, Zhang J, Zong NC, Lam MP, Ping P (2012) Metabolic labeling reveals proteome dynamics of mouse mitochondria. Mol Cell Proteomics 11:1586–1594. https://doi.org/10.1074/mcp.M112.021162
Kirkham AA, Beka V, Prado CM (2021) The effect of caloric restriction on blood pressure and cardiovascular function: a systematic review and meta-analysis of randomized controlled trials. Clin Nutr 40:728–739. https://doi.org/10.1016/j.clnu.2020.06.029
Kleinbongard P, Lieder HR, Skyschally A, Alloosh M, Godecke A, Rahmann S, Sturek M, Heusch G (2022) Non-responsiveness to cardioprotection by ischaemic preconditioning in Ossabaw minipigs with genetic predisposition to, but without the phenotype of the metabolic syndrome. Basic Res Cardiol 117:58. https://doi.org/10.1007/s00395-022-00965-0
Kleinbongard P, Skyschally A, Heusch G (2017) Cardioprotection by remote ischemic conditioning and its signal transduction. Pflugers Arch 469:159–181. https://doi.org/10.1007/s00424-016-1922-6
Klementieva NV, Lukyanov KA, Markina NM, Lukyanov SA, Zagaynova EV, Mishin AS (2016) Green-to-red primed conversion of Dendra2 using blue and red lasers. Chem Commun (Camb) 52:13144–13146. https://doi.org/10.1039/c6cc05599k
Kraus F, Ryan MT (2017) The constriction and scission machineries involved in mitochondrial fission. J Cell Sci 130:2953–2960. https://doi.org/10.1242/jcs.199562
Kuznetsov AV, Veksler V, Gellerich FN, Saks V, Margreiter R, Kunz WS (2008) Analysis of mitochondrial function in situ in permeabilized muscle fibers, tissues and cells. Nat Protoc 3:965–976. https://doi.org/10.1038/nprot.2008.61
Lahera V, de Las HN, Lopez-Farre A, Manucha W, Ferder L (2017) Role of mitochondrial dysfunction in hypertension and obesity. Curr Hypertens Rep 19:11. https://doi.org/10.1007/s11906-017-0710-9
Le Page S, Niro M, Fauconnier J, Cellier L, Tamareille S, Gharib A, Chevrollier A, Loufrani L, Grenier C, Kamel R, Sarzi E, Lacampagne A, Ovize M, Henrion D, Reynier P, Lenaers G, Mirebeau-Prunier D, Prunier F (2016) Increase in cardiac ischemia-reperfusion injuries in Opa1+/- mouse model. PLoS One 11:e0164066. https://doi.org/10.1371/journal.pone.0164066
Lecour S, Andreadou I, Botker HE, Davidson SM, Heusch G, Ruiz-Meana M, Schulz R, Zuurbier CJ, Ferdinandy P, Hausenloy DJ, on behalf of the European Union CCAC (2021) Improving preclinical assessment of cardioprotective therapies (IMPACT) criteria: guidelines of the EU-CARDIOPROTECTION COST action. Basic Res Cardiol 116:52. https://doi.org/10.1007/s00395-021-00893-5
Legros F, Lombes A, Frachon P, Rojo M (2002) Mitochondrial fusion in human cells is efficient, requires the inner membrane potential, and is mediated by mitofusins. Mol Biol Cell 13:4343–4354. https://doi.org/10.1091/mbc.e02-06-0330
Lesnefsky EJ, Gudz TI, Moghaddas S, Migita CT, Ikeda-Saito M, Turkaly PJ, Hoppel CL (2001) Aging decreases electron transport complex III activity in heart interfibrillar mitochondria by alteration of the cytochrome c binding site. J Mol Cell Cardiol 33:37–47. https://doi.org/10.1006/jmcc.2000.1273
Lesnefsky EJ, Slabe TJ, Stoll MS, Minkler PE, Hoppel CL (2001) Myocardial ischemia selectively depletes cardiolipin in rabbit heart subsarcolemmal mitochondria. Am J Physiol Heart Circ Physiol 280:H2770-2778. https://doi.org/10.1152/ajpheart.2001.280.6.H2770
Lesnefsky EJ, Tandler B, Ye J, Slabe TJ, Turkaly J, Hoppel CL (1997) Myocardial ischemia decreases oxidative phosphorylation through cytochrome oxidase in subsarcolemmal mitochondria. Am J Physiol 273:H1544-1554. https://doi.org/10.1152/ajpheart.1997.273.3.H1544
Li H, Feng J, Zhang Y, Feng J, Wang Q, Zhao S, Meng P, Li J (2019) Mst1 deletion attenuates renal ischaemia-reperfusion injury: the role of microtubule cytoskeleton dynamics, mitochondrial fission and the GSK3beta-p53 signalling pathway. Redox Biol 20:261–274. https://doi.org/10.1016/j.redox.2018.10.012
Li YJ, Cao YL, Feng JX, Qi Y, Meng S, Yang JF, Zhong YT, Kang S, Chen X, Lan L, Luo L, Yu B, Chen S, Chan DC, Hu J, Gao S (2019) Structural insights of human mitofusin-2 into mitochondrial fusion and CMT2A onset. Nat Commun 10:4914. https://doi.org/10.1038/s41467-019-12912-0
Liang P, Ye F, Hou CC, Pi L, Chen F (2021) Mesenchymal stem cell therapy for patients with ischemic heart failure- past, present, and future. Curr Stem Cell Res Ther 16:608–621. https://doi.org/10.2174/1574888X15666200309144906
Lin X, Li H (2021) Obesity: epidemiology, pathophysiology, and therapeutics. Front Endocrinol (Lausanne) 12:706978. https://doi.org/10.3389/fendo.2021.706978
Liu M, Lv J, Pan Z, Wang D, Zhao L, Guo X (2022) Mitochondrial dysfunction in heart failure and its therapeutic implications. Front Cardiovasc Med 9:945142. https://doi.org/10.3389/fcvm.2022.945142
Ljubicic V, Menzies KJ, Hood DA (2010) Mitochondrial dysfunction is associated with a pro-apoptotic cellular environment in senescent cardiac muscle. Mech Ageing Dev 131:79–88. https://doi.org/10.1016/j.mad.2009.12.004
Logan DC (2006) The mitochondrial compartment. J Exp Bot 57:1225–1243. https://doi.org/10.1093/jxb/erj151
Lu X, Thai PN, Lu S, Pu J, Bers DM (2019) Intrafibrillar and perinuclear mitochondrial heterogeneity in adult cardiac myocytes. J Mol Cell Cardiol 136:72–84. https://doi.org/10.1016/j.yjmcc.2019.08.013
Makarewich CA, Zhang H, Davis J, Correll RN, Trappanese DM, Hoffman NE, Troupes CD, Berretta RM, Kubo H, Madesh M, Chen X, Gao E, Molkentin JD, Houser SR (2014) Transient receptor potential channels contribute to pathological structural and functional remodeling after myocardial infarction. Circ Res 115:567–580. https://doi.org/10.1161/CIRCRESAHA.115.303831
Mamuya WS, Brecher P (1992) Fibronectin expression in the normal and hypertrophic rat heart. J Clin Invest 89:392–401. https://doi.org/10.1172/JCI115598
Maneechote C, Palee S, Kerdphoo S, Jaiwongkam T, Chattipakorn SC, Chattipakorn N (2019) Balancing mitochondrial dynamics via increasing mitochondrial fusion attenuates infarct size and left ventricular dysfunction in rats with cardiac ischemia/reperfusion injury. Clin Sci (Lond) 133:497–513. https://doi.org/10.1042/CS20190014
Maneechote C, Palee S, Kerdphoo S, Jaiwongkam T, Chattipakorn SC, Chattipakorn N (2018) Differential temporal inhibition of mitochondrial fission by Mdivi-1 exerts effective cardioprotection in cardiac ischemia/reperfusion injury. Clin Sci (Lond) 132:1669–1683. https://doi.org/10.1042/CS20180510
Maneechote C, Palee S, Kerdphoo S, Jaiwongkam T, Chattipakorn SC, Chattipakorn N (2022) Modulating mitochondrial dynamics attenuates cardiac ischemia-reperfusion injury in prediabetic rats. Acta Pharmacol Sin 43:26–38. https://doi.org/10.1038/s41401-021-00626-3
Maneechote C, Palee S, Kerdphoo S, Jaiwongkam T, Chattipakorn SC, Chattipakorn N (2020) Pharmacological inhibition of mitochondrial fission attenuates cardiac ischemia-reperfusion injury in pre-diabetic rats. Biochem Pharmacol 182:114295. https://doi.org/10.1016/j.bcp.2020.114295
Marsboom G, Toth PT, Ryan JJ, Hong Z, Wu X, Fang YH, Thenappan T, Piao L, Zhang HJ, Pogoriler J, Chen Y, Morrow E, Weir EK, Rehman J, Archer SL (2012) Dynamin-related protein 1-mediated mitochondrial mitotic fission permits hyperproliferation of vascular smooth muscle cells and offers a novel therapeutic target in pulmonary hypertension. Circ Res 110:1484–1497. https://doi.org/10.1161/CIRCRESAHA.111.263848
Martelli PL, Savojardo C, Fariselli P, Tasco G, Casadio R (2015) Computer-based prediction of mitochondria-targeting peptides. Methods Mol Biol 1264:305–320. https://doi.org/10.1007/978-1-4939-2257-4_27
Mattie S, Riemer J, Wideman JG, McBride HM (2018) A new mitofusin topology places the redox-regulated C terminus in the mitochondrial intermembrane space. J Cell Biol 217:507–515. https://doi.org/10.1083/jcb.201611194
McWilliams TG, Prescott AR, Allen GF, Tamjar J, Munson MJ, Thomson C, Muqit MM, Ganley IG (2016) mito-QC illuminates mitophagy and mitochondrial architecture in vivo. J Cell Biol 214:333–345. https://doi.org/10.1083/jcb.201603039
Mei Y, Hu H, Deng L, Sun X, Tan W (2022) Isosteviol sodium attenuates high fat/high cholesterol-induced myocardial dysfunction by regulating the Sirt1/AMPK pathway. Biochem Biophys Res Commun 621:80–87. https://doi.org/10.1016/j.bbrc.2022.06.044
Mei Y, Liu B, Su H, Zhang H, Liu F, Ke Q, Sun X, Tan W (2020) Isosteviol sodium protects the cardiomyocyte response associated with the SIRT1/PGC-1alpha pathway. J Cell Mol Med 24:10866–10875. https://doi.org/10.1111/jcmm.15715
Melo DS, Costa-Pereira LV, Santos CS, Mendes BF, Costa KB, Santos CF, Rocha-Vieira E, Magalhaes FC, Esteves EA, Ferreira AJ, Guatimosim S, Dias-Peixoto MF (2016) Severe calorie restriction reduces cardiometabolic risk factors and protects rat hearts from ischemia/reperfusion injury. Front Physiol 7:106. https://doi.org/10.3389/fphys.2016.00106
Molina AJ, Wikstrom JD, Stiles L, Las G, Mohamed H, Elorza A, Walzer G, Twig G, Katz S, Corkey BE, Shirihai OS (2009) Mitochondrial networking protects beta-cells from nutrient-induced apoptosis. Diabetes 58:2303–2315. https://doi.org/10.2337/db07-1781
Montaigne D, Marechal X, Coisne A, Debry N, Modine T, Fayad G, Potelle C, El Arid JM, Mouton S, Sebti Y, Duez H, Preau S, Remy-Jouet I, Zerimech F, Koussa M, Richard V, Neviere R, Edme JL, Lefebvre P, Staels B (2014) Myocardial contractile dysfunction is associated with impaired mitochondrial function and dynamics in type 2 diabetic but not in obese patients. Circulation 130:554–564. https://doi.org/10.1161/CIRCULATIONAHA.113.008476
Morrow DA, Givertz MM (2005) Modulation of myocardial energetics: emerging evidence for a therapeutic target in cardiovascular disease. Circulation 112:3218–3221. https://doi.org/10.1161/CIRCULATIONAHA.105.581819
Mouton AJ, Li X, Hall ME, Hall JE (2020) Obesity, hypertension, and cardiac dysfunction: novel roles of immunometabolism in macrophage activation and inflammation. Circ Res 126:789–806. https://doi.org/10.1161/CIRCRESAHA.119.312321
Muller W (1976) Subsarcolemmal mitochondria and capillarization of soleus muscle fibers in young rats subjected to an endurance training. A morphometric study of semithin sections. Cell Tissue Res 174:367–389. https://doi.org/10.1007/BF00220682
Naon D, Hernandez-Alvarez MI, Shinjo S, Wieczor M, Ivanova S, Martins de Brito O, Quintana A, Hidalgo J, Palacin M, Aparicio P, Castellanos J, Lores L, Sebastian D, Fernandez-Veledo S, Vendrell J, Joven J, Orozco M, Zorzano A, Scorrano L (2023) Splice variants of mitofusin 2 shape the endoplasmic reticulum and tether it to mitochondria. Science 380:eadh9351. https://doi.org/10.1126/science.adh9351
Napoli E, Song G, Liu S, Espejo A, Perez CJ, Benavides F, Giulivi C (2017) Zdhhc13-dependent Drp1 S-palmitoylation impacts brain bioenergetics, anxiety, coordination and motor skills. Sci Rep 7:12796. https://doi.org/10.1038/s41598-017-12889-0
Niemann B, Chen Y, Issa H, Silber RE, Rohrbach S (2010) Caloric restriction delays cardiac ageing in rats: role of mitochondria. Cardiovasc Res 88:267–276. https://doi.org/10.1093/cvr/cvq273
Niemann B, Li L, Simm A, Molenda N, Kockskamper J, Boening A, Rohrbach S (2021) Caloric restriction reduces sympathetic activity similar to beta-blockers but conveys additional mitochondrio-protective effects in aged myocardium. Sci Rep 11:1931. https://doi.org/10.1038/s41598-021-81438-7
Niemann B, Pan R, Teschner M, Boening A, Silber RE, Rohrbach S (2013) Age and obesity-associated changes in the expression and activation of components of the AMPK signaling pathway in human right atrial tissue. Exp Gerontol 48:55–63. https://doi.org/10.1016/j.exger.2012.04.005
Nishida M, Kurose H (2008) Roles of TRP channels in the development of cardiac hypertrophy. Naunyn Schmiedebergs Arch Pharmacol 378:395–406. https://doi.org/10.1007/s00210-008-0321-8
Nishimura A, Shimauchi T, Tanaka T, Shimoda K, Toyama T, Kitajima N, Ishikawa T, Shindo N, Numaga-Tomita T, Yasuda S, Sato Y, Kuwahara K, Kumagai Y, Akaike T, Ide T, Ojida A, Mori Y, Nishida M (2018) Hypoxia-induced interaction of filamin with Drp1 causes mitochondrial hyperfission-associated myocardial senescence. Sci Signal. https://doi.org/10.1126/scisignal.aat5185
Noone J, O’Gorman DJ, Kenny HC (2022) OPA1 regulation of mitochondrial dynamics in skeletal and cardiac muscle. Trends Endocrinol Metab 33:710–721. https://doi.org/10.1016/j.tem.2022.07.003
Numaga-Tomita T, Oda S, Shimauchi T, Nishimura A, Mangmool S, Nishida M (2017) TRPC3 channels in cardiac fibrosis. Front Cardiovasc Med 4:56. https://doi.org/10.3389/fcvm.2017.00056
Nyolczas N, Dekany M, Muk B, Szabo B (2018) Combination of hydralazine and isosorbide-dinitrate in the treatment of patients with heart failure with reduced ejection fraction. Adv Exp Med Biol 1067:31–45. https://doi.org/10.1007/5584_2017_112
Okada Y, Okada T, Sato-Numata K, Islam MR, Ando-Akatsuka Y, Numata T, Kubo M, Shimizu T, Kurbannazarova RS, Marunaka Y, Sabirov RZ (2019) Cell volume-activated and volume-correlated anion channels in mammalian cells: their biophysical, molecular, and pharmacological properties. Pharmacol Rev 71:49–88. https://doi.org/10.1124/pr.118.015917
Ong SB, Hausenloy DJ (2010) Mitochondrial morphology and cardiovascular disease. Cardiovasc Res 88:16–29. https://doi.org/10.1093/cvr/cvq237
Ong SB, Hernandez-Resendiz S, Crespo-Avilan GE, Mukhametshina RT, Kwek XY, Cabrera-Fuentes HA, Hausenloy DJ (2018) Inflammation following acute myocardial infarction: multiple players, dynamic roles, and novel therapeutic opportunities. Pharmacol Ther 186:73–87. https://doi.org/10.1016/j.pharmthera.2018.01.001
Ong SB, Kalkhoran SB, Cabrera-Fuentes HA, Hausenloy DJ (2015) Mitochondrial fusion and fission proteins as novel therapeutic targets for treating cardiovascular disease. Eur J Pharmacol 763:104–114. https://doi.org/10.1016/j.ejphar.2015.04.056
Ong SB, Kalkhoran SB, Hernandez-Resendiz S, Samangouei P, Ong SG, Hausenloy DJ (2017) Mitochondrial-shaping proteins in cardiac health and disease—the long and the short of it! Cardiovasc Drugs Ther 31:87–107. https://doi.org/10.1007/s10557-016-6710-1
Ong SB, Subrayan S, Lim SY, Yellon DM, Davidson SM, Hausenloy DJ (2010) Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation 121:2012–2022. https://doi.org/10.1161/CIRCULATIONAHA.109.906610
Ortega FB, Lavie CJ, Blair SN (2016) Obesity and cardiovascular disease. Circ Res 118:1752–1770. https://doi.org/10.1161/CIRCRESAHA.115.306883
Owens GC, Edelman DB (2015) Photoconvertible fluorescent protein-based live imaging of mitochondrial fusion. Methods Mol Biol 1313:237–246. https://doi.org/10.1007/978-1-4939-2703-6_18
Pakhomov AA, Martynov VI, Orsa AN, Bondarenko AA, Chertkova RV, Lukyanov KA, Petrenko AG, Deyev IE (2017) Fluorescent protein Dendra2 as a ratiometric genetically encoded pH-sensor. Biochem Biophys Res Commun 493:1518–1521. https://doi.org/10.1016/j.bbrc.2017.09.170
Palmer JW, Tandler B, Hoppel CL (1977) Biochemical properties of subsarcolemmal and interfibrillar mitochondria isolated from rat cardiac muscle. J Biol Chem 252:8731–8739
Parameyong A, Govitrapong P, Chetsawang B (2015) Melatonin attenuates the mitochondrial translocation of mitochondrial fission proteins and Bax, cytosolic calcium overload and cell death in methamphetamine-induced toxicity in neuroblastoma SH-SY5Y cells. Mitochondrion 24:1–8. https://doi.org/10.1016/j.mito.2015.07.004
Parim B, Sathibabu Uddandrao VV, Saravanan G (2019) Diabetic cardiomyopathy: molecular mechanisms, detrimental effects of conventional treatment, and beneficial effects of natural therapy. Heart Fail Rev 24:279–299. https://doi.org/10.1007/s10741-018-9749-1
Patterson G, Davidson M, Manley S, Lippincott-Schwartz J (2010) Superresolution imaging using single-molecule localization. Annu Rev Phys Chem 61:345–367. https://doi.org/10.1146/annurev.physchem.012809.103444
Patterson GH, Lippincott-Schwartz J (2002) A photoactivatable GFP for selective photolabeling of proteins and cells. Science 297:1873–1877. https://doi.org/10.1126/science.1074952
Pei H, Du J, Song X, He L, Zhang Y, Li X, Qiu C, Zhang Y, Hou J, Feng J, Gao E, Li D, Yang Y (2016) Melatonin prevents adverse myocardial infarction remodeling via Notch1/Mfn2 pathway. Free Radic Biol Med 97:408–417. https://doi.org/10.1016/j.freeradbiomed.2016.06.015
Pennanen C, Parra V, Lopez-Crisosto C, Morales PE, Del Campo A, Gutierrez T, Rivera-Mejias P, Kuzmicic J, Chiong M, Zorzano A, Rothermel BA, Lavandero S (2014) Mitochondrial fission is required for cardiomyocyte hypertrophy mediated by a Ca2+-calcineurin signaling pathway. J Cell Sci 127:2659–2671. https://doi.org/10.1242/jcs.139394
Penpargkul S, Fein F, Sonnenblick EH, Scheuer J (1981) Depressed cardiac sarcoplasmic reticular function from diabetic rats. J Mol Cell Cardiol 13:303–309. https://doi.org/10.1016/0022-2828(81)90318-7
Perkins GA, Frey TG (2000) Recent structural insight into mitochondria gained by microscopy. Micron 31:97–111. https://doi.org/10.1016/s0968-4328(99)00065-7
Pham AH, McCaffery JM, Chan DC (2012) Mouse lines with photo-activatable mitochondria to study mitochondrial dynamics. Genesis 50:833–843. https://doi.org/10.1002/dvg.22050
Picard M, Wright KJ, Ritchie D, Thomas MM, Hepple RT (2012) Mitochondrial function in permeabilized cardiomyocytes is largely preserved in the senescent rat myocardium. PLoS One 7:e43003. https://doi.org/10.1371/journal.pone.0043003
Pigazzini ML, Kirstein J (2020) In vivo quantification of protein turnover in aging C. elegans using Photoconvertible dendra2. J Vis Exp. https://doi.org/10.3791/61196
Powers SK, Smuder AJ, Kavazis AN, Quindry JC (2014) Mechanisms of exercise-induced cardioprotection. Physiology (Bethesda) 29:27–38. https://doi.org/10.1152/physiol.00030.2013
Prince FP (2002) Lamellar and tubular associations of the mitochondrial cristae: unique forms of the cristae present in steroid-producing cells. Mitochondrion 1:381–389. https://doi.org/10.1016/s1567-7249(01)00038-1
Qi Z, Huang Z, Xie F, Chen L (2019) Dynamin-related protein 1: a critical protein in the pathogenesis of neural system dysfunctions and neurodegenerative diseases. J Cell Physiol 234:10032–10046. https://doi.org/10.1002/jcp.27866
Qin Y, Gao M, Li A, Sun J, Liu B, Gong G (2018) Mitoflash lights single mitochondrial dynamics events in mature cardiomyocytes. Biochem Biophys Res Commun 503:729–736. https://doi.org/10.1016/j.bbrc.2018.06.068
Quiles JM, Gustafsson AB (2022) The role of mitochondrial fission in cardiovascular health and disease. Nat Rev Cardiol 19:723–736. https://doi.org/10.1038/s41569-022-00703-y
Rachdaoui N, Austin L, Kramer E, Previs MJ, Anderson VE, Kasumov T, Previs SF (2009) Measuring proteome dynamics in vivo: as easy as adding water? Mol Cell Proteomics 8:2653–2663. https://doi.org/10.1074/mcp.M900026-MCP200
Ramachandra CJA, Hernandez-Resendiz S, Crespo-Avilan GE, Lin YH, Hausenloy DJ (2020) Mitochondria in acute myocardial infarction and cardioprotection. EBioMedicine 57:102884. https://doi.org/10.1016/j.ebiom.2020.102884
Raven JA (2021) Determinants, and implications, of the shape and size of thylakoids and cristae. J Plant Physiol 257:153342. https://doi.org/10.1016/j.jplph.2020.153342
Ren S, Zhang H, Mu Y, Sun M, Liu P (2013) Pharmacological effects of Astragaloside IV: a literature review. J Tradit Chin Med 33:413–416. https://doi.org/10.1016/s0254-6272(13)60189-2
Riva A, Tandler B, Loffredo F, Vazquez E, Hoppel C (2005) Structural differences in two biochemically defined populations of cardiac mitochondria. Am J Physiol Heart Circ Physiol 289:H868-872. https://doi.org/10.1152/ajpheart.00866.2004
Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, Tuft RA, Pozzan T (1998) Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 280:1763–1766. https://doi.org/10.1126/science.280.5370.1763
Robert P, Nguyen PMC, Richard A, Grenier C, Chevrollier A, Munier M, Grimaud L, Proux C, Champin T, Lelievre E, Sarzi E, Vessieres E, Henni S, Prunier D, Reynier P, Lenaers G, Fassot C, Henrion D, Loufrani L (2021) Protective role of the mitochondrial fusion protein OPA1 in hypertension. FASEB J 35:e21678. https://doi.org/10.1096/fj.202000238RRR
Rocha AG, Franco A, Krezel AM, Rumsey JM, Alberti JM, Knight WC, Biris N, Zacharioudakis E, Janetka JW, Baloh RH, Kitsis RN, Mochly-Rosen D, Townsend RR, Gavathiotis E, Dorn GW 2nd (2018) MFN2 agonists reverse mitochondrial defects in preclinical models of Charcot-Marie-Tooth disease type 2A. Science 360:336–341. https://doi.org/10.1126/science.aao1785
Rohrbach S, Aslam M, Niemann B, Schulz R (2014) Impact of caloric restriction on myocardial ischaemia/reperfusion injury and new therapeutic options to mimic its effects. Br J Pharmacol 171:2964–2992. https://doi.org/10.1111/bph.12650
Rosano GM, Vitale C (2018) Metabolic modulation of cardiac metabolism in heart failure. Card Fail Rev 4:99–103. https://doi.org/10.15420/cfr.2018.18.2
Rosdah AA, Abbott BM, Langendorf CG, Deng Y, Truong JQ, Waddell HMM, Ling NXY, Smiles WJ, Delbridge LMD, Liu GS, Oakhill JS, Lim SY, Holien JK (2022) A novel small molecule inhibitor of human Drp1. Sci Rep 12:21531. https://doi.org/10.1038/s41598-022-25464-z
Rubler S, Dlugash J, Yuceoglu YZ, Kumral T, Branwood AW, Grishman A (1972) New type of cardiomyopathy associated with diabetic glomerulosclerosis. Am J Cardiol 30:595–602. https://doi.org/10.1016/0002-9149(72)90595-4
Ryan JJ, Marsboom G, Fang YH, Toth PT, Morrow E, Luo N, Piao L, Hong Z, Ericson K, Zhang HJ, Han M, Haney CR, Chen CT, Sharp WW, Archer SL (2013) PGC1alpha-mediated mitofusin-2 deficiency in female rats and humans with pulmonary arterial hypertension. Am J Respir Crit Care Med 187:865–878. https://doi.org/10.1164/rccm.201209-1687OC
Seferovic PM, Vardas P, Jankowska EA, Maggioni AP, Timmis A, Milinkovic I, Polovina M, Gale CP, Lund LH, Lopatin Y, Lainscak M, Savarese G, Huculeci R, Kazakiewicz D, Coats AJS, National Heart Failure Societies of the ESCmc (2021) The Heart failure association atlas: heart failure epidemiology and management statistics 2019. Eur J Heart Fail 23:906–914. https://doi.org/10.1002/ejhf.2143
Semenzato M, Kohr MJ, Quirin C, Menabo R, Alanova P, Alan L, Pellattiero A, Murphy E, Di Lisa F, Scorrano L (2023) Oxidization of optic atrophy 1 cysteines occurs during heart ischemia-reperfusion and amplifies cell death by oxidative stress. Redox Biol 63:102755. https://doi.org/10.1016/j.redox.2023.102755
Serasinghe MN, Chipuk JE (2017) Mitochondrial fission in human diseases. Handb Exp Pharmacol 240:159–188. https://doi.org/10.1007/164_2016_38
Serna JDC, Caldeira da Silva CC, Kowaltowski AJ (2020) Functional changes induced by caloric restriction in cardiac and skeletal muscle mitochondria. J Bioenerg Biomembr 52:269–277. https://doi.org/10.1007/s10863-020-09838-4
Sharp WW, Fang YH, Han M, Zhang HJ, Hong Z, Banathy A, Morrow E, Ryan JJ, Archer SL (2014) Dynamin-related protein 1 (Drp1)-mediated diastolic dysfunction in myocardial ischemia-reperfusion injury: therapeutic benefits of Drp1 inhibition to reduce mitochondrial fission. FASEB J 28:316–326. https://doi.org/10.1096/fj.12-226225
Shimada T, Horita K, Murakami M, Ogura R (1984) Morphological studies of different mitochondrial populations in monkey myocardial cells. Cell Tissue Res 238:577–582. https://doi.org/10.1007/BF00219874
Shinmura K, Tamaki K, Bolli R (2005) Short-term caloric restriction improves ischemic tolerance independent of opening of ATP-sensitive K+ channels in both young and aged hearts. J Mol Cell Cardiol 39:285–296. https://doi.org/10.1016/j.yjmcc.2005.03.010
Shitara H, Kaneda H, Sato A, Iwasaki K, Hayashi J, Taya C, Yonekawa H (2001) Non-invasive visualization of sperm mitochondria behavior in transgenic mice with introduced green fluorescent protein (GFP). FEBS Lett 500:7–11. https://doi.org/10.1016/s0014-5793(01)02574-1
Shou J, Huo Y (2022) PINK1 phosphorylates Drp 1(S616) to improve mitochondrial fission and inhibit the progression of hypertension-induced HFpEF. Int J Mol Sci. https://doi.org/10.3390/ijms231911934
Shutt T, Geoffrion M, Milne R, McBride HM (2012) The intracellular redox state is a core determinant of mitochondrial fusion. EMBO Rep 13:909–915. https://doi.org/10.1038/embor.2012.128
Sivakumar A, Subbiah R, Balakrishnan R, Rajendhran J (2017) Cardiac mitochondrial dynamics: miR-mediated regulation during cardiac injury. J Mol Cell Cardiol 110:26–34. https://doi.org/10.1016/j.yjmcc.2017.07.003
Sloat SR, Whitley BN, Engelhart EA, Hoppins S (2019) Identification of a mitofusin specificity region that confers unique activities to Mfn1 and Mfn2. Mol Biol Cell 30:2309–2319. https://doi.org/10.1091/mbc.E19-05-0291
Song M, Chen Y, Gong G, Murphy E, Rabinovitch PS, Dorn GW 2nd (2014) Super-suppression of mitochondrial reactive oxygen species signaling impairs compensatory autophagy in primary mitophagic cardiomyopathy. Circ Res 115:348–353. https://doi.org/10.1161/CIRCRESAHA.115.304384
Song Z, Chen H, Fiket M, Alexander C, Chan DC (2007) OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J Cell Biol 178:749–755. https://doi.org/10.1083/jcb.200704110
Sotomayor-Flores C, Rivera-Mejias P, Vasquez-Trincado C, Lopez-Crisosto C, Morales PE, Pennanen C, Polakovicova I, Aliaga-Tobar V, Garcia L, Roa JC, Rothermel BA, Maracaja-Coutinho V, Ho-Xuan H, Meister G, Chiong M, Ocaranza MP, Corvalan AH, Parra V, Lavandero S (2020) Angiotensin-(1–9) prevents cardiomyocyte hypertrophy by controlling mitochondrial dynamics via miR-129-3p/PKIA pathway. Cell Death Differ 27:2586–2604. https://doi.org/10.1038/s41418-020-0522-3
Soundararajan R, Hernandez-Cuervo H, Stearns TM, Griswold AJ, Patil SS, Fukumoto J, Narala VR, Galam L, Lockey R, Kolliputi N (2022) A-Kinase Anchor Protein 1 deficiency causes mitochondrial dysfunction in mouse model of hyperoxia induced acute lung injury. Front Pharmacol 13:980723. https://doi.org/10.3389/fphar.2022.980723
Sridharan K, Sequeira RP (2018) Drugs for treating severe hypertension in pregnancy: a network meta-analysis and trial sequential analysis of randomized clinical trials. Br J Clin Pharmacol 84:1906–1916. https://doi.org/10.1111/bcp.13649
Stanley WC, Recchia FA, Lopaschuk GD (2005) Myocardial substrate metabolism in the normal and failing heart. Physiol Rev 85:1093–1129. https://doi.org/10.1152/physrev.00006.2004
Stotland A, Gottlieb RA (2016) α-MHC MitoTimer mouse: in vivo mitochondrial turnover model reveals remarkable mitochondrial heterogeneity in the heart. J Mol Cell Cardiol 90:53–58. https://doi.org/10.1016/j.yjmcc.2015.11.032
Subach FV, Patterson GH, Manley S, Gillette JM, Lippincott-Schwartz J, Verkhusha VV (2009) Photoactivatable mCherry for high-resolution two-color fluorescence microscopy. Nat Methods 6:153–159. https://doi.org/10.1038/nmeth.1298
Sun X, Yang Y, Xie Y, Shi X, Huang L, Tan W (2018) Protective role of STVNa in myocardial ischemia reperfusion injury by inhibiting mitochondrial fission. Oncotarget 9:1898–1905. https://doi.org/10.18632/oncotarget.22969
Sun YL, Li SH, Yang L, Wang Y (2018) miR-376b-3p attenuates mitochondrial fission and cardiac hypertrophy by targeting mitochondrial fission factor. Clin Exp Pharmacol Physiol 45:779–787. https://doi.org/10.1111/1440-1681.12938
Suwanjang W, Abramov AY, Charngkaew K, Govitrapong P, Chetsawang B (2016) Melatonin prevents cytosolic calcium overload, mitochondrial damage and cell death due to toxically high doses of dexamethasone-induced oxidative stress in human neuroblastoma SH-SY5Y cells. Neurochem Int 97:34–41. https://doi.org/10.1016/j.neuint.2016.05.003
Szulik MW, Valdez S, Walsh M, Davis K, Bia R, Horiuchi E, O’Very S, Laxman AK, Sandaklie-Nicolova L, Eberhardt DR, Durrant JR, Sheikh H, Hickenlooper S, Creed M, Brady C, Miller M, Wang L, Garcia-Llana J, Tracy C, Drakos SG, Funai K, Chaudhuri D, Boudina S, Franklin S (2023) SMYD1a protects the heart from ischemic injury by regulating OPA1-mediated cristae remodeling and supercomplex formation. Basic Res Cardiol 118:20. https://doi.org/10.1007/s00395-023-00991-6
Tanaka A, Cleland MM, Xu S, Narendra DP, Suen DF, Karbowski M, Youle RJ (2010) Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol 191:1367–1380. https://doi.org/10.1083/jcb.201007013
Tsushima K, Bugger H, Wende AR, Soto J, Jenson GA, Tor AR, McGlauflin R, Kenny HC, Zhang Y, Souvenir R, Hu XX, Sloan CL, Pereira RO, Lira VA, Spitzer KW, Sharp TL, Shoghi KI, Sparagna GC, Rog-Zielinska EA, Kohl P, Khalimonchuk O, Schaffer JE, Abel ED (2018) Mitochondrial reactive oxygen species in lipotoxic hearts induce post-translational modifications of AKAP121, DRP1, and OPA1 that promote mitochondrial fission. Circ Res 122:58–73. https://doi.org/10.1161/CIRCRESAHA.117.311307
Varanita T, Soriano ME, Romanello V, Zaglia T, Quintana-Cabrera R, Semenzato M, Menabo R, Costa V, Civiletto G, Pesce P, Viscomi C, Zeviani M, Di Lisa F, Mongillo M, Sandri M, Scorrano L (2015) The OPA1-dependent mitochondrial cristae remodeling pathway controls atrophic, apoptotic, and ischemic tissue damage. Cell Metab 21:834–844. https://doi.org/10.1016/j.cmet.2015.05.007
Wai T, Garcia-Prieto J, Baker MJ, Merkwirth C, Benit P, Rustin P, Ruperez FJ, Barbas C, Ibanez B, Langer T (2015) Imbalanced OPA1 processing and mitochondrial fragmentation cause heart failure in mice. Science 350:aad0116. https://doi.org/10.1126/science.aad0116
Wang D, Wang J, Bonamy GM, Meeusen S, Brusch RG, Turk C, Yang P, Schultz PG (2012) A small molecule promotes mitochondrial fusion in mammalian cells. Angew Chem Int Ed Engl 51:9302–9305. https://doi.org/10.1002/anie.201204589
Wang Y, Liu Q, Cai J, Wu P, Wang D, Shi Y, Huyan T, Su J, Li X, Wang Q, Wang H, Zhang F, Bae ON, Tie L (2022) Emodin prevents renal ischemia-reperfusion injury via suppression of CAMKII/DRP1-mediated mitochondrial fission. Eur J Pharmacol 916:174603. https://doi.org/10.1016/j.ejphar.2021.174603
Watanabe T, Saotome M, Nobuhara M, Sakamoto A, Urushida T, Katoh H, Satoh H, Funaki M, Hayashi H (2014) Roles of mitochondrial fragmentation and reactive oxygen species in mitochondrial dysfunction and myocardial insulin resistance. Exp Cell Res 323:314–325. https://doi.org/10.1016/j.yexcr.2014.02.027
Weindruch R (1996) The retardation of aging by caloric restriction: studies in rodents and primates. Toxicol Pathol 24:742–745. https://doi.org/10.1177/019262339602400618
Wiedenmann J, Ivanchenko S, Oswald F, Schmitt F, Rocker C, Salih A, Spindler KD, Nienhaus GU (2004) EosFP, a fluorescent marker protein with UV-inducible green-to-red fluorescence conversion. Proc Natl Acad Sci U S A 101:15905–15910. https://doi.org/10.1073/pnas.0403668101
Wild S, Roglic G, Green A, Sicree R, King H (2004) Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care 27:1047–1053. https://doi.org/10.2337/diacare.27.5.1047
Writing Committee M, Members AAJC (2022) 2022 AHA/ACC/HFSA guideline for the management of heart failure. J Card Fail 28:e1–e167. https://doi.org/10.1016/j.cardfail.2022.02.010
Wu S, Lu Q, Ding Y, Wu Y, Qiu Y, Wang P, Mao X, Huang K, Xie Z, Zou MH (2019) Hyperglycemia-driven inhibition of AMP-activated protein kinase α2 induces diabetic cardiomyopathy by promoting mitochondria-associated endoplasmic reticulum membranes in vivo. Circulation 139:1913–1936. https://doi.org/10.1161/CIRCULATIONAHA.118.033552
Wu S, Lu Q, Wang Q, Ding Y, Ma Z, Mao X, Huang K, Xie Z, Zou MH (2017) Binding of FUN14 domain containing 1 with inositol 1,4,5-trisphosphate receptor in mitochondria-associated endoplasmic reticulum membranes maintains mitochondrial dynamics and function in hearts in vivo. Circulation 136:2248–2266. https://doi.org/10.1161/CIRCULATIONAHA.117.030235
Xu LX, Wang RX, Jiang JF, Yi GC, Chang JJ, He RL, Jiao HX, Zheng B, Gui LX, Lin JJ, Huang ZH, Lin MJ, Wu ZJ (2023) TRPC6 promotes daunorubicin-induced mitochondrial fission and cell death in rat cardiomyocytes with the involvement of ERK1/2-DRP1 activation. Toxicol Appl Pharmacol 470:116547. https://doi.org/10.1016/j.taap.2023.116547
Xu S, Cherok E, Das S, Li S, Roelofs BA, Ge SX, Polster BM, Boyman L, Lederer WJ, Wang C, Karbowski M (2016) Mitochondrial E3 ubiquitin ligase MARCH5 controls mitochondrial fission and cell sensitivity to stress-induced apoptosis through regulation of MiD49 protein. Mol Biol Cell 27:349–359. https://doi.org/10.1091/mbc.E15-09-0678
Xu S, Wang P, Zhang H, Gong G, Gutierrez Cortes N, Zhu W, Yoon Y, Tian R, Wang W (2016) CaMKII induces permeability transition through Drp1 phosphorylation during chronic β-AR stimulation. Nat Commun 7:13189. https://doi.org/10.1038/ncomms13189
Yach D, Stuckler D, Brownell KD (2006) Epidemiologic and economic consequences of the global epidemics of obesity and diabetes. Nat Med 12:62–66. https://doi.org/10.1038/nm0106-62
Yamaguchi J, Nishiyama S, Shimanuki M, Ono T, Sato A, Nakada K, Hayashi J, Yonekawa H, Shitara H (2012) Comprehensive application of an mtDsRed2-Tg mouse strain for mitochondrial imaging. Transgenic Res 21:439–447. https://doi.org/10.1007/s11248-011-9539-1
Yellon DM, Beikoghli Kalkhoran S, Davidson SM (2023) The RISK pathway leading to mitochondria and cardioprotection: how everything started. Basic Res Cardiol 118:22. https://doi.org/10.1007/s00395-023-00992-5
Yu LM, Dong X, Xue XD, Xu S, Zhang X, Xu YL, Wang ZS, Wang Y, Gao H, Liang YX, Yang Y, Wang HS (2021) Melatonin attenuates diabetic cardiomyopathy and reduces myocardial vulnerability to ischemia-reperfusion injury by improving mitochondrial quality control: role of SIRT6. J Pineal Res 70:e12698. https://doi.org/10.1111/jpi.12698
Yu R, Liu T, Ning C, Tan F, Jin SB, Lendahl U, Zhao J, Nister M (2019) The phosphorylation status of Ser-637 in dynamin-related protein 1 (Drp1) does not determine Drp1 recruitment to mitochondria. J Biol Chem 294:17262–17277. https://doi.org/10.1074/jbc.RA119.008202
Yu X, Jia L, Yu W, Du H (2019) Dephosphorylation by calcineurin regulates translocation of dynamin-related protein 1 to mitochondria in hepatic ischemia reperfusion induced hippocampus injury in young mice. Brain Res 1711:68–76. https://doi.org/10.1016/j.brainres.2019.01.018
Yuan L, Yang J, Li Y, Yuan L, Liu F, Yuan Y, Tang X (2022) Matrine alleviates cisplatin-induced acute kidney injury by inhibiting mitochondrial dysfunction and inflammation via SIRT3/OPA1 pathway. J Cell Mol Med 26:3702–3715. https://doi.org/10.1111/jcmm.17398
Zacharioudakis E, Agianian B, Kumar Mv V, Biris N, Garner TP, Rabinovich-Nikitin I, Ouchida AT, Margulets V, Nordstrom LU, Riley JS, Dolgalev I, Chen Y, Wittig AJH, Pekson R, Mathew C, Wei P, Tsirigos A, Tait SWG, Kirshenbaum LA, Kitsis RN, Gavathiotis E (2022) Modulating mitofusins to control mitochondrial function and signaling. Nat Commun 13:3775. https://doi.org/10.1038/s41467-022-31324-1
Zerihun M, Sukumaran S, Qvit N (2023) The Drp1-mediated mitochondrial fission protein interactome as an emerging core player in mitochondrial dynamics and cardiovascular disease therapy. Int J Mol Sci. https://doi.org/10.3390/ijms24065785
Zhai M, Li B, Duan W, Jing L, Zhang B, Zhang M, Yu L, Liu Z, Yu B, Ren K, Gao E, Yang Y, Liang H, Jin Z, Yu S (2017) Melatonin ameliorates myocardial ischemia reperfusion injury through SIRT3-dependent regulation of oxidative stress and apoptosis. J Pineal Res. https://doi.org/10.1111/jpi.12419
Zhang H, Sun X, Xie Y, Tian F, Hu H, Tan W (2018) Isosteviol sodium inhibits astrogliosis after cerebral ischemia/reperfusion injury in rats. Biol Pharm Bull 41:575–584. https://doi.org/10.1248/bpb.b17-00921
Zhang H, Wang P, Bisetto S, Yoon Y, Chen Q, Sheu SS, Wang W (2017) A novel fission-independent role of dynamin-related protein 1 in cardiac mitochondrial respiration. Cardiovasc Res 113:160–170. https://doi.org/10.1093/cvr/cvw212
Zhang LX, Li CX, Kakar MU, Khan MS, Wu PF, Amir RM, Dai DF, Naveed M, Li QY, Saeed M, Shen JQ, Rajput SA, Li JH (2021) Resveratrol (RV): a pharmacological review and call for further research. Biomed Pharmacother 143:112164. https://doi.org/10.1016/j.biopha.2021.112164
Zhang X, Lu Z, Abdul KSM, Changping MA, Tan KS, Jovanovi A, Tan W (2020) Isosteviol sodium protects heart embryonic H9c2 cells against oxidative stress by activating Akt/GSK-3β signaling pathway. Pharmazie 75:36–40. https://doi.org/10.1691/ph.2020.9851
Zhang Y, Wang Y, Xu J, Tian F, Hu S, Chen Y, Fu Z (2019) Melatonin attenuates myocardial ischemia-reperfusion injury via improving mitochondrial fusion/mitophagy and activating the AMPK-OPA1 signaling pathways. J Pineal Res 66:e12542. https://doi.org/10.1111/jpi.12542
Zhao L, Zou X, Feng Z, Luo C, Liu J, Li H, Chang L, Wang H, Li Y, Long J, Gao F, Liu J (2014) Evidence for association of mitochondrial metabolism alteration with lipid accumulation in aging rats. Exp Gerontol 56:3–12. https://doi.org/10.1016/j.exger.2014.02.001
Zheng M, Bai Y, Sun X, Fu R, Liu L, Liu M, Li Z, Huang X (2022) Resveratrol reestablishes mitochondrial quality control in myocardial ischemia/reperfusion injury through Sirt1/Sirt3-Mfn2-Parkin-PGC-1α pathway. Molecules. https://doi.org/10.3390/molecules27175545
Zhou H, Hu S, Jin Q, Shi C, Zhang Y, Zhu P, Ma Q, Tian F, Chen Y (2017) Mff-dependent mitochondrial fission contributes to the pathogenesis of cardiac microvasculature ischemia/reperfusion injury via induction of mROS-mediated cardiolipin oxidation and HK2/VDAC1 disassociation-involved mPTP opening. J Am Heart Assoc. https://doi.org/10.1161/JAHA.116.005328
Acknowledgements
DJH is supported by the Duke-NUS Signature Research Programme funded by the Ministry of Health, Singapore Ministry of Health’s National Medical Research Council under its Singapore Translational Research Investigator Award (MOH-STaR21jun-0003), Centre Grant scheme (NMRC CG21APR1006), and Collaborative Centre Grant scheme (NMRC/CG21APRC006), and A*STAR under its PREVENT-HF Industry Alignment Fund—Pre-Positioning Programme (IAF-PP, H23J2a0033). This article is based upon work supported by the COST Action EU-CARDIOPROTECTION IG16225 supported by COST (European Cooperation in Science and Technology). LS is supported by Fondation Leducq TNE15004, by Ministry of Education, University and Research (MIUR) FIRB RBAP11Z3YA_005, by Ministry of University and Research (MUR) 2020PKLEPN and by National Recovery and Resilience Plan (NRRP), Mission 4, Component 2, Investment 1.4, funded by the European Union – NextGenerationEU. This work was supported by Naresuan University (NU), and National Science, Research and Innovation Fund (NSRF) (Grant No. R2566B045), partially supported by Reinventing University Program 2023, The Office of the Permanent Secretary of the Ministry of Higher Education, Science, Research and Innovation and Naresuan University, Thailand (Grant No. R2566A046). SL is supported by the Singapore Ministry of Health’s National Medical Research Council under Open Fund–Young Individual Research Grant (OF-YIRG, MOH-000230).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All authors have no relevant conflicts or disclosures.
Additional information
This article is part of the topical collection “Mitochondria at the heart of cardioprotection”.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hernandez-Resendiz, S., Prakash, A., Loo, S.J. et al. Targeting mitochondrial shape: at the heart of cardioprotection. Basic Res Cardiol 118, 49 (2023). https://doi.org/10.1007/s00395-023-01019-9
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00395-023-01019-9