
Abstract
Heart failure with preserved ejection fraction (HFpEF) is a complex heterogeneous disease for which our pathophysiological understanding is still limited and specific prevention and treatment strategies are lacking. HFpEF is characterised by diastolic dysfunction and cardiac remodelling (fibrosis, inflammation, and hypertrophy). Recently, microvascular dysfunction and chronic low-grade inflammation have been proposed to participate in HFpEF development. Furthermore, several recent studies demonstrated the occurrence of generalized lymphatic dysfunction in experimental models of risk factors for HFpEF, including obesity, hypercholesterolaemia, type 2 diabetes mellitus (T2DM), hypertension, and aging. Here, we review the evidence for a combined role of coronary (micro)vascular dysfunction and lymphatic vessel alterations in mediating key pathological steps in HFpEF, including reduced cardiac perfusion, chronic low-grade inflammation, and myocardial oedema, and their impact on cardiac metabolic alterations (oxygen and nutrient supply/demand imbalance), fibrosis, and cardiomyocyte stiffness. We focus primarily on HFpEF caused by metabolic risk factors, such as obesity, T2DM, hypertension, and aging.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
More than half of the patients with heart failure (HF), notably women, suffer from HF with preserved ejection fraction (HFpEF; EF > 50%), a complex cardiovascular syndrome characterised by diastolic dysfunction and cardiac stiffening, fibrosis, inflammation, and hypertrophy. As a consequence of population aging, as well as the increases in common comorbidities, such as obesity, type 2 diabetes mellitus (T2DM), and hypertension, the prevalence of HFpEF is rising [115]. Alarmingly, there are no specific treatments for HFpEF, likely due to incomplete pathophysiological understanding of the underlying mechanisms, patient population heterogeneity, and inadequate diagnosis [110]. In this review, we summarize current evidence of coronary microvascular dysfunction linked to chronic low-grade inflammation, oxidative stress, and microvascular wall barrier dysfunction in HFpEF. Furthermore, we discuss the potential role of lymphatic dysfunction in HFpEF development. We review the role of microvascular and lymphatic dysfunction in mediating reduced left ventricular (LV) compliance (both cardiomyocyte stiffness and cardiac fibrosis) and cardiac metabolic changes occurring during HFpEF development. In each case, we will focus on what is known about these topics in both HFpEF and the commonly associated comorbidities. Finally, we highlight possible therapeutic approaches for HFpEF.
Coronary microvascular dysfunction in HFpEF
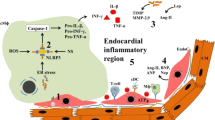
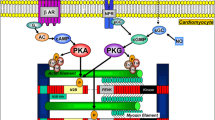
Vascular endothelial cells constitute the majority of the non-cardiomyocyte population in the healthy heart, therefore cardiac endothelial structural and/or functional abnormalities have major impacts on cardiac health. Coronary macrovascular dysfunction has been scarcely investigated in HFpEF. However, the current microvascular paradigm proposes endothelial dysfunction as the central mediator connecting chronic systemic low-grade inflammation with myocardial dysfunction and remodelling in HFpEF (Fig. 1) [94]. In this model, metabolic syndrome (MetS)-related comorbidities, such as obesity, T2DM, and hypertension, trigger chronic systemic low-grade inflammation, characterised by elevated levels of circulating immune cells and pro-inflammatory cytokines and upregulation of endothelial adhesion molecules, such as intercellular and vascular cellular adhesion molecule-1 (ICAM-1 and VCAM-1), and corresponding ligands on circulating leucocytes. The resultant increased myocardial infiltration of leucocytes, especially monocytes, elevates cardiac transforming growth factor beta (TGFβ) levels, thereby inducing cardiac fibrosis. Furthermore, the systemic pro-inflammatory state causes coronary microvascular endothelial cells to produce excessive reactive oxygen species (ROS), contributing to cardiac oxidative stress resulting in oxidation of nitric oxide (NO). Consequently, the reduced NO bioavailability leads to impaired nitric oxide/cyclic guanosine monophosphate/protein kinase G (NO/cGMP/PKG) signalling, causing vascular endothelial dysfunction and cardiomyocyte hypertrophy and stiffening. Decreased NO bioavailability, increased leucocyte infiltration, oxidative stress, and/or neurohormonal activation trigger coronary microvascular endothelial dysfunction and reduced flow-mediated dilatation, which adversely impact cardiac perfusion, as observed in most HFpEF comorbidities (Table 1) [34, 94].

Cardiac and vascular oxidative stress and chronic low-grade inflammation in HFpEF. The metabolic syndrome (obesity, type 2 diabetes mellitus, hypercholesterolaemia, and hypertension) induces chronic systemic low-grade inflammation, as well as direct deleterious effects in the heart (left) and in its coronary endothelium (right). Chronic cardiac low-grade inflammation develops due to increased transmigration of immune cells across activated endothelial cells (EC). Furthermore, endothelial and cardiomyocyte (CM) oxidative stress result from an imbalance between antioxidant defences and reactive oxygen species (ROS) production. Immune mediators, e.g. tumor necrosis factor (TNF)-α, interferon (IFN)-γ, and interleukin 1 (IL)-1β, further increase ROS production. Prolonged ROS-mediated inflammasome activation and the resultant increased transforming growth factor (TGF)-β levels alter the expression of pro-fibrotic genes, contributing to cardiac fibrosis. Furthermore, severe oxidative stress causes lipid, protein, and DNA alterations, leading to mitochondrial dysfunction ultimately resulting in poor cardiomyocyte ATP production, calcium handling, and contractility. In addition, ROS-induced protein modifications (e.g. S-nitrosylation) lead to sarcomeric myofilament dysfunction and reduced endothelial nitric oxide synthase (eNOS)-mediated nitric oxide (NO) production. In parallel, oxidative stress leads to eNOS uncoupling, contributing to poor flow-mediated vasodilation and cardiac perfusion. This further aggravates the cardiomyocyte energy supply-demand imbalance. Furthermore, increased myocardial activation of inducible nitric oxide synthase (iNOS) leads to increased nitrosative stress. Finally, persistent vascular pro-inflammatory activation and oxidative stress may induce endothelial cell death, contributing to vascular rarefaction and reduced cardiac perfusion
Coronary microvascular dysfunction may be determined by endothelium-dependent and/or -independent mechanisms. Endothelium-dependent dysfunction develops due to an imbalance between endothelium-derived relaxing factors (e.g. NO) and constrictors (e.g. endothelin 1) [127]. In HFpEF patients, plasma levels of NO metabolites were lower compared to HFrEF subjects, indicating a reduced NO bioavailability [19]. In contrast, in moderate regional ischemia, the NO production is increased due to elevated inducible nitric oxide synthase (iNOS) expression, thereby reducing endothelium-dependent vasodilation [46]. Furthermore, HFpEF patients showed increased levels of endothelin-1, a predictor of 1-year HF hospitalisation associated with long-term mortality [21]. Endothelium-dependent coronary microvascular dysfunction was present in 29% of the HFpEF patients, notably those presenting with a greater burden of T2DM and lower high-density lipoprotein (HDL) levels [133]. On the other hand, endothelium-independent dysfunction is the result of changes in vascular tone mediated by an imbalance between vasoconstrictors (e.g. angiotensin II) and vasodilators (e.g. adenosine) acting on vascular smooth muscle cells. A recent study showed that 33% of the HFpEF patients, mostly older, hypertensive subjects, displayed endothelium-independent dysfunction, as reflected by reduced coronary flow reserve (CFR) [133]. Interestingly, this endothelium-independent microvascular dysfunction was associated with a worsened diastolic function and increased mortality [133]. In experimental swine models and diabetic patients, metabolic risk factors (e.g., hypercholesterolaemia, T2DM, and chronic kidney disease) reduced CFR by increasing the basal blood flow following perturbations in myocardial efficiency [9, 41, 96, 121]. Of note, increased basal myocardial blood flow correlated with diastolic dysfunction in female T2DM patients, while CFR did not [41]. As such, basal myocardial blood flow could represent a superior marker of coronary microvascular dysfunction in certain pathological settings [8]. Nevertheless, while administration of the smooth muscle cell relaxants, such as sodium nitroprusside, improves endothelium-independent vasodilation in HFrEF [62], its use in HFpEF is debated [109].
In parallel to these functional vascular alterations, a reduction in myocardial microvascular density, called microvascular rarefaction, is observed in HFpEF patients (Table 1) [84]. Capillary rarefaction contributes to insufficient cardiac perfusion by impairing myocardial oxygen delivery in HFpEF patients [123]. Rarefaction of resistance vessels, including small arteries and arterioles, increases microvascular coronary resistance, resulting in reduced cardiac perfusion. Interestingly, in a multiple comorbidity swine model, experimental reductions in myocardial blood flow led to increased myocardial oxygen extraction [121]. This increase occurred despite a reduction in coronary capillary density [121]. As such, rarefaction or dysfunction of coronary resistance vessels was proposed to be responsible for the observed impairment of myocardial blood flow and oxygen delivery [121]. Notably, reduced cardiac perfusion leads to local blood supply-demand imbalance and energy metabolite deficiency, causing cardiac metabolic reprogramming and dysfunction. More than 50% of the patients with coronary microvascular dysfunction had an impaired CFR, which was independently associated with a worsened diastolic function and increased hospitalisation for HFpEF [119]. Microvascular rarefaction may precede disease development, as HFpEF-associated comorbidities show microvascular rarefaction (Table 1). For example, microvascular rarefaction is suggested to impede insulin delivery to muscles and adipose tissue, contributing to poor insulin uptake [7]. In addition, in young adults with familial predisposition to hypertension and in patients with borderline or established hypertension, a reduced dermal capillary density has been shown [3, 88]. In obese patients, increased LV filling pressure correlated with lower coronary microvascular density, potentially contributing to impaired cardiac metabolism underlying diastolic dysfunction [16]. Furthermore, increased subepicardial and pericoronary adipose tissue, as observed in obese, T2DM, and elderly patients, correlated with an impaired CFR, microvasculature, and coronary function, leading to deteriorated diastolic function [85]. However, despite accumulating evidence for coronary microvascular dysfunction and rarefaction during HFpEF development, its exact role in disease progression is still unknown.
Inflammation as a trigger of coronary vascular dysfunction
The most frequent HFpEF-associated comorbidities are all associated with chronic systemic low-grade inflammation (Table 1 and Fig. 1) [15, 51, 98, 116]. HFpEF patients showed elevated systemic inflammatory markers, such as acute inflammatory C-reactive protein (CRP), which increased with the number of comorbidities, and raised circulating levels of neutrophils and monocytes [24, 30, 38, 49]. Additionally, in vitro culture of healthy donor monocytes with serum from HFpEF patients promoted alternative anti-inflammatory/pro-fibrotic macrophage differentiation [38].
Both chronic systemic low-grade inflammation and activation of the renin–angiotensin–aldosterone axis (RAAS) lead to endothelial cell activation by upregulating adhesion molecules. Elevated advanced glycation end products (AGEs)/AGE receptor (RAGE) signalling in T2DM stimulates the nuclear factor kappa-B (NFкB) signalling pathway, inducing pro-inflammatory genes and RAGE, forming a vicious cycle of self-renewing pro-inflammatory signals [10]. HFpEF patients showed increased expression of adhesion molecules on the coronary endothelium, together with elevated myocardial infiltration of CD45+ leucocytes and CD3+ T-lymphocytes [129]. Furthermore, there was a positive correlation between echocardiographic indices of diastolic dysfunction (E/e′) and splenic activation, suggesting a role of increased splenic myeloid cell oversupply in HFpEF patients [49]. While both systemic and cardiac inflammation have been observed in HFpEF patients, the causal involvement of cardiac inflammation in coronary microvascular dysfunction in HFpEF has never been investigated.
Given the plethora of cardiac detrimental effect triggered by chronic low-grade inflammation, the use of cytokine inhibitors has been extensively investigated in HF patients [77]. The ability to translate this to a drug has not met success and in some cases has even led to worsening of HF and/or death [77]. Nevertheless, IL-1β blockage (Anakinra), for example, improved aerobic exercise capacity in HFpEF patients and is currently investigated in a phase 2 clinical trial (NCT02173548) [126]. Furthermore, the multi-cytokine blocker Pentoxifylline reduced vascular events, systemic inflammation, all-cause mortality, and improved the prognosis in HFrEF patients [18]. In addition to anti-cytokine therapies, lipid-lowering statins have anti-inflammatory effects and are associated with a reduced re-hospitalisation and mortality in HFpEF patients [78]. Despite this, currently no approved effective anti-inflammatory drug has been approved for the treatment or prevention of HFpEF.
Oxidative stress as a trigger of coronary vascular dysfunction
Oxidative stress is induced by increased ROS production and/or reduced antioxidant enzyme levels, leading to both endothelial and cardiac dysfunction (Fig. 1). As cardiomyocytes are rich in mitochondria, they have an elevated baseline ROS production compared to other cell types. Thus, altered mitochondrial function and/or reduced antioxidant enzyme levels lead to cardiac oxidative stress. Of note, risk factors for HFpEF further stimulate ROS production (Table 1) [17, 34, 37, 53, 91]. For example, AGE-RAGE signalling in T2DM induces oxidative stress by directly activating nicotinamide adenine dinucleotide phosphate oxidases (NOX), decreasing the activity of enzymatic antioxidant defences, and indirectly by reducing cellular antioxidant systems [100]. Consequently, chronic systemic low-grade inflammation is proposed as a major trigger, together with oxidative stress and NO dysregulation, for the development of coronary microvascular dysfunction in HFpEF [94].
Within vascular endothelial cells, elevated ROS production triggers canonical NFкB signalling, leading to cytokine production and proteasome and inflammasome activation, which may cause endothelial cell apoptosis and pyroptosis (Fig. 1) [34]. Endothelial oxidative stress accelerates NO degradation by superoxide anion (O2−)-mediated peroxynitrite (ONOO−) formation, thereby promoting protein nitrosylation, resulting in endothelial dysfunction and cell death (Fig. 1) [52]. Increased cardiac levels of hydrogen peroxide (H2O2) and reactive oxidative metabolites, endothelial nitric oxide synthase (eNOS) uncoupling, and macrophage and endothelial NOX2 expression and reduced NO levels all indicate the presence of myocardial oxidative stress in HFpEF patients (Table 1) [35]. Beyond oxidation, inhibition of NO production could reduce NO bioavailability, by for example AGE-induced elevation of asymmetric dimethyl-l-arginine (ADMA) levels, an eNOS inhibitor, thereby contributing to endothelium-dependent dysfunction associated with a worsened prognosis of HFpEF [5]. Despite the signs of cardiac oxidative stress in HFpEF patients, the causal involvement of cardiac oxidative stress in the development or aggravation of coronary microvascular dysfunction is not well understood.
In HF patients and experimental mouse models, several anti-oxidative stress therapies have been investigated that either (1) inhibit oxidative stress producers, (2) improve endogenous antioxidant capacity, or (3) supplement exogeneous antioxidants. Mitochondria Szeto-Schiller-31 (SS-31; Elamipretide) attenuated cardiac remodelling in hypertensive cardiomyopathy and is currently investigated as a novel therapeutic in phase II trials for HFpEF (NCT02814097). Furthermore, treatment with the mitochondria-targeted antioxidant MitoTEMPO or N-acetylcysteine (NAC) prevented diastolic dysfunction in rodent models of diabetes and hypertensive cardiomyopathy [74, 130]. Supplementation with an antioxidant cocktail, containing alpha lipoic acid, vitamin C, and E, also reduced systemic inflammation and improved conduit artery endothelium-dependent vasodilation in HFpEF patients [101]. In summary, therapies targeting oxidative stress could be a potential therapy for HFpEF. However, their underlying mechanism needs to be further elucidated.
Microvessel wall barrier dysfunction in HFpEF
The microvascular endothelium is a barrier opposing free exchange between blood and tissues, which tightly regulates transport of plasma constituents and immune cells in most organs. Events such as ischaemia cause increased vascular endothelial growth factor (VEGF)-A levels, leading to vascular barrier breakdown, increased extravasation of immune cells, and oedema. In addition to its well-known occurrence in HFrEF, there is accumulating evidence of microvascular wall barrier dysfunction in HFpEF-associated comorbidities [11, 55, 79, 128], while its role in the development of coronary microvascular dysfunction has been scarcely addressed in HFpEF (Table 1).
Vascular barrier function is controlled on several levels by different cell types. For example, the endothelial glycocalyx covers the luminal surface of vascular endothelial cells and together with cell–cell junctions serves as a barrier for solute and macromolecule exchanges. It is also a mechanotransducer, which senses endothelial shear stress, attenuates coagulation and leucocyte adhesion to the endothelium, and affects vasoregulatory responses to flow. A damaged glycocalyx induces the production of pro-adhesion mediators, thereby triggering the adherence of neutrophils to the endothelium (Fig. 2) [54]. Thinning of the glycocalyx occurs in several HFpEF-associated comorbidities (Table 1 and Fig. 2) [31, 50, 75, 86]. In HFpEF patients, increased circulating levels of syndecan-1, a glycocalyx shedding biomarker, were associated with endothelial dysfunction and a doubling of plasma syndecan-1 levels increased risk of all-cause mortality and rehospitalisation [120]. Interestingly, exogenous NO administration during reperfusion preserved vascular integrity and attenuated cardiac oedema formation though protection of the glycocalyx in guinea pigs subjected to ischemia/reperfusion injury [14].

Microvessel wall barrier dysfunction in HFpEF. The metabolic syndrome induces via chronic systemic low-grade inflammation deleterious effects in coronary endothelial cells (EC). It leads to the degradation of the endothelial glycocalyx layer, thereby promoting endothelial immune cell adhesion and transmigration. Furthermore, metabolic syndrome-induced cellular oxidative stress may lead to glycocalyx damage and cell death of both endothelial and mural cells. In addition, pro-inflammatory mediators, such as tumor necrosis factor (TNF)-α and interleukin (IL)-1β, together with oxidative stress, increase vascular growth factor (VEGF)-A levels. Increased VEGF-A signalling weakens vascular barriers (e.g. loss of cell–cell junctions), which facilitates paracellular passage of immune cells and trans-vascular transport by transcellular vesiculo-vascular organ (VVO) formation. VEGF-A also stimulates vascular basement membrane remodelling through extracellular matrix (ECM) proteases activation, leading to reduced vascular stability and vascular regression
Vascular endothelial hyperpermeability has been shown to occur in the microvasculature of peripheral tissues of hypertensive or diabetic patients, as well as obese or elderly rodent models [11, 55, 79, 128]. Disruption and remodelling of cell-cell junctions is a major cause of vascular barrier integrity loss and occurs in murine models of diastolic dysfunction and MetS and in aged endothelial cells (Table 1 and Fig. 2) [55, 64, 89, 106]. A number of pro-permeabilizing stimuli, including VEGF-A and inflammatory agents (e.g. histamine and bradykinin), stimulate hyperphosphorylation of vascular endothelial (VE)-cadherin, resulting in the breakdown of junctional contacts [23]. Moreover, the VEGFA-, histamine-, and serotonin-stimulated formation of trans-endothelial channels from coalesced vesicles or vacuoles, called vesiculo-vacuolar organelles (VVOs) is another proposed route of transcytosis-mediated extravasation during vascular leakage (Fig. 2). Vascular hyperpermeability by trans- or paracellular pathways leads to increased influx of solutes, macromolecules, and immune cells to the interstitium.
Beyond endothelial cells, pericytes, the main mural cell type of microvessels, are crucial for regulating vascular blood flow and microvascular stability. Cardiac pericytes are involved in many processes regulating cardiac homeostasis, such as vascular maturation, supply of trophic substances, fibrosis, and blood flow. HFpEF-diseased ZSF1 rats showed disorganized accumulation of vascular pericytes in subendocardial hyperproliferative (inflamed) foci, while vascular pericyte coverage was reduced compared to controls [122]. Further investigations of microvascular barrier dysfunction in HFpEF are warranted with the aim to develop novel tools for diagnosis or target-specific therapy to limit vascular dysfunction.
Lymphatic dysfunction in HF
The heart has an extensive lymphatic network that relies on cardiac contractions to propel lymph fluid towards draining cardiac lymph nodes [33]. Cardiac lymphatic drainage is essential for cardiac fluid balance [82]. Insufficient cardiac lymphangiogenesis contributes to myocardial oedema, inflammation, and fibrosis following experimental myocardial infarction in rodents, and it has been shown that cardiac oedema subseqeuntly adversely affects heart function [13, 47]. However, it is unknown whether the same is true for HFpEF. HFpEF patients show increased interstitial water in the lungs, suggesting impaired lymphatic drainage [102]. In addition, decreased left ventricular contractility and relaxation was observed in a canine model of acute lymphatic obstruction, suggesting a role for lymphatic dysfunction in diastolic dysfunction [73]. Furthermore, the generalized oedema that accompanies advanced HFpEF further indicates that lymphatic dysfunction may be involved in HFpEF development and/or progression.
Lymphatic dysfunction and remodelling has been shown to occur in experimental models of multiple HFpEF-associated comorbidities (Table 1 and Fig. 3) [25, 134, 135]. For example, murine genetic- or diet-induced obesity models showed reduced dermal lymphatic collecting vessel pumping rates, as well as reduced lymphatic capillary density with accumulation of pro-inflammatory cells around lymphatic vessels [69, 87]. These lymphatic-associated immune cells may further reduce collector vessel pumping rates by excessive production of NO via iNOS [69, 87]. Another example is hypercholesterolaemia, which was associated with lymphatic capillary regression, dermal backflow of lymph, dilation of initial lymphatics, and reduced muscular layer coverage in collecting ducts in mice, which all were reversible upon cholesterol-lowering treatment [70]. Rodent models of T2DM showed reduced lymphangiogenesis and lymphatic vascular integrity and function, with insufficient NO bioavailability and poor lymphangiogenesis, contributing to lymphatic permeability and delayed wound healing, respectively [20, 108]. In contrast, T2DM patients had increased dermal lymphatic density and lymphatic endothelial proliferation [42]. Furthermore, canine models of chronic hypertension showed increased myocardial blood capillary permeability accompanied by elevated cardiac lymphatic transport [65]. Interestingly, spontaneously hypertensive rats showed increased VEGF-C-mediated cardiac lymphangiogenesis accompanying LV remodelling [132]. Collectively, these studies indicate an increased demand on the lymphatic system in these HFpEF-associated comorbidities, resulting in maladaptive lymphangiogenesis, as well as hyperpermeability and integrity loss in lymphatics, together leading to lymphatic transport dysfunction. Several studies have shown that immune cells actively participate in lymphangiogenesis and alter lymphatic function [1, 61, 117]. Pro-inflammatory immune cells have been shown to accumulate around lymphatic vessels and reduce lymphatic transport during acute inflammation in models of obesity [69, 87]. Furthermore, while B-cells and neutrophils stimulate lymphangiogenesis, Th2-skewed CD4+ helper T-cells have been found to inhibit lymphangiogenesis in tissues such as lymph nodes, skin, and lung [2, 4, 59, 112, 118]. The complexity of immune-lymphatic interactions is further highlighted by the role of the lymphatic system in limiting immune cell residence in tissues [6, 40, 56]. Accordingly, many studies have shown that inhibition of lymphangiogenesis prolongs the inflammatory response and increases oedema at the inflammatory site [6, 40, 56], suggesting that increasing lymphangiogenesis might be a potential novel therapeutic option to reduce chronic inflammation in HFpEF.

Lymphatic vasculature in the metabolic syndrome. The lymphatic system is composed of highly permeable blunt-ended lymphatic capillaries, which drain into larger collecting lymphatic vessels endowed with valves to prevent backflow and a muscular layer that propulses the lymph towards draining lymph nodes. Experimental models of metabolic syndrome components showed lymphatic dysfunction, including rarefaction, and dilation of initial lymphatic capillaries, but also enlargement, hyperpermeability, and poor contraction of collecting ducts, together resulting in reduced lymphatic transport capacity
Therapeutic lymphangiogenesis induced by lymphangiogenic growth factors VEGF-C and -D has shown some potential in terms of treating HFpEF-associated comorbidities. For example, overexpression of VEGF-D increased renal lymphangiogenesis and prevented salt- and NOS inhibition-induced hypertension in mice [72]. Similarly, systemic delivery of VEGF-C lowered blood pressure and preserved cardiac function in salt-sensitive hypertensive rats, while neutralization of both VEGF-C and -D aggravated hypertension and cardiac dysfunction [131]. Finally, therapeutic lymphangiogenesis has been shown to accelerate the resolution of both acute and chronic inflammation in various settings [61]. This shows the potential of therapeutic lymphangiogenesis in treating certain HFpEF-related comorbidities but also highlights lymphatic function and lymphangiogenesis as one of the most underappreciated and unstudied therapeutic opportunities.
Reduced LV compliance in HFpEF: cardiac fibrosis and cardiomyocyte passive stiffness
Diastolic dysfunction is caused by impaired active relaxation and/or reduced compliance of the ventricle. Both increases in extracellular matrix deposition (fibrosis) and cardiomyocyte passive stiffness (titin modifications) reduce LV compliance, as observed in HFpEF patients [36].
Increased cardiomyocyte stiffness: titin post-translational modifications and isotype switching
The giant sarcomeric protein titin, a force-transducing bidirectional spring, is the main determinant of cardiomyocyte stiffness. Alternative splicing of titin mRNA creates isoforms with differential stiffnesses: the short, stiffer N2B isoform, and the longer, more compliant N2BA isoform. Extension of the elastic I-band region in both titin isoforms supports myocardial passive relaxation during diastole. Post-translational modifications of titin rapidly alter cardiomyocyte stiffness. In HFpEF patients, hypophosphorylation of N2B and ex vivo administration of PKG are associated with increased LV stiffness [12, 125]. Interestingly, PKG activity is reduced in HFpEF as a result of reduced NO bioavailability due to oxidative stress, leading to impaired NO/cGMP/PKG signalling [94]. This suggests that oxidative stress, NO bioavailability, and PKG play a crucial role in regulating cardiomyocyte stiffness. Furthermore, other protein kinases, such as PKA, PKC, extracellular signal-regulated kinase-2 (ERK2), and Ca2+/calmodulin-dependent kinase-II (CAMKII) also modify passive stiffness [36]. HFpEF patients showed increased site-specific titin phosphorylation on PEVK (a region rich in specific amino acids) S11878 and reduced phosphorylation on N2B unique sequence (N2Bus) S4185, which was associated with increased LV stiffness [136]. In addition to these post-translational modifications of titin, alteration of the N2BA/N2B isoform ratio occurs in pathology [76]. However, HFpEF patients do not consistently show isoform changes, with only some exhibiting increased levels of the stiff N2B isoform [124]. Altogether, increased cardiomyocyte stiffness in HFpEF may be mediated by short term post-translational modifications, which are interconnected with the oxidative state, and by isotype switching in the long term.
Collagen quantity, type, and cross-linking are altered during HFpEF
Myocardial fibrosis and extracellular matrix accumulation are hallmarks of adverse cardiac remodelling associated with HFpEF and its comorbidities (Table 1) [58, 84, 90, 99, 103, 113, 136]. The quantity, type, and degree of crosslinking of collagen influences tissue stiffness. Increased collagen deposition, switching from more flexible collagen III to stiffer collagen I, and collagen cross-linking is associated with worse diastolic function in HFpEF patients [58, 84, 136]. Furthermore, collagen cross-linking also correlates with increased LV filling pressures and elevated risk of hospitalisation in hypertensive HF patients [39].
Lysyl oxidases (LOXs) are the main enzymes involved in collagen and elastin cross-linking during cardiac remodelling [105]. Upregulation of LOX expression and collagen cross-linking is associated with impaired diastolic tissue Doppler parameters (e.g. E/E′) in HFpEF patients [58]. Furthermore, AGE-mediated cross-linking of collagen was also observed in aged and T2DM patients, resulting in reduced vascular elasticity and myocardial flexibility, contributing to vascular and myocardial stiffness, and ultimately diastolic dysfunction [29]. Cardiac fibrosis is also associated with cardiac microvascular rarefaction in HFpEF patients, indicating that insufficient cardiac perfusion may cause excessive collagen deposition [84]. Pro-inflammatory mediators (e.g. IL-6, TNFα, CCL2, and TGFβ) participate in fibrosis regulation, and pharmacological prevention of cardiac infiltration of pro-inflammatory monocytes attenuates fibrosis development in murine hypertension [32]. The role of specific cytokines in cardiac fibrosis is, however, debated. Interestingly, cardiac oedema increased collagen production, whereas reduction of cardiac oedema attenuated interstitial cardiac fibrosis in rodent myocardial infarction, suggesting a potential role of oedema and poor lymphatic transport in regulating cardiac interstitial fibrosis [47].
Several anti-fibrotic therapeutics, focused on inhibiting aldosterone signalling with mineralocorticoid receptor antagonists (MRA), have been trialled in patients with in HFpEF [63]. Promisingly, MRA improved cardiac fibrosis markers and diastolic function [92]. Another anti-fibrotic therapy is the AGE-crosslink breaker alagebrium chloride (ALT-711), which improved diastolic function by reducing cardiac stiffness in elderly HFpEF patients (NCT01014572) [71]. While antifibrotic therapies show clinical efficacy in HFrEF patients [93], their effects are more limited in HFpEF patients. One reason for this discrepancy could be the variation in fibrosis pathophysiology found between HFrEF and HFpEF. Indeed, while HFrEF patients present with scar development or “reparative fibrosis” [83], HFpEF patients are much more likely to show interstitial “reactive fibrosis” and perivascular fibrosis [27]. Differences in the mechanisms underlying fibrosis deposition could thus explain why interventions targeting the type of fibrosis that occurs in HFrEF have had limited success in HFpEF patients [57]. Innovative approaches to reduce cardiac stiffening, however, are expected to provide significant benefit in HFpEF.
Impact of microvascular and lymphatic dysfunction on cardiac metabolism
The healthy heart requires a large, constant supply of energy of which 30% is provided by carbohydrate oxidation (mainly glucose and lactate) and 70% by beta-oxidation of free fatty acids (FFAs) at rest. Cardiac metabolic alterations in HFpEF patients have been poorly investigated and current hypotheses are largely based on metabolic alterations observed in the separate comorbidities (Table 1). For example, in aging and hypertensive LV hypertrophy, there is a decreased reliance on cardiac FFA utilization and beta-oxidation observed [28, 60]. In contrast, insulin resistance and increased circulating triglyceride and FFA levels make cardiac ATP synthesis more dependent on FFA oxidation [43, 66]. Indeed, animal models of obesity and T2DM with diastolic dysfunction show reduced cardiac glucose uptake and increased FFA beta-oxidation [22]. The increased dependence on FFA oxidation in obese and T2DM patients in combination with cardiac microvascular dysfunction and rarefaction might result in a mismatch between oxygen supply and demand [95, 104]. Some studies reported a maintained cardiac creatine phosphate/ATP ratio in T2DM patients, whereas others demonstrated a reduction, suggesting that cardiac energy depletion may occur in T2DM [67, 104]. Impaired cardiac lymphatic transport, as seen in experimental models of HFrEF [47], could cause a build-up of waste products influencing energy metabolism. Intriguingly, whereas angiogenesis depends on active glycolysis, lymphangiogenesis depends on FFA beta-oxidation. It is conceivable that metabolic impairment and increased FFA beta-oxidation in cardiomyocytes during HFpEF may limit substrate availability in cardiac lymphatic endothelial cells, limiting lymphangiogenic responses during cardiac hypertrophy.
Interventions, such as caloric restriction, and insulin-sensitizing or glucose-lowering agents, such as metformin, dipeptidyl peptidase-4 (DPP-4) inhibitors, and glucagon-like peptide-1 (GLP-1) receptor agonists, all improve murine diastolic function [45]. In HFrEF patients, metabolic therapies that limit cardiac FFA beta-oxidation and promote glucose oxidation are promising [45]. Till now only sodium-glucose transport protein 2 (SGLT2) inhibitors (Dapagliflozin) have been proven to be beneficial in HFrEF patients, whereas its therapeutic benefit in HFpEF is currently being investigated [81]. To develop innovative metabolic therapies, further investigations are required to delineate the nature and cause of the alterations in cardiac metabolism in HFpEF and their links to other cardiac changes, such as insufficient perfusion and chronic low-grade inflammation.
Conclusion
Currently, effective and specific therapies for HFpEF are lacking due to incomplete understanding of disease mechanisms. Advances in this area are hindered in part by the limited number of adequate animal models. Indeed, the disease mechanisms that were discovered in experimental animal studies may be influenced by the short-comings to account for comorbidities, age, gender, and hormonal status of current experimental animal models. Given that HFpEF is such a clinically heterogeneous disease, there may not be one single uniting pathological mechanism driving the disease and, therefore, having multiple pre-clinical animal models that can be compared to human pathophysiology is critical. Due to their similarity to human disease progression, large animal models, such as the multiple comorbidity swine model [114], are especially attractive.
Symptoms of HFpEF are not disease-specific and are often difficult to interpret in patients, leading to inadequate or late diagnosis [26, 44, 107]. Echocardiography alone often fails to demonstrate diastolic dysfunction, resulting in the need for invasive cardiac functional assessment. While multiple algorithms and clinical definitions have been proposed, risk stratification of patients in several HFpEF subgroup has not yet been achieved [97]. As such, there is an urgent need for an improved clinical definition of HFpEF, likely to be driven by the development of novel diagnostic tools, such as cardiac and non-cardiac imaging strategies and early biomarkers for risk stratification.
Nevertheless, it is envisaged that therapies targeting generalized vascular dysfunction, such as (1) anti-inflammatory drugs, (2) antioxidants, (3) anti-vascular permeability drugs (such as glycocalyx, pericyte, and cell–cell junction stabilizers), and/or (4) anti-vascular rarefaction drugs (such as angiogenic therapy) might prevent or treat HFpEF by restoring cardiac perfusion and attenuating cardiac inflammation. The translation of vascular drugs to clinical practice in HFpEF, as well as HFrEF, is still difficult due to the complex and multifactorial pathophysiology of the coronary circulation [48, 68]. In parallel, treatments targeting systemic lymphatic dysfunction, such as VEGF-C or -D lymphangiogenic therapy, might be considered to limit oedema and chronic low-grade inflammation in HFpEF. These vascular therapies are expected to complement other current major therapeutic targets in HFpEF, such as cardiac fibrosis targeting with aldosterone pathway inhibitors, or directly targeting chronic low-grade inflammation and oxidative stress (e.g. pirfenidone). Finally, it remains to be determined if cardiac metabolism in HFpEF patients could benefit from (1) FFA beta-oxidation antagonists (2) glucose uptake agonists, or (3) AMPK agonists.
Notably, as HFpEF is a multifactorial disease, combined personalized medicine targeting cardiac vascular and lymphatic dysfunction, fibrosis, inflammation, and metabolic flux alterations may be needed to limit diastolic dysfunction and improve quality of life.
References
Alitalo K (2011) The lymphatic vasculature in disease. Nat Med 17:1371–1380. https://doi.org/10.1038/nm.2545
Angeli V, Ginhoux F, Llodra J, Quemeneur L, Frenette PS, Skobe M, Jessberger R, Merad M, Randolph GJ (2006) B cell-driven lymphangiogenesis in inflamed lymph nodes enhances dendritic cell mobilization. Immunity 24:203–215. https://doi.org/10.1016/j.immuni.2006.01.003
Antonios TF, Rattray FE, Singer DR, Markandu ND, Mortimer PS, MacGregor GA (1999) Maximization of skin capillaries during intravital video-microscopy in essential hypertension: comparison between venous congestion, reactive hyperaemia and core heat load tests. Clin Sci (Lond) 97:523–528
Avraham T, Zampell JC, Yan A, Elhadad S, Weitman ES, Rockson SG, Bromberg J, Mehrara BJ (2013) Th2 differentiation is necessary for soft tissue fibrosis and lymphatic dysfunction resulting from lymphedema. FASEB J 27:1114–1126. https://doi.org/10.1096/fj.12-222695
Babes EE, Babes VV (2013) ADMA and prognosis in patients with heart failure and preserved ejection fraction. Eur Heart J 34:P1491. https://doi.org/10.1093/eurheartj/eht308.P1491
Baluk P, Tammela T, Ator E, Lyubynska N, Achen MG, Hicklin DJ, Jeltsch M, Petrova TV, Pytowski B, Stacker SA, Yla-Herttuala S, Jackson DG, Alitalo K, McDonald DM (2005) Pathogenesis of persistent lymphatic vessel hyperplasia in chronic airway inflammation. J Clin Invest 115:247–257. https://doi.org/10.1172/JCI22037
Baron AD, Tarshoby M, Hook G, Lazaridis EN, Cronin J, Johnson A, Steinberg HO (2000) Interaction between insulin sensitivity and muscle perfusion on glucose uptake in human skeletal muscle: evidence for capillary recruitment. Diabetes 49:768–774
Bender SB (2019) Linking coronary microvascular and cardiac diastolic dysfunction in diabetes: are women more vulnerable? Diabetes 68:474–475. https://doi.org/10.2337/dbi18-0053
Bender SB, de Beer VJ, Tharp DL, Bowles DK, Laughlin MH, Merkus D, Duncker DJ (2016) Severe familial hypercholesterolemia impairs the regulation of coronary blood flow and oxygen supply during exercise. Basic Res Cardiol 111:61. https://doi.org/10.1007/s00395-016-0579-9
Bierhaus A, Humpert PM, Morcos M, Wendt T, Chavakis T, Arnold B, Stern DM, Nawroth PP (2005) Understanding RAGE, the receptor for advanced glycation end products. J Mol Med (Berl) 83:876–886. https://doi.org/10.1007/s00109-005-0688-7
Blau CW, Cowley TR, O'Sullivan J, Grehan B, Browne TC, Kelly L, Birch A, Murphy N, Kelly AM, Kerskens CM, Lynch MA (2012) The age-related deficit in LTP is associated with changes in perfusion and blood-brain barrier permeability. Neurobiol Aging 33(1005):e1023–1035. https://doi.org/10.1016/j.neurobiolaging.2011.09.035
Borbely A, Falcao-Pires I, van Heerebeek L, Hamdani N, Edes I, Gavina C, Leite-Moreira AF, Bronzwaer JG, Papp Z, van der Velden J, Stienen GJ, Paulus WJ (2009) Hypophosphorylation of the Stiff N2B titin isoform raises cardiomyocyte resting tension in failing human myocardium. Circ Res 104:780–786. https://doi.org/10.1161/CIRCRESAHA.108.193326
Brakenhielm E, Alitalo K (2019) Cardiac lymphatics in health and disease. Nat Rev Cardiol 16:56–68. https://doi.org/10.1038/s41569-018-0087-8
Bruegger D, Rehm M, Jacob M, Chappell D, Stoeckelhuber M, Welsch U, Conzen P, Becker BF (2008) Exogenous nitric oxide requires an endothelial glycocalyx to prevent postischemic coronary vascular leak in guinea pig hearts. Crit Care 12:R73. https://doi.org/10.1186/cc6913
Bruunsgaard H, Ladelund S, Pedersen AN, Schroll M, Jorgensen T, Pedersen BK (2003) Predicting death from tumour necrosis factor-alpha and interleukin-6 in 80-year-old people. Clin Exp Immunol 132:24–31. https://doi.org/10.1046/j.1365-2249.2003.02137.x
Campbell DJ, Somaratne JB, Prior DL, Yii M, Kenny JF, Newcomb AE, Kelly DJ, Black MJ (2013) Obesity is associated with lower coronary microvascular density. PLoS ONE 8:e81798. https://doi.org/10.1371/journal.pone.0081798
Chabrashvili T, Kitiyakara C, Blau J, Karber A, Aslam S, Welch WJ, Wilcox CS (2003) Effects of ANG II type 1 and 2 receptors on oxidative stress, renal NADPH oxidase, and SOD expression. Am J Physiol Regul Integr Comp Physiol 285:R117–124. https://doi.org/10.1152/ajpregu.00476.2002
Champion S, Lapidus N, Cherie G, Spagnoli V, Oliary J, Solal AC (2014) Pentoxifylline in heart failure: a meta-analysis of clinical trials. Cardiovasc Ther 32:159–162. https://doi.org/10.1111/1755-5922.12076
Chirinos JA, Akers SR, Trieu L, Ischiropoulos H, Doulias PT, Tariq A, Vasim I, Koppula MR, Syed AA, Soto-Calderon H, Townsend RR, Cappola TP, Margulies KB, Zamani P (2016) Heart failure, left ventricular remodeling, and circulating nitric oxide metabolites. J Am Heart Assoc. https://doi.org/10.1161/JAHA.116.004133
Cho CH, Sung HK, Kim KT, Cheon HG, Oh GT, Hong HJ, Yoo OJ, Koh GY (2006) COMP-angiopoietin-1 promotes wound healing through enhanced angiogenesis, lymphangiogenesis, and blood flow in a diabetic mouse model. Proc Natl Acad Sci USA 103:4946–4951. https://doi.org/10.1073/pnas.0506352103
Chowdhury MA, Moukarbel GV, Gupta R, Frank SM, Anderson AM, Liu LC, Khouri SJ (2019) Endothelin 1 is associated with heart failure hospitalization and long-term mortality in patients with heart failure with preserved ejection fraction and pulmonary hypertension. Cardiology 143:124–133. https://doi.org/10.1159/000501100
Christoffersen C, Bollano E, Lindegaard ML, Bartels ED, Goetze JP, Andersen CB, Nielsen LB (2003) Cardiac lipid accumulation associated with diastolic dysfunction in obese mice. Endocrinology 144:3483–3490. https://doi.org/10.1210/en.2003-0242
Claesson-Welsh L (2015) Vascular permeability–the essentials. Ups J Med Sci 120:135–143. https://doi.org/10.3109/03009734.2015.1064501
Collier P, Watson CJ, Voon V, Phelan D, Jan A, Mak G, Martos R, Baugh JA, Ledwidge MT, McDonald KM (2011) Can emerging biomarkers of myocardial remodelling identify asymptomatic hypertensive patients at risk for diastolic dysfunction and diastolic heart failure? Eur J Heart Fail 13:1087–1095. https://doi.org/10.1093/eurjhf/hfr079
Conway WC, Faries MB, Nicholl MB, Terando AM, Glass EC, Sim M, Morton DL (2009) Age-related lymphatic dysfunction in melanoma patients. Ann Surg Oncol 16:1548–1552. https://doi.org/10.1245/s10434-009-0420-x
Daniels LB, Clopton P, Bhalla V, Krishnaswamy P, Nowak RM, McCord J, Hollander JE, Duc P, Omland T, Storrow AB, Abraham WT, Wu AH, Steg PG, Westheim A, Knudsen CW, Perez A, Kazanegra R, Herrmann HC, McCullough PA, Maisel AS (2006) How obesity affects the cut-points for B-type natriuretic peptide in the diagnosis of acute heart failure. Results from the Breathing Not Properly Multinational Study. Am Heart J 151:999–1005. https://doi.org/10.1016/j.ahj.2005.10.011
de Boer RA, De Keulenaer G, Bauersachs J, Brutsaert D, Cleland JG, Diez J, Du XJ, Ford P, Heinzel FR, Lipson KE, McDonagh T, Lopez-Andres N, Lunde IG, Lyon AR, Pollesello P, Prasad SK, Tocchetti CG, Mayr M, Sluijter JPG, Thum T, Tschope C, Zannad F, Zimmermann WH, Ruschitzka F, Filippatos G, Lindsey ML, Maack C, Heymans S (2019) Towards better definition, quantification and treatment of fibrosis in heart failure. A scientific roadmap by the Committee of Translational Research of the Heart Failure Association (HFA) of the European Society of Cardiology. Eur J Heart Fail 21:272–285. https://doi.org/10.1002/ejhf.1406
de las Fuentes L, Soto PF, Cupps BP, Pasque MK, Herrero P, Gropler RJ, Waggoner AD, Davila-Roman VG (2006) Hypertensive left ventricular hypertrophy is associated with abnormal myocardial fatty acid metabolism and myocardial efficiency. J Nucl Cardiol 13:369–377. https://doi.org/10.1016/j.nuclcard.2006.01.021
Deluyker D, Ferferieva V, Noben JP, Swennen Q, Bronckaers A, Lambrichts I, Rigo JM, Bito V (2016) Cross-linking versus RAGE: How do high molecular weight advanced glycation products induce cardiac dysfunction? Int J Cardiol 210:100–108. https://doi.org/10.1016/j.ijcard.2016.02.095
DuBrock HM, AbouEzzeddine OF, Redfield MM (2018) High-sensitivity C-reactive protein in heart failure with preserved ejection fraction. PLoS ONE 13:e0201836. https://doi.org/10.1371/journal.pone.0201836
Eskens BJ, Leurgans TM, Vink H, Vanteeffelen JW (2014) Early impairment of skeletal muscle endothelial glycocalyx barrier properties in diet-induced obesity in mice. Physiol Rep 2:e00194. https://doi.org/10.1002/phy2.194
Falkenham A, de Antueno R, Rosin N, Betsch D, Lee TD, Duncan R, Legare JF (2015) Nonclassical resident macrophages are important determinants in the development of myocardial fibrosis. Am J Pathol 185:927–942. https://doi.org/10.1016/j.ajpath.2014.11.027
Flaht-Zabost A, Gula G, Ciszek B, Czarnowska E, Jankowska-Steifer E, Madej M, Niderla-Bielinska J, Radomska-Lesniewska D, Ratajska A (2014) Cardiac mouse lymphatics: developmental and anatomical update. Anat Rec (Hoboken) 297:1115–1130. https://doi.org/10.1002/ar.22912
Forrester SJ, Kikuchi DS, Hernandes MS, Xu Q, Griendling KK (2018) Reactive oxygen species in metabolic and inflammatory signaling. Circ Res 122:877–902. https://doi.org/10.1161/CIRCRESAHA.117.311401
Franssen C, Chen S, Unger A, Korkmaz HI, De Keulenaer GW, Tschope C, Leite-Moreira AF, Musters R, Niessen HW, Linke WA, Paulus WJ, Hamdani N (2016) Myocardial microvascular inflammatory endothelial activation in heart failure with preserved ejection fraction. JACC Heart Fail 4:312–324. https://doi.org/10.1016/j.jchf.2015.10.007
Franssen C, Gonzalez Miqueo A (2016) The role of titin and extracellular matrix remodelling in heart failure with preserved ejection fraction. Neth Heart J 24:259–267. https://doi.org/10.1007/s12471-016-0812-z
Furukawa S, Fujita T, Shimabukuro M, Iwaki M, Yamada Y, Nakajima Y, Nakayama O, Makishima M, Matsuda M, Shimomura I (2004) Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest 114:1752–1761. https://doi.org/10.1172/JCI21625
Glezeva N, Voon V, Watson C, Horgan S, McDonald K, Ledwidge M, Baugh J (2015) Exaggerated inflammation and monocytosis associate with diastolic dysfunction in heart failure with preserved ejection fraction: evidence of M2 macrophage activation in disease pathogenesis. J Card Fail 21:167–177. https://doi.org/10.1016/j.cardfail.2014.11.004
Gonzalez A, Lopez B, Ravassa S, San Jose G, Diez J (2019) The complex dynamics of myocardial interstitial fibrosis in heart failure. Focus on collagen cross-linking. Biochim Biophys Acta Mol Cell Res 1866:1421–1432. https://doi.org/10.1016/j.bbamcr.2019.06.001
Guo R, Zhou Q, Proulx ST, Wood R, Ji RC, Ritchlin CT, Pytowski B, Zhu Z, Wang YJ, Schwarz EM, Xing L (2009) Inhibition of lymphangiogenesis and lymphatic drainage via vascular endothelial growth factor receptor 3 blockade increases the severity of inflammation in a mouse model of chronic inflammatory arthritis. Arthritis Rheum 60:2666–2676. https://doi.org/10.1002/art.24764
Haas AV, Rosner BA, Kwong RY, Rao AD, Garg R, Di Carli MF, Adler GK (2019) Sex differences in coronary microvascular function in individuals with Type 2 diabetes. Diabetes 68:631–636. https://doi.org/10.2337/db18-0650
Haemmerle M, Keller T, Egger G, Schachner H, Steiner CW, Stokic D, Neumayer C, Brown MK, Kerjaschki D, Hantusch B (2013) Enhanced lymph vessel density, remodeling, and inflammation are reflected by gene expression signatures in dermal lymphatic endothelial cells in type 2 diabetes. Diabetes 62:2509–2529. https://doi.org/10.2337/db12-0844
Hammer S, van der Meer RW, Lamb HJ, Schar M, de Roos A, Smit JW, Romijn JA (2008) Progressive caloric restriction induces dose-dependent changes in myocardial triglyceride content and diastolic function in healthy men. J Clin Endocrinol Metab 93:497–503. https://doi.org/10.1210/jc.2007-2015
Hawkins NM, Petrie MC, Jhund PS, Chalmers GW, Dunn FG, McMurray JJ (2009) Heart failure and chronic obstructive pulmonary disease: diagnostic pitfalls and epidemiology. Eur J Heart Fail 11:130–139. https://doi.org/10.1093/eurjhf/hfn013
Heggermont WA, Papageorgiou AP, Heymans S, van Bilsen M (2016) Metabolic support for the heart: complementary therapy for heart failure? Eur J Heart Fail 18:1420–1429. https://doi.org/10.1002/ejhf.678
Heinzel FR, Gres P, Boengler K, Duschin A, Konietzka I, Rassaf T, Snedovskaya J, Meyer S, Skyschally A, Kelm M, Heusch G, Schulz R (2008) Inducible nitric oxide synthase expression and cardiomyocyte dysfunction during sustained moderate ischemia in pigs. Circ Res 103:1120–1127. https://doi.org/10.1161/CIRCRESAHA.108.186015
Henri O, Pouehe C, Houssari M, Galas L, Nicol L, Edwards-Levy F, Henry JP, Dumesnil A, Boukhalfa I, Banquet S, Schapman D, Thuillez C, Richard V, Mulder P, Brakenhielm E (2016) Selective stimulation of cardiac lymphangiogenesis reduces myocardial edema and fibrosis leading to improved cardiac function following myocardial infarction. Circulation 133:1484–1497. https://doi.org/10.1161/CIRCULATIONAHA.115.020143(discussion 1497)
Heusch G (2016) The coronary circulation as a target of cardioprotection. Circ Res 118:1643–1658. https://doi.org/10.1161/CIRCRESAHA.116.308640
Hulsmans M, Sager HB, Roh JD, Valero-Munoz M, Houstis NE, Iwamoto Y, Sun Y, Wilson RM, Wojtkiewicz G, Tricot B, Osborne MT, Hung J, Vinegoni C, Naxerova K, Sosnovik DE, Zile MR, Bradshaw AD, Liao R, Tawakol A, Weissleder R, Rosenzweig A, Swirski FK, Sam F, Nahrendorf M (2018) Cardiac macrophages promote diastolic dysfunction. J Exp Med 215:423–440. https://doi.org/10.1084/jem.20171274
Ikonomidis I, Voumvourakis A, Makavos G, Triantafyllidi H, Pavlidis G, Katogiannis K, Benas D, Vlastos D, Trivilou P, Varoudi M, Parissis J, Iliodromitis E, Lekakis J (2018) Association of impaired endothelial glycocalyx with arterial stiffness, coronary microcirculatory dysfunction, and abnormal myocardial deformation in untreated hypertensives. J Clin Hypertens (Greenwich) 20:672–679. https://doi.org/10.1111/jch.13236
Illan-Gomez F, Gonzalvez-Ortega M, Orea-Soler I, Alcaraz-Tafalla MS, Aragon-Alonso A, Pascual-Diaz M, Perez-Paredes M, Lozano-Almela ML (2012) Obesity and inflammation: change in adiponectin, C-reactive protein, tumour necrosis factor-alpha and interleukin-6 after bariatric surgery. Obes Surg 22:950–955. https://doi.org/10.1007/s11695-012-0643-y
Incalza MA, D'Oria R, Natalicchio A, Perrini S, Laviola L, Giorgino F (2018) Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vascul Pharmacol 100:1–19. https://doi.org/10.1016/j.vph.2017.05.005
Inoguchi T, Li P, Umeda F, Yu HY, Kakimoto M, Imamura M, Aoki T, Etoh T, Hashimoto T, Naruse M, Sano H, Utsumi H, Nawata H (2000) High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C–dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes 49:1939–1945
Jacob M, Paul O, Mehringer L, Chappell D, Rehm M, Welsch U, Kaczmarek I, Conzen P, Becker BF (2009) Albumin augmentation improves condition of guinea pig hearts after 4 hr of cold ischemia. Transplantation 87:956–965. https://doi.org/10.1097/TP.0b013e31819c83b5
Jia G, Habibi J, Aroor AR, Hill MA, DeMarco VG, Lee LE, Ma L, Barron BJ, Whaley-Connell A, Sowers JR (2018) Enhanced endothelium epithelial sodium channel signaling prompts left ventricular diastolic dysfunction in obese female mice. Metabolism 78:69–79. https://doi.org/10.1016/j.metabol.2017.08.008
Kajiya K, Detmar M (2006) An important role of lymphatic vessels in the control of UVB-induced edema formation and inflammation. J Invest Dermatol 126:919–921. https://doi.org/10.1038/sj.jid.5700126
Kanwar M, Walter C, Clarke M, Patarroyo-Aponte M (2016) Targeting heart failure with preserved ejection fraction: current status and future prospects. Vasc Health Risk Manag 12:129–141. https://doi.org/10.2147/VHRM.S83662
Kasner M, Westermann D, Lopez B, Gaub R, Escher F, Kuhl U, Schultheiss HP, Tschope C (2011) Diastolic tissue Doppler indexes correlate with the degree of collagen expression and cross-linking in heart failure and normal ejection fraction. J Am Coll Cardiol 57:977–985. https://doi.org/10.1016/j.jacc.2010.10.024
Kataru RP, Kim H, Jang C, Choi DK, Koh BI, Kim M, Gollamudi S, Kim YK, Lee SH, Koh GY (2011) T lymphocytes negatively regulate lymph node lymphatic vessel formation. Immunity 34:96–107. https://doi.org/10.1016/j.immuni.2010.12.016
Kates AM, Herrero P, Dence C, Soto P, Srinivasan M, Delano DG, Ehsani A, Gropler RJ (2003) Impact of aging on substrate metabolism by the human heart. J Am Coll Cardiol 41:293–299. https://doi.org/10.1016/s0735-1097(02)02714-6
Kim H, Kataru RP, Koh GY (2012) Regulation and implications of inflammatory lymphangiogenesis. Trends Immunol 33:350–356. https://doi.org/10.1016/j.it.2012.03.006
Kleinbongard P, Bose D, Baars T, Mohlenkamp S, Konorza T, Schoner S, Elter-Schulz M, Eggebrecht H, Degen H, Haude M, Levkau B, Schulz R, Erbel R, Heusch G (2011) Vasoconstrictor potential of coronary aspirate from patients undergoing stenting of saphenous vein aortocoronary bypass grafts and its pharmacological attenuation. Circ Res 108:344–352. https://doi.org/10.1161/CIRCRESAHA.110.235713
Kosmas CE, Silverio D, Sourlas A, Montan PD, Guzman E (2018) Role of spironolactone in the treatment of heart failure with preserved ejection fraction. Ann Transl Med 6:461. https://doi.org/10.21037/atm.2018.11.16
Krouwer VJ, Hekking LH, Langelaar-Makkinje M, Regan-Klapisz E, Post JA (2012) Endothelial cell senescence is associated with disrupted cell-cell junctions and increased monolayer permeability. Vasc Cell 4:12. https://doi.org/10.1186/2045-824X-4-12
Laine GA (1988) Microvascular changes in the heart during chronic arterial hypertension. Circ Res 62:953–960
Leichman JG, Aguilar D, King TM, Vlada A, Reyes M, Taegtmeyer H (2006) Association of plasma free fatty acids and left ventricular diastolic function in patients with clinically severe obesity. Am J Clin Nutr 84:336–341. https://doi.org/10.1093/ajcn/84.1.336
Levelt E, Mahmod M, Piechnik SK, Ariga R, Francis JM, Rodgers CT, Clarke WT, Sabharwal N, Schneider JE, Karamitsos TD, Clarke K, Rider OJ, Neubauer S (2016) Relationship between left ventricular structural and metabolic remodeling in Type 2 diabetes. Diabetes 65:44–52. https://doi.org/10.2337/db15-0627
Levy BI, Heusch G, Camici PG (2019) The many faces of myocardial ischaemia and angina. Cardiovasc Res 115:1460–1470. https://doi.org/10.1093/cvr/cvz160
Liao S, Cheng G, Conner DA, Huang Y, Kucherlapati RS, Munn LL, Ruddle NH, Jain RK, Fukumura D, Padera TP (2011) Impaired lymphatic contraction associated with immunosuppression. Proc Natl Acad Sci USA 108:18784–18789. https://doi.org/10.1073/pnas.1116152108
Lim HY, Rutkowski JM, Helft J, Reddy ST, Swartz MA, Randolph GJ, Angeli V (2009) Hypercholesterolemic mice exhibit lymphatic vessel dysfunction and degeneration. Am J Pathol 175:1328–1337. https://doi.org/10.2353/ajpath.2009.080963
Little WC, Zile MR, Kitzman DW, Hundley WG, O'Brien TX, Degroof RC (2005) The effect of alagebrium chloride (ALT-711), a novel glucose cross-link breaker, in the treatment of elderly patients with diastolic heart failure. J Card Fail 11:191–195. https://doi.org/10.1016/j.cardfail.2004.09.010
Lopez Gelston CA, Balasubbramanian D, Abouelkheir GR, Lopez AH, Hudson KR, Johnson ER, Muthuchamy M, Mitchell BM, Rutkowski JM (2018) Enhancing renal lymphatic expansion prevents hypertension in mice. Circ Res 122:1094–1101. https://doi.org/10.1161/CIRCRESAHA.118.312765
Ludwig LL, Schertel ER, Pratt JW, McClure DE, Ying AJ, Heck CF, Myerowitz PD (1997) Impairment of left ventricular function by acute cardiac lymphatic obstruction. Cardiovasc Res 33:164–171. https://doi.org/10.1016/s0008-6363(96)00177-0
Luo M, Guan X, Luczak ED, Lang D, Kutschke W, Gao Z, Yang J, Glynn P, Sossalla S, Swaminathan PD, Weiss RM, Yang B, Rokita AG, Maier LS, Efimov IR, Hund TJ, Anderson ME (2013) Diabetes increases mortality after myocardial infarction by oxidizing CaMKII. J Clin Invest 123:1262–1274. https://doi.org/10.1172/JCI65268
Machin DR, Bloom SI, Campbell RA, Phuong TTT, Gates PE, Lesniewski LA, Rondina MT, Donato AJ (2018) Advanced age results in a diminished endothelial glycocalyx. Am J Physiol Heart Circ Physiol 315:H531–H539. https://doi.org/10.1152/ajpheart.00104.2018
Makarenko I, Opitz CA, Leake MC, Neagoe C, Kulke M, Gwathmey JK, del Monte F, Hajjar RJ, Linke WA (2004) Passive stiffness changes caused by upregulation of compliant titin isoforms in human dilated cardiomyopathy hearts. Circ Res 95:708–716. https://doi.org/10.1161/01.RES.0000143901.37063.2f
Mann DL (2015) Innate immunity and the failing heart: the cytokine hypothesis revisited. Circ Res 116:1254–1268. https://doi.org/10.1161/CIRCRESAHA.116.302317
Marume K, Takashio S, Nagai T, Tsujita K, Saito Y, Yoshikawa T, Anzai T (2019) Effect of statins on mortality in heart failure with preserved ejection fraction without coronary artery disease—report from the JASPER study. Circ J 83:357–367. https://doi.org/10.1253/circj.CJ-18-0639
Mathews MK, Merges C, McLeod DS, Lutty GA (1997) Vascular endothelial growth factor and vascular permeability changes in human diabetic retinopathy. Invest Ophthalmol Vis Sci 38:2729–2741
Matsubara J, Sugiyama S, Nozaki T, Sugamura K, Konishi M, Ohba K, Matsuzawa Y, Akiyama E, Yamamoto E, Sakamoto K, Nagayoshi Y, Kaikita K, Sumida H, Kim-Mitsuyama S, Ogawa H (2011) Pentraxin 3 is a new inflammatory marker correlated with left ventricular diastolic dysfunction and heart failure with normal ejection fraction. J Am Coll Cardiol 57:861–869. https://doi.org/10.1016/j.jacc.2010.10.018
McMurray JJV, Solomon SD, Inzucchi SE, Kober L, Kosiborod MN, Martinez FA, Ponikowski P, Sabatine MS, Anand IS, Belohlavek J, Bohm M, Chiang CE, Chopra VK, de Boer RA, Desai AS, Diez M, Drozdz J, Dukat A, Ge J, Howlett JG, Katova T, Kitakaze M, Ljungman CEA, Merkely B, Nicolau JC, O'Meara E, Petrie MC, Vinh PN, Schou M, Tereshchenko S, Verma S, Held C, DeMets DL, Docherty KF, Jhund PS, Bengtsson O, Sjostrand M, Langkilde AM, Committees D-HT, Investigators (2019) Dapagliflozin in patients with heart failure and reduced ejection fraction. N Engl J Med 381:1995–2008. https://doi.org/10.1056/NEJMoa1911303
Mehlhorn U, Geissler HJ, Laine GA, Allen SJ (2001) Myocardial fluid balance. Eur J Cardiothorac Surg 20:1220–1230. https://doi.org/10.1016/s1010-7940(01)01031-4
Michels da Silva D, Langer H, Graf T (2019) Inflammatory and molecular pathways in heart failure-ischemia, HFpEF and transthyretin cardiac amyloidosis. Int J Mol Sci. https://doi.org/10.3390/ijms20092322
Mohammed SF, Hussain S, Mirzoyev SA, Edwards WD, Maleszewski JJ, Redfield MM (2015) Coronary microvascular rarefaction and myocardial fibrosis in heart failure with preserved ejection fraction. Circulation 131:550–559. https://doi.org/10.1161/CIRCULATIONAHA.114.009625
Nakanishi K, Fukuda S, Tanaka A, Otsuka K, Taguchi H, Shimada K (2017) Relationships between periventricular epicardial adipose tissue accumulation, coronary microcirculation, and left ventricular diastolic dysfunction. Can J Cardiol 33:1489–1497. https://doi.org/10.1016/j.cjca.2017.08.001
Nieuwdorp M, van Haeften TW, Gouverneur MC, Mooij HL, van Lieshout MH, Levi M, Meijers JC, Holleman F, Hoekstra JB, Vink H, Kastelein JJ, Stroes ES (2006) Loss of endothelial glycocalyx during acute hyperglycemia coincides with endothelial dysfunction and coagulation activation in vivo. Diabetes 55:480–486
Nitti MD, Hespe GE, Kataru RP, Garcia Nores GD, Savetsky IL, Torrisi JS, Gardenier JC, Dannenberg AJ, Mehrara BJ (2016) Obesity-induced lymphatic dysfunction is reversible with weight loss. J Physiol 594:7073–7087. https://doi.org/10.1113/JP273061
Noon JP, Walker BR, Webb DJ, Shore AC, Holton DW, Edwards HV, Watt GC (1997) Impaired microvascular dilatation and capillary rarefaction in young adults with a predisposition to high blood pressure. J Clin Invest 99:1873–1879. https://doi.org/10.1172/JCI119354
Oku H, Kodama T, Sakagami K, Puro DG (2001) Diabetes-induced disruption of gap junction pathways within the retinal microvasculature. Invest Ophthalmol Vis Sci 42:1915–1920
Oliveira-Junior SA, Martinez PF, Guizoni DM, Campos DH, Fernandes T, Oliveira EM, Okoshi MP, Okoshi K, Padovani CR, Cicogna AC (2014) AT1 receptor blockade attenuates insulin resistance and myocardial remodeling in rats with diet-induced obesity. PLoS ONE 9:e86447. https://doi.org/10.1371/journal.pone.0086447
Oliver CN, Ahn BW, Moerman EJ, Goldstein S, Stadtman ER (1987) Age-related changes in oxidized proteins. J Biol Chem 262:5488–5491
Pandey A, Garg S, Matulevicius SA, Shah AM, Garg J, Drazner MH, Amin A, Berry JD, Marwick TH, Marso SP, de Lemos JA, Kumbhani DJ (2015) Effect of mineralocorticoid receptor antagonists on cardiac structure and function in patients with diastolic dysfunction and heart failure with preserved ejection fraction: a meta-analysis and systematic review. J Am Heart Assoc 4:e002137. https://doi.org/10.1161/JAHA.115.002137
Papademetriou V, Toumpourleka M, Imprialos KP, Alataki S, Manafis A, Stavropoulos K (2018) The role of mineralocorticoid receptor antagonists in heart failure with reduced ejection fraction. Curr Pharm Des 24:5517–5524. https://doi.org/10.2174/1381612825666190219141326
Paulus WJ, Tschope C (2013) A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol 62:263–271. https://doi.org/10.1016/j.jacc.2013.02.092
Peterson LR, Herrero P, Schechtman KB, Racette SB, Waggoner AD, Kisrieva-Ware Z, Dence C, Klein S, Marsala J, Meyer T, Gropler RJ (2004) Effect of obesity and insulin resistance on myocardial substrate metabolism and efficiency in young women. Circulation 109:2191–2196. https://doi.org/10.1161/01.CIR.0000127959.28627.F8
Picchi A, Limbruno U, Focardi M, Cortese B, Micheli A, Boschi L, Severi S, De Caterina R (2011) Increased basal coronary blood flow as a cause of reduced coronary flow reserve in diabetic patients. Am J Physiol Heart Circ Physiol 301:H2279–2284. https://doi.org/10.1152/ajpheart.00615.2011
Pieske B, Tschope C, de Boer RA, Fraser AG, Anker SD, Donal E, Edelmann F, Fu M, Guazzi M, Lam CSP, Lancellotti P, Melenovsky V, Morris DA, Nagel E, Pieske-Kraigher E, Ponikowski P, Solomon SD, Vasan RS, Rutten FH, Voors AA, Ruschitzka F, Paulus WJ, Seferovic P, Filippatos G (2019) How to diagnose heart failure with preserved ejection fraction: the HFA-PEFF diagnostic algorithm: a consensus recommendation from the Heart Failure Association (HFA) of the European Society of Cardiology (ESC). Eur Heart J 40:3297–3317. https://doi.org/10.1093/eurheartj/ehz641
Pitsavos C, Tampourlou M, Panagiotakos DB, Skoumas Y, Chrysohoou C, Nomikos T, Stefanadis C (2007) Association between low-grade systemic inflammation and Type 2 diabetes mellitus among men and women from the ATTICA study. Rev Diabet Stud 4:98–104. https://doi.org/10.1900/RDS.2007.4.98
Querejeta R, Lopez B, Gonzalez A, Sanchez E, Larman M, Martinez Ubago JL, Diez J (2004) Increased collagen type I synthesis in patients with heart failure of hypertensive origin: relation to myocardial fibrosis. Circulation 110:1263–1268. https://doi.org/10.1161/01.CIR.0000140973.60992.9A
Ramasamy R, Vannucci SJ, Yan SS, Herold K, Yan SF, Schmidt AM (2005) Advanced glycation end products and RAGE: a common thread in aging, diabetes, neurodegeneration, and inflammation. Glycobiology 15:16R–28R. https://doi.org/10.1093/glycob/cwi053
Ratchford SM, Clifton HL, Gifford J, LaSalle DT, Bunsawat K, Theisen J, Richardson RS, Wright JB, Ryan JJ, Wray W (2019) Impact of acute antioxidant administration on inflammation and vascular function in heart failure with preserved ejection fraction. FASEB J 33:No. 1_supplement
Reddy YNV, Obokata M, Wiley B, Koepp KE, Jorgenson CC, Egbe A, Melenovsky V, Carter RE, Borlaug BA (2019) The haemodynamic basis of lung congestion during exercise in heart failure with preserved ejection fraction. Eur Heart J 40:3721–3730. https://doi.org/10.1093/eurheartj/ehz713
Regan TJ, Lyons MM, Ahmed SS, Levinson GE, Oldewurtel HA, Ahmad MR, Haider B (1977) Evidence for cardiomyopathy in familial diabetes mellitus. J Clin Invest 60:884–899. https://doi.org/10.1172/JCI108843
Rijzewijk LJ, van der Meer RW, Lamb HJ, de Jong HW, Lubberink M, Romijn JA, Bax JJ, de Roos A, Twisk JW, Heine RJ, Lammertsma AA, Smit JW, Diamant M (2009) Altered myocardial substrate metabolism and decreased diastolic function in nonischemic human diabetic cardiomyopathy: studies with cardiac positron emission tomography and magnetic resonance imaging. J Am Coll Cardiol 54:1524–1532. https://doi.org/10.1016/j.jacc.2009.04.074
Rodriguez C, Martinez-Gonzalez J (2019) The role of Lysyl oxidase enzymes in cardiac function and remodeling. Cells. https://doi.org/10.3390/cells8121483
Rummery NM, Hill CE (2004) Vascular gap junctions and implications for hypertension. Clin Exp Pharmacol Physiol 31:659–667. https://doi.org/10.1111/j.1440-1681.2004.04071.x
Rutten FH, Moons KG, Cramer MJ, Grobbee DE, Zuithoff NP, Lammers JW, Hoes AW (2005) Recognising heart failure in elderly patients with stable chronic obstructive pulmonary disease in primary care: cross sectional diagnostic study. BMJ 331:1379. https://doi.org/10.1136/bmj.38664.661181.55
Scallan JP, Hill MA, Davis MJ (2015) Lymphatic vascular integrity is disrupted in type 2 diabetes due to impaired nitric oxide signalling. Cardiovasc Res 107:89–97. https://doi.org/10.1093/cvr/cvv117
Schwartzenberg S, Redfield MM, From AM, Sorajja P, Nishimura RA, Borlaug BA (2012) Effects of vasodilation in heart failure with preserved or reduced ejection fraction implications of distinct pathophysiologies on response to therapy. J Am Coll Cardiol 59:442–451. https://doi.org/10.1016/j.jacc.2011.09.062
Senni M, Paulus WJ, Gavazzi A, Fraser AG, Diez J, Solomon SD, Smiseth OA, Guazzi M, Lam CS, Maggioni AP, Tschope C, Metra M, Hummel SL, Edelmann F, Ambrosio G, Stewart Coats AJ, Filippatos GS, Gheorghiade M, Anker SD, Levy D, Pfeffer MA, Stough WG, Pieske BM (2014) New strategies for heart failure with preserved ejection fraction: the importance of targeted therapies for heart failure phenotypes. Eur Heart J 35:2797–2815. https://doi.org/10.1093/eurheartj/ehu204
Shah KB, Kop WJ, Christenson RH, Diercks DB, Henderson S, Hanson K, Li SY, deFilippi CR (2011) Prognostic utility of ST2 in patients with acute dyspnea and preserved left ventricular ejection fraction. Clin Chem 57:874–882. https://doi.org/10.1373/clinchem.2010.159277
Shin K, Kataru RP, Park HJ, Kwon BI, Kim TW, Hong YK, Lee SH (2015) TH2 cells and their cytokines regulate formation and function of lymphatic vessels. Nat Commun 6:6196. https://doi.org/10.1038/ncomms7196
Song Y, Yao Q, Zhu J, Luo B, Liang S (1999) Age-related variation in the interstitial tissues of the cardiac conduction system; and autopsy study of 230 Han Chinese. Forensic Sci Int 104:133–142. https://doi.org/10.1016/s0379-0738(99)00103-6
Sorop O, Heinonen I, van Kranenburg M, van de Wouw J, de Beer VJ, Nguyen ITN, Octavia Y, van Duin RWB, Stam K, van Geuns RJ, Wielopolski PA, Krestin GP, van den Meiracker AH, Verjans R, van Bilsen M, Danser AHJ, Paulus WJ, Cheng C, Linke WA, Joles JA, Verhaar MC, van der Velden J, Merkus D, Duncker DJ (2018) Multiple common comorbidities produce left ventricular diastolic dysfunction associated with coronary microvascular dysfunction, oxidative stress, and myocardial stiffening. Cardiovasc Res 114:954–964. https://doi.org/10.1093/cvr/cvy038
Steinberg BA, Zhao X, Heidenreich PA, Peterson ED, Bhatt DL, Cannon CP, Hernandez AF, Fonarow GC, Get With the Guidelines Scientific Advisory C, Investigators (2012) Trends in patients hospitalized with heart failure and preserved left ventricular ejection fraction: prevalence, therapies, and outcomes. Circulation 126:65–75. https://doi.org/10.1161/CIRCULATIONAHA.111.080770
Sung KC, Suh JY, Kim BS, Kang JH, Kim H, Lee MH, Park JR, Kim SW (2003) High sensitivity C-reactive protein as an independent risk factor for essential hypertension. Am J Hypertens 16:429–433. https://doi.org/10.1016/s0895-7061(03)00566-1
Tammela T, Alitalo K (2010) Lymphangiogenesis: molecular mechanisms and future promise. Cell 140:460–476. https://doi.org/10.1016/j.cell.2010.01.045
Tan KW, Chong SZ, Wong FH, Evrard M, Tan SM, Keeble J, Kemeny DM, Ng LG, Abastado JP, Angeli V (2013) Neutrophils contribute to inflammatory lymphangiogenesis by increasing VEGF-A bioavailability and secreting VEGF-D. Blood 122:3666–3677. https://doi.org/10.1182/blood-2012-11-466532
Taqueti VR, Solomon SD, Shah AM, Desai AS, Groarke JD, Osborne MT, Hainer J, Bibbo CF, Dorbala S, Blankstein R, Di Carli MF (2018) Coronary microvascular dysfunction and future risk of heart failure with preserved ejection fraction. Eur Heart J 39:840–849. https://doi.org/10.1093/eurheartj/ehx721
Tromp J, van der Pol A, Klip IT, de Boer RA, Jaarsma T, van Gilst WH, Voors AA, van Veldhuisen DJ, van der Meer P (2014) Fibrosis marker syndecan-1 and outcome in patients with heart failure with reduced and preserved ejection fraction. Circ Heart Fail 7:457–462. https://doi.org/10.1161/CIRCHEARTFAILURE.113.000846
van de Wouw J, Sorop O, van Drie RWA, van Duin RWB, Nguyen ITN, Joles JA, Verhaar MC, Merkus D, Duncker DJ (2020) Perturbations in myocardial perfusion and oxygen balance in swine with multiple risk factors: a novel model of ischemia and no obstructive coronary artery disease. Basic Res Cardiol 115:21. https://doi.org/10.1007/s00395-020-0778-2
van Dijk CG, Oosterhuis NR, Xu YJ, Brandt M, Paulus WJ, van Heerebeek L, Duncker DJ, Verhaar MC, Fontoura D, Lourenco AP, Leite-Moreira AF, Falcao-Pires I, Joles JA, Cheng C (2016) Distinct endothelial cell responses in the heart and kidney microvasculature characterize the progression of heart failure with preserved ejection fraction in the obese ZSF1 rat with cardiorenal metabolic syndrome. Circ Heart Fail 9:e002760. https://doi.org/10.1161/CIRCHEARTFAILURE.115.002760
van Empel VP, Mariani J, Borlaug BA, Kaye DM (2014) Impaired myocardial oxygen availability contributes to abnormal exercise hemodynamics in heart failure with preserved ejection fraction. J Am Heart Assoc 3:e001293. https://doi.org/10.1161/JAHA.114.001293
van Heerebeek L, Borbely A, Niessen HW, Bronzwaer JG, van der Velden J, Stienen GJ, Linke WA, Laarman GJ, Paulus WJ (2006) Myocardial structure and function differ in systolic and diastolic heart failure. Circulation 113:1966–1973. https://doi.org/10.1161/CIRCULATIONAHA.105.587519
van Heerebeek L, Hamdani N, Falcao-Pires I, Leite-Moreira AF, Begieneman MP, Bronzwaer JG, van der Velden J, Stienen GJ, Laarman GJ, Somsen A, Verheugt FW, Niessen HW, Paulus WJ (2012) Low myocardial protein kinase G activity in heart failure with preserved ejection fraction. Circulation 126:830–839. https://doi.org/10.1161/CIRCULATIONAHA.111.076075
Van Tassell BW, Arena R, Biondi-Zoccai G, Canada JM, Oddi C, Abouzaki NA, Jahangiri A, Falcao RA, Kontos MC, Shah KB, Voelkel NF, Dinarello CA, Abbate A (2014) Effects of interleukin-1 blockade with anakinra on aerobic exercise capacity in patients with heart failure and preserved ejection fraction (from the D-HART pilot study). Am J Cardiol 113:321–327. https://doi.org/10.1016/j.amjcard.2013.08.047
Versari D, Daghini E, Virdis A, Ghiadoni L, Taddei S (2009) Endothelium-dependent contractions and endothelial dysfunction in human hypertension. Br J Pharmacol 157:527–536. https://doi.org/10.1111/j.1476-5381.2009.00240.x
Viazzi F, Leoncini G, Ratto E, Parodi A, Falqui V, Conti N, Tomolillo C, Ravera G, Deferrari G, Pontremoli R (2008) Vascular permeability, blood pressure, and organ damage in primary hypertension. Hypertens Res 31:873–879. https://doi.org/10.1291/hypres.31.873
Westermann D, Lindner D, Kasner M, Zietsch C, Savvatis K, Escher F, von Schlippenbach J, Skurk C, Steendijk P, Riad A, Poller W, Schultheiss HP, Tschope C (2011) Cardiac inflammation contributes to changes in the extracellular matrix in patients with heart failure and normal ejection fraction. Circ Heart Fail 4:44–52. https://doi.org/10.1161/CIRCHEARTFAILURE.109.931451
Wilder T, Ryba DM, Wieczorek DF, Wolska BM, Solaro RJ (2015) N-acetylcysteine reverses diastolic dysfunction and hypertrophy in familial hypertrophic cardiomyopathy. Am J Physiol Heart Circ Physiol 309:H1720–1730. https://doi.org/10.1152/ajpheart.00339.2015
Yang GH, Zhou X, Ji WJ, Liu JX, Sun J, Dong Y, Jiang TM, Li YM (2017) VEGF-C-mediated cardiac lymphangiogenesis in high salt intake accelerated progression of left ventricular remodeling in spontaneously hypertensive rats. Clin Exp Hypertens 39:740–747. https://doi.org/10.1080/10641963.2017.1324478
Yang GH, Zhou X, Ji WJ, Zeng S, Dong Y, Tian L, Bi Y, Guo ZZ, Gao F, Chen H, Jiang TM, Li YM (2014) Overexpression of VEGF-C attenuates chronic high salt intake-induced left ventricular maladaptive remodeling in spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol 306:H598–609. https://doi.org/10.1152/ajpheart.00585.2013
Yang JH, Obokata M, Reddy YNV, Redfield MM, Lerman A, Borlaug BA (2020) Endothelium-dependent and independent coronary microvascular dysfunction in patients with heart failure with preserved ejection fraction. Eur J Heart Fail 22:432–441. https://doi.org/10.1002/ejhf.1671
Zawieja SD, Gasheva O, Zawieja DC, Muthuchamy M (2016) Blunted flow-mediated responses and diminished nitric oxide synthase expression in lymphatic thoracic ducts of a rat model of metabolic syndrome. Am J Physiol Heart Circ Physiol 310:H385–393. https://doi.org/10.1152/ajpheart.00664.2015
Zawieja SD, Wang W, Chakraborty S, Zawieja DC, Muthuchamy M (2016) Macrophage alterations within the mesenteric lymphatic tissue are associated with impairment of lymphatic pump in metabolic syndrome. Microcirculation 23:558–570. https://doi.org/10.1111/micc.12307
Zile MR, Baicu CF, Ikonomidis JS, Stroud RE, Nietert PJ, Bradshaw AD, Slater R, Palmer BM, Van Buren P, Meyer M, Redfield MM, Bull DA, Granzier HL, LeWinter MM (2015) Myocardial stiffness in patients with heart failure and a preserved ejection fraction: contributions of collagen and titin. Circulation 131:1247–1259. https://doi.org/10.1161/CIRCULATIONAHA.114.013215
Acknowledgements
This work was supported by an European Research Area Network Cardiovascular disease [LYMIT-DIS 2016] consisting of funding from Fonds Wetenschappelijk Onderzoek [GOH7716N to E.A.V.J.], Agence National de la Recherche [16-ECVD-0004 to E.B. and P.M.], Nederlandse organisatie voor Wetenschappelijk Onderzoek [ZonMw 2016T091 to M.v.B.], and Instituto de Salud Carlos III [AC16/00020 to A.G.], National Centre for Research and Development [ERA-CVD/LyMitDis/1/2017 to A.R. and E.C.]. I.C. and A.G. were supported by the Fonds Wetenschappelijk Onderzoek [1160718N] and the Instituto de Salud Carlos III [CB16/11/00483 and PI18/01469], respectively. E.A.V.J., S.H., and S.J.S. were supported by Fonds Wetenschappelijk Onderzoek [G091018N]. S.H. acknowledges the support of the European Research Area Network Cardiovascular disease [Macro ERA 2016], Nederlandse organisatie voor Wetenschappelijk Onderzoek [Vidi 91796338], Dutch Heart Foundation [CVON2016-Early HFPEF, 2015-10, CVON She-PREDICTS, Grant 2017-21, and Arena-PRIME, 2017-18]. E.B. and P.M. acknowledge the support of the FHU REMOD-VHF and generalized institutional funds from INSERM, and the European Regional Development Fund [CPER/FEDER 2015 (DO-IT) and CPER/FEDER 2016 (PACT-CBS)].
Author information
Authors and Affiliations
Contributions
IC and SS researched data for the article, wrote the manuscript, and edited the manuscript for critical comments and changes suggested by the other authors. Most essential contributors were for the microvascular section (inflammation/oxidative stress/microvessel wall barrier); IC, SJS, EAVJ, AR, SH and EC; for the lymphatic section; SS, IC, AR, and EB; for the LV compliance section (cardiomyocyte passive stiffness/fibrosis); IC, SJS, AG, AK, and MB; for the metabolism section SS, IC, MB, and AK; for therapeutic section all authors contributed equally.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cuijpers, I., Simmonds, S.J., van Bilsen, M. et al. Microvascular and lymphatic dysfunction in HFpEF and its associated comorbidities. Basic Res Cardiol 115, 39 (2020). https://doi.org/10.1007/s00395-020-0798-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00395-020-0798-y