
Abstract
Restoration of myocardial blood flow after ischemia triggers an inflammatory response involving toll-like receptors (TLRs). TLR2−/−-mice show short-term advantages upon reperfusion injury as compared with WT controls. Accordingly, it has been shown that transient TLR2-blockade prior to reperfusion is associated with improved left-ventricular performance after myocardial scar formation. We present here adverse myocardial remodeling due to a chronic lack of TLR2 expression. Myocardial ischemia/reperfusion (MI/R) was surgically induced in C3HeN-mice by ligation of the left anterior descending coronary artery for 20 min, followed by 24 h or 28 days of reperfusion. TLR2−/−-mice and TLR2-Ab treated (T2.5) WT-mice displayed a reduction of infarct size, plasma troponin T concentrations, and leukocyte infiltration as compared with untreated controls after 24 h of reperfusion. After 28 days, however, magnetic resonance imaging revealed a marked left ventricular dilation in TLR2−/−-animals, which was associated with pronounced matrix remodeling characterized by reduced collagen and decorin density in the infarct scar. Our data show adverse effects on myocardial remodeling in TLR2−/−-mice. Although interception with TLR2 signaling is a promising concept for the prevention of reperfusion injury after myocardial ischemia, these data give cause for serious concern with respect to the time-point and duration of the potential treatment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Myocardial ischemia is one of the leading causes of death and morbidity in the western industrialized world. The consequences of myocardial ischemia and subsequent reperfusion (MI/R) can be modified by different preconditioning and therapeutic strategies, albeit the gross of compounds that have been reported to be cardioprotective in animals have not yet been transferred into human therapy [3, 5, 20].
This is also true for preconditioning, blockade, or genetic targeting of toll-like receptors (TLRs). TLRs are receptors of the innate immune system, which in addition to their recognition of bacterial wall fragments and other pathogen-associated patterns (PAMPs) [1, 4, 26] are responsible for the inflammatory response to a number of “danger associated patterns” (DAMPs) [25] that are released from host tissue itself upon sterile insult.
Of the family of TLRs specifically TLR2 and TLR4 are expressed in the heart. Pharmacologic preconditioning by application of TLR2 agonists such as Pam3CSK4 or lipoteichoic acid (LTA) or TLR4 agonist lipopolysaccharide (LPS) has been shown to provide cardioprotection in terms of reduction of infarct size or leukocyte infiltration [6, 17, 23, 30, 31]. Comparable effects have been observed in TLR2- or TLR4-deficient animals [7, 11, 19]. In the case of TLR4−/− mice, however, the initially observed infarct size reduction does not transfer into improved ventricular function after myocardial wound healing [15]. TLR2−/− mice show a blunted endothelial dysfunction and a reduction in infarct size after MI/R [11]. However, it is not known whether chronic TLR2 deficiency affects myocardial wound healing.
In a recent study, Arslan et al. [2] applied a TLR2-blocking antibody (T2.5) and reported subsequent infarct size reduction that was associated with an improved left ventricular function after 28 days.
In the present study, we provide evidence for an adverse course of myocardial wound healing in TLR2−/− mice despite the presence of analogous short-term effects in comparison with T2.5-treated animals. To identify potential underlying mechanisms we targeted extracellular matrix deposition in the infarct scar.
Materials and methods
Animals
C3HeN adult male wild-type (WT) and TLR2−/− mice [16] were used for the experiments. Animals were housed under specific pathogen-free conditions and given a standard diet and water ad libitum. All procedures were performed in accordance with the national guidelines on animal care and approved by the local government body.
Anti-TLR2 monoclonal antibody
A blocking monoclonal antibody (T2.5) directed against the extracellular domain of murine TLR2 cross-reacting with human TLR2 has been used as previously described [16]. Mice received an intravenous injection of 16 μg/g bodyweight T2.5 in 300 μl phosphate-buffered saline (PBS) 30 min prior to coronary artery ligation.
Myocardial ischemia/reperfusion (MI/R)
Anesthesia was induced by intra-peritoneal (i.p.) injection of pentobarbital (90 mg/kg). For postoperative analgesia buprenorphine (0.05 mg/kg) was administered subcutaneously. Mice were subjected to myocardial ischemia (20 min) and reperfusion (24 h, 3, 7, or 28 days) via occlusion of the left anterior descending coronary artery (LAD) as previously described [17, 20]. Successful occlusion was confirmed by the myocardium turning notably pale. After 20 min, the ligation was loosened and blood flow was restored. The chest was closed using two circular 6–0 polyvinyl sutures. When spontaneous breathing was sufficient, animals were disconnected from the ventilator and were allowed to breathe 100% oxygen. All animals received saline (16 μL/g bodyweight i.p.) to prevent dehydration.
Area at risk and infarct size determination
Hearts were perfused with 1% Evans blue dye (Sigma–Aldrich) to detect the area at risk (AR) as previously reported [20]. Heart slices were incubated for 25 min in 0.5% p-nitro-blue tetrazolium (NBT, Sigma–Aldrich), resulting in viable myocardium staining purple. IS, AR, and the total area of the left ventricle (LV) were planimetrically assessed using SigmaScan Pro (SPSS Inc., Chicago, IL, USA) image measurement software. Surgical procedures and planimetric measurements were performed by an investigator blinded for genotype or treatment.
Troponin T
Heparinized blood was centrifuged to obtain plasma and stored at −30°C until assayed. Troponin T (TnT) was measured using the Cardiac reader system (Roche Diagnostics, Basel, Switzerland) according to the manufacturer’s instructions.
Histology
Heart slices were fixed in 4% paraformaldehyde overnight, embedded in paraffin and cut into 5-μm sections. Hematoxylin and Eosin (H&E) staining was performed using standard protocols. Leukocyte accumulation in the AR was quantified on H&E stained sections. A total of ten microscopic fields covering 1 mm2 of the AR were photographed and leukocytes were counted by a blinded investigator.
Sirius red (SR) staining was performed according to standard protocols. Photomicrographs were taken from bright field and under polarized light from identical regions of each section. Biglycan (Bcn) and decorin (Dcn) were detected after chondroitinase ABC digestion with polyclonal rabbit antisera against murine biglycan (1:1,000, LF 106) and murine decorin (1:1,000, LF 113) kindly provided by Larry Fisher (National Institute of Dental and Craniofacial Research, National Institutes of Health, Bethesda, MD, USA).
Using SigmaScan Pro (SPSS Inc., Chicago, IL, USA) image measurement software the intensity of SR staining or Bgn and Dcn immuno-staining was quantified in regions of myocardial infarct scar formation, in which no cardiomyocytes were interposed. The average intensity of the region of interest was calculated and subtracted from the background intensity.
PCR and real-time PCR
Three and seven days after MI/R total RNA was isolated from snap-frozen tissue collected from the ischemic and remote area of WT (n = 7) and TLR2−/− mice (n = 7), delineated as described above by standard Evans Blue staining, using Tri Reagent (Sigma–Aldrich, Deisenhofen, Germany).
Reverse transcription of RNA was performed with SuperScript II Reverse Transcriptase (Invitrogen, Heidelberg, Germany), followed by amplification of the resulting cDNA (Taq PCR Core Kit; Qiagen, Hilden, Germany) according to the manufacturer’s instructions. Primers used were synthesized by Sigma. Cycling parameters were as following: denaturation at 95°C for 3 min, followed by 24 cycles (18S) or 30 cycles (all other genes) of 30 s at 95°C, 45 s at 58°C and 1 min at 72°C with a final extension of 5 min at 72°C. Amplification products were separated in 1–2% agarose gels and visualized with SYBR safe DNA gel stain (Invitrogen). Primers used were CGATCGCTACCCGGCGTTCC (forward) and GGGGTTCGGGCACTGCTTCC (reverse) for transforming growth factor β (TGF-β), GAGTTTCCGTGCCTGGCCCC (forward) and ACCTCGGGGACCCATCTGGC (reverse) for collagen α1, ACGAACTTCACCTGGACCAC (forward) and CAGAGGATAAGGGCAGCAAG (reverse) for biglycan, TAAAAGGTCGTGAAAATACAT (forward) and GAAGTCAAATAAGCCTCTCTG (reverse) for decorin, CCCAGGTGTGGGGTGCCTGA (forward) and TGGTGTTGTTGCACCTGTTGGCT (reverse) for matrix metalloproteinase 1 (MMP-1), CCGGTCCTGCTGTGGCTGTG (forward) and TGCGCCAAAAGTGCCTGTCT (reverse) for MMP-3, CCCTACCCGAGTGGACGCGA (forward) and AAGGCGGAGTCCAGCGTTGC (reverse) for MMP-9, and GGCCCCCTTTGCATCTCTGGC (forward) and CGTTCCTTAGGCGGCCCGTG (reverse) for tissue inhibitor of matrix metalloproteinase 1 (TIMP1).
The resulting 18S values were used as standard for presentation of the mRNA data of the Cnx43 transcript.
In addition, real-time PCR for decorin was performed from the same samples (n = 6 for each group due limitations of the 96-well plate) using SYBR Green incorporation on a StepOnePlus Real-Time PCR system (Applied Biosystems, Weiterstadt, Germany) according to the manufacturer’s instructions.
Magnetic resonance imaging (MRI)
Images were recorded with a 400-MHz DRX 9.4-T wide-bore nuclear magnetic resonance spectrometer (Bruker Biospin Corporation, Billerica, MA, USA) as previously described [13]. Mice were subjected to 20 min of ischemia and 28 days of reperfusion. For MRI mice were anesthetized with 1.2% isoflurane in 30% oxygen and 70% nitrogen. Animals were breathing spontaneously throughout the procedure. End-systolic and end-diastolic volume (ESV/EDV), mean wall diameter (mWD), and heart rate (HR) were measured. From these parameters stroke volume (SV), ejection fraction (EF), cardiac output (CO), and left ventricular mass per bodyweight (LV-mass/bw) were calculated. The mean wall diameter was computed as the average distance between the inner and outer circumference of the left ventricular wall on every virtual section of the heart.
Pre-existing differences in left ventricular dimensions or function have been excluded by echocardiography. Animals were anesthetized with isoflurane (2% in room air) and echocardiography was performed with a Vevo 770 High-Resolution In vivo Micro-Imaging System (Visual Sonics, Amsterdam, The Netherlands).
Statistics
Data are presented as mean ± SEM of n observations. Comparisons between groups were made using Prism 5.02 (GraphPad Software Inc., USA) after datasets passed normality testing—using the t test or ANOVA followed by Bonferroni’s post-hoc test. Comparisons between groups that failed to pass normality testing were made using the Mann–Whitney test. P values <0.05 were considered statistically significant. Correlations between parameters were estimated using the Pearson method. Best-fit lines in Figs. 5 and 6 were calculated by linear regression.
Results
TLR2 interception leads to reduced myocardial damage and inflammation
The area at risk [AR(%LV)] was not different between study groups (Fig. 1a). 24 h after subjecting TLR2−/− mice and WT animals treated with a TLR2-blocking monoclonal antibody (T2.5) to MI/R the mice hearts showed a significant reduction in infarct size in relation to the area at risk [IS(%AR)] as compared with untreated WT mice (Fig. 1b). Plasma troponin T (TnT) levels as an indicator of cardiomyocyte damage were also significantly lower in TLR2−/− and T2.5-treated WT animals (Fig. 1c). Since accumulation of inflammatory cells is one of the hallmarks of reperfusion injury, we quantified leukocytes in the AR. Both TLR2−/− and T2.5-treated WT mice displayed a significantly reduced tissue infiltration 24 h after MI/R compared with untreated WT mice (Fig. 1d).

Comparative analysis of MI/R effects in WT mice, TLR2 deficient mice (TLR2−/−), or WT mice treated with anti-TLR2 mAb (T2.5). Area at risk (a), infarct size (b), TnT plasma levels (c), and leukocyte recruitment (d) were analyzed 24 h upon experimental MI/R. 28 days after MI/R no significant differences could be detected in area at risk (e) or infarct size (f) between WT and TLR2−/− animals. * P < 0.05, ** P < 0.01, *** P < 0.001 compared with WT; one-way ANOVA/Bonferroni’s, n-numbers are depicted in the columns
LV-dilation of TLR2−/− hearts upon completion of infarct scar formation
In order to address whether TLR2 deficiency also positively affects remodeling processes and cardiac function, LV function was determined by magnetic resonance imaging after a prolonged follow-up period. Surprisingly, TLR2−/−-mice showed a marked dilation of the left ventricle 28 days after MI/R (Fig. 2a). End-systolic and end-diastolic volumes were significantly increased compared with WT hearts (Fig. 2b, c). The mean wall diameter was not different between groups (Fig. 2d), while the calculated LV mass/bodyweight was significantly higher in TLR2−/−-animals (Fig. 2e) as compared to WT controls.

Magnetic resonance imaging 28 days after MI/R. a Depicts representative MR images during end-systole and end-diastole. For video please see the online supplemental material. End-systolic (b) and end-diastolic (c) volumes were significantly decreased in TLR2−/− hearts as compared with WT hearts, whereas no difference in mean wall diameter (d) was detected. Ventricular wall hypertrophy is revealed by a significant increase in LV-mass in relation to bodyweight (e) in TLR2−/− animals. Mann–Whitney test, n = 6
Although the ejection fraction was significantly reduced in TLR2−/−-hearts, stroke volume was not affected by LV dilation (Fig. 3a, b). Under anesthetic conditions no difference in heart rate between groups could be observed and therefore cardiac output was preserved (Fig. 3c, d).

Cardiac function. The ejection fraction (EF) of the left ventricle was significantly decreased in TLR2−/− hearts (a), which—due to the observed dilation—results in equal stroke volumes (b) compared with WT. Since heart rates under isoflurane anesthesia were similar (c), cardiac output was also not affected by LV-dilation (d). Mann–Whitney test, n = 6
Baseline characterization of LV function in healthy WT and TLR2−/− mice was performed by echocardiography in a different set of experiments (n = 12). No differences regarding left-ventricular dimensions, ejection fraction, or heart rate could be detected (data not shown).
Collagen, biglycan, and decorin expression within the infarct scar
The intensity of SR-stained collagen fibers in the infarct scars of WT animals was significantly higher as compared with TLR2−/−-hearts (Fig. 4a, b). This finding was accompanied by a significantly increased intensity signal of decorin immuno-staining in infarct scars of WT animals (Fig. 4c). Biglycan deposition, however, did not significantly differ between groups (Fig. 4d). Hypertrophy of left-ventricular cardiomyocytes is aggravated in TLR2−/− hearts. Cardiomyocyte diameter in TLR2−/− left ventricles was significantly higher compared to WT in both ischemic and remote myocardium (Fig. 5).

Scar formation 28 days after MI/R. Intensity of collagen staining was quantified in infarcts scars in bright field (SR sirius red, a) and polarized light (pol, b). The intensity of immunostaining of the small leucine-rich proteoglycans decorin (Dcn, c) and biglycan (Bgn, d) was measured in regions of scar formation. TLR2−/− mice show a decreased density of collagen and decorin deposition in infarct scars, while biglycan deposition was not significantly affected. Mann–Whitney test, n = 6. e Depicts two examples from each group

Cardiomyocyte hypertrophy is more pronounced TLR2−/− hearts. Mean cardiomyocyte diameter was determined 28 days after MI/R (n = 6) and was significantly higher in TLR2−/− hearts in both ischemic and remote myocardium (a). A significant correlation between cardiomyocyte diameter and left ventricular mass/bodyweight could be observed (b). TLR2−/− animals are represented by open circles, WT animals by filled circles. CM cardiomyocyte, LV left ventricle
Decorin deposition correlates with collagen and biglycan density
As expected, collagen and decorin deposition correlated with each other in infarct scars, and likewise biglycan and decorin (Fig. 6a, b). No significant correlation was observed between collagen and biglycan deposition (r 2 = 0.2288, P = ns, not shown).

Correlations between collagen, biglycan, and decorin deposition, and LV dilation. Collagen and decorin deposition correlated in infarct scars (a). Likewise was the deposition of decorin correlated with biglycan (b). No significant correlation can be found between collagen and biglycan (not shown). Of note, a strong correlation between collagen deposition and end-systolic and –diastolic volumes and a weaker correlation with LV-mass/bodyweight was detected (c–e). Decorin deposition also correlates to a smaller degree with LV dilation but strongly with LV-mass/bodyweight (f–h). No significant correlation was found between biglycan and LV dilation or hypertrophy (not shown). TLR2−/− animals are represented by open circles, WT animals by filled circles
Collagen and decorin deposition correlate with LV dilation and hypertrophy
We observed a strong correlation between collagen deposition and end-systolic and end-diastolic volumes and a weaker correlation with LV-mass/bodyweight (Fig. 6c–e). Decorin deposition also correlated with LV dilation and even more strongly with LV-mass/bodyweight (F–H). No significant correlation could be found between biglycan and LV dilation or hypertrophy (r 2 = 0.1265 vs. end-systolic volume, r 2 = 0.1713 vs. end-diastolic volume, r 2 = 0.3301 vs. LV-mass/bodyweight; all P = ns, not shown).
No differences in mRNA expression of matrix regulators after 3 and 7 days of reperfusion
PCR analysis did not reveal significant differences in TGF-β, MMP-1, -3 and -9, TIMP1, biglycan, decorin, and collagen α1 chain mRNA expression between WT and TLR2−/− animals in both ischemic and remote myocardium 3 and 7 days after MI/R.
Discussion
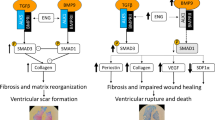
Here, we demonstrate a transient cardioprotection capacity of TLR2 deficiency in experimental MI/R. However, timely limitation of TLR2 blockade might be critical to prevent adverse myocardial scar formation and remodeling resulting in LV-dilation.
We have reported previously that TLR2−/− mice are protected against lethal brady-arrhythmias within the first 24 h of reperfusion [18].
A report by Arslan et al. [2] demonstrated cardioprotection by means of TLR2 pathway interception through TLR2 blockade. These authors demonstrated a short-term and long-term benefit when the antibody was injected shortly prior to reperfusion. Due to a limited half-life of the antibody used [16] it is reasonable to assume that TLR2 blockade was effective during the initial 4 h of reperfusion within which the oxidative burst causing inflammation and promoting infarct formation occurs. During the later phase of reperfusion when myocardial wound healing is initiated, TLR2 function had been recovered to support myocardial wound healing and scar formation. The observed improved left ventricular function 28 days after MI/R is interpreted by us as a result of a smaller infarct size as compared with controls.
The short-term effects of TLR2 antibody blockade was confirmed in the present study. Furthermore, TLR2−/− mice developed infarcts, troponin T levels, and leukocyte infiltration of similar sizes and degrees as compared with antibody-treated mice. However, contrasting the improved LV-function reported for antibody-treated animals [2], LV-function in TLR2−/− animals was impaired.
Subsequent to myocardial ischemia/reperfusion injury, cardiac fibroblasts are activated to produce extracellular matrix molecules to preserve mechanical stability of the ventricular wall [29]. The most prominent extracellular matrix molecules are collagens which are subject to post-translational modification by small leucine-rich proteoglycans such as biglycan and decorin. The latter mediate collagen fibril organization thereby increasing the mechanical strength of the infarct scar. Decorin null-mutant mice display loosely packed and less organized collagen fibrils in myocardial infarct scars leading to ventricular dilation and impaired left ventricular systolic function [27]. We have previously shown that biglycan deficiency leads to disturbed collagen deposition after myocardial infarction with subsequent ventricular rupture [28]. TLR2 signaling has been connected to biglycan by Schaefer et al. [22] who identified biglycan as a ligand of TLR2 in macrophages inducing the production of inflammatory cytokines via the NF-κB pathway. A more recent study provided evidence for a regulatory role of TLR2 on phospholipid transfer protein expression after co-localization with biglycan in human aortic valve stenosis [10]. However, Csont et al. [8] observed cardioprotective/anti-inflammatory properties of biglycan when primary rat cardiomyocytes were treated with biglycan prior to hypoxia/reoxygenation. To our knowledge no direct link between TLR2 and decorin has been established to date.
In the present study—in parallel with impaired hemodynamics—decorin and collagen deposition within the infarct scar were reduced in TLR2−/−-animals. In fact, decorin and collagen density negatively correlated with left ventricular dilation (end-systolic and end–diastolic volume). Decorin binds to collagen fibrils and is thought to control the lateral fusion of fibrils and the structure of three-dimensional collagen networks [9, 12, 14]. We therefore assume that the mechanical strength of the ventricular wall is impaired due to down-regulation of decorin and a subsequently decreased collagen network compaction in post-ischemic TLR2−/− hearts, and that dilation may be a direct result of this. Subsequently, cardiomyocyte hypertrophy compensates for the loss of ventricular wall integrity, which might be the reason for the observed negative correlation between LV-mass/bodyweight and collagen or decorin. Of note, no significant difference in biglycan density could be observed between TLR2−/− and WT infarct scars. But although biglycan and decorin are thought to synergistically facilitate collagen organization, one does not compensate for the lack of the other [27, 28].
TGF-β being an early activator of extracellular matrix production [21, 24] was not differentially regulated in TLR2−/− animals. Nor could differences in gene expression of matrix metalloproteinases 1, 3, and 9, and their inhibitor TIMP1 be detected, which mediate collagen turnover after deposition. MMPs are, however, subject to posttranslational activation. So the finding of equal MMP and TIMP synthesis does not exclude different activities with a subsequent increased degradation or turnover of collagen fibrils in TLR2−/− hearts. This might furthermore be supported by the fact that the transcription products of collagen α1, decorin and biglycan were also not changed in TLR2−/− hearts, besides the observed differences in collagen and decorin deposition in the infarct scar.
In summary, the results presented here underline that interference with TLR2 signaling in the reperfused myocardium is capable of limiting infarct size and initial cardiomyocyte necrosis. Furthermore, they imply undisturbed TLR2 signaling as mandatory for scar formation. TLR2 pathway interception should therefore be tightly limited in time in order to minimize myocardial damage and abrogate lethal arrhythmias while permitting TLR2 signaling during myocardial wound healing.
References
Akira S, Uematsu S, Takeuchi O (2006) Pathogen recognition and innate immunity. Cell 124:783–801
Arslan F, Smeets MB, O’Neill LA, Keogh B, McGuirk P, Timmers L, Tersteeg C, Hoefer IE, Doevendans PA, Pasterkamp G, de Kleijn DP (2010) Myocardial ischemia/reperfusion injury is mediated by leukocytic toll-like receptor-2 and reduced by systemic administration of a novel anti-toll-like receptor-2 antibody. Circulation 121:80–90
Atar D, Petzelbauer P, Schwitter J, Huber K, Rensing B, Kasprzak JD, Butter C, Grip L, Hansen PR, Suselbeck T, Clemmensen PM, Marin-Galiano M, Geudelin B, Buser PT (2009) Effect of intravenous FX06 as an adjunct to primary percutaneous coronary intervention for acute ST-segment elevation myocardial infarction results of the F.I.R.E. (Efficacy of FX06 in the Prevention of Myocardial Reperfusion Injury) trial. J Am Coll Cardiol 53:720–729
Beutler B, Hoebe K, Du X, Ulevitch RJ (2003) How we detect microbes and respond to them: the Toll-like receptors and their transducers. J Leukoc Biol 74:479–485
Bolli R, Becker L, Gross G, Mentzer R Jr, Balshaw D, Lathrop DA (2004) Myocardial protection at a crossroads: the need for translation into clinical therapy. Circ Res 95:125–134
Brown JM, Grosso MA, Terada LS, Whitman GJ, Banerjee A, White CW, Harken AH, Repine JE (1989) Endotoxin pretreatment increases endogenous myocardial catalase activity and decreases ischemia-reperfusion injury of isolated rat hearts. Proc Natl Acad Sci USA 86:2516–2520
Chong AJ, Shimamoto A, Hampton CR, Takayama H, Spring DJ, Rothnie CL, Yada M, Pohlman TH, Verrier ED (2004) Toll-like receptor 4 mediates ischemia/reperfusion injury of the heart. J Thorac Cardiovasc Surg 128:170–179
Csont T, Görbe A, Bereczki E, Szunyog A, Aypar E, Tóth ME, Varga ZV, Csonka C, Fülöp F, Sántha M, Ferdinandy P (2010) Biglycan protects cardiomyocytes against hypoxia/reoxygenation injury: Role of nitric oxide. J Mol Cell Cardiol 48:649–652
Danielson KG, Baribault H, Holmes DF, Graham H, Kadler KE, Iozzo RV (1997) Targeted disruption of decorin leads to abnormal collagen fibril morphology and skin fragility. J Cell Biol 136:729–743
Derbali H, Bosse Y, Cote N, Pibarot P, Audet A, Pepin A, Arsenault B, Couture C, Despres JP, Mathieu P (2010) Increased biglycan in aortic valve stenosis leads to the overexpression of phospholipid transfer protein via Toll-like receptor 2. Am J Pathol 176:2638–2645
Favre J, Musette P, Douin-Echinard V, Laude K, Henry JP, Arnal JF, Thuillez C, Richard V (2007) Toll-like receptors 2-deficient mice are protected against postischemic coronary endothelial dysfunction. Arterioscler Thromb Vasc Biol 27:1064–1071
Fischer JW, Kinsella MG, Clowes MM, Lara S, Clowes AW, Wight TN (2000) Local expression of bovine decorin by cell-mediated gene transfer reduces neointimal formation after balloon injury in rats. Circ Res 86:676–683
Flogel U, Laussmann T, Godecke A, Abanador N, Schafers M, Fingas CD, Metzger S, Levkau B, Jacoby C, Schrader J (2005) Lack of myoglobin causes a switch in cardiac substrate selection. Circ Res 96:e68–e75
Jarvelainen H, Vernon RB, Gooden MD, Francki A, Lara S, Johnson PY, Kinsella MG, Sage EH, Wight TN (2004) Overexpression of decorin by rat arterial smooth muscle cells enhances contraction of type I collagen in vitro. Arterioscler Thromb Vasc Biol 24:67–72
Kim SC, Ghanem A, Stapel H, Tiemann K, Knuefermann P, Hoeft A, Meyer R, Grohe C, Knowlton AA, Baumgarten G (2007) Toll-like receptor 4 deficiency: smaller infarcts, but no gain in function. BMC Physiol 7:5–5
Meng G, Rutz M, Schiemann M, Metzger J, Grabiec A, Schwandner R, Luppa PB, Ebel F, Busch DH, Bauer S, Wagner H, Kirschning CJ (2004) Antagonistic antibody prevents toll-like receptor 2-driven lethal shock-like syndromes. J Clin Invest 113:1473–1481
Mersmann J, Berkels R, Zacharowski P, Tran N, Koch A, Iekushi K, Dimmeler S, Granja TF, Boehm O, Claycomb WC, Zacharowski K (2010) Preconditioning by toll-like receptor 2 agonist Pam3CSK4 reduces CXCL1-dependent leukocyte recruitment in murine myocardial ischemia/reperfusion injury. Crit Care Med 38:903–909
Mersmann J, Koch A, Tran N, Zimmermann R, Granja TF, Larmann J, Herzog C, Theilmeier G, Bornstein SR, Kirschning CJ, Zacharowski K (2010) Toll-like receptor 2 signaling triggers fatal arrhythmias upon myocardial ischemia-reperfusion. Crit Care Med 38:1927–1932
Oyama J, Blais C Jr, Liu X, Pu M, Kobzik L, Kelly RA, Bourcier T (2004) Reduced myocardial ischemia-reperfusion injury in toll-like receptor 4-deficient mice. Circulation 109:784–789
Petzelbauer P, Zacharowski PA, Miyazaki Y, Friedl P, Wickenhauser G, Castellino FJ, Groger M, Wolff K, Zacharowski K (2005) The fibrin-derived peptide B beta15–42 protects the myocardium against ischemia-reperfusion injury. Nat Med 11:298–304
Sakata Y, Chancey A, Divakaran V, Sekiguchi K, Sivasubramanian N, Mann D (2008) Transforming growth factor-beta receptor antagonism attenuates myocardial fibrosis in mice with cardiac-restricted overexpression of tumor necrosis factor. Basic Res Cardiol 103:60–68
Schaefer L, Babelova A, Kiss E, Hausser HJ, Baliova M, Krzyzankova M, Marsche G, Young MF, Mihalik D, Gotte M, Malle E, Schaefer RM, Grone HJ (2005) The matrix component biglycan is proinflammatory and signals through Toll-like receptors 4 and 2 in macrophages. J Clin Invest 115:2223–2233
Schober P, Oprea G, Mersmann J, Nebert A, Zacharowski K, Zacharowski PA (2008) Lipoteichoic acid induces delayed myocardial protection in isolated rat hearts: a comparison with endotoxin. Resuscitation 79:311–315
Tiede K, Melchior-Becker A, Fischer J (2010) Transcriptional and posttranscriptional regulators of biglycan in cardiac fibroblasts. Basic Res Cardiol 105:99–108
Vabulas RM, Ahmad-Nejad P, da Costa C, Miethke T, Kirschning CJ, Hacker H, Wagner H (2001) Endocytosed HSP60s use toll-like receptor 2 (TLR2) and TLR4 to activate the toll/interleukin-1 receptor signaling pathway in innate immune cells. J Biol Chem 276:31332–31339
Valeur H, Valen G (2009) Innate immunity and myocardial adaptation to ischemia. Basic Res Cardiol 104:22–32
Weis SM, Zimmerman SD, Shah M, Covell JW, Omens JH, Ross J Jr, Dalton N, Jones Y, Reed CC, Iozzo RV, McCulloch AD (2005) A role for decorin in the remodeling of myocardial infarction. Matrix Biol 24:313–324
Westermann D, Mersmann J, Melchior A, Freudenberger T, Petrik C, Schaefer L, Lullmann-Rauch R, Lettau O, Jacoby C, Schrader J, Brand-Herrmann SM, Young MF, Schultheiss HP, Levkau B, Baba HA, Unger T, Zacharowski K, Tschope C, Fischer JW (2008) Biglycan is required for adaptive remodeling after myocardial infarction. Circulation 117:1269–1276
Whittaker P (1997) Collagen and ventricular remodeling after acute myocardial infarction: concepts and hypotheses. Basic Res Cardiol 92:79–81
Zacharowski K, Frank S, Otto M, Chatterjee PK, Cuzzocrea S, Hafner G, Pfeilschifter J, Thiemermann C (2000) Lipoteichoic acid induces delayed protection in the rat heart: a comparison with endotoxin. Arterioscler Thromb Vasc Biol 20:1521–1528
Zacharowski K, Otto M, Hafner G, Chatterjee PK, Thiemermann C (1999) Endotoxin induces a second window of protection in the rat heart as determined by using p-nitro-blue tetrazolium staining, cardiac troponin T release, and histology. Arterioscler Thromb Vasc Biol 19:2276–2280
Acknowledgment
This work was supported by a grant from the Deutsche Forschungsgemeinschaft (SFB 815/A17) to K.Z.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Mersmann, J., Habeck, K., Latsch, K. et al. Left ventricular dilation in toll-like receptor 2 deficient mice after myocardial ischemia/reperfusion through defective scar formation. Basic Res Cardiol 106, 89–98 (2011). https://doi.org/10.1007/s00395-010-0127-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00395-010-0127-y