
Abstract
The present study investigates the effects of the first mutation of troponin C (hcTnCL29Q) found in a patient with hypertrophic cardiomyopathy (HCM) on force–pCa relations and the interplay with phosphorylation of sarcomeric PKA substrates. In triton-skinned murine cardiac fibers, the endogenous mcTnC was extracted and the fibers were subsequently reconstituted with recombinant wild-type and mutant hcTnC. Force–pCa relations of preparations containing hcTnCL29Q or hcTnCWT were similar. Incubation of fibers reconstituted with the recombinant proteins with phosphatase to dephosphorylate sarcomeric PKA substrates induced an increase in Ca2+ sensitivity, slightly more pronounced (0.04 pCa units) in hcTnCL29Q-containing fibers. Incubation of the dephosphorylated fibers with PKA induced significant rightward shifts of force–pCa relations of similar magnitude with both, hcTnCL29Q and hcTnCWT. No significant effects of hcTnCL29Q on the velocity of unloaded shortening were observed. In conclusion, no major differences in contractile parameters of preparations containing hcTnCL29Q compared to hcTnCWT were observed. Therefore, it appears unlikely that hcTnCL29Q induces the development of HCM by affecting the regulation of Ca2+-activated force and interference with PKA-mediated modulation of the Ca2+ sensitivity of contraction.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cardiac troponin C (cTnC) is the Ca2+-binding subunit of the heterotrimeric troponin complex (cTn), which further consists of the inhibitory subunit, troponin I (cTnI), and the tropomyosin binding subunit, troponin T (cTnT), and is essential in initiating contraction of the heart. As an EF-hand protein, cTnC consists of an N-terminal and a C-terminal lobe, which each contain two metal-binding sites. In cardiac muscle only the reversible Ca2+ binding to a single site, the regulatory site II in the N-terminal lobe of cTnC (cNTnC), is essential for the regulation of contraction, while regulatory site I is non-functional, however, its amino acid composition may influence the Ca2+-binding properties of site II [17, 28]. Sites III and IV of the C-terminal lobe are permanently metal-bound under physiological conditions and are thought to serve a structural function (reviewed in [22]).
During the systolic elevation of cytosolic Ca2+, Ca2+ binding to the regulatory site II of cTnC opens a hydrophobic pocket in cNTnC, thereby exposing strong interaction sites for the switch peptide of cTnI (~residues 147–163). This interaction pulls the inhibitory region of cTnI (~residues 128–147) away from its binding site on actin, allowing strong binding of myosin crossbridges to actin and thus providing the basis for Ca2+-induced cardiac contraction (reviewed in [22]). Specific to the heart, the “open” conformation is stabilized by binding of the N-terminal arm of cTnI (residues 16–29) to cNTnC [42]. This interaction is weakened by PKA-mediated phosphorylation of Ser23/24 of cTnI giving rise to the well-known decrease in Ca2+ sensitivity of actomyosin interaction and contraction which is thought to contribute to the lusitropic effect of β-adrenergic receptor stimulation (reviewed in [26 and references therein; 30]). Given the prominent role cTnC plays in the regulation of cardiac contraction, it is intuitively apparent that an impaired function of cTnC will likely lead to inefficient regulation of contraction and subsequently to systolic or diastolic dysfunction which may result in cardiomyopathy.
Hypertrophic cardiomyopathy (HCM) is considered to be the most frequent inherited disease of the myocardium and thought to be caused by point mutations in genes encoding proteins of the cardiac sarcomere (reviewed in [39]). More recently it was also recognized that about 30% of idiopathic dilated cardiomyopathies can be ascribed to mutations of sarcomeric proteins (reviewed in [1]). However, despite the fact that, in many sarcomeric proteins, a large number of HCM- or DCM-linked mutations were found, relatively few mutations were found so far in human cTnC [4, 21, 25, 31]. Whereas HCM is characterized by diastolic dysfunction and normal systolic function [29], DCM is characterized by systolic dysfunction [6]. The tendency of HCM-linked mutations in cTn subunits to increase and of DCM-linked mutations to decrease the Ca2+ sensitivity of contraction [9, 38] may reflect the fact that sarcomeric processes contribute to impaired relaxation (HCM) or contraction (DCM) of the myocardium.
The HCM-associated mutation hcTnCL29Q was found in a 60-year-old male patient who presented with dyspnea on exertion and echocardiographic examination revealed a concentric hypertrophy of the left ventricle [21]. Leu-29 is located at the transition of helix A to the non-functional Ca2+-binding site I. Amino acid substitutions in this region may impact on the structure and the Ca2+ affinity of site II of cNTnC. Indeed, in cold water fish, the L29Q substitution belongs to a series of amino acid exchanges that increase Ca2+ sensitivity of the heart at low temperatures [10]. Further, L29Q is located adjacent to the residues which bind to the non-phosphorylated N-terminal arm of cTnI. Studies with isolated proteins indicate that the mutation interferes with binding of the N-terminal arm of cTnI to cNTnC, which in effect would mimic the modulatory effect of cTnI phosphorylation at Ser23/24 on Ca2+ activation [4, 13, 35]. These studies give rise to the interesting possibility that the mutation will alter Ca2+ activation and affect the functional transduction of PKA-dependent phosphorylation of cTnI to Ca2+-activated force; however, functional studies yielded highly contradictory results as to its effects on Ca2+-dependent force and on acto-S1 ATPase activity [15, 28, 35].
The effects of PKA-mediated phosphorylation of cTnI are critical for the response of the heart to β-adrenergic stimulation, and alterations in the beta-adrenergic response are thought to be a major mechanism contributing to the pathogenesis of heart failure [40, 41]. In accordance with the structural studies [4, 13, 35] the modulatory effect of cTnI phosphorylation by PKA on the Ca2+ sensitivity of acto-S1 MgATPase activity and of sliding velocity of actin filaments in the in vitro motility assay was abrogated [35]. However, it remains unknown whether the PKA-induced decrease in Ca2+ sensitivity of contraction is affected in the myofilament lattice of myofibrils.
The aim of the present study was therefore to investigate the effect of recombinant hcTnCL29Q on isometric force and on unloaded shortening velocity of triton-skinned myocardium. Such preparations are devoid of functional cell membranes and a functional sarcoplasmatic reticulum, while the myofilament lattice of the myofibrils remains structurally intact. Biochemical exchange protocols allow to incorporate recombinant troponin subunits into the cardiac sarcomeres [12, 15, 28]. In particular, we investigated whether the mutation abrogates the PKA-induced decrease in Ca2+ sensitivity of contraction in myofibrils by dephosphorylating skinned fibers containing hcTnCL29Q with the catalytic subunit of Mn2+-PP1c and rephosphorylating them with PKA [33].
Materials and methods
Skinned cardiac fibers
The ‘‘Principle of laboratory animal care’’ (NIH publication No. 86–23, revised 1985) were followed, and the study was approved by the responsible Animal Care and Use Committee. Adult male mice of the strain HIM:OF1 were sacrificed by cervical dislocation. The hearts were removed and immediately transferred to ice-cold preparation buffer containing 132 mM NaCl, 5 mM KCl, 1 mM MgCl2, 10 mM Tris, 5 mM EGTA, 1 mM NaN3, pH 7.1 (20°C). Fibers of a diameter of ~200 μm were prepared on ice from left ventricular papillary muscles. For skinning, the fibers were fixed isometrically and incubated in skinning solution on ice for 4 h. The skinning solution contained 5 mM KH2PO4, 5 mM NaN3, 3 mM magnesium acetate, 5 mM K2EGTA, 3 mM Na2MgATP, 47 mM sodium creatine phosphate, 2 mM dithiothreitol (DTT), 0.2 mM 4-(2-aminoethyl)benzenesulfonyl-fluoride (AEBSF), 10 μM leupeptin, 10 μM antipain, 5 mg/l aprotinin, and 1% (m/v) Triton X-100. After skinning, the fibers were stored on ice in skinning solution without Triton X-100 over night.
Isometric tension and force data analysis
Activating and relaxing solution contained either 3 mM (Ca)K2EGTA (activating solution) or 3 mM K2EGTA (relaxing solution), 10 mM imidazole, 10 mM Na2MgATP, 3 mM MgCl2, 32.7 mM sodium creatine phosphate, 2 mM DTT, pH 7.0 (10°C), μ = 178 mM. Skinned fibers were mounted between a force transducer (KG7A with bridge-amplifier DUBAM 7C, Scientific Instruments, Heidelberg) and a fixed clamp in skinning solution without Triton X-100 and pre-stretched by 10% of their initial length. Before recording force–pCa relations, a test contraction was elicited in activating solution (pCa 4.6). The fibers were then transferred to relaxing solution (pCa 8). Force–pCa relations were recorded by incubating the fibers in solutions with cumulatively rising Ca2+ concentrations which were mixed from activating and relaxing solutions, and pCa50 (pCa required for half-maximal tension) and n H (Hill coefficient) were calculated by fitting the data to the Hill equation as described [36]. Unless noted otherwise, the experiments were performed at 10°C controlled by thermo-electric coolers to preserve the structural integrity of fibers during activation [36].
Unloaded shortening velocity
After mounting, the preparations were pre-stretched by 10% of their initial length. The fibers were activated (pCa 4.6) at 10°C and force was allowed to stabilize for ~5 min. Unloaded shortening velocities were determined with two different methods using a commercial setup (Scientific Instruments, Heidelberg, Germany):
-
1.
Force–velocity relations were recorded using the technique of isotonic load clamping: briefly, shortening velocities were recorded at constant loads clamped to values between 5 and 30% of the force during activation. The unloaded shortening velocity (V MAX) was calculated for each preparation according to Hill [20] using Sigma Plot 4.0 (Systat Software Inc., Richmond, California, USA).
-
2.
Unloaded shortening velocities were determined using the “Edman slack test” [16]: briefly, a series of nine releases (ramp time ~1 ms) of an amplitude between 12 and 17% of the fiber length was applied onto the preparation. The fiber was stretched back to its initial length 0.2 s after each release. Force was monitored continuously during this procedure. The duration of unloaded shortening was determined for each release, and the velocity of unloaded shortening was calculated according to Edman [16] using Sigma Plot 4.0 (Systat Software Inc.).
Extraction of endogenous troponin C and reconstitution with recombinant human cardiac troponin C
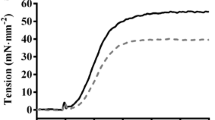
Recombinant human cardiac troponin C (hcTnCL29Q and hcTnCWT) was generously provided by K. Jaquet (Bochum, Germany). For extraction of endogenous murine cTnC (mcTnC) and reconstitution with hcTnC, the method of Dohet et al. [12] was modified (cf. Fig. 1). The complete procedure was performed at 10°C. Skinned fibers were incubated for 10 min in buffer 1 containing 10 mM imidazole, 2.5 mM EGTA, 7.5 mM EDTA, 135 mM potassium propionate, pH 6.8 (20°C). After this, the fibers were incubated for 180 min in extraction buffer containing 0.1 mM trifluoperazine, 2 mM trans-1,2-diaminocyclohexane-N,N,N,N-tetraacetic acid (CDTA), 2 mM imidazole, 1 mM NaN3, 2 mM DTT, 0.5 mM AEBSF, 0.01 mM leupeptin, 0.01 mM antipain, 5 mg/l aprotinin, pH 7.4 (20°C). Then the fibers were equilibrated in relaxing solution for 5 min. To check the efficiency of mcTnC extraction, the fibers were Ca2+-activated (pCa 4.6) for 15 min. Only fibers which did not develop force in activating solution any more were used for cTnC reconstitution and incubated in relaxing solution containing either 2 g/l hcTnCL29Q or hcTnCWT for 60 min. Following reconstitution with recombinant hcTnC, force–pCa relations or unloaded shortening were measured as described above either directly, or after application of the protocols for dephosphorylation or PKA phosphorylation.

Force transient recorded during cTnC extraction and reconstitution. Maximal Ca2+-activated force (F MAX) is determined at pCa 4.6, then mcTnC is extracted by incubation in “buffer 1” and extraction buffer (B1, EB, see methods). Ca2+-activated force is lost after 180 min incubation in EB. Following reconstitution with recombinant hcTnC Ca2+-dependent force is partially restored
Phosphatase treatment
Phosphatase treatment was performed as described [33]. Briefly, skinned fibers were equilibrated for 10 min at 10°C with PP1c buffer containing 20 mM Na2MnATP, 3 mM MnCl2, 3 mM K2EGTA, 10 mM imidazole, 5 mM MgCl2, 10 mM sodium creatine phosphate and 2 mM DTT, pH 6.85 (20°C), followed by incubation in the same buffer with 0.5 kU/ml PP1c (Sigma, Deisenhofen, Germany) at 10°C for 90 min. Following PP1c treatment the fibers were incubated in relaxing solution containing 50 mM DTT at 20°C for 30 min.
PKA phosphorylation
Recombinant human catalytic subunit (C-subunit) of PKA (14 MU/ml, generously provided by K. Jaquet, Bochum, Germany) was stored at 4°C in 1 mM EDTA, 2 mM DTT, 5 mM β-mercaptoethanol, 150 mM K2HPO4/KH2PO4, pH 6.5 [19]. The C-subunit was diluted in relaxing solution to give a final activity of 0.5 MU/ml. The fibers were incubated in this solution at 20°C for 60 min. Control fibers were subjected to the same protocol with appropriate control buffer (PKA buffer) without C-subunit. Phosphorylation was verified by incubating fibers with PKA in the presence of [γ32P]-ATP and autoradiography using X-ray films as described [33].
Statistics
All data were calculated using SIGMA PLOT 4.0 (Systat Software Inc., Richmond, California, USA) and are given as mean ± SEM. “n” in figures and tables refers to the number of animals. Differences among groups was tested by Student’s t-test or one way ANOVA and Newman–Keuls test as post-test using graph pad software, a P value <0.05 was considered as statistically significant.
Results
Effect of hcTnCL29Q on isometric force
To assess the effect of the hcTnCL29Q mutation on Ca2+-activated steady state force and on shortening velocity, endogenous murine cardiac troponin C (mcTnC) was extracted from skinned fibers by incubation in extraction buffer for 3 h followed by reconstitution with either wild-type or mutant hcTnC. Incubation in extraction buffer induced a complete loss of Ca2+-dependent force in activating solution (pCa 4.6, see Fig. 1). Ca2+-regulated force recovered after reconstitution of the fibers in relaxing solution containing recombinant hcTnC. However, as reported by others [3, 12, 15, 28], maximal Ca2+-activated force (pCa 4.6) in the reconstituted fibers was reduced (Fig. 1), which was similar in fibers exchanged for hcTnCWT or hcTnCL29Q (Fig. 2; Table 1).

Force–pCa relations of fibers reconstituted with hcTnCL29Q or hcTnCWT. Force–pCa relations were measured before extraction of mcTnC (dashed lines, open circle) and after reconstitution with hcTnCL29Q (a) or hcTnCWT (b), respectively (solid lines, open triangle). Symbols represent mean ±SEM for n = 6 preparations
To assess the effect of the mutation on the force–pCa relation, a full force–pCa relation was obtained before extraction and after reconstitution. Thus, each fiber served as its own control. As a measure of Ca2+ sensitivity the pCa50 value, and as a measure of cooperativity the Hill coefficient was calculated for each curve. Before extraction, the force–pCa relations were similar between fibers to be exchanged for either wild-type or mutant hcTnC. Following reconstitution, the Ca2+ sensitivity slightly decreased by a similar extent in both cases, which did not reach the level of statistical significance (Fig. 2; Table 1). The shifts in pCa50 were calculated for each fiber, giving similar results for the exchange procedure with hcTnCL29Q or hcTnCWT (cf. Table 4). The steepness of the force–pCa relation, which is indicative of the cooperativity, was not significantly different between hcTnCL29Q- and hcTnCWT-containing fibers (Fig. 2; Table 1). Passive force, i.e. force in the absence of Ca2+, also remained similar in fibers exchanged with wild-type or mutant hcTnC (Figs. 2, 3, 4; Tables 1, 2, 3).

Dephosphorylation of PKA targets by Mn2+-dependent PP1c. Fibers were either treated or not treated with PP1c and then incubated with PKA in the presence of [γ32P]-ATP as in [32]. Lysates were subjected to SDS-PAGE. Gels were Coomassie-stained, dried, and exposed to X-ray films. 32P incorporation into cTnI was enhanced by ~twofold in fibers pretreated with PP1c

Phosphatase and PKA treatment of cTnC-exchanged fibers. Force–pCa relations were measured before extraction of endogenous mcTnC (dashed lines, open triangle) and after reconstitution with hcTnCL29Q (a) or hcTnCWT (b) and treatment with PP1c (solid lines, open circle). After phosphatase treatment, the pCa50 values were not significantly different between fibers reconstituted with hcTnCL29Q or hcTnCWT, respectively. Symbols represent mean ±SEM for n = 5 preparations. After phosphatase treatment, PKA was added (dotted lines, open square), resulting in significant rightward shifts of the force–pCa relations of similar magnitude. Symbols represent mean ±SEM for n = 3 preparations
Effect of hcTnCL29Q on isometric force after treatment with phosphatase and PKA
We have previously shown that, in triton-skinned murine cardiac fibers, sarcomeric PKA substrates are phosphorylated in a functionally relevant manner [24, 33], which means that hcTnCL29Q and hcTnCWT were exchanged into fibers containing phosphorylated cTnI. This may influence the effect of the mutation on the force–pCa relation [4, 14, 35]. We previously reported [24, 33] that the commercially available Mn2+-dependent catalytic subunit of PP1c which has a different substrate specificity than the type 1 phosphatases can be used as a tool to dephosphorylated these proteins. To verify that cTnI in the fibers of the present study was phosphorylated, fibers were incubated with or without PP1c and then treated with PKA in the presence of [γ32P]-ATP. Similar to our previous study [33], PKA incorporated ~twofold more 32P into cTnI after pretreatment with phosphatase (Fig. 3). We note, that in the present series of experiment, 32P incorporation into cMyBP-C is less than in our previous report [33] which is probably due to the lower specific activity of [γ32P]-ATP used here as detection of 32P incorporation into cMyBP-C requires a higher specific activity (unpublished observations). It remains unknown whether PP1c treatment also dephosphorylates other proteins; however, it is unlikely that dephosphorylation of PKA-independent sites contributes to the PP1c-induced changes of the force–pCa relation because PKA quantitatively reverses the effect of PP1c [33].
In both, hcTnCL29Q- or hcTnCWT-exchanged fibers, treatment with Mn2+-dependent catalytic subunit of PP1c resulted in an increase in Ca2+ sensitivity. Passive force and pCa50 increased and n H slightly decreased compared to the force–pCa relations before mcTnC extraction, while the reduction of maximal force was similar to the fibers not treated with phosphatase (see Fig. 4; Table 2). The increase in Ca2+ sensitivity was more pronounced in fibers exchanged for hcTnCL29Q compared to the fibers exchanged for hcTnCWT (ΔpCa50 = 0.04 pCa units; see Tables 2, 4). However, this difference did not reach the level of statistical significance.
In a reconstituted system, the PKA-induced decrease in Ca2+ sensitivity of acto-S1 ATPase activity was abrogated by the mutation [35]. To investigate whether in hcTnCL29Q-exchanged skinned fibers the PKA-induced decrease in Ca2+ sensitivity on contraction is also lost (cf. [35]), the phosphatase-treated fibers were incubated with PKA. As previously reported [33], this resulted in a significant rightward shift of the force–pCa relation of similar magnitude in both, fibers exchanged with hcTnCL29Q (ΔpCa50 = 0.20 pCa units) and hcTnCWT (ΔpCa50 = 0.18 pCa units; cf. Fig. 4; Tables 3, 4), whereas no significant changes were observed after incubation with PKA buffer only (data not shown). Hence, hcTnCL29Q does not blunt the PKA-induced Ca2+-desensitization of force.
Effect of hcTnCL29Q on the velocity of unloaded shortening
To investigate whether hcTnCL29Q affects the velocity of unloaded shortening at high [Ca2+], endogenous mcTnC was extracted and force reconstituted with either hcTnCL29Q or hcTnCWT as above. The velocity of unloaded shortening was determined before and after extraction of mcTnC and reconstitution with hcTnCL29Q or hcTnCWT from load–velocity relations. After reconstitution, the velocity of unloaded shortening was reduced by 37% (hcTnCL29Q) or 35% (hcTnCWT) compared to unloaded shortening before mcTnC extraction (see Fig. 5). These findings were confirmed by data from the “Edman slack test”. Here, the velocity of unloaded shortening was reduced by 37% (hcTnCL29Q) or 39% (hcTnCWT, see Fig. 5). In summary, hcTnCL29Q compared to hcTnCWT did not influence the velocity of unloaded shortening in a significantly different way.

Unloaded shortening velocity. The velocity of unloaded shortening was determined before extraction of mcTnC (control, gray bars) and after reconstitution (transparent bars) with hcTnCL29Q (n = 6) or hcTnCWT (n = 6) using the method of isotonic load clamping and the Edman slack test
Discussion
L29Q was the first mutation of cTnC described in one patient with hypertrophic cardiomyopathy and no available family history [21]. However, since this mutation is in an area of cTnC which has the potential to affect the activation properties of cTnC [4, 13, 28, 35], its functional effects were investigated by several groups resulting in rather divergent observations. However, none of the studies in fibers investigated whether this mutation blunts the response to PKA as suggested by structural investigations and observations with reconstituted proteins in solution [4, 13, 35]. We report here, that the mutation has no significant effect on Ca2+ sensitivity of contraction confirming a recent report [15]. More importantly, we show here for the first time, that the mutation does not interfere with the PKA-induced decrease in Ca2+ sensitivity of contraction. In addition, there was no effect on unloaded shortening of skinned murine myocardium.
In fibers not treated with phosphatase to dephosphorylate sarcomeric PKA substrates or PKA to phosphorylate them, hcTnCL29Q had no significant effect on the force–pCa relation or on unloaded shortening velocity compared to hcTnCWT. This is in keeping with the observation of Dweck et al. [15] who found no significant changes in the Ca2+ sensitivities of ATPase activity of reconstituted regulated myofilaments, of ATPase activity of cardiac myofibrils, and of force of skinned porcine myocardium [15]. In contrast, Liang et al. [28] reported an increase in pCa50 by 0.06 pCa units in skinned murine cardiomyocytes exchanged with hcTnCL29Q compared to hcTnCWT at a sarcomere length of 2.3 μm. Interestingly the Ca2+-sensitizing effect of the mutation increased to a difference in pCa50 of 0.11 pCa units at a sarcomere length of 1.9 μm [28]. It is not immediately apparent why in our hands and in the study of Dweck et al. [15] the increase in Ca2+ sensitivity is smaller or negligible compared to the investigation of Liang and co-workers [28]. Dweck investigated the effects of the mutation in skinned porcine cardiac fibers and proposed that the difference between their results and those of Liang possibly reflected the difference between lower rodents and higher mammals. However, since we like Liang et al. [28] used murine cardiac muscle, a species difference most likely does not account for the observed differences. In any case, our results in conjunction with those of Dweck et al. [15] could well explain the rather benign course of the disease in the patient who experienced his first HCM-related symptoms at the rather high age of 59 years [21].
In the study of Liang et al. [28], the enhanced Ca2+ sensitivity of force correlated with an enhanced Ca2+-binding affinity of cTnCL29Q, measured as Ca2+-dependent changes in fluorescence of isolated F27W-cTnCs [28]. As an explanation it was proposed that (1) the L29Q mutation may decrease the stability of the non-functional Ca2+-binding loop of cTnC which in turn impacts on the transition to the “open” conformation of the N-domain, and (2) that the exposure of the hydrophobic surface, to which the switch region of cTnI binds upon activation, is increased by the L29Q mutation, which is important for propagating the signal along the thin filament to release inhibition of actomyosin interaction [28]. Dweck et al. [15] also investigated Ca2+ binding to mutant and wild-type cTnCs, reporting unclear findings towards hcTnCL29Q. The mutant cTnC induced a sensitization of Ca2+ binding to the regulatory site in hcTnCL29Q labeled only at Cys-84, however, a desensitization of Ca2+ binding was observed in double-labeled hcTnCL29Q. Interestingly, the tendency of isolated hcTnCL29Q to induce a significant change of Ca2+ binding to the regulatory site was abolished when hcTnCL29Q was incorporated into regulated thin filaments [15].
Since the L29Q substitution could also interfere with the binding of cNTnC to the N-terminal arm of cTnI [4, 13, 35], a decrease in Ca2+ sensitivity could also be envisioned. This is because binding of the N-terminal arm of cTnI is required to stabilize the “open” conformation of cNTnC [14, 27, 42]. The substitution of the non-polar with a polar residue in the L29Q mutation interrupts binding of the N-terminal cTnI peptide to a peptide derived from the Leu-29 region of cTnC in a peptide array assay [35]. Likewise, this interaction is weakened by phosphorylation of Ser23/24 of cTnI, causing a shift of the distribution of the “open/closed” conformation of the N-domain towards the latter [34]. In addition, phosphorylation of Ser23/24 accelerates the closing rate of cTnC induced by Ca2+ dissociation [13]. Because the affinity of the Ca2+-bound N-domain of cTnCL29Q for the switch peptide (aa 147–163) of cTnI was the same in the presence of the dephosphorylated or phosphorylated N-terminal arm of cTnI (residues 1–29), it was proposed that the impact of phosphorylation was abolished [4].
After treatment with phosphatase, we observed an increase in Ca2+ sensitivity in both, hcTnCL29Q- and hcTnCWT-exchanged fibers, whereby Ca2+ sensitivity, although not statistically significant, was somewhat higher in hcTnCL29Q- compared to hcTnCWT-exchanged fibers (ΔpCa50 = 0.04 pCa units). These experiments suggest that dephosphorylation of PKA substrates unmasks a Ca2+-sensitizing effect of the hcTnCL29Q mutation. The PKA-induced rightward shift of the force–pCa relation was not affected by hcTnCL29Q.
In contrast to the fiber studies [our study, 15, 28], which found no change or an increase in Ca2+ sensitivity of force, and in contrast to the findings of Dweck et al. [15], who reported a non-significant increase in the Ca2+ sensitivity of ATPase activity of regulated thin filaments, Schmidtmann and colleagues [35], using a reconstituted system with skeletal actin and tropomyosin together with cardiac troponin, indeed reported that the mutation decreased the Ca2+ sensitivity of actomyosin subfragment 1 (actoS1)-ATPase activity and of sliding velocity of thin filaments in an in vitro velocity assay by about 0.1 pCa units and abolished the phosphorylation-induced decrease in Ca2+ sensitivity. It should be noted that the mutation-induced decrease in Ca2+ sensitivity was much smaller than that induced by phosphorylation of cTnI [35]. In this system, the mutation blunts the effect of PKA-induced Ca2+ desensitization which may not be surprising based on the peptide binding and NMR-studies [13, 35], which is in contrast to the findings of the present study with skinned myocardium. There are several possible explanations for these divergent results:
First
An important difference between in vitro assays as in Schmidtmann et al. [35] and skinned myocardium is the number of actin-myosin interactions which is load dependent and low in in vitro ATPase and motility assays but high under isometric conditions in fibers. Thus, in the in vitro assays Ca2+-dependent activation of actomyosin interaction is dominated by mechanisms intrinsic to the thin filaments whereas in fibers it is in addition influenced by cooperative mechanisms of cross-bridge activation (reviewed in [18]). The latter notion is supported by the much higher cooperativity of pCa-force relations in the present study (n H = 4.33-5.56) compared to Schmidtmann et al.’s [35] pCa-ATPase relation (n H = 1.0–1.9) and pCa–velocity relation (n H = 1.0–1.4). For this reason, in vitro assays may be more sensitive to capture functional changes resulting from altered TnI–TnC interactions which may be blunted by additional, cross-bridge dependent mechanisms in isometrically contracting fibers. However, we note that fibers are closer to the physiological situation in the heart.
Second
The protein composition of the reconstituted thin filaments and the thin filaments in the fibers differ. In the reconstituted system, skeletal tropomyosin was used and the system lacks cardiac myosin binding protein C (cMyBP-C). While in assays with proteins in solution, phosphorylation of Ser23/24 of cTnI is sufficient to decrease the Ca2+ sensitivity of contraction, recent experiments with cMyBP-C null mice demonstrate that cMyBP-C is required together with cTnI phosphorylation for PKA effects on contractile parameters [5, 8]; however, it is not known whether this requires the protein itself or its phosphorylation. Future experiments have to clarify the interplay between the L29Q mutation, cTnI phosphorylation and cMyBP-C.
Mutation-induced effects in in vitro systems also appear to be sensitive to the source of tropomyosin [11] as exemplified by the cTnI G203S mutation. In one study [7], this mutation induced a decrease in Ca2+ sensitivity of isometric force without affecting sliding velocity of thin filaments. In contrast, another study [23] reported a significant enhancement of the sliding velocity. A major methodological difference is the use of human cardiac tropomyosin [7] or tropomyosin prepared from rabbit skeletal muscle [23]. Taken together, these results indicate that the effect of a mutation seen in in vitro systems may strongly depend not only on the experimental conditions such as protein composition but also on pH and temperature, both of which have a significant impact on the Ca2+ activation of cTnC [17]. Differences in the protein composition (skeletal vs. cardiac) may also account for the contradictory findings between the works of Schmidtmann et al. [35] and of Dweck et al. [15].
To conclude, the effects on Ca2+ sensitivity induced by hcTnCL29Q are small to negligible compared to the alterations in Ca2+ sensitivity observed with other HCM-linked mutations of troponin subunits, and the magnitude and the direction of the effect of the mutation—decrease or increase or no change in Ca2+ sensitivity—appears to depend on the experimental system. Furthermore, the comparatively small changes in cTnC structure and interaction with cTnI are not reflected in altered contractile parameters, confirming other findings that switch kinetics of troponin are modulated by the myofilament lattice [37]. If hcTnCL29Q does not alter Ca2+-dependent force, how can the development of HCM in the patient be explained? The possibility remains that the mutation influences other PKA-dependent parameters of Ca2+-dependent force, such as relaxation kinetics or tension cost. In fact, an impaired energy homeostasis has been suggested to underlie the hypertrophy in HCM [2]. A note of caution is warranted. The mutation has only been described in one patient who experienced first HCM-related symptoms at the high age of 59 years [21]. The authors of the mutation report emphasize that a familial incidence was not investigated and that they could “not exclude that the mutation is simply a rare polymorphism without any phenotypical relevance” [21]. Still, the mutation could be a de novo mutation. De novo mutations of sarcomeric proteins have been reported [32]. It is for these cases which lack a familial background that criteria need to be established for judging the potential severity of a mutation. Future studies should show whether skinned myocardium which allows to investigate the functional effects of troponin mutations within the cardiac sarcomere could be a building block in such a diagnostic scheme. In any case, our results obtained in skinned murine myocardium are consistent with the findings of Dweck et al. [15] with skinned porcine myocardium, showing consistency in cardiac preparations from two different species. Together with the clinical data [21] a likely explanation for the lack of effects is that the hcTnCL29Q mutation is a benign polymorphism accidentally found in an HCM patient. Finally, it is puzzling that mutations in cTnC are rare. Perhaps this reflects the key role of this highly conserved molecule for cardiac function [17].
References
Arad M, Lahat H, Freimark D (2005) Genetic ideology of dilated cardiomyopathy. Isr Med Assoc J 7:392–396
Ashrafian H, Redwood C, Blair E, Watkins H (2003) Hypertrophic cardiomyopathy: a paradigm for myocardial energy depletion. Trends Genet 19:263–268
Babu A, Scordilis SP, Sonnenblick EH, Gulati J (1987) The control of myocardial contraction with skeletal fast muscle troponin C. J Biol Chem 262:5815–5822
Baryshnikova OK, Li MX, Sykes BD (2008) Modulation of cardiac troponin C function by the cardiac-specific N-terminus of troponin I: influence of PKA phosphorylation and involvement in cardiomyopathies. J Mol Biol 375:735–751
Brickson S, Fitzsimons DP, Pereira L, Hacker T, Valdivia H, Moss RL (2007) In vivo left ventricular functional capacity is compromised in cMyBP-C null mice. Am J Physiol Heart Circ Physiol 292:H1747–H1754
Burkett EL, Hershberger RE (2005) Clinical and genetic issues in familial dilated cardiomyopathy. J Am Coll Cardiol 45:969–981
Burton D, Abdulrazzak H, Knott A, Elliott K, Redwood C, Watkins H, Marston S, Ashley C (2002) Two mutations in troponin I that cause hypertrophic cardiomyopathy have contrasting effects on cardiac muscle contractility. Biochem J 362:443–451
Cazorla O, Szilagyi S, Vignier N, Salazar G, Krämer E, Vassort G, Carrier L, Lacampagne A (2006) Length and protein kinase A modulations of myocytes in cardiac myosin binding protein C-deficient mice. Cardiovasc Res 69:370–380
Chang AN, Potter JD (2005) Sarcomeric protein mutations in dilated cardiomyopathy. Heart Fail Rev 10:225–235
Churcott CS, Moyes CD, Bressler BH, Baldwin KM, Tibbits GF (1994) Temperature and pH effects on Ca2+ sensitivity of cardiac myofibrils: a comparison of trout with mammals. Am J Physiol 267:R62–R70
Clemmens EW, Entezari M, Martyn DA, Regnier M (2005) Different effects of cardiac versus skeletal muscle regulatory proteins on in vitro measures of actin filament speed and force. J Physiol 566:737–746
Dohet C, al-Hillawi E, Trayer IP, Rüegg JC (1995) Reconstitution of skinned cardiac fibres with human recombinant cardiac troponin-I mutants and troponin-C. FEBS Lett 377:131–134
Dong WJ, Xing J, Ouyang Y, An J, Cheung HC (2008) Structural kinetics of cardiac troponin C mutants linked to familial hypertrophic and dilated cardiomyopathy in troponin complexes. J Biol Chem 283:3424–3432
Dong WJ, Xing J, Villain M, Hellinger M, Robinson JM, Chandra M, Solaro RJ, Umeda PK, Cheung HC (1999) Conformation of the regulatory domain of cardiac muscle troponin C in its complex with cardiac troponin I. J Biol Chem 274:31382–31390
Dweck D, Hus N, Potter JD (2008) Challenging current paradigms related to cardiomyopathies: are changes in the Ca2+ sensitivity of myofilaments containing cardiac troponin C mutations (G159D and L29Q) good predictors of the phenotypic outcomes? J Biol Chem 283:33119–33128
Edman KA (1979) The velocity of unloaded shortening and its relation to sarcomere length and isometric force in vertebrate muscle fibres. J Physiol 291:143–159
Gillis TE, Marshall CR, Tibbits GF (2007) Functional and evolutionary relationships of troponin C. Physiol Genomics 32:16–27
Gordon AM, Homsher E, Regnier M (2000) Regulation of contraction in striated muscle. Physiol Rev 80:853–924
Herberg FW, Bell SM, Taylor SS (1993) Expression of the catalytic subunit of cAMP-dependent protein kinase in Escherichia coli: multiple isozymes reflect different phosphorylation states. Protein Eng 6:771–777
Hill AV (1938) The heat of shortening and the dynamic constants of muscle. Proc R Soc Lond B Biol Sci 126:136–195
Hoffmann B, Schmidt-Traub H, Perrot A, Osterziel KJ, Gessner R (2001) First mutation in cardiac troponin C, L29Q, in a patient with hypertrophic cardiomyopathy. Hum Mutat 17:524
Kobayashi T, Solaro RJ (2005) Calcium, thin filaments, and the integrative biology of cardiac contractility. Annu Rev Physiol 67:39–67
Köhler J, Chen Y, Brenner B, Gordon AM, Kraft T, Martyn DA, Regnier M, Rivera AJ, Wang CK, Chase PB (2003) Familial hypertrophic cardiomyopathy mutations in troponin I (K183D, G203S, K206Q) enhance filament sliding. Physiol Genomics 14:117–128
Krüger M, Linke WA (2006) Protein kinase-A phosphorylates titin in human heart muscle and reduces myofibrillar passive tension. J Muscle Res Cell Motil 27:435–444
Landstrom AP, Parvatiyar MS, Pinto JR, Marquardt ML, Bos JM, Tester DJ, Ommen SR, Potter JD, Ackerman MJ (2008) Molecular and functional characterization of novel hypertrophic cardiomyopathy susceptibility mutations in TNNC1-encoded troponin C. J Mol Cell Cardiol 45:281–288
Layland J, Solaro RJ, Shah AM (2005) Regulation of cardiac contractile function by troponin I phosphorylation. Cardiovasc Res 66:12–21
Li MX, Spyracopoulos L, Sykes BD (1999) Binding of cardiac troponin-I147–163 induces a structural opening in human cardiac troponin-C. Biochemistry 38:8289–8298
Liang B, Chung F, Qu Y, Pavlov D, Gillis TE, Tikunova SB, Davis JP, Tibbits GF (2008) Familial hypertrophic cardiomyopathy-related cardiac troponin C mutation L29Q affects Ca2+ binding and myofilament contractility. Physiol Genomics 33:257–266
Maron BJ (2002) Hypertrophic cardiomyopathy: a systematic review. JAMA 287:1308–1320
Metzger JM, Westfall MV (2004) Covalent and noncovalent modification of thin filament action: the essential role of troponin in cardiac muscle regulation. Circ Res 94:146–158
Mogensen J, Murphy RT, Shaw T, Bahl A, Redwood C, Watkins H, Burke M, Elliott PM, McKenna WJ (2004) Severe disease expression of cardiac troponin C and T mutations in patients with idiopathic dilated cardiomyopathy. J Am Coll Cardiol 44:2033–2040
Morita H, Rehm HL, Menesses A, McDonough B, Roberts AE, Kucherlapati R, Towbin JA, Seidman JG, Seidman CE (2008) Shared genetic causes of cardiac hypertrophy in children and adults. N Engl J Med 358:1899–1908
Neulen A, Blaudeck N, Zittrich S, Metzler D, Pfitzer G, Stehle R (2007) Mn2+-dependent protein phosphatase 1 enhances protein kinase A-induced Ca2+ desensitisation in skinned murine myocardium. Cardiovasc Res 74:124–132
Sakthivel S, Finley NL, Rosevear PR, Lorenz JN, Gulick J, Kim S, VanBuren P, Martin LA, Robbins J (2005) In vivo and in vitro analysis of cardiac troponin I phosphorylation. J Biol Chem 280:703–714
Schmidtmann A, Lindow C, Villard S, Heuser A, Mügge A, Gessner R, Granier C, Jaquet K (2005) Cardiac troponin C-L29Q, related to hypertrophic cardiomyopathy, hinders the transduction of the protein kinase A dependent phosphorylation signal from cardiac troponin I to C. FEBS J 272:6087–6097
Siedner S, Krüger M, Schroeter M, Metzler D, Roell W, Fleischmann BK, Hescheler J, Pfitzer G, Stehle R (2003) Developmental changes in contractility and sarcomeric proteins from the early embryonic to the adult stage in the mouse heart. J Physiol 548:493–505
Solzin J, Iorga B, Sierakowski E, Gomez Alcazar DP, Ruess DF, Kubacki T, Zittrich S, Blaudeck N, Pfitzer G, Stehle R (2007) Kinetic mechanism of the Ca2+-dependent switch-on and switch-off of cardiac troponin in myofibrils. Biophys J 93:3917–3931
Tardiff JC (2005) Sarcomeric proteins and familial hypertrophic cardiomyopathy: linking mutations in structural proteins to complex cardiovascular phenotypes. Heart Fail Rev 10:237–248
Taylor MR, Carniel E, Mestroni L (2004) Familial hypertrophic cardiomyopathy: clinical features, molecular genetics and molecular genetic testing. Expert Rev Mol Diagn 4:99–113
van der Velden J, Papp Z, Zaremba R, Boontje NM, de Jong JW, Owen VJ, Burton PB, Goldmann P, Jaquet K, Stienen GJ (2003) Increased Ca2+ sensitivity of the contractile apparatus in end-stage human heart failure results from altered phosphorylation of contractile proteins. Cardiovasc Res 57:37–47
Verduyn SC, Zaremba R, van der Velden J, Stienen GJ (2007) Effects of contractile protein phosphorylation on force development in permeabilized rat cardiac myocytes. Basic Res Cardiol 102:476–487
Ward DG, Brewer SM, Cornes MP, Trayer IP (2003) Cross-linking study of the N-terminal extension of human cardiac troponin I. Biochemistry 42:10324–10332
Acknowledgments
We thank K. Jaquet (Bochum, Germany) for suggesting the experiments, for helpful discussions, and for the generous gifts of recombinant human cardiac troponin C (hcTnCL29Q and hcTnCWT) and of recombinant catalytic subunit of PKA. The authors also thank M. Krüger (Münster, Germany) for stimulating discussions and critically reading the manuscript. This work was supported by the DFG (SFB 612-A2), Koeln Fortune (Faculty of Medicine, Cologne), and the Center of Molecular Medicine, University of Cologne.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Neulen, A., Stehle, R. & Pfitzer, G. The cardiac troponin C mutation Leu29Gln found in a patient with hypertrophic cardiomyopathy does not alter contractile parameters in skinned murine myocardium. Basic Res Cardiol 104, 751–760 (2009). https://doi.org/10.1007/s00395-009-0038-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00395-009-0038-y