
Abstract
CONSTITUTIVE PHOTOMORPHOGENIC 1 (COP1), an E3 ubiquitin ligase, functions as a central repressor of light signaling and regulates various light-mediated developmental and metabolic processes in plants. However, detailed mechanisms underlying COP1-regulated flavonoid biosynthesis and embryogenesis in rice seeds remain largely unknown. Here, we performed transcriptome analysis of the rice cop1 (yellowish-pericarp embryo lethal [yel]) null mutant, characterized by flavonoid accumulation in pericarp and abnormal development of embryo, to identify and profile the expression genes involved in flavonoid biosynthesis and embryo development. Comparative transcriptome analysis of yel-hc and wild-type seeds revealed 979 differentially expressed genes (DEGs), of which 577 were upregulated and 402 were downregulated in yel-hc seeds. Functional annotation of DEGs revealed that DEGs were mainly enriched in ‘metabolism’, ‘transcription factors’, ‘secondary metabolites’, and ‘flavonoid biosynthesis’. The DEGs encoding AP2-EREBP, MYB, and bZIP transcription factors (TFs) were predominantly upregulated, whereas those encoding HB, bHLH, and ABI3VP1 TFs were downregulated in yel-hc seeds. Comparative gene expression analysis revealed that genes involved in the C-glycosyl flavone biosynthesis pathway, including OsP1, were activated, whereas anthocyanin biosynthesis genes showed no significant change in expression. In addition, transcript levels of embryo development-related genes, especially homeobox auxin regulation genes, as well as somatic embryogenesis-related genes, were significantly downregulated in yel-hc. Taken together, these results indicate that OsCOP1 plays a crucial role in regulation of flavonoid biosynthesis and embryo structure formation, and changes in the expression of light signal transduction-related genes could have a significant impact on flavonoid biosynthesis and embryogenesis in rice seed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Light is one of the most important environmental factors affecting flavonoid biosynthesis in plants. Several different types of plant photoreceptors receive light signals and activate various signal transduction pathways to regulate flavonoid biosynthesis under the light condition (Peng et al. 2013; Jaakola 2013). CONSTITUTIVE PHOTOMORPHOGENIC1 (COP1), an E3 ubiquitin ligase, acts as a central regulator in light signal transduction and light-induced anthocyanin biosynthesis pathways downstream of multiple light receptors by promoting the ubiquitination and degradation of various transcription factors (TFs), such as myeloblastosis (MYB), ELONGATED HYPOCOTYL5 (HY5), and HY5 HOMOLOG (HYH); repressing the expression of structural genes involved in the anthocyanin biosynthesis pathway; and thus suppressing anthocyanin biosynthesis (Maier et al. 2013; Ang et al. 1998; Holm et al. 2002). The mechanisms and regulation patterns associated with COP1-mediated or light-induced anthocyanin biosynthesis have been extensively studied in many plant species, including the model plant Arabidopsis thaliana (Maier et al. 2013), eggplant (Solanum melongena L.) (Jiang et al. 2016), apple (Malus domestica) (Li et al. 2012), pear (Pyrus bretschneideri) (Wu et al. 2019), litchi (Litchi chinensis Sonn.) (Zhang et al. 2016), and sweet cherry (Prunus avium L.) (Liang et al. 2020). However, the COP1-mediated light signaling pathway that regulates flavonoid accumulation in rice (Oryza sativa L.) grain has not yet been fully elucidated.
Glycosylation is one of the most common modifications of flavonoids (Alseekh et al. 2020), and some cereal crops, including rice and wheat (Triticum aestivum L.), predominantly accumulate flavonoids in the C-glycosylated form (Brazier-Hicks et al. 2009). The biosynthetic pathways of various flavonoids, including C-glycosyl flavonoids, have been defined in many plant species. In higher plants, flavonoids are usually synthesized via the general phenylpropanoid pathway, in which phenylalanine is converted into 4-coumaroyl-CoA by three enzymes including phenylalanine ammonia lyase (PAL), cinnamate 4-hydroxylase (C4H), and 4-coumarate: coenzyme A (CoA) ligase (4CL). The first committed step in the flavonoid biosynthetic pathway is catalyzed by chalcone synthase (CHS), which produces naringenin chalcone. Subsequently, chalcone isomerase (CHI) catalyzes the isomerization of chalcone to form naringenin, the common precursor of various classes of flavonoids (Winkel-Shirley 2001). In the following reaction, flavanone-3′-hydroxylase (F3’H) generates eriodictyol using naringenin as a substrate, and then flavanone 2‐hydroxylase (F2H) converts naringenin or eriodictyol to the corresponding 2-hydroxyflavanones. In the C-glycosyl flavone biosynthesis pathway, C-glycosyl-2-hydroxyflavanones are generated by C-glycosyltransferase (CGT) which catalyze the UDP-glucose-dependent C-glycosylation. In the last step, C-glycosyl flavones are produced by dehydratase, which selectively converts C-glucosyl-2-hydroxyflavanones to C-glycosyl flavones (Du et al. 2010; Brazier-Hicks et al. 2009).
It is well-known that the flavonoid biosynthetic pathway is mainly regulated by TFs, including a R2R3-MYB, a basic helix-loop-helix (bHLH), and a WD40-repeat protein (Ramsay and Glover 2005). Together, these proteins form the MYB-bHLH-WD40 (MBW) complex, which binds to the promoters of structural genes that play a crucial role in the biosynthesis of flavonoids including anthocyanin and proanthocyanidin (Gonzalez et al. 2008; Xu et al. 2015; Li 2014). In rice, several regulatory genes encoding the components of the MBW complex have been identified. The R2R3-type MYB TFs, OsC1 (maize C1 homolog) is involved in anthocyanin accumulation in the leaf sheath and apiculus (Chin et al. 2016), and another MYB TF, OsKala3, is associated with anthocyanin biosynthesis in black rice pericarp (Kim et al. 2021b; Maeda et al. 2014). Among the bHLH-type TFs, Rc regulates proanthocyanidin biosynthesis in the pericarp of red rice grain (Sweeney et al. 2006), and OsB1 and OsB2/OsKala4 are responsible for anthocyanin biosynthesis in the purple leaf and black rice pericarp, respectively (Sakamoto et al. 2001; Oikawa et al. 2015). Oryza sativa TRANSPARENT TESTA GLABRA1 (OsTTG1), a WD40-repeat TF, is a crucial determinant of anthocyanin biosynthesis in various organs, such as hull, root, culm, grain, and leaf (Yang et al. 2021). Although these individual regulators have been identified, the comprehensive system regulated by MBW complexes for flavonoid biosynthesis in rice remains to be established.
The genetic regulation and molecular mechanism of embryo development have been intensively investigated in Arabidopsis, a dicot plant (ten Hove et al. 2015; Lau et al. 2012). Although many essential processes responsible for embryo development, including the asymmetric division of cells, establishment of apical-basal and radical patterns, and differentiation of epidermal and vascular tissues, are conserved between monocots and dicots, a number of embryo developmental characteristics are distinct between monocots and dicots (Zhao et al. 2017). Unlike dicot embryo, the monocot embryo exhibits morphologically distinct embryonic organ types (such as scutellum, coleoptile, and coleorhiza) and organ number (such as cotyledons). Furthermore, the development of monocot embryo does not show a stereotypic cell division pattern and is not typically organized compared with that of dicot embryo. Thus, the morphology of suspensor is unclear, and apical and basal cell lineages are difficult to distinguish in monocot embryos. In addition to apical-basal polarity, the monocot embryo shows dorsal–ventral polarity during early embryogenesis (ten Hove et al. 2015; Zhao et al. 2017; Radoeva et al. 2019). For these reasons, rice embryogenesis exhibits biologically novel features compared with Arabidopsis embryogenesis. Consistent with the differences in embryo development between monocots and dicots, obvious differences in embryonic development and lethality have been observed between rice and Arabidopsis cop1 null mutants. While the Arabidopsis cop1 null mutant shows seedling lethality, the rice cop1 null mutant exhibits failed seed germination and unstructured embryo development (Kim et al. 2021a; Castle and Meinke 1994). Thus, the elucidation of genes that affect transcriptomic changes in the developing rice embryo will provide novel insights into embryogenesis in monocots or grasses, and will expand our understanding of the molecular mechanisms of COP1-related embryogenesis in rice.
Genes and their interactions responsible for photomorphogenesis and anthocyanin accumulation have been well-studied in the Arabidopsis cop1 mutant. However, we recently found that the newly isolated rice cop1 mutant exhibits distinct phenotypic characteristics, such as high-level C-glycosyl flavone accumulation and embryo lethality, compared with the previously reported Arabidopsis cop1 mutants (Kim et al. 2021a). Additionally, comprehensive transcriptomic changes caused by the mutation of OsCOP1, which is associated with seed pigmentation and embryogenic processes, remain still unknown. Here, we performed comparative transcriptome analysis of the oscop1 null mutant to investigate the gene families involved in C-glycosyl flavone biosynthesis and abnormal embryo formation during early seed development. This study provides transcriptomic evidence of how OsCOP1 mediates gene expression to regulate flavonoid biosynthesis and embryogenesis during seed development in rice.
Materials and Methods
Plant Materials and Sample Preparation
The yel-hc mutant was derived from japonica rice cultivar Hwacheong by N-methyl-N-nitrosourea (MNU) mutagenesis. The yel-hc mutant was fixed and maintained as a heterozygote, since the mutant allele is embryonic lethal in the homozygous state. Wild-type and yel-hc mutant plants were grown in the paddy field at the Experimental Farm of Seoul National University, Suwon, Korea. Harvested rice grains were air-dried, and the moisture content was reduced to approximately 13%. Samples were stored in an environmentally controlled room at 11 °C for 2 months, and then de-husked and hand-selected to eliminate cracked or discolored seeds. Each seed sample was ground using a mill (IKA A11B, Staufen, Germany) and sieved bypassing through a 300 μm filter prior to further experiments.
Measurement of Dehulled Grain, Embryo, and Endosperm Weights
Hundred-grain, -endosperm, and -embryo weights were measured (100 seeds × 3 replicates; 10% water content) using an analytical balance (CAS Corporation, NJ, USA). To measure the weight of embryos and endosperms, the embryos were dissected from grains and weighed separately. Phenotypic data collected from the yel-hc mutant and wild-type materials were statistically analyzed using SAS version 9.4 (SAS Institute Inc., Cary, NC, USA).
Embryo Microscopy
Dehulled yel-hc and wild-type mature seeds were incubated in water for 3 h at room temperature to soften the embryos. Then, the whole seed was longitudinally sliced in half, and the embryo was photographed using the HD’MEASURE software (HANA Vision Systems, Korea).
Total Flavonoid Content
Total flavonoid content was determined as described previously (Jia et al. 1999), with a slight modification. Briefly, 250 μL of crude extract was mixed with 1,250 μL of distilled H2O and 75 μL of NaNO2 (5%, w/v). After 5 min, 150 μL of 10% AlCl3·6H2O was added, and the mixture was left to settle for 6 min. Then, 500 μL of 1 M NaOH and 275 μL of distilled H2O were added to the solution, mixed well, and incubated at room temperature for 15 min. Then, the absorbance of the mixture was measured at 510 nm. A calibration curve was prepared using a standard solution of (+)-catechin hydrate, and the total flavonoid content was expressed as milligrams of catechin equivalents (CE) per 100 g of tissue dry weight (DW). Three independent biological extracts, each with three technical replicates, were analyzed.
RNA Isolation, Library Preparation, and Transcriptome Sequencing
Total RNA was extracted from 7-day-old seeds (7 days after pollination) of the wild-type and yel-hc mutant, with three biological replicates, using TaKaRa MiniBEST Plant RNA Extraction Kit (Takara, Japan), according to the manufacturer’s instructions. The cDNA libraries were prepared for 150 bp paired-end sequencing using TruSeq Stranded mRNA Sample Prep Kit (Illumina, CA, USA). Briefly, mRNA molecules were purified from 1 μg of total RNA using oligo (dT) magnetic beads. The purified mRNAs were fragmented, and first-strand cDNA was synthesized using random hexamer primers. Then, using the first-strand cDNA as a template, the second strand of cDNA was synthesized, generating double-stranded cDNAs. After the sequential steps of end repair, A-tailing, and adapter ligation, the cDNA libraries were amplified by PCR, and their quality was evaluated with Agilent 2100 BioAnalyzer (Agilent, CA, USA). The cDNA libraries were then quantified using the KAPA library quantification kit (Kapa Biosystems, MA, USA), according to the manufacturer’s protocol. Following cluster amplification of denatured templates, sequencing was performed using the Illumina NovaSeq 6000 platform (Illumina, CA, USA) in paired-end (2×150 bp) mode.
Transcriptome Analysis
Paired-end RNA-seq reads were initially aligned to the IRGSP-1.0 pseudomolecule/MSU7 (http://rice.plantbiology.msu.edu/annotation_pseudo_current.shtml) version of rice reference transcriptome sequences to determine inner distance between mate pairs (mate inner distance) using a homemade Python script. The RNA-seq reads were then realigned to the same version of rice reference genome using Tophat2 (v2.1.1) and Bowtie2 (v2.3.4.1), considering the reference gene annotation and distribution of mate inner distance to increase accuracy in transcriptome sequence alignment. The number of reads mapped to the coding sequence (CDS) of each gene was counted using HTSeq-count (v0.12.4) under the ‘union’ mode. Statistical significance of the differences in gene expression levels between the triplicate ‘wild-type’ and ‘yel-hc’ samples was examined using DESeq2 (v4.1), considering adjusted p-value (padj) less than 0.05. Genes differentially expressed between yel-hc and wild-type samples (log2 fold change [FC] > 2) were defined as differentially expressed genes (DEGs). Gene ontology (GO) enrichment analysis of the DEGs was implemented through the web-based Comprehensive Annotation of Rice Multi-Omics (CARMO) platform (http://bioinfo.sibs.ac.cn/carmo/) (Wang et al. 2015), with false discovery rate (FDR) < 0.05. Kyoto Encyclopedia of Genes and Genomes (KEGG; https://www.genome.jp/kegg/) (Kanehisa and Goto 2000) pathway enrichment analysis of DEGs was performed using the TBtools software (Chen et al. 2020), with q-value < 0.05, to test the enrichment of DEGs in particular KEGG pathways. The categories of TFs in rice were obtained from the Plant Transcription Factor Database (PlnTFDB; http://plntfdb.bio.uni-potsdam.de/v3.0/ (Perez-Rodriguez et al. 2010) to analyze TF-encoding DEGs. The MapMan tool (http://MapMan.gabipd.org; version 3.5.1R2) was used to visualize the functional categories of DEGs in the pathways (Thimm et al. 2004). Heatmap visualization was carried out using the Clustvis tool (https://biit.cs.ut.ee/clustvis/) (Metsalu and Vilo 2015).
Quantitative Real-Time PCR (qRT-PCR) Analysis
Total RNA samples were subjected to first-strand cDNA synthesis using M-MLV reverse transcriptase (Promega, Madison, WI, USA), and qRT-PCR was performed using TB Green® Premix Ex Taq™ II (Tli RNaseH Plus) (Takara Bio, Japan) on a CFX96™ Real-time PCR Detection System (Bio-Rad, Hercules, CA, USA), according to the manufacturer’s instructions. Primers used for qRT-PCR analysis are listed in Table S1. Expression levels of genes were normalized relative to that of ACTIN, a housekeeping gene. Data were analyzed using the comparative Ct method. Expression levels were compared using two-tailed Student’s t-test.
Results
Characterization of the Phenotypic Traits of yel-hc Seeds
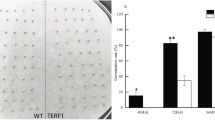
The Hwacheong (japonica rice)-derived yellowish-pericarp embryo lethal (yel) mutant, yel-hc, displayed yellow and dark-brown to black coloration in the pericarp and embryo, respectively, because of high-level flavonoid accumulation (Fig. 1)a–d. Embryonic lethality was one of the most distinct characteristics of the yel-hc mutant compared with wild-type Hwacheong. To observe the intact mature embryo, the imbibed yel-hc and wild-type seeds were sliced longitudinally. Mature wild-type seeds showed normally developed embryos (Fig. 1)e, whereas yel-hc seeds contained abnormally differentiated embryos and indistinguishable embryonic organs including coleoptile, epiblast, radicle, and shoot apex, resulting in the appearance of irregular and unstructured organs in the embryo (Fig. 1f–i). In addition, dark green pigmentation was observed in yel-hc embryos, and the hundred-grain weight was significantly lower in yel-hc than in the wild-type (Fig. 1)j. Consistently, the hundred-endosperm and -embryo weights were also significantly lower in the yel-hc mutant than in the wild-type (Fig. 1)k and l. However, the relative endosperm weight, defined as the weight of the endosperm relative to that of the grain, was higher in yel-hc (97.8%) than in the wild-type (96.5%), whereas the relative embryo weight, defined as the weight of the embryo relative to that of the grain, was lower in yel-hc (2.2%) than in the wild-type (3.5%) (Fig. 1) k and l. These results indicate that the yel-hc mutation influences the overall development of seed; color of embryo and pericarp and weight of grain, endosperm, and embryo in rice.

Characterization of the phenotypic traits and total flavonoid contents of Hwacheong (wild-type) and yel-hc seeds. a, b Seeds of the wild-type (a) and yel-hc (b). c, d Embryos of the wild-type (c) and yel-hc (d). Scale bars = 1 mm. e–i Longitudinal sections of imbibed wild-type (e) and yel-hc (f–i) seeds. j–l Evaluation of 100 grain weight (j), 100 endosperm weight (k), and 100 embryo weight (l). m Total flavonoid contents of wild-type and yel-hc seeds. Cp coleoptile; Ep epiblast; Rd radicle; Sc scutellum; Sa shoot apex. Error bars represent standard deviation (SD) of three technical repeats. Asterisks indicate statistical significance, as determined by Student’s t-test (**p < 0.01; ***p < 0.001)
Total Flavonoid Content of yel-hc Grain
Total flavonoid contents of yel-hc and wild-type grain extracts were assessed. The yel-hc grains showed a remarkably high total flavonoid content compared with wild-type grains (Fig. 1)m. The yel-hc grain contained 157.2 mg CE/100 g DW, which was approximately 60-fold higher than the total flavonoid content of wild-type grain, a significant difference (p < 0.001) (Fig. 1)m.
RNA-Seq and DEG Identification
To investigate the impact of mutation in the OsCOP1 gene on the transcriptome dynamics of rice seed, 7-day-old developing seeds of the wild-type and yel-hc mutant were subjected to RNA-seq. An average of 81.5 million paired-end reads, representing 96.6% of the total raw reads, were obtained from each sample, among which an average of 81.9% properly paired reads were mapped to the Nipponbare reference genome. A total of 27,810–28,493 expressed genes were detected in wild-type and yel-hc samples (Table S2). DEGs were selected based on two thresholds: | log2FC |> 2 and padj < 0.05. A total of 979 DEGs, including 577 upregulated and 402 downregulated genes, were identified (Fig. 2a and b).

Functional classification of differentially expressed genes (DEGs). a Volcano plot of genes differently expressed between wild-type and yel-hc seeds. The DEGs were identified based on two thresholds: |log2FC|> 2 and padj < 0.05. Red and blue dots represent significantly upregulated and significantly downregulated genes, respectively, and gray dots represent non-significant DEGs. b Numbers of upregulated and downregulated DEGs. c The 20 most enriched GO terms of DEGs (FDR < 0.05). d KEGG pathway analysis of DEGs. The size of each point represents the number of DEGs in the pathway, and the color of the point represents the -log10FDR or -log10q-value. Rich factor indicates the ratio of the number of DEGs to the number of genes annotated in the GO term or KEGG pathway
GO Term and KEGG Pathway Enrichment Analyses of DEGs
GO term enrichment analysis was performed to classify the DEGs into three categories: biological process (BP), cellular component (CC), and molecular function (MF). The results showed that DEGs were enriched in 15 GO terms in the BP category, four CC terms, and 16 MF terms (FDR < 0.05; Table S3). The top 20 enriched GO terms are shown in Fig. 2c. In the BP category, the most representative GO terms were ‘response to abiotic stimulus,’ ‘metabolic process,’ ‘oxidation–reduction process,’ and ‘biosynthetic process.’ In the CC category, DEGs were highly enriched in ‘cytoplasmic membrane-bounded vesicle,’ ‘cell,’ and ‘nucleus’ subcategories. In the MF category, the most significantly enriched subcategories were ‘sequence-specific DNA binding transcription factor activity,’ ‘oxidoreductase activity,’ ‘DNA binding,’ and ‘catalytic activity’ (Fig. 2c and Table S3). To further identify the functional pathways potentially involved in seed development in the rice cop1 null mutant, pathway enrichment analysis was performed based on the KEGG database. The results showed that 914 DEGs were assigned to 125 KEGG pathways (Table S4). Among them, DEGs were significantly enriched in seven pathways including ‘biosynthesis of other secondary metabolites,’ ‘transcription factors,’ ‘metabolism,’ ‘flavonoid biosynthesis,’ ‘cytochrome P450,’ ‘phenylpropanoid biosynthesis,’ and ‘prodigiosin biosynthesis’ (Fig. 2 d and Table S4). Most of the DEGs involved in KEGG pathways such as ‘biosynthesis of other secondary metabolites,’ ‘transcription factors,’ and ‘metabolism’ were assigned to the GO terms ‘metabolic process,’ ‘biosynthetic process,’ and ‘sequence-specific DNA binding transcription factor activity.’ Thus, KEGG pathway and GO term enrichment analyses provided further insight into the potential function of DEGs, which were predominantly associated with ‘biosynthesis of other secondary metabolites’ and ‘transcription factors’.
Metabolic Pathway Analysis of DEGs Using MapMan
To gain further insight into the metabolic and molecular factors affected by the mutation of OsCOP1, DEGs were analyzed using the MapMan tool. In the metabolism overview, a total of 76 DEGs were mainly assigned to cell walls, lipids, and secondary metabolisms. Most of the DEGs in the secondary metabolism pathway, which related to flavonoid and phenolic compound biosynthesis, were upregulated (Fig. 3a and Table S5). The predominance of secondary metabolism, cell wall, and lipid metabolism terms in MapMan analysis was consistent with the results of GO term and KEGG pathway enrichment analyses (Fig. 2c and d). In the regulation overview, the enriched DEGs were associated with TF, protein degradation, IAA and ethylene regulation, and receptor kinases. Notably, many genes related to ethylene signal transduction were upregulated. In particular, genes encoding MYB, bZIP, bHLH, AP2/EREBP, and homeobox TFs were the most abundant, indicating that these genes are largely involved in regulating flavonoid biosynthesis and embryogenesis in rice seeds (Fig. 3b and Table S6). The expression of genes belonging to large enzyme families, such as cytochrome P450, oxidases, UDP-glycosyltransferases, glutathione-S-transferases, GDSL-lipases, and peroxidases, showed predominant changes in yel-hc seeds. Many of the genes encoding cytochrome P450 proteins, oxidases, and UDP-glycosyltransferases were upregulated, whereas most of the DEGs encoding peroxidases were downregulated (Fig. S1a and Table S7). In addition, most of the DEGs involved in the biosynthesis of secondary metabolites, such as phenylpropanoids, lignin, lignans, and flavonoids, were significantly upregulated (Fig. S1b and Table S8). Taken together, these results suggest that flavonoid biosynthesis and embryo development are complex regulatory processes, involving transcriptional regulation and multiple metabolic pathways, in rice.

MapMan overview of changes in the expression of differentially expressed genes (DEGs) in yel-hc developing seeds. a Metabolism overview. b Regulation overview. The DEGs were binned into MapMan functional categories. Data represent log2FC values. Red and blue colors represent upregulated and downregulated genes, respectively
Identification of Differentially Expressed TF Genes
Numerous molecular genetic studies have revealed that TFs play crucial roles in plant growth and development. In this study, we investigated TF genes associated with flavonoid biosynthesis and embryo development. Diverse families of TF-encoding genes were found to be differentially expressed; among 979 DEGs, 120 TF genes (66 upregulated and 54 downregulated) belonging to 33 different families were identified (Fig. 4a). Among the 66 upregulated TF genes, AP2-EREBP genes were the most abundant, followed by MYB, Orphans, bHLH, MYB-related, and WRKY. On the other hand, the TF gene family members, such as HB, bHLH, C2H2, OFP, and ABI3VP1, were mainly downregulated in the developing yel-hc seeds (Fig. 4a and Table S9). Among the identified TF genes, those encoding components of the MBW complex, which plays a central role in regulating flavonoid biosynthesis, were identified. The result revealed that all 10 differentially expressed TF genes putatively belonging to the MYB gene family were upregulated. Among these, two MYB TF genes, OsC1 and OsP1 (homolog of maize pericarp color 1), which act as crucial regulatory genes in anthocyanin and flavonoid biosynthesis pathways, were significantly upregulated (Fig. 4b and Table S9). Furthermore, 11 putative bHLH TF genes were identified, of which seven were downregulated and three were upregulated. In addition, expression levels of two WD40 genes, including OsRUP2 (LOC_Os02g02380), were significantly increased, while that of a putative WD40 gene was downregulated in yel-hc seeds (Fig. 4b). These results are consistent with the MapMan pathway analysis of DEGs, suggesting that the mutation of OsCOP1 affects the transcript levels of TF genes playing important roles in flavonoid biosynthesis and plant development.

Analysis of transcription factor (TF) family genes and MYB-bHLH-WD40 (MBW) complex genes among the differentially expressed genes (DEGs). a Differentially expressed TF genes. Numbers indicate the number of genes. b Heat map of the expression patterns of MBW complex genes. The color scale represents the log-transformed fragments per kilobase transcript per million mapped reads (FPKM) values. Gene IDs and names are shown on the right
Analysis of Flavonoid Biosynthesis-Related Genes in yel-hc Seeds
The expression of genes related to main flavonoid biosynthesis was analyzed to explore genes responsible for the distinctive coloration of yel-hc seeds. First, we screened genes that function in the phenylpropanoid pathway to convert phenylalanine into 4-coumaroyl-CoA, which is ultimately utilized for flavonoid biosynthesis. Certain genes, including those encoding PAL (LOC_Os04g43800, LOC_Os02g41670), C4H (LOC_Os01g60450, LOC_Os02g26810), and 4CL (LOC_Os02g46970), were significantly upregulated in the developing yel-hc seeds (Fig. 5). Additionally, genes encoding CHS (LOC_Os11g32650) and CHI (LOC_Os11g02440, LOC_Os12g02370, LOC_Os03g60509), which are necessary to produce naringenin, were predominantly expressed in yel-hc. Interestingly, the expression level of genes involved in the C-glycosyl flavone biosynthesis pathway was significantly upregulated in yel-hc. For example, genes encoding F3'H (LOC_Os10g17260), F2H (LOC_Os06g01250), and CGT (LOC_Os06g18010) showed high expression levels in yel-hc. In particular, the expression level of OsCGT which catalyzes C-glycosylation to produce C-glycosyl flavones such as isoorientin and vitexin in the last step of the pathway, was dramatically increased in yel-hc. Genes encoding key flavonol biosynthetic enzymes, including F3H (LOC_Os03g03034) and FLS (LOC_Os02g52840 and LOC_Os01g61610), were also activated. By contrast, several major genes involved in anthocyanin and proanthocyanidins biosynthesis downstream of the flavonoid biosynthetic pathway, such as DIHYDROFLAVONOL 4-REDUCTASE (DFR), ANTHOCYANIDIN SYNTHASE (ANS), UDP-GLUCOSE FLAVONOID 3-O-GLYCOSYLTRANSFERASE (UFGT), LEUCOANTHOCYANIDIN REDUCTASE (LAR), and ANTHOCYANIN REDUCTASE (ANR), were downregulated or showed no significant difference in expression between wild-type and yel-hc seeds (Fig. 5). These results suggest that the mutation of OsCOP1 highly upregulated expression of genes involved in the flavonoid and C-glycosyl flavone biosynthetic pathways, resulting in the high-level accumulation of C-glycosyl flavones including isoorientin in yel-hc seeds.

Transcriptome profiling of genes involved in the flavonoid biosynthetic pathway. Heat map shows the changes in transcript levels of a representative subset of genes. Orange and blue colors indicate upregulation and downregulation, respectively. The number in the rectangle represents the log2FC value. Red and blue circles indicate significant (FDR < 0.05) and non-significant differences, respectively. PAL phenylalanine ammonia lyase; C4H cinnamate 4-hydroxylase; 4CL 4-coumarate: CoA ligase; CHS chalcone synthase; CHI chalcone isomerase; F3'H flavonoid 3'-hydroxylase; F2H flavanone 2-hydroxylase; CGT C-glycosyltransferase; DH dehydratase; F3H flavonoid 3-hydroxylase; FLS flavonol synthase; DFR dihydroflavonol 4-reductase; ANS anthocyanidin synthase; UFGT UDP-glucose: flavonoid 3-O-glucosyltransferase; LAR leucoanthocyanidin reductase; ANR anthocyanidin reductase; FNSII flavone synthase II; OMT O-methyltransferase; C5'H chrysoeriol 5'-hydroxylase
Analysis of Embryo Development-Related Genes in yel-hc Seeds
Next, we analyzed the expression levels of previously reported embryogenesis-related genes. Some of the selected genes essential for embryo formation and development showing significant changes in expression (FDR < 0.05) are shown in Fig. 6. Consistent with the characteristics of yel-hc embryo, such as developmental defects and lethality, the expression level of developmental regulatory genes, such as auxin-signaling genes (OsPIN2, OsPIN1d, OsYUC7, and OsYUC9) and homeobox genes (OSH1, OSH15, OsWOX6, and OsWOX9C), were significantly downregulated in yel-hc (Fig. 6 and Table S10). Interestingly, transcript levels of genes involved in somatic embryogenesis (OsVP1, OsLEC1, OsLEC2, OsBBM1, OsBBM2, and OsBBM3) were significantly downregulated in yel-hc. In particular, expression levels of OsBBM3 and OsLEC1 were dramatically reduced in yel-hc (Fig. 6 and Table S10). Among the reported embryogenesis-related TF genes, the expression of NAC DOMAIN-CONTAINING PROTEIN 7 (OsNAC7), GROWTH-REGULATING FACTOR 10 (OsGRF10), and BZIP PROTEIN 8 (OsBZ8) was significantly downregulated in developing yel-hc seeds. Among the 15 differentially expressed late embryogenesis abundant (LEA) genes, 12 including OsEM were downregulated, and three were upregulated. Furthermore, some embryo development-related genes, such as AMINOCYCLOPROPANE-1-CARBOXYLIC ACID OXIDASE 2 (OsACO2), LIPID TRANSFER PROTEIN 2.1 (OsLTP2.1), and ZINC FINGER CCCH DOMAIN-CONTAINING PROTEIN 43 (OsC3H43), were downregulated, whereas GIBBERELLIN 20-OXIDASE 1 (OsGA20ox1) and ALPHA-AMYLASE 1A (OsAmy1A) were significantly upregulated in yel-hc (Fig. 6 and Table S10). These results indicate that mutation in OsCOP1 influences the overall embryo formation and development processes in the developing seed.

Heat map of the expression patterns of embryogenesis-related genes. The color scale represents log-transformed fragments per kilobase transcript per million mapped reads (FPKM) values. Gene IDs and names are shown on the right
Validation of RNA-Seq Data
The transcriptional profiles of 23 putative genes involved in flavonoid and anthocyanin biosynthesis, embryogenesis, and COP1-mediated signal transduction pathway were further validated by qRT-PCR. The dramatic upregulation of flavonoid/C-glycosyl flavone biosynthetic genes and downregulation of all zygotic/somatic embryogenesis-related genes, except OsAmy1A, observed in RNA-seq was confirmed by qRT-PCR. Similarly, the highly upregulated expression of OsCOP1-mediated light signaling transduction genes, OsHY5 and OsRUP2 observed in the RNA-seq was validated by qRT-PCR analysis (Fig. 7a). Thus, most of the genes, except OsANR, exhibited similar expression patterns as both RNA-seq and qRT-PCR analyses, with a strong correlation (Pearson correlation coefficient; R = 0.97) (Fig. 7b). However, among the four anthocyanin biosynthesis genes including OsDFR, OsANS, OsLAR, and OsANR, which showed no significant difference between WT and yel-hc in the RNA-seq analysis, OsANR was slightly but significantly downregulated in yel-hc in the qRT-PCR analysis (Fig. 7b). These results indicate that the transcriptomic profiling was accurately conducted and offered reliable explanation to correspond the response of mutation in OsCOP1 in transcriptome level.

Validation of the RNA-seq data of crucial genes by quantitative real-time PCR (qRT-PCR). a Comparison of log2FC values of genes between qRT-PCR and RNA-seq results. In RT-qPCR analysis, log2FC values were calculated using the 2−ΔΔCt method, and data were expressed as mean ± SD of three biological replicates. Asterisks indicate statistical significance, as determined by Student’s t-test (*p < 0.05; **p < 0.01). NS, not significant. b Correlation between qRT-PCR and RNA-seq data. The log2FC values of each gene obtained using the two methods were compared
Discussion
COP1 is an E3 ubiquitin ligase and plays an important role in flavonoid biosynthesis by regulating light signal transduction in plants. A high level of anthocyanin accumulation has been reported in the cotyledons and leaves of Arabidopsis cop1 mutants including cop1-5 (Castle and Meinke 1994; Deng et al. 1991). In the cop1 mutant, the two R2R3-MYB TFs, PRODUCTION OF ANTHOCYANIN PIGMENT1 (PAP1) and PAP2, are required for anthocyanin biosynthesis and the expression of structural genes. The activation of PAP1 and PAP2 is regulated by the COP1/SPA E3 ubiquitin ligase complex via HY5 in a light-dependent manner (Maier et al. 2013; Shin et al. 2013). Homologs of AtPAP1 (AtMYB75) gene have been identified in other plant species, such as tomato, sweet potato, grapevine, and apple, but no AtMYB75 homolog has yet been identified in rice, since AtMYB75 is a dicot-specific gene (Zhao and Bartley 2014). In the current study, ten MYB genes were identified as DEGs, and two of these genes, OsC1 (LOC_Os06g10350) and OsP1 (LOC_Os03g19120), which are well-defined as flavonoid regulatory genes, were highly upregulated in 7-day-old yel-hc seeds (Fig. 4 and Table S9). The P1 gene, which encodes an R2R3-MYB TF, is known to participate in the regulation of flavonoid biosynthesis in plants. Characterization of rice R2R3-MYB genes revealed that OsP1 (LOC_Os03g19120) is most closely related to subgroup 7 (SG7) MYBs including the maize gene ZmP1 and Arabidopsis genes AtMYB11, AtMYB12, and AtMYB111 (Zheng et al. 2019). In maize, ZmP1 primarily expresses in floral tissues, such as pericarps, cob glumes, and silks, and promotes the accumulation of phlobaphenes, C-glycosyl flavones, and maysin by activating the expression of a subset of flavonoid biosynthesis genes (Casas et al. 2016; Zhang et al. 2003; Grotewold et al. 1994; Falcone Ferreyra et al. 2013).
Another R2R3-MYB TF gene, C1, is known as a crucial regulator of anthocyanin biosynthesis in plants. In rice, several studies demonstrated that OsC1, a homolog of ZmC1, mainly regulates anthocyanin accumulation but not flavonoid accumulation in apiculus, stigma, hull, and leaf sheath by coregulating anthocyanin biosynthesis together with other anthocyanin biosynthesis genes such as OsDFR, OsPa, and OsRb (Saitoh et al. 2004; Sun et al. 2018; Chin et al. 2016; Meng et al. 2021; Zheng et al. 2019). However, it is likely that OsC1 is not associated with anthocyanin biosynthesis in rice pericarp (Zheng et al. 2019). According to Zheng et al. (2019), most cultivated rice lost their ability to synthesize anthocyanins because of negative selection during domestication. Cultivated rice accessions, which possess null mutations in certain anthocyanin biosynthesis genes, such as OsC1 and OsRb, were artificially selected by humans, although the reasons for selection against anthocyanin biosynthesis are unclear (Zheng et al. 2019). Hwacheong, the progenitor of yel-hc, is a typical japonica cultivar with white pericarp and non-pigmented leaves. It is likely that Hwacheong, like many other japonica cultivars, harbors non-functional alleles of the anthocyanin biosynthesis genes OsC1 and OsRb, which may explain the absence of anthocyanin biosynthesis in yel-hc seeds despite the high-level expression of OsC1. Therefore, these findings may suggest that OsP1 regulates the activation of an alternative flavonoid biosynthesis pathway in yel-hc seeds. Taken together, these results suggest that OsP1, rather than OsC1, may be a crucial regulator of a set of genes involved in the C-glycosyl flavone biosynthetic pathway in rice, and anthocyanin and C-glycosyl flavone biosynthetic pathways may be regulated by different regulatory genes or TFs in a tissue-specific manner in monocots and dicots.
In this study, we found that the biosynthesis of flavonoids, including C-glycosyl flavones, is regulated by MYB TFs and multiple regulatory factors. The expression of flavonoid biosynthetic genes, including OsCHS (LOC_Os11g32650), OsCHI (LOC_Os11g02440, LOC_Os12g02370, and LOC_Os03g60509), OsF3’H (LOC_Os10g17260), and OsF2H (LOC_Os06g01250), and a C-glycosylation pathway gene, OsCGT (LOC_Os06g18010), was significantly induced by the mutation of OsCOP1 in yel-hc. In particular, expression of OsCGT, which uses 2-hydroxyflavanones as substrates to generate C-glycosyl flavones, was dramatically upregulated (Fig. 5). By contrast, the expression level of structural genes involved in the biosynthesis of anthocyanins (OsDFR, OsANS, and OsUFGTs) and proanthocyanidins (OsLAR and OsANR) was downregulated in yel-hc, or there was no significant difference between WT and yel-hc seeds (Figs. 5 and 7). These results are consistent with our previous metabolite analysis, which showed that C-glycosyl flavones and their derivatives, including luteolin 6-C-glucoside (isoorientin), isoorientin 2"-O-glucoside, vitexin 2"-O-glucoside, isoscoparin 2"-O-glucoside, chrysoeriol 6, 8-di-C-hexoside, chrysoeriol 6-C-glucoside (isoscoparin), and apigenin 6-C-glucoside (isovitexin), accumulated to high levels in the embryo and endosperm of yel-hc. Among these, isoorientin, a CGT-catalyzed C-glycosyl flavone, was the most abundant flavone both in the embryo and endosperm of yel-hc; however, anthocyanins were not detectable in the embryo and pericarp of yel-hc (Kim et al. 2018). In addition, considerable research on flavonoid biosynthetic genes, controlled by regulatory genes, has been conducted in maize and rice, and the results of this research are consistent with those of this study. In maize, ZmP1 regulates the transcription of F3'H, which catalyzes the conversion of naringenin to eriodictyol by binding to F3'H promoter (Sharma et al. 2012), and directly targets F2H, which converts naringenin and eriodictyol into 2-hydroxynaringenin and 2-hydroxyeriodictyol, respectively, resulting in the accumulation of flavones (Morohashi et al. 2012). Similarly, OsP1 specifically activates upstream biosynthetic genes such as OsCHS, OsCHI, and OsF3'H, whereas OsC1 activates all anthocyanin biosynthetic genes including OsCHS, OsCHI, OsF3'H, OsF3H, OsDFR, and OsANS via the ternary MYB-bHLH-WD40 complex (OsC1–OsRb–OsPAC1) in rice leaves (Zheng et al. 2019). Therefore, our transcriptome profiling results demonstrate that the expression of flavonoid biosynthetic genes is upregulated by regulatory genes such as OsP1, and these biosynthetic and regulatory genes play an important role in flavonoid (C-glycosyl flavone) biosynthesis, but not anthocyanin biosynthesis, in the embryo and pericarp of rice cop1 null mutant. Further investigation is needed to elucidate a more accurate regulation of the different branches of flavonoid biosynthesis by MYBs or MBW complexes in the rice pericarp.
RNA-seq provides a comprehensive overview of several gene families involved in embryogenesis. Homeobox (HB) genes, which encode TFs that regulate the expression of genes involved in pattern formation, cell specification, and cell differentiation during embryogenesis, are thought to play roles in plant development. Sato et al. demonstrated that Oryza sativa homeobox1 (OSH1) and OSH15 encode KNOTTED1 (KN1)-like homeobox (KNOX) protein involved in the formation and maintenance of shoot apical meristem (SAM) (Sato et al. 1996, 1998), while the osh1 osh15 (d6) double mutant embryo failed to form SAM (Tsuda et al. 2011). Other KNOX genes, such as OSH6 and OSH71, were highly expressed in the central part of the embryo at the globular embryo stage during rice embryogenesis (Sentoku et al. 1999). In the present study, the expression level of OSH1 and OSH15 was strongly reduced in the 7-day-old developing seeds of yel-hc. In addition, the expression levels of OSH6 and OSH71 were also significantly downregulated in yel-hc, although not to the same extent as that of OSH1 and OSH15 (Fig. 6 and Table S10). These results imply that the downregulated expression of these HB genes is one of the main causes of the abnormal differentiation of embryo organs, especially in the SAM region of yel-hc.
WUSCHEL-related homeobox (WOX) proteins, which comprise a plant-specific subclade of the HB TF family, play critical roles in early embryogenesis. WOX2 and WOX8 induce distinct cell fates after the asymmetric zygotic division by restoring their expression to the apical (WOX2) and basal (WOX8) lineages. In addition, WOX8 and its closest homolog, WOX9, redundantly affect normal development in both the basal and apical embryo lineages and promote PIN-FORMED 1 (PIN1) expression in the proembryo of Arabidopsis (Breuninger et al. 2008). The maize homologs of AtWOX8 and AtWOX9, ZmWOX9A and ZmWOX9B, respectively, are expressed at higher levels in the basal part than in the apical part, indicating that these genes are potentially involved in axis formation during early embryo patterning, as in Arabidopsis (Chen et al. 2017). In rice, WOX genes are involved in root apical meristem (RAM) specification and maintenance (QHB/OsWOX5) (Kamiya et al. 2003), meristem differentiation (OsWOX4) (Ohmori et al. 2013), crown root development (OsWOX11) (Zhao et al. 2009), and tiller growth (DWT1/OsWOX7) (Wang et al. 2014). In the present study, the expression of OsWOX6, OsWOX9C, and OsWOX10 was significantly downregulated in yel-hc (Fig. 6 and Table S10). These three WOX genes support the results of a previous study, which showed that WOX genes are involved in developmental regulation during early embryogenesis in rice. Among them, OsWOX9C, which is closely related to AtWOX8 and AtWOX9, showed biased expression along the apical-basal axis, and OsWOX6 and OsWOX10, which are phylogenetically close to AtWOX11 and AtWOX12 (Lian et al. 2014), exhibited biased expression along the dorsal–-ventral axis in rice embryo at 3 days after pollination (DAP) (Itoh et al. 2016).
PIN genes, involved in auxin efflux, are necessary for establishing apical-basal polarity during early embryogenesis. PIN1, PIN3, PIN4, and PIN7 are expressed in the embryo in Arabidopsis (Friml et al. 2003; Adamowski and Friml 2015). Analysis of the spatial expression pattern of PIN-like genes revealed that OsPIN1, OsPIN2, OsPIN5, and OsPIN8 showed polarized expression along the apical-basal axis in 3-DAP rice embryo (Itoh et al. 2016). In our study, the expression of OsPIN1b, OsPIN1d, and OsPIN2 was significantly downregulated in yel-hc (Fig. 6 and Table S10). These findings serve as reliable evidence supporting the role of OsPINs in auxin-dependent polarity determination during early embryogenesis in rice. Altogether, these results indicate that COP1 has a wide influence on embryo development, and that the embryo defects of yel-hc originate during embryogenesis, when SAM differentiation, axis formation, and polarity determination are not established correctly. This is because of the insufficient expression of HBs, WOXs, and PINs, although the encoded proteins are functionally redundant.
Aside from zygotic embryogenesis-related genes, several somatic embryogenesis-related genes, such as BABY BOOM (BBM) (Boutilier et al. 2002), LEAFY COTYLEDON (LEC) (Lotan et al. 1998; Stone et al. 2008), FUSCA3 (FUS3) (Luerssen et al. 1998; Ledwon and Gaj 2011), ABA INSENSITIVE3 (ABI3) (Parcy et al. 1994), WUSCHEL (WUS) (Zuo et al. 2002; Lou et al. 2022), WOX (van der Graaff et al. 2009), EMBRYOMAKER (Tsuwamoto et al. 2010), SOMATIC EMBRYOGENESIS RECEPTOR-LIKE KINASE (SERK) (Hecht et al. 2001), and AGAMOUS-LIKE 15 (AGL15) (Thakare et al. 2008), which regulate plant cell identity and development, somatic embryogenesis, and seed development, have been identified in plants. However, some of these genes, including BBM and LEC, exhibit overlapping functions during embryo formation, such as polar transport and auxin gradient, and are directly or indirectly linked to zygotic embryogenesis-related genes in the molecular network (Dodeman et al. 1997; Horstman et al. 2017). In this study, OsBBM1/OsPLT6, OsBBM2/OsPLT3, and OsBBM3/OsPLT5 genes were significantly downregulated in yel-hc. Other somatic embryogenesis marker genes, such as OsVP1, OsLEC1/OsHAP3E, and OsLEC2/OsFUS3-like 1, also exhibited lower expression in yel-hc than in the wild-type (Fig. 6). These results coincide with the functional characterization of the genes, which demonstrated that triple knockout of the three rice BBM genes (OsBBM1, OsBBM2, and OsBBM3) leads to embryo arrest and abortion (Khanday et al. 2019), and the knockout mutation of OsLEC1/OsNF-YB7 caused abnormal embryo development and embryonic lethality in rice (Niu et al. 2021). Additionally, Khanday et al. demonstrated that OsBBM1 directly regulates the expression of auxin biosynthesis YUCCA genes (OsYUC6, OsYUC7, and OsYUC9), and the formation of embryogenic calli was severely reduced in the Osyuc7 Osyuc9 double mutant and Osyuc6 Osyuc7 Osyuc9 triple mutant rice seeds (Khanday et al. 2020). In the present study, the expression of OsYUC7 and OsYUC9 was significantly downregulated in yel-hc. In our previous study, we tried to generate cop1 loss-of-function plants through anther culture; however, no homozygous recessive plants could be recovered from 293 doubled-haploid plants derived by anther culture (data not shown). This result suggests that recessive mutation of OsCOP1 is deleterious and hinders callus formation. Taken together, these results indicate that OsCOP1 is responsible for somatic embryogenesis in rice, and that its function is intertwined with embryo development and embryonic lethality in plants. In addition, a class of potential flavonoids, such as flavone, flavanone, and flavonol, and their corresponding genes responsible for somatic embryogenesis were identified in cotton, implying that the biosynthesis and accumulation of flavonoids might contribute to transdifferentiation during somatic embryogenesis (Guo et al. 2019). Evidence has also been found for the interaction of microRNA with its target gene (GhmiR157a-GhSPL10), which regulates cellular dedifferentiation and callus proliferation by modulating ethylene-mediated flavonoid biosynthesis during somatic embryogenesis in cotton (Wang et al. 2018). However, the signaling between flavonoid biosynthesis and somatic embryogenesis remains unclear. Thus, further research is needed to understand the regulatory or biochemical mechanism underlying somatic embryogenesis at the molecular level.
Several studies have shown that HY5, a bZIP TF, positively regulates anthocyanin biosynthesis by directly or indirectly regulating the transcription of genes involved in both early and late biosynthetic steps (Maier et al. 2013; Shin et al. 2013; An et al. 2017). In addition, it was reported that HY5 is required for the transcriptional activation of MYB12 and MYB111 genes, which activate FLS under UV-B, to produce flavonol glycosides in Arabidopsis (Stracke et al. 2010). In the present study, the transcription of OsHY5 (LOC_Os06g39960) and OsHY5-like (LOC_Os02g10860) was dramatically increased in developing yel-hc seeds (Table S9). The yel-hc phenotype caused by the mutation of OsCOP1, which inhibits HY5 ubiquitination, supports the regulatory role of COP1–HY5 interaction that COP1 negatively regulates HY5, a positive regulator of flavonoid biosynthesis, resulting in embryo and pericarp coloration in rice seed. In other words, upregulation of HY5 results in the activation of MYB genes, such as OsP1, which regulates flavonoid biosynthesis and subsequently regulates downstream genes to convert the light signal into flavonoids.
COP1 and UV RESISTANCE LOCUS 8 (UVR8) are central regulators of UV-B-induced photomorphogenesis and stress (Favory et al. 2009). The UVR8 monomer, generated by UV-B, directly interacts with the COP1–SPA1 complex, which is separated from the CULLIN 4-DAMAGED DNA BINDING PROTEIN 1 (CUL4-DDB1) E3 ligase complex and disrupts the COP1-mediated ubiquitination and degradation of HY5 (Huang et al. 2013). The activated UVR8-COP1-HY5 signaling pathway induces the expression of genes encoding two WD40-repeat proteins, REPRESSOR OF UV-B PHOTOMORPHOGENESIS 1 (RUP1) and RUP2, enabling negative feedback regulation by binding to the UVR8 monomer and mediating UVR8 redimerization in the UVR8-mediated signal transduction pathway (Gruber et al. 2010). In our study, the OsRUP2 (LOC_Os02g02380) gene was highly upregulated in developing yel-hc seeds (Fig. 4). Consistent with the findings in Arabidopsis, it seems that the expression of RUP2 is mediated by COP1 in rice and the RUP2 protein directly interacts with UVR8-COP1. In addition, COP1 is able to target RUP proteins for ubiquitination and degradation, resulting in the regulation of RUP accumulation and the stabilization of HY5 proteins (Ren et al. 2019; Gruber et al. 2010). Besides UV-B-induced photomorphogenesis, flavonoid and anthocyanin biosynthesis pathways are closely related to the UV-B signaling pathway (Jaakola 2013; Peng et al. 2013); therefore, it is important to further study the biological function of RUP proteins and their interaction with COP1 and HY5 in rice.
Conclusion
In this study, transcriptome profiling was conducted to identify key genes involved in COP1-mediated flavonoid (C-glycosyl flavone) biosynthesis and embryogenesis in the developing seeds of yel, cop1 null mutant in rice, and to elucidate the underlying molecular mechanisms. Genes involved in C-glycosyl flavone biosynthesis were significantly upregulated and potentially play a crucial role in the pigmentation of embryo and pericarp in yel-hc. Mutation in OsCOP1 reduced the expression level of genes responsible for somatic embryogenesis as well as zygotic embryogenesis. The identified putative genes that govern flavonoid biosynthesis and embryogenesis could be used to increase the content of the medicinally beneficial C-glycosyl flavones in rice seeds through metabolic engineering approaches and provide pivotal information on embryo formation and development.
Data Availability
The RNA-seq data generated in this study were deposited in the NCBI Sequence Read Archive (SRA) database (https://www.ncbi.nlm.nih.gov/sra/docs/) under the accession numbers SRR17615112, SRR17617328, SRR17616196, SRR17617339, SRR17616327, and SRR17616287; BioProject accession number, PRJNA797189.
References
Adamowski M, Friml J (2015) PIN-dependent auxin transport: action, regulation, and evolution. Plant Cell 27(1):20–32. https://doi.org/10.1105/tpc.114.134874
Alseekh S, Perez de Souza L, Benina M, Fernie AR (2020) The style and substance of plant flavonoid decoration; towards defining both structure and function. Phytochemistry 174:112347. https://doi.org/10.1016/j.phytochem.2020.112347
An JP, Qu FJ, Yao JF, Wang XN, You CX, Wang XF, Hao YJ (2017) The bZIP transcription factor MdHY5 regulates anthocyanin accumulation and nitrate assimilation in apple. Hortic Res 4:17023. https://doi.org/10.1038/hortres.2017.23
Ang LH, Chattopadhyay S, Wei N, Oyama T, Okada K, Batschauer A, Deng XW (1998) Molecular interaction between COP1 and HY5 defines a regulatory switch for light control of Arabidopsis development. Mol Cell 1(2):213–222. https://doi.org/10.1016/S1097-2765(00)80022-2
Boutilier K, Offringa R, Sharma VK, Kieft H, Ouellet T, Zhang L, Hattori J, Liu CM, van Lammeren AA, Miki BL, Custers JB, van Lookeren Campagne MM (2002) Ectopic expression of BABY BOOM triggers a conversion from vegetative to embryonic growth. Plant Cell 14(8):1737–1749. https://doi.org/10.1105/tpc.001941
Brazier-Hicks M, Evans KM, Gershater MC, Puschmann H, Steel PG, Edwards R (2009) The C-glycosylation of flavonoids in cereals. J Biol Chem 284(27):17926–17934. https://doi.org/10.1074/jbc.M109.009258
Breuninger H, Rikirsch E, Hermann M, Ueda M, Laux T (2008) Differential expression of WOX genes mediates apical-basal axis formation in the Arabidopsis embryo. Dev Cell 14(6):867–876. https://doi.org/10.1016/j.devcel.2008.03.008
Casas MI, Falcone-Ferreyra ML, Jiang N, Mejia-Guerra MK, Rodriguez E, Wilson T, Engelmeier J, Casati P, Grotewold E (2016) Identification and characterization of maize salmon silks genes involved in insecticidal maysin biosynthesis. Plant Cell 28(6):1297–1309. https://doi.org/10.1105/tpc.16.00003
Castle LA, Meinke DW (1994) A FUSCA gene of Arabidopsis encodes a novel protein essential for plant development. Plant Cell 6(1):25–41. https://doi.org/10.1105/tpc.6.1.25
Chen C, Chen H, Zhang Y, Thomas HR, Frank MH, He Y, Xia R (2020) TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol Plant 13(8):1194–1202. https://doi.org/10.1016/j.molp.2020.06.009
Chen J, Strieder N, Krohn NG, Cyprys P, Sprunck S, Engelmann JC, Dresselhaus T (2017) Zygotic genome activation occurs shortly after fertilization in maize. Plant Cell 29(9):2106–2125. https://doi.org/10.1105/tpc.17.00099
Chin HS, Wu YP, Hour AL, Hong CY, Lin YR (2016) Genetic and evolutionary analysis of purple leaf sheath in rice. Rice (n y) 9(1):8. https://doi.org/10.1186/s12284-016-0080-y
Deng XW, Caspar T, Quail PH (1991) cop1: a regulatory locus involved in light-controlled development and gene expression in Arabidopsis. Genes Dev 5(7):1172–1182. https://doi.org/10.1101/gad.5.7.1172
Dodeman VL, Ducreux G, Kreis M (1997) Zygotic embryogenesis versus somatic embryogenesis. J Exp Bot 48(313):1493–1509. https://doi.org/10.1093/jxb/48.8.1493
Du YG, Chu H, Chu IK, Lo C (2010) CYP93G2 Is a flavanone 2-hydroxylase required for C-glycosylflavone biosynthesis in rice. Plant Physiol 154(1):324–333. https://doi.org/10.1104/pp.110.161042
Falcone Ferreyra ML, Rodriguez E, Casas MI, Labadie G, Grotewold E, Casati P (2013) Identification of a bifunctional maize C- and O-glucosyltransferase. J Biol Chem 288(44):31678–31688. https://doi.org/10.1074/jbc.M113.510040
Favory JJ, Stec A, Gruber H, Rizzini L, Oravecz A, Funk M, Albert A, Cloix C, Jenkins GI, Oakeley EJ, Seidlitz HK, Nagy F, Ulm R (2009) Interaction of COP1 and UVR8 regulates UV-B-induced photomorphogenesis and stress acclimation in Arabidopsis. EMBO J 28(5):591–601. https://doi.org/10.1038/emboj.2009.4
Friml J, Vieten A, Sauer M, Weijers D, Schwarz H, Hamann T, Offringa R, Jurgens G (2003) Efflux-dependent auxin gradients establish the apical-basal axis of Arabidopsis. Nature 426(6963):147–153. https://doi.org/10.1038/nature02085
Gonzalez A, Zhao M, Leavitt JM, Lloyd AM (2008) Regulation of the anthocyanin biosynthetic pathway by the TTG1/bHLH/Myb transcriptional complex in Arabidopsis seedlings. Plant J 53(5):814–827. https://doi.org/10.1111/j.1365-313X.2007.03373.x
Grotewold E, Drummond BJ, Bowen B, Peterson T (1994) The myb-homologous P gene controls phlobaphene pigmentation in maize floral organs by directly activating a flavonoid biosynthetic gene subset. Cell 76(3):543–553. https://doi.org/10.1016/0092-8674(94)90117-1
Gruber H, Heijde M, Heller W, Albert A, Seidlitz HK, Ulm R (2010) Negative feedback regulation of UV-B-induced photomorphogenesis and stress acclimation in Arabidopsis. Proc Natl Acad Sci U S A 107(46):20132–20137. https://doi.org/10.1073/pnas.0914532107
Guo H, Guo H, Zhang L, Tang Z, Yu X, Wu J, Zeng F (2019) Metabolome and transcriptome association analysis reveals dynamic regulation of purine metabolism and flavonoid synthesis in transdifferentiation during somatic embryogenesis in cotton. Int J Mol Sci 20:9. https://doi.org/10.3390/ijms20092070
Hecht V, Vielle-Calzada JP, Hartog MV, Schmidt EDL, Boutilier K, Grossniklaus U, de Vries SC (2001) The Arabidopsis SOMATIC EMBRYOGENESIS RECEPTOR KINASE 1 gene is expressed in developing ovules and embryos and enhances embryogenic competence in culture. Plant Physiol 127(3):803–816. https://doi.org/10.1104/pp.127.3.803
Holm M, Ma LG, Qu LJ, Deng XW (2002) Two interacting bZIP proteins are direct targets of COP1-mediated control of light-dependent gene expression in Arabidopsis. Genes Dev 16(10):1247–1259. https://doi.org/10.1101/gad.969702
Horstman A, Bemer M, Boutilier K (2017) A transcriptional view on somatic embryogenesis. Regeneration (oxf) 4(4):201–216. https://doi.org/10.1002/reg2.91
Huang X, Ouyang X, Yang P, Lau OS, Chen L, Wei N, Deng XW (2013) Conversion from CUL4-based COP1-SPA E3 apparatus to UVR8-COP1-SPA complexes underlies a distinct biochemical function of COP1 under UV-B. Proc Natl Acad Sci U S A 110(41):16669–16674. https://doi.org/10.1073/pnas.1316622110
Itoh J, Sato Y, Sato Y, Hibara K, Shimizu-Sato S, Kobayashi H, Takehisa H, Sanguinet KA, Namiki N, Nagamura Y (2016) Genome-wide analysis of spatiotemporal gene expression patterns during early embryogenesis in rice. Development 143(7):1217–1227. https://doi.org/10.1242/dev.123661
Jaakola L (2013) New insights into the regulation of anthocyanin biosynthesis in fruits. Trends Plant Sci 18(9):477–483. https://doi.org/10.1016/j.tplants.2013.06.003
Jia Z, Tang MC, Wu JM (1999) The determination of flavonoid contents in mulberry and their scavenging effects on superoxide radicals. Food Chem 64(4):555–559. https://doi.org/10.1016/S0308-8146(98)00102-2
Jiang MM, Ren L, Lian HL, Liu Y, Chen HY (2016) Novel insight into the mechanism underlying light-controlled anthocyanin accumulation in eggplant (Solanum melongena L.). Plant Sci 249:46–58. https://doi.org/10.1016/j.plantsci.2016.04.001
Kamiya N, Nagasaki H, Morikami A, Sato Y, Matsuoka M (2003) Isolation and characterization of a rice WUSCHEL-type homeobox gene that is specifically expressed in the central cells of a quiescent center in the root apical meristem. Plant J 35(4):429–441. https://doi.org/10.1046/j.1365-313X.2003.01816.x
Kanehisa M, Goto S (2000) KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28(1):27–30. https://doi.org/10.1093/nar/28.1.27
Khanday I, Santos-Medellín C, Sundaresan V (2020) Rice embryogenic trigger BABY BOOM1 promotes somatic embryogenesis by upregulation of auxin biosynthesis genes. Biorxiv. https://doi.org/10.1101/2020.08.24.265025
Khanday I, Skinner D, Yang B, Mercier R, Sundaresan V (2019) A male-expressed rice embryogenic trigger redirected for asexual propagation through seeds. Nature 565(7737):91. https://doi.org/10.1038/s41586-018-0785-8
Kim B, Piao R, Lee G, Koh E, Lee Y, Woo S, Reflinur Jiang W, Septiningsih EM, Thomson MJ, Koh HJ (2021) OsCOP1 regulates embryo development and flavonoid biosynthesis in rice (Oryza sativa L.). Theor Appl Genet. 134(8):2587–2601. https://doi.org/10.1007/s00122-021-03844-9
Kim B, Woo S, Kim MJ, Kwon SW, Lee J, Sung SH, Koh HJ (2018) Identification and quantification of flavonoids in yellow grain mutant of rice (Oryza sativa L.). Food Chem 241:154–162. https://doi.org/10.1016/j.foodchem.2017.08.089
Kim D, Yang J, Ha SH, Kim JK, Lee JY, Lim SH (2021) An OsKala3, R2R3 MYB TF, Is a common key player for black rice pericarp as main partner of an OsKala4, bHLH TF. Fronti Plant Sci. https://doi.org/10.3389/fpls.2021b.765049
Lau S, Slane D, Herud O, Kong J, Jurgens G (2012) Early embryogenesis in flowering plants: setting up the basic body pattern. Annu Rev Plant Biol 63:483–506. https://doi.org/10.1146/annurev-arplant-042811-105507
Ledwon A, Gaj MD (2011) LEAFY COTYLEDON1, FUSCA3 expression and auxin treatment in relation to somatic embryogenesis induction in Arabidopsis. Plant Growth Regul 65(1):157–167. https://doi.org/10.1007/s10725-011-9585-y
Li S (2014) Transcriptional control of flavonoid biosynthesis: fine-tuning of the MYB-bHLH-WD40 (MBW) complex. Plant Signal Behav 9(1):e27522. https://doi.org/10.4161/psb.27522
Li YY, Mao K, Zhao C, Zhao XY, Zhang HL, Shu HR, Hao YJ (2012) MdCOP1 ubiquitin E3 ligases interact with MdMYB1 to regulate light-induced anthocyanin biosynthesis and red fruit coloration in apple. Plant Physiol 160(2):1011–1022. https://doi.org/10.1104/pp.112.199703
Lian G, Ding Z, Wang Q, Zhang D, Xu J (2014) Origins and evolution of WUSCHEL-related homeobox protein family in plant kingdom. Sci World J 2014:534140. https://doi.org/10.1155/2014/534140
Liang D, Zhu T, Deng Q, Lin L, Tang Y, Wang J, Wang X, Luo X, Zhang H, Lv X, Xia H (2020) PacCOP1 negatively regulates anthocyanin biosynthesis in sweet cherry (Prunus avium L.). J Photochem Photobiol B 203:111779. https://doi.org/10.1016/j.jphotobiol.2020.111779
Lotan T, Ohto M, Yee KM, West MAL, Lo R, Kwong RW, Yamagishi K, Fischer RL, Goldberg RB, Harada JJ (1998) Arabidopsis LEAFY COTYLEDON1 is sufficient to induce embryo development in vegetative cells. Cell 93(7):1195–1205. https://doi.org/10.1016/S0092-8674(00)81463-4
Lou H, Huang YT, Wang WZ, Cai ZY, Cai HY, Liu ZQ, Sun L, Xu QJ (2022) Overexpression of the AtWUSCHEL gene promotes somatic embryogenesis and lateral branch formation in birch (Betula platyphylla Suk). Plant Cell Tiss Org 150(2):371–383. https://doi.org/10.1007/s11240-022-02290-9
Luerssen H, Kirik V, Herrmann P, Misera S (1998) FUSCA3 encodes a protein with a conserved VP1/AB13-like B3 domain which is of functional importance for the regulation of seed maturation in Arabidopsis thaliana. Plant J 15(6):755–764. https://doi.org/10.1046/j.1365-313x.1998.00259.x
Maeda H, Yamaguchi T, Omoteno M, Takarada T, Fujita K, Murata K, Iyama Y, Kojima Y, Morikawa M, Ozaki H, Mukaino N, Kidani Y, Ebitani T (2014) Genetic dissection of black grain rice by the development of a near isogenic line. Breed Sci 64(2):134–141. https://doi.org/10.1270/jsbbs.64.134
Maier A, Schrader A, Kokkelink L, Falke C, Welter B, Iniesto E, Rubio V, Uhrig JF, Hulskamp M, Hoecker U (2013) Light and the E3 ubiquitin ligase COP1/SPA control the protein stability of the MYB transcription factors PAP1 and PAP2 involved in anthocyanin accumulation in Arabidopsis. Plant J 74(4):638–651. https://doi.org/10.1111/tpj.12153
Meng L, Qi C, Wang C, Wang S, Zhou C, Ren Y, Cheng Z, Zhang X, Guo X, Zhao Z, Wang J, Lin Q, Zhu S, Wang H, Wang Z, Lei C, Wan J (2021) Determinant factors and regulatory systems for anthocyanin biosynthesis in rice apiculi and stigmas. Rice (NY) 14(1):37. https://doi.org/10.1186/s12284-021-00480-1
Metsalu T, Vilo J (2015) ClustVis: a web tool for visualizing clustering of multivariate data using principal component analysis and heatmap. Nucleic Acids Res 43(W1):W566–W570. https://doi.org/10.1093/nar/gkv468
Morohashi K, Casas MI, Falcone Ferreyra ML, Falcone Ferreyra L, Mejia-Guerra MK, Pourcel L, Yilmaz A, Feller A, Carvalho B, Emiliani J, Rodriguez E, Pellegrinet S, McMullen M, Casati P, Grotewold E (2012) A genome-wide regulatory framework identifies maize pericarp color1 controlled genes. Plant Cell 24(7):2745–2764. https://doi.org/10.1105/tpc.112.098004
Niu B, Zhang Z, Zhang J, Zhou Y, Chen C (2021) The rice LEC1-like transcription factor OsNF-YB9 interacts with SPK, an endosperm-specific sucrose synthase protein kinase, and functions in seed development. Plant J 106(5):1233–1246. https://doi.org/10.1111/tpj.15230
Ohmori Y, Tanaka W, Kojima M, Sakakibara H, Hirano HY (2013) WUSCHEL-RELATED HOMEOBOX4 is involved in meristem maintenance and is negatively regulated by the CLE gene FCP1 in rice. Plant Cell 25(1):229–241. https://doi.org/10.1105/tpc.112.103432
Oikawa T, Maeda H, Oguchi T, Yamaguchi T, Tanabe N, Ebana K, Yano M, Ebitani T, Izawa T (2015) The birth of a black rice gene and its local spread by introgression. Plant Cell 27(9):2401–2414. https://doi.org/10.1105/tpc.15.00310
Parcy F, Valon C, Raynal M, Gaubier-Comella P, Delseny M, Giraudat J (1994) Regulation of gene expression programs during Arabidopsis seed development: roles of the ABI3 locus and of endogenous abscisic acid. Plant Cell 6(11):1567–1582. https://doi.org/10.1105/tpc.6.11.1567
Peng T, Saito T, Honda C, Ban Y, Kondo S, Liu JH, Hatsuyama Y, Moriguchi T (2013) Screening of UV-B-induced genes from apple peels by SSH: possible involvement of MdCOP1-mediated signaling cascade genes in anthocyanin accumulation. Physiol Plant 148(3):432–444. https://doi.org/10.1111/ppl.12002
Perez-Rodriguez P, Riano-Pachon DM, Correa LG, Rensing SA, Kersten B, Mueller-Roeber B (2010) PlnTFDB: updated content and new features of the plant transcription factor database. Nucleic Acids Res 38:822–827. https://doi.org/10.1093/nar/gkp805
Radoeva T, Vaddepalli P, Zhang Z, Weijers D (2019) Evolution, initiation, and diversity in early plant embryogenesis. Dev Cell 50(5):533–543. https://doi.org/10.1016/j.devcel.2019.07.011
Ramsay NA, Glover BJ (2005) MYB-bHLH-WD40 protein complex and the evolution of cellular diversity. Trends Plant Sci 10(2):63–70. https://doi.org/10.1016/j.tplants.2004.12.011
Ren H, Han J, Yang P, Mao W, Liu X, Qiu L, Qian C, Liu Y, Chen Z, Ouyang X, Chen X, Deng XW, Huang X (2019) Two E3 ligases antagonistically regulate the UV-B response in Arabidopsis. Proc Natl Acad Sci U S A 116(10):4722–4731. https://doi.org/10.1073/pnas.1816268116
Saitoh K, Onishi K, Mikami I, Thidar K, Sano Y (2004) Allelic diversification at the C (OsC1) locus of wild and cultivated rice: Nucleotide changes associated with phenotypes. Genetics 168(2):997–1007. https://doi.org/10.1534/genetics.103.018390
Sakamoto W, Ohmori T, Kageyama K, Miyazaki C, Saito A, Murata M, Noda K, Maekawa M (2001) The purple leaf (Pl) locus of rice: the Pl(w) allele has a complex organization and includes two genes encoding basic helix-loop-helix proteins involved in anthocyanin biosynthesis. Plant Cell Physiol 42(9):982–991. https://doi.org/10.1093/pcp/pce128
Sato Y, Hong SK, Tagiri A, Kitano H, Yamamoto N, Nagato Y, Matsuoka M (1996) A rice homeobox gene, OSH1, is expressed before organ differentiation in a specific region during early embryogenesis. Proc Natl Acad Sci USA 93(15):8117–8122. https://doi.org/10.1073/pnas.93.15.8117
Sato Y, Sentoku N, Nagato Y, Matsuoka M (1998) Isolation and characterization of a rice homeobox gene, OSH15. Plant Mol Biol 38(6):983–998. https://doi.org/10.1023/A:1006065622251
Sentoku N, Sato Y, Kurata N, Ito Y, Kitano H, Matsuoka M (1999) Regional expression of the rice KN1-type homeobox gene family during embryo, shoot, and flower development. Plant Cell 11(9):1651–1664. https://doi.org/10.1105/tpc.11.9.1651
Sharma M, Chai CL, Morohashi K, Grotewold E, Snook ME, Chopra S (2012) Expression of flavonoid 3 ’-hydroxylase is controlled by P1, the regulator of 3-deoxyflavonoid biosynthesis in maize. Bmc Plant Biol. https://doi.org/10.1186/1471-2229-12-196
Shin DH, Choi M, Kim K, Bang G, Cho M, Choi SB, Choi G, Park YI (2013) HY5 regulates anthocyanin biosynthesis by inducing the transcriptional activation of the MYB75/PAP1 transcription factor in Arabidopsis. FEBS Lett 587(10):1543–1547. https://doi.org/10.1016/j.febslet.2013.03.037
Stone SL, Braybrook SA, Paula SL, Kwong LW, Meuser J, Pelletier J, Hsieh TF, Fischer RL, Goldberg RB, Harada JJ (2008) Arabidopsis LEAFY COTYLEDON2 induces maturation traits and auxin activity: implications for somatic embryogenesis. Proc Natl Acad Sci U S A 105(8):3151–3156. https://doi.org/10.1073/pnas.0712364105
Stracke R, Favory JJ, Gruber H, Bartelniewoehner L, Bartels S, Binkert M, Funk M, Weisshaar B, Ulm R (2010) The Arabidopsis bZIP transcription factor HY5 regulates expression of the PFG1/MYB12 gene in response to light and ultraviolet-B radiation. Plant Cell Environ 33(1):88–103. https://doi.org/10.1111/j.1365-3040.2009.02061.x
Sun XM, Zhang ZY, Chen C, Wu W, Ren NN, Jiang CH, Yu JP, Zhao Y, Zheng XM, Yang QW, Zhang HL, Li JJ, Li ZC (2018) The C-S-A gene system regulates hull pigmentation and reveals evolution of anthocyanin biosynthesis pathway in rice. J Exp Bot 69(7):1485–1498. https://doi.org/10.1093/jxb/ery001
Sweeney MT, Thomson MJ, Pfeil BE, McCouch S (2006) Caught red-handed: Rc encodes a basic helix-loop-helix protein conditioning red pericarp in rice. Plant Cell 18(2):283–294. https://doi.org/10.1105/tpc.105.038430
ten Hove CA, Lu KJ, Weijers D (2015) Building a plant: cell fate specification in the early Arabidopsis embryo. Development 142(3):420–430. https://doi.org/10.1242/dev.111500
Thakare D, Tang W, Hill K, Perry SE (2008) The MADS-domain transcriptional regulator AGAMOUS-LIKE15 promotes somatic embryo development in Arabidopsis and soybean. Plant Physiol 146(4):1663–1672. https://doi.org/10.1104/pp.108.115832
Thimm O, Blasing O, Gibon Y, Nagel A, Meyer S, Kruger P, Selbig J, Muller LA, Rhee SY, Stitt M (2004) MAPMAN: a user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J 37(6):914–939. https://doi.org/10.1111/j.1365-313x.2004.02016.x
Tsuda K, Ito Y, Sato Y, Kurata N (2011) Positive autoregulation of a KNOX gene is essential for shoot apical meristem maintenance in rice. Plant Cell 23(12):4368–4381. https://doi.org/10.1105/tpc.111.090050
Tsuwamoto R, Yokoi S, Takahata Y (2010) Arabidopsis EMBRYOMAKER encoding an AP2 domain transcription factor plays a key role in developmental change from vegetative to embryonic phase. Plant Mol Biol 73(4–5):481–492. https://doi.org/10.1007/s11103-010-9634-3
van der Graaff E, Laux T, Rensing SA (2009) The WUS homeobox-containing (WOX) protein family. Genome Biol 10(12):248. https://doi.org/10.1186/gb-2009-10-12-248
Wang J, Qi M, Liu J, Zhang Y (2015) CARMO: a comprehensive annotation platform for functional exploration of rice multi-omics data. Plant J 83(2):359–374. https://doi.org/10.1111/tpj.12894
Wang L, Liu N, Wang T, Li J, Wen T, Yang X, Lindsey K, Zhang X (2018) The GhmiR157a-GhSPL10 regulatory module controls initial cellular dedifferentiation and callus proliferation in cotton by modulating ethylene-mediated flavonoid biosynthesis. J Exp Bot 69(5):1081–1093. https://doi.org/10.1093/jxb/erx475
Wang WF, Li G, Zhao J, Chu HW, Lin WH, Zhang DB, Wang ZY, Liang WQ (2014) DWARF TILLER1, a WUSCHEL-related homeobox transcription factor, is required for tiller growth in rice. Plos Genet 10:3. https://doi.org/10.1371/journal.pgen.1004154
Winkel-Shirley B (2001) Flavonoid biosynthesis. A colorful model for genetics, biochemistry, cell biology and biotechnology. Plant Physiol 126(2):485–493. https://doi.org/10.1104/pp.126.2.485
Wu M, Si M, Li X, Song L, Liu J, Zhai R, Cong L, Yue R, Yang C, Ma F, Xu L, Wang Z (2019) PbCOP11 contributes to the negative regulation of anthocyanin biosynthesis in pear. Plants (Basel) 8:2. https://doi.org/10.3390/plants8020039
Xu W, Dubos C, Lepiniec L (2015) Transcriptional control of flavonoid biosynthesis by MYB-bHLH-WDR complexes. Trends Plant Sci 20(3):176–185. https://doi.org/10.1016/j.tplants.2014.12.001
Yang X, Wang J, Xia X, Zhang Z, He J, Nong B, Luo T, Feng R, Wu Y, Pan Y, Xiong F, Zeng Y, Chen C, Guo H, Xu Z, Li D, Deng G (2021) OsTTG1, a WD40 repeat gene, regulates anthocyanin biosynthesis in rice. Plant J 107(1):198–214. https://doi.org/10.1111/tpj.15285
Zhang HN, Li WC, Wang HC, Shi SY, Shu B, Liu LQ, Wei YZ, Xie JH (2016) Transcriptome profiling of light-regulated anthocyanin biosynthesis in the pericarp of litchi. Front Plant Sci 7:963. https://doi.org/10.3389/fpls.2016.00963
Zhang PF, Wang YB, Zhang JB, Maddock S, Snook M, Peterson T (2003) A maize QTL for silk maysin levels contains duplicated Myb-homologous genes which jointly regulate flavone biosynthesis. Plant Mol Biol 52(1):1–15. https://doi.org/10.1023/A:1023942819106
Zhao KM, Bartley LE (2014) Comparative genomic analysis of the R2R3 MYB secondary cell wall regulators of Arabidopsis, poplar, rice, maize, and switchgrass. Bmc Plant Biol. https://doi.org/10.1186/1471-2229-14-135
Zhao P, Begcy K, Dresselhaus T, Sun MX (2017) Does early embryogenesis in eudicots and monocots involve the same mechanism and molecular players? Plant Physiol 173(1):130–142. https://doi.org/10.1104/pp.16.01406
Zhao Y, Hu YF, Dai MQ, Huang LM, Zhou DX (2009) The WUSCHEL-related homeobox gene WOX11 Is required to activate shoot-borne crown root development in rice. Plant Cell 21(3):736–748. https://doi.org/10.1105/tpc.108.061655
Zheng J, Wu H, Zhu H, Huang C, Liu C, Chang Y, Kong Z, Zhou Z, Wang G, Lin Y, Chen H (2019) Determining factors, regulation system, and domestication of anthocyanin biosynthesis in rice leaves. New Phytol 223(2):705–721. https://doi.org/10.1111/nph.15807
Zuo J, Niu QW, Frugis G, Chua NH (2002) The WUSCHEL gene promotes vegetative-to-embryonic transition in Arabidopsis. Plant J 30(3):349–359. https://doi.org/10.1046/j.1365-313x.2002.01289.x
Acknowledgements
This work was funded by “Cooperative Research Program for Agriculture Science and Technology Development (Project No. PJ01572901)”, Rural Development Administration, Republic of Korea.
Author information
Authors and Affiliations
Contributions
BK: designed and performed the research, analyzed the data, and wrote the manuscript. SS, HZ, and ZJ: analyzed the RNA-seq data. CL, SJ, and JS: validated the RNA-seq data. SWK: revised the manuscript. HJK: designed and supervised the experiment and revised the manuscript. All authors have read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no potential competing interest.
Additional information
Handling Editor: Pramod Kumar Nagar.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kim, B., Shim, S., Zhang, H. et al. Dynamic Transcriptome Changes Driven by the Mutation of OsCOP1 Underlie Flavonoid Biosynthesis and Embryogenesis in the Developing Rice Seed. J Plant Growth Regul 42, 4436–4452 (2023). https://doi.org/10.1007/s00344-023-10909-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00344-023-10909-0