
Abstract
Key message
Novel mutations of OsCOP1 were identified to be responsible for yellowish pericarp and embryo lethal phenotype, which revealed that OsCOP1 plays a crucial role in flavonoid biosynthesis and embryogenesis in rice seed.
Abstract
Successful production of viable seeds is a major component of plant life cycles, and seed development is a complex, highly regulated process that affects characteristics such as seed viability and color. In this study, three yellowish-pericarp embryo lethal (yel) mutants, yel-hc, yel-sk, and yel-cc, were produced from three different japonica cultivars of rice (Oryza sativa L). Mutant seeds had yellowish pericarps and exhibited embryonic lethality, with significantly reduced grain size and weight. Morphological aberrations were apparent by 5 days after pollination, with abnormal embryo development and increased flavonoid accumulation observed in the yel mutants. Genetic analysis and mapping revealed that the phenotype of the three yel mutants was controlled by a single recessive gene, LOC_Os02g53140, an ortholog of Arabidopsis thaliana CONSTITUTIVE PHOTOMORPHOGENIC 1 (COP1). The yel-hc, yel-sk, and yel-cc mutants carried mutations in the RING finger, coiled-coil, and WD40 repeat domains, respectively, of OsCOP1. CRISPR/Cas9-targeted mutagenesis was used to knock out OsCOP1 by targeting its functional domains, and transgenic seed displayed the yel mutant phenotype. Overexpression of OsCOP1 in a homozygous yel-hc mutant background restored pericarp color, and the aberrant flavonoid accumulation observed in yel-hc mutant was significantly reduced in the embryo and endosperm. These results demonstrate that OsCOP1 is associated with embryo development and flavonoid biosynthesis in rice grains. This study will facilitate a better understanding of the functional roles of OsCOP1 involved in early embryogenesis and flavonoid biosynthesis in rice seeds.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Light is a crucial signaling component that controls a variety of physiological and developmental processes in morphogenesis, germination, and flowering in plants. Light signaling is also involved in metabolic alterations, including the biosynthesis of chlorophyll (Ilag et al. 1994; Pattanayak et al. 2005) and several types of pigments (Takos et al. 2006; von Lintig et al. 1997; Zoratti et al. 2014). These light signaling pathways are triggered upon perception of light signals by multiple photoreceptors, such as red/far red light-sensing phytochromes (phyA to phyE), UV-A/blue light-sensing cryptochromes (cry1 and cry2), phototropins (phot1 and phot2), Zeitlupes (ZTL), and the UV-B-sensing photoreceptor (UVR8), which transmit signals to genes that regulate developmental and metabolic processes such as anthocyanin biosynthesis in higher plants (Jiao et al. 2007; Kami et al. 2010).
The light signaling regulator CONSTITUTIVELY PHOTOMORPHOGENIC 1 (COP1) is an essential negative regulator of light-mediated plant development. In darkness, COP1 acts as an E3 ubiquitin ligase to repress light signaling by targeting numerous photomorphogenic-promoting transcription factors, including ELONGATED HYPOCOTYL5 (HY5), HY5-HOMOLOG (HYH), LONG AFTER FAR-RED1 (LAF1), and LONG HYPOCOTYL IN FAR-RED1 (HFR1), for ubiquitination and degradation in the nucleus (Holm et al. 2002; Osterlund et al. 2000; Seo et al. 2003; Yang et al. 2005). In addition to acting as a repressor of photomorphogenesis (Ang et al. 1998; Deng et al. 1991; Ma et al. 2002; Seo et al. 2003), COP1 functions as a crucial regulator in several biological processes, such as flowering time (Jang et al. 2015; Tanaka et al. 2011; Xu et al. 2016a), circadian regulation (Yu et al. 2008), stomatal development (Kang et al. 2009; Lee et al. 2017), UV-B signaling (Favory et al. 2009; Huang et al. 2014; Yin et al. 2015), abiotic stress responses (Kim et al. 2016), and hormone signaling (Pacin et al. 2016; Zheng et al. 2017). In particular, COP1 plays a central role in regulating anthocyanin biosynthesis and fruit coloration by complexing with other proteins (Li et al. 2012; Liang et al. 2020; Maier et al. 2013).
The COP1 protein comprises three distinct structural domains that are conserved in both higher plants and vertebrates: a zinc-binding RING finger motif, a coiled-coil domain, and a WD40 repeat domain (Stacey et al. 1999). These highly modulated domains of COP1 are involved in self-association, dimerization, and protein interactions with other proteins (Holm et al. 2001; Torii et al. 1998). The RING finger domain plays a critical role in mediating the recognition of substrates as well as the transfer of ubiquitin to substrates and to the RING finger domain itself (Joazeiro and Weissman 2000; Kosarev et al. 2002). The coiled-coil domain is involved in dimerization and interactions with other proteins, including CIP1, SPA1, and CSU2 (Hoecker and Quail 2001; Lee et al. 2018; Matsui et al. 1995; Xu et al. 2015). In COP1, the WD40 domain is found at the C-terminus and consists of seven WD40 repeats that mediate protein–protein interactions. COP1 directly interacts with several regulators, such as HY5 and HYH, and photoreceptors (cytochromes and UVR8) via its WD40 domain (Holm et al. 2001, 2002; Ponnu et al. 2019; Yin et al. 2015).
Mutagenesis is an important strategy to create mutations which are valuable sources for functional genomics as well as crop improvement. In particular, relatively small genome size of rice has enabled a genome-wide saturation mutagenesis and revelation of full spectrum of gene functions by generating allelic series of mutations with small population (Henikoff and Comai 2003). Over the past several decades, numerous mutagenesis methods have been developed for rice, but chemical and physical mutagens have been widely applied to induce genetic variability and to study gene function due to its simple and low cost strategy (Viana et al. 2019; Wang et al. 2013). The genome changes between wild-type and mutants caused by chemical and physical mutagen have been identified efficiently in recent years using Targeting Induced Local Lesions IN Genomes (TILLING) (Till et al. 2007) and MutMap (Abe et al. 2012), next-generation sequencing (NGS)-based gene mapping tool in rice. These researches through mutagenesis would be helpful to identify unknown pathways or genes and could be directly applied to the rice breeding.
Genome editing technology using the Clustered Regularly Interspaced Short Palindromic Repeats/CRISPR-associated Cas9 (CRISPR/Cas9) system is a powerful and promising tool for precise modification of genome sequences in crop plants. In recent years, the CRISPR/Cas9 system has been widely adopted for crop improvement due to its simplicity, specificity, efficiency, and feasibility compared with zinc finger nuclease and transcription activator-like effector nuclease editing technologies (Khan 2019). Studies employing CRISPR/Cas9 genome editing tools have been used to validate gene functions and improve useful traits in rice, such as herbicide resistance (Li et al. 2016a; Sun et al. 2016), rice blast disease resistance (Wang et al. 2016), cold tolerance (Zeng et al. 2019), salt tolerance (Zhang et al. 2019), amylose content (Sun et al. 2017), and grain size and/or yield (Li et al. 2016b; Xu et al. 2016b). Ongoing improvements in genome editing tools will accelerate the pace of crop improvement and gene validation experiments.
The function of COP1 in photomorphogenic development and anthocyanin biosynthesis has been extensively studied in Arabidopsis. However, little is known regarding the role of COP1 in flavonoid biosynthesis and embryo development in rice. In this study, novel OsCOP1 mutants that exhibited a yellowish pericarp color and seed lethality were identified, and CRISPR/Cas9 gene editing tool was used to validate the mutant phenotype. These findings provide new insights into the regulation of flavonoid biosynthesis and embryo development in rice.
Materials and methods
Plant materials and growth conditions
Three yellowish-pericarp embryo lethal (yel) mutants, yel-hc, yel-sk, and yel-cc, were derived from japonica rice cultivars Hwacheong, Samkwang, and Chucheong, respectively, by induction with N-methyl-N-nitrosourea. Due to their embryonic lethality, the mutants were maintained as heterozygotes. F2 mapping populations were developed from crosses between the three heterozygous yel mutants and Milyang 23 (M.23, Korean indica-type rice). F2 populations derived from crosses between the three heterozygous yel mutants and their respective wild types, Hwacheong, Samkwang, and Chucheong, were used to calculate segregation ratios. F2 populations, wild-type varieties, and yel mutants were cultivated in a paddy field at the Experimental Farm of Seoul National University, Suwon, Korea.
Phenotypic analysis
Dimensions of dehulled rice including length, width, and thickness were measured in a total of 90 matured grains (30 seeds × 3 replicates) using digimatic calipers (Mitutoyo, Japan). Hundred-grain weights were measured (100 seeds × 3 replicates; 10% water content) using an analytical balance (CAS Corporation, NJ, USA). Phenotypic data collected from the three yel mutants and their respective wild types were statistically analyzed using SAS version 9.4 (SAS Institute Inc., Cary, NC, USA).
Morphological and histological observations of developing seed
Caryopses of Hwacheong and the yel-hc mutant were collected at several stages of seed development and imaged using HD’MEASURE software (HANA Vision Systems, Korea). For histological examination, samples were collected at 3 days after pollination (DAP), 4 DAP, 5 DAP, 7 DAP, 10 DAP, and 40 DAP (matured), and fixed in formalin-acetic acid-alcohol fixative (3.7% formaldehyde, 5% acetic acid, and 50% ethanol) at 4 °C. Seeds were dehydrated by soaking in a series of ethanol solutions of increasing concentration (50%, 70%, 80%, 90%, 95%, and 100%) for 3 h each, followed by soaking in 100% ethanol overnight at room temperature. The dehydrated samples were soaked in a series of mixtures of absolute ethanol and Histo-Clear (3:1, 1:1, 1:3) for 3 h each, followed by soaking in 100% Histo-Clear overnight at room temperature. For paraffin infiltration, Paraplast® (Sigma, USA) was gradually added to Histo-Clear (3:1, 1:1, 1:3) at 60 °C. Finally, samples were stored in 100% melted paraffin for 24 h at 60 °C. Paraffin-infiltrated samples were embedded in an embed block and cut into 10-μm sections with an HM 340 E Rotary Microtome (Microm Lab, Germany). The sections were stained with 1% Safranin O solution and observed under a CX31 Microscope (Olympus, Japan).
Map-based cloning of yel gene
Genomic DNAs were extracted from a total of 898 matured mutant-type seeds and young heterozygous and wild-type leaves of F2 individuals derived from the yel-hc x Milyang23 population. To determine the locus of the gene underlying the yel-hc mutant, bulked segregant analysis (BSA) (Michelmore et al. 1991) was performed with the first batch of STS sequence-tagged-site (STS) markers, which were developed by designing primers based on the DNA sequence differences between indica and japonica rice cultivars (Chin et al. 2007). Next, additional STS primers were designed with Primer3 version 0.4.0 (http://frodo.wi.mit.edu/primer3) based on available rice genome sequence data (http://www.ncbi.nlm.nih.gov) and were used to refine the region of the yel-hc locus. Additionally, a total of 192 F2 individuals were used for each of the yel-sk and yel-cc mutant gene mapping. The overall mapping strategy and primers were the same as for yel-hc. Primers designed and used in this study are detailed in Table S1.
Sequence analysis of candidate genes
The full-length genomic DNA sequence of candidate genes in Hwacheong and yel-hc was divided into several overlapping segments and then amplified. PCR products were purified using a PCR purification kit (iNtRON Biotechnology, Korea), TA-cloned into the pGEM-T Easy Vector (Promega, USA), and transformed into E. coli strain DH5α. The obtained sequences were compared using CodonCode Aligner software (version 1.6.3; CodonCode Corporation, MA, USA).
Vector constructs and rice transformation
CRISPR/Cas9 vectors to knock out COP1 were constructed as described previously (Lowder et al. 2015). Briefly, two guide RNAs (gRNAs) for each target were designed using the web-based tools CRISPR RGEN Tools (http://www.rgenome.net/cas-designer/) (Park et al. 2015) and CRISPRdirect (https://crispr.dbcls.jp/) (Naito et al. 2015). The two individual gRNAs were cloned into two gRNA expression vectors, pYPQ131C (Addgene plasmid #69,284) and pYPQ132C (Addgene plasmid #69,285), under the expression of the OsU6 promoter. The gRNA expression cassettes were then assembled into the Golden Gate recipient vector pYPQ142 (Addgene plasmid #69,294). For the Cas9 entry vector, pYPQ165 (Addgene #109,327), which is driven by an egg cell-specific promoter, was used. Final binary T-DNA vectors were prepared using a Gateway assembly LR reaction with the Cas9 entry vector (pYPQ165), gRNA cassettes (pYPQ142), and pMDC99 (binary vector). The full-length OsCOP1 cDNA was amplified from Hwacheong cDNA and used for construction of the overexpression vector. The amplified fragment was transferred into the pMDC32 Gateway binary vector containing the cauliflower mosaic virus (CaMV) 35S promoter using a pCR™ 8/GW/TOPO® TA Cloning Kit (Invitrogen, USA) and Gateway™ LR Clonase™ II Enzyme mix (Invitrogen). The resulting OsCOP1 cDNA overexpression construct was denoted as 35S::OsCOP1. For the promoter-GUS (β-glucuronidase) assay, the genomic sequence containing the putative promoter region (− 2215 to − 1 bp upstream of the start codon) of OsCOP1 was amplified from genomic DNA and cloned into the binary vector pHGWFS7 using Gateway® BP and LR clonase enzyme mixes (Invitrogen).
The final constructs were transformed into seeds of the japonica cultivar Dongjin, via Agrobacterium-mediated transformation using the LBA4404 strain, as described previously (Nishimura et al. 2006), with slight modifications. Primers used for vector construction and genotyping are listed in Table S1.
Histochemical GUS assay
Histochemical GUS-staining was performed as described previously (Jefferson et al. 1987). X-Gluc buffer solution was vacuum-infiltrated into tissue samples, which were incubated overnight at 37 °C, after which the staining solution was serially replaced with 95% and 70% (w/v) ethanol to remove the chlorophyll. Staining patterns in tissue samples were examined using HD’MEASURE software (HANA Vision, Korea).
Quantitative real-time PCR analysis
Total RNA was extracted from 7-day-old seeds of the three yel mutants and their respective wild types, 35S::OsCOP1, knockout lines, and Dongjin using a TaKaRa MiniBEST Plant RNA Extraction Kit (Takara Bio, Japan). Total RNA samples were converted into first-strand cDNA using M-MLV reverse transcriptase (Promega, USA), and qRT-PCR was performed using SYBR Premix Ex Taq (Takara Bio, Japan) on a CFX96™ Real-Time PCR Detection System (Bio-Rad, USA), according to the manufacturer’s instructions. Primers used for qRT-PCR analysis are listed in Table S1. Expression levels of OsCOP1 were normalized relative to ACTIN, a housekeeping gene. Data were analyzed using the comparative Ct method. Expression levels were compared using a two-tailed Student’s t test.
Quantitative analysis of major flavonoids using HPLC
HPLC sample preparation and quantitative analysis were performed as described previously (Kim et al. 2018). Whole grain, embryo, and endosperm tissues from yel-hc, Hwacheong, and transgenic overexpression seeds were powdered, and three technical replicates were prepared for quantitative analysis of six representative flavonoid compounds (isoorientin, isoorientin 2″-O-glucoside, vitexin 2″-O-glucoside, isoscoparin 2″-O-glucoside, isoscoparin, and isovitexin). HPLC was performed on a XBridge C18 column (250 × 4.6 mm, 5 μm; Waters, Milford, MA, USA) using an Ultimate 3000 UHPLC system (Thermo Fisher Scientific, Inc., MA, USA) equipped with an autosampler, a gradient system, and a diode array detector (DAD). Chromatograms were acquired at 365 nm, and photodiode array spectra were recorded from 190 to 800 nm. The injection volume of each sample was 5 μL.
Results
Morphological characterization of yel mutants
Three yellowish-pericarp embryo lethal (yel) mutants, yel-hc, yel-sk, and yel-cc, were derived from japonica rice cultivars Hwacheong, Samkwang, and Chucheong, respectively. The most prominent characteristic of the mutants was a bicolor grain phenotype. Embryos were black in all three yel mutants, and the pericarp was yellowish in yel-hc and yel-sk grains and mixed yellow-purple in yel-cc grains (Fig. 1a). The black pigmentation in yel mutant embryos was first observed at approximately 5 DAP and the yellowish pericarp color was clearly visible at 35 DAP, the late-maturing stage (Fig. 1b). All three yel mutants exhibited significantly reduced grain length and thickness compared with wild type, and grain width was also significantly lower in the yel-sk and yel-cc mutants. Correspondingly, hundred-grain-weight was also significantly lower in yel mutants than in wild type. However, there was no significant difference between yel mutants and wild type in the length-to-width ratio of de-hulled rice grains (Table 1). These results indicated that the yel mutations influenced embryo and pericarp color, grain size, and grain weight in rice.

Comparison of grain morphology and development between wild-type and yel mutant. a Phenotype of wild-type and yel mutant seeds. Upper and lower panels represent wild-type and yel mutants, respectively. b Morphological differences during seed development between wild-type (Hwacheong) and yel-hc mutant. Upper and lower panels show seed development of wild-type (Hwacheong) and yel-hc mutant, respectively. Numbers at the bottom of the subpanel indicate days after pollination. Scale bar = 2 mm
Embryo development of yel mutants
Homozygous yel mutant seeds failed to germinate under both standard germination testing and on MS medium (data not shown). Longitudinal sections of yel-hc seeds were examined to determine when defects occured during yel mutant embryo development. Normal embryonic development was observed in both yel-hc and wild-type embryos during early embryogenesis, including at the globular stage (Fig. 2a and f). The coleoptile primordium was differentiated in both the wild-type and yel-hc mutant, and no morphological differences were observed at 4 DAP (Fig. 2b and g). In wild-type embryos, the coleoptile, epiblast, and scutellum had formed, and the shoot and root meristems had begun to differentiate, at 5 DAP. However, the overall morphology of yel-hc embryos differed from that of wild type at 5 DAP. It was not possible to determine organ differentiation in the yel-hc embryo as the shape and structure of organs were unclear (Fig. 2c and h). At more advanced developmental stages, all organs were normally differentiated and developed in wild-type embryos, whereas abnormally differentiated organs were observed in yel-hc embryos (Fig. 2d, e and i, j). In the mature embryo, all of the organs were evident in wild type, but, despite completion of embryogenesis, irregular and unstructured organs were observed in the yel-hc mutant (Fig. S1). These results demonstrated that the mutation of YEL caused defects in embryonic differentiation and development, resulting in seed lethality and failure to germinate in yel mutants.

Embryo development in wild-type and yel mutant. Upper and lower panels represent wild-type (Hwacheong) and yel-hc embryo development, respectively. a and f Embryo at 3 DAP (bar = 20 μm), b and g 4 DAP (bar = 100 μm), c and h 5 DAP (bar = 200 μm), d and i 7 DAP (bar = 300 μm), and e and j 10 DAP (Bar = 300 μm). Cp, coleoptile; Ep, epiblast; Rd, radicle; Sc, scutellum; Sm, shoot meristem; Rm, root meristem; Sa, shoot apex
Genetic analysis and map-based cloning of the YEL gene
As homozygous yel mutants were lethal, heterozygotes of each yel mutant and their respective wild type were used in crosses. The segregation ratios of wild-type and heterozygous plants for yel-hc, yel-sk, and ye1-cc in F1 progenies were 12:14, 11:10, and 14:11, respectively. These ratios were close to a 1:1 ratio, and the segregation ratios of wild-type and yel mutant grains produced by F1 plants fitted a 3:1 ratio (WT:yel-hc = 479:145, χ2 = 1.034 < χ2 0.05(1) = 3.841, p = 0.31; WT:yel-sk = 496:154, χ2 = 0.593 < χ2 0.05(1) = 3.841, p = 0.44; WT:yel-cc = 418:128, χ2 = 0.706 < χ2 0.05(1) = 3.841, p = 0.40). These results indicated that the yel phenotype was controlled by a single recessive gene.
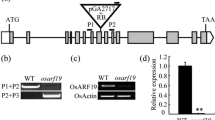
An F2 population derived from a cross between a yel-hc heterozygous mutant and Milyang23 (M.23) was used to map the locus responsible for the yel-hc phenotype. For the first step of genetic mapping, BSA was performed using 84 polymorphic STS markers, and the yel-hc locus was localized between two flanking markers, S02135 and S02140, on chromosome 2 (Fig. 3a). To refine the flanking region, BSA was again conducted using an F2 population of 840 individuals with newly designed STS markers between the two flanking markers (Table S1). The yel-hc locus was finally mapped to a region of approximately 55 kb between the S02K-1 and S02K markers (Fig. 3a). Seven predicted open reading frames (ORFs) were located within this candidate region, and analysis of wild-type and yel-hc mutant sequences revealed a 706 bp deletion in the yel-hc mutant at LOC_Os02g53140 (Os02g0771100), an ortholog of AtCOP1 (Fig. 3b). This 706 bp deletion included part of the 5′ untranslated region (− 19 to − 1, where the A of ATG is + 1), the entire coding sequence (CDS) of exon 1 (+ 1 to + 342), and part of intron 1 (+ 343 to + 687), resulting in the loss of the conserved RING finger motif of LOC_Os02g53140 (Fig. 3b). A similar mapping strategy and markers confirmed that yel-sk and yel-cc harbored mutations in the same gene as yel-hc. In the yel-sk mutant, a single base change (G to A) was discovered at the splicing donor site of the 5′ end of intron 2, within the coding region for the coiled-coil motif (Fig. 3b). In the yel-cc mutant, a single point mutation (G to T) was identified in exon 7 that generated a premature stop codon (GAG to TAG; Glu409 to stop) in the WD40 repeat motif (Fig. 3b). Three transcripts were identified in homozygous yel-sk seed. The first, most abundant, transcript contained the entire 473 bp of intron 2; the second included 64 bp of intron 2; and the third transcript type was the wild-type form. The insertion of the intron 2 sequence was predicted to introduce a premature termination codon at residue 145, resulting in the loss of the coiled-coil and WD40 functional motifs (Figure S2). These results indicated that the mutations identified in LOC_Os02g53140 were responsible for the yellowish pericarp and embryo lethal phenotypes in yel mutants.

Map-based cloning of the YEL gene. a Schematic diagram of YEL locus mapping. b Gene and protein domain structure of YEL. Black lines, white solid boxes, and black solid boxes indicate introns, untranslated regions, and exons, respectively. Mutation positions are marked with black arrows. ATG and TGA indicate the initiation and termination codons, respectively. The position and direction of gRNAs are marked by arrows above the gene structure
Validation of mutations causing the yel phenotype
To assess the functions of the individual OsCOP1 domains and the effects of the yel mutations, sequences for each domain were disrupted using the CRISPR/Cas9 system. Two gRNAs for each of the three constructs were designed to target crucial mutation points or domains, including the RING finger, coiled-coil, and WD40 repeat domains (Fig. 3b). An egg cell-specific Cas9 promoter was used to overcome lethality during tissue culture and increase the frequency of mutated homozygous T1 seeds generated from positive T0 transgenic plants. T1 seeds exhibiting the yel mutant phenotype were observed when the RING finger and WD40 repeat motifs of the OsCOP1 gene were targeted (Fig. 4a), but no T1 seeds exhibiting the yel phenotype were generated upon targeting of the coiled-coil motif, even though several positive T0 transgenic events occurred. The mutations were confirmed by Sanger sequencing the target region of each domain in T1 seeds displaying the yel mutant phenotype. The most frequently identified mutations were single base pair insertion and deletion mutations. A large fragment deletion between two gRNAs induced in transgenic seeds targeting the WD40 repeat domain was also observed (Fig. 4b). The insertions and deletions introduced into the OsCOP1 gene in seeds with yel phenotypes were all predicted to lead to frameshifts and premature stops, and thereby to produce aberrant proteins. These results demonstrated that the yel mutant phenotype was caused by loss-of-function of crucial domains in OsCOP1.

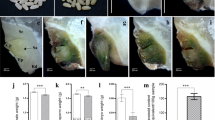
CRISPR/Cas9 mediated knockout and overexpression for complementation of yel mutations. a Grain phenotype of wild-type rice (Dongjin) and transgenic seeds. b Sequence comparison of target regions in OsCOP1. The start codon/RING finger domain and WD40 repeat domain were targeted for the yel-hc and yel-cc mutations, respectively. Red dashes and letters indicate deletions and insertions, respectively, in transgenic lines. The start codon, target sequence and protospacer adjacent motif are indicated by blue underline, gray boxes, and green boxes/black underline, respectively. Mutation types are shown to the right of each mutated sequence (−; deletion, + ; insertion). Numbers in parentheses indicate the number of mutated individuals identified from transgenic T1 seeds. c Phenotype of grains segregated from F2 progenies with the 35S::OsCOP1 transgene in the homozygous yel-hc background. Normal-type (left), yel mutant-type (middle), and partial recovery-type (right) seeds (bar = 1 mm). d–f Contents of six major flavonoids in whole grain (d), embryo (e), and endosperm (f) of the wild-type (Hwacheong), yel-hc, and partial recovery-type grains (35S::OsCOP1). ND; not detected. Error bars represent SD for three technical experiments. Asterisks indicate statistical significance, as determined by Student’s t test (***p < 0.001)
To complement the yel phenotype, the full-length OsCOP1 cDNA was introduced into wild-type Dongjin rice under the control of the 35S promoter. Eight positive transgenic lines were produced and crossed with a heterozygous yel-hc mutant to introduce the overexpression construct (35S::OsCOP1) into the homozygous yel-hc genetic background. F1 plants harboring 35S::OsCOP1 were selected, and the phenotypes of F2 progenies from the self-pollinated F1 plant were observed. As shown in Fig. 4c, three different types of seed were segregated from the F2 progenies: normal-type (normal pericarp and embryo color), yel mutant-type (yellowish-pericarp and black colored embryo), and partial recovery-type (normal pericarp color but black colored embryo). A total of 93 F2 seeds were genotyped to find the fully recovered seeds containing 35S::OsCOP1 transgene in homozygous yel-hc mutant background. All 20 yel mutant-type seeds showed yel-hc homozygous genotype (yel/yel), and all the partial recovery-type seeds contained 35S::OsCOP1 transgene in homozygous yel-hc mutant background (yel/yel + 35S_OsCOP1). However, among the normal-type seeds, none of the fully recovered transgenic yel seeds carrying 35S::OsCOP1 transgene were detected. (Table S2).
In our previous study, we demonstrated that high levels of flavonoids accumulated in the embryo and endosperm of yel-hc mutant grains (Kim et al. 2018). The contents of six major flavonoids were estimated in the embryo, endosperm, and whole grain of wild-type, yel-hc and partial recovery-type seeds. The total amounts of all six flavonoids were significantly reduced by approximately 46%, 23%, and 98% in the whole grain, embryo, and endosperm of partial recovery-type (35S::OsCOP1) seeds, respectively, compared with the yel-hc mutant seeds (Fig. 4d–f). The lack of accumulation of flavonoids was observed in the endosperm of partial recovery-type seeds overexpressing the 35S::OsCOP1 transgene, resulting in reduced flavonoid levels and restoration of wild type pericarp color. However, although the total amounts of the six main flavonoids were significantly reduced in the partial recovery-type seeds, high levels of flavonoids still accumulated in the embryo. In addition, partial recovery-type seeds still failed to germinate on both MS media and under standard germination conditions (data not shown). These results suggested that overexpression of the OsCOP1 gene in yel-hc seeds partially restored the yel phenotype by reducing flavonoid accumulation in the embryo and pericarp.
Relative expression of YEL
Quantitative real-time PCR (qRT-PCR) analysis using developing seeds (7 DAP) showed that expression of OsCOP1 was significantly higher in the yel-sk mutant than in wild type, but no significant differences were observed between wild-type and yel-hc and yel-cc mutants (Fig. 5a). No statistically significant differences in relative OsCOP1 expression level were observed between wild-type and CRISPR/Cas9 knockout seeds, which was consistent with the expression pattern of the yel-hc and yel-cc mutants. By contrast, OsCOP1 transcript levels were significantly higher in 35S::COP1 transgenic T2 seeds than in wild-type and yel mutant seeds or knockout lines (Fig. 5b).

Expression patterns of the YEL gene. a Relative expression levels of OsCOP1 in yel mutant and corresponding wild-type seeds using qRT-PCR. HC, Hwacheong; SK, Samkwang; CC, Chucheong. b Relative expression levels of OsCOP1 in wild-type (Dongjin), knockout, and partial recovery-type (35S::OSCOP1) seeds. Expression levels of OsCOP1 were normalized relative to ACTIN. Error bars represent SD for three technical experiments. Asterisks indicate statistical significance, as determined by Student’s t test (*p < 0.05; **p < 0.01). c–j GUS expression patterns in various organs in OsCOP1 promoter::GUS transgenic plants. c Spikelet; d, floral organ; e, pollen; f, leaf; g, seed; h, embryo; i, pericarp; j, node
OsCOP1 promoter–GUS expression pattern
Transgenic plants were generated containing the GUS reporter gene under the control of the OsCOP1 promoter region, and GUS expression was observed in several organs. GUS signals were detected primarily in pollen, node, embryo, and pericarp tissues, but no expression was detected in leaf, spikelet, stigma, or endosperm tissues (Fig. 5c–j). Interestingly, GUS staining in seed was concentrated in the embryo and pericarp, consistent with the flavonoid accumulation pattern in yel mutant seed.
Discussion
COP1 has been extensively studied as a repressor of plant photomorphogenesis, alongside DET (DE-ETIOLATED) and FUS (FUSCA), which also exhibit light-grown phenotypes in darkness. Many cop/det/fus mutants have been screened and identified in Arabidopsis (McNellis et al. 1994; Wei and Deng 1996), with the majority shown to cause constitutive photomorphogenesis. However, some cop/det/fus mutant alleles, such as cop1-5, cop1-8, det1-6, and fus7-1, had high levels of anthocyanin accumulation in cotyledons and leaves, and exhibited seedling lethal phenotypes under dark conditions (Castle and Meinke 1994; Ma et al. 2003; Stacey et al. 2000). Many of these phenotypically similar mutants exhibiting the ‘fusca’ phenotype were revealed to be allelic to each mutant group (Castle and Meinke 1994; Chory et al. 1989; Misera et al. 1994). However, unlike Arabidopsis, few cop1 mutants have been reported in rice. A loss-of-function mutation in PETER PAN SYNDROME (PPS), an ortholog of Arabidopsis COP1, produced dwarfed dark-green plants that showed a prolonged juvenile phase and early flowering in both short-day and long-day conditions (Tanaka et al. 2011). In this study, three allelic cop1 mutants were isolated that exhibited embryonic lethality and accumulated flavonoids in the pericarp and embryo (Figs. 1, 3). The three cop1 mutants are likely to be complete loss-of-function mutants and, based on the severity of the phenotype, the yel phenotype likely represents the cop1 null phenotype in rice. To the best of our knowledge, cop1 null mutants exhibiting a fusca phenotype similar to those of Arabidopsis mutants have not yet been reported in rice.
Two distinctive differences were apparent between rice yel mutants and Arabidopsis cop1 null mutants with the fusca phenotype, such as cop1-5, namely, differences in embryonic development and lethality and differences in pigmentation. First, despite germination delays, Arabidopsis cop1 null mutant seeds completed embryogenesis and germinated successfully, although seedling growth was severely retarded and viable plants did not develop (Castle and Meinke 1994; McNellis et al. 1994; Stacey et al. 2000). In addition, despite the lethal nature of the Arabidopsis cop1 null mutants, morphological abnormalities in embryogenesis were not observed for all the lethal alleles (McNellis et al. 1994). By contrast, in rice, all the homozygous recessive yel mutant seeds failed to germinate, and all showed defective embryo morphologies during embryogenesis (Fig. 2 and Fig. S1). Due to this embryonic lethality, it was impossible to observe the photomorphogenic phenotype in homozygous yel mutant plants. Studies on embryogenesis in plants indicate that there are crucial developmental events in early embryogenesis that are important for establishing seedling architecture after germination, such as embryo pattern formation, polarity, positioning, and cell fate determination. Although these embryogenic processes are similar among plant species, there are some differences in the patterns of cell division and organ differentiation positioning (Radoeva et al. 2019; Zhao et al. 2017). Embryogenesis in Arabidopsis and rice differ in their cell division patterns, position of shoot apical meristem with respect to embryo polarity, position of radicle differentiation, and the effects of embryo–endosperm interactions on embryo development (Itoh et al. 2005; Nagasawa et al. 2013). Rice and Arabidopsis also differ in their embryogenesis-related gene expression patterns and regulatory networks (Itoh et al. 2016). Taken together, this suggests that the differences in morphological defects between rice and Arabidopsis embryos in cop1 null mutants may be due to differences in developmental processes between monocots and dicots during early embryogenesis.
Second, the seed coat and cotyledons in Arabidopsis cop1 null mutants were dark purple as a consequence of anthocyanin accumulation (Castle and Meinke 1994; McNellis et al. 1994; Stacey et al. 2000). In rice, yel mutants displayed yellowish pericarps and black embryos in dried seeds. Our previous study showed that the accumulated pigments in the embryo and pericarp were mainly C-glycosyl flavones, such as isoorientin, isovitexin, and isoscoparin, and were not anthocyanins or their derivatives (Kim et al. 2018). Flavonoid biosynthesis is controlled by structural and regulatory genes, and the genes involved in the flavonoid pathway are differentially regulated in monocot and dicot species, as indicated by the regulation patterns of regulatory genes (Petroni and Tonelli 2011), functional similarities of homologous genes (Spelt et al. 2000), and species-specific enzymes (Tohge et al. 2017). Taken together, this suggests that the biosynthetic pathways of anthocyanins and C-glycosyl flavones are regulated by transcription factors that interact with COP1, but that these transcription factors and/or their interactions with COP1 may differ among plant species. This also suggests that the transcription factors and genes downstream of COP1 that are involved in the flavonoid biosynthesis pathway may be differentially regulated in monocots and dicots.
In Arabidopsis, cop1 mutants have been separated into three classes, weak, strong, and lethal, according to their light reactivity (McNellis et al. 1994). Homozygous cop1 mutant seedlings in both the weak and strong categories exhibited light-grown characteristics when grown under dark conditions, albeit with a strongly reduced seed set. Homozygous lethal cop1 mutant seedlings exhibited a similar light-grown phenotype in darkness, but also had dark purple seeds and were seedling lethal. Mutations in the C-terminal region of the COP1 protein, containing the WD40 repeat regions, produced a much more severe phenotype, similar to that of complete loss-of-function mutants, than mutations in the N-terminal region. Many cop mutants have mutations within the WD40 repeat region, including cop1-5, cop1-7, and cop1-8, all of which display a lethal phenotype (Ma et al. 2003; McNellis et al. 1994; Zhou et al. 1998). In rice, the pps-1 mutation is located between the coiled-coil and WD40 domains, similar to weak cop1 alleles in Arabidopsis such as cop1-4 and cop1-6, which contain mutations in the N-terminal (Stacey et al. 2000). The rice pps-1 mutation was dark-green in color and had a dwarf phenotype, contrasting with pps-2, which had a premature stop codon in the WD40 domain and exhibited ~ 5% germination (Tanaka et al. 2011). Of the two pps mutants, it is likely that the pps-2 mutant allele, which has a mutation in the C-terminal of COP1, represents a more severe phenotype than pps-1. In this study, three different mutations in the RING finger, coiled-coil, and WD40 repeat domains of YEL caused similar phenotypic changes: embryonic lethality and accumulation of flavonoids in the pericarp and embryo. Interestingly, although the pps-2 mutation was located between the mutations in the CPR_WD-23 and CPR_WD-13 knockout lines (Fig. 4a and b), which generated premature stop codons three amino acids before and eight amino acids after the pps-2 mutation point, respectively, pps-2 did not exhibit the same phenotype as the yel mutants, although the pps-2 germination rate was extremely low and plants died within a week. These results suggest that the yel phenotype and other severe phenotypes can be induced by knocking out the crucial domain of COP1 in rice, irrespective of whether the mutation is in the N-terminal or C-terminal.
Knockout of essential genes generally leads to lethality or sterility, and knockout approaches are therefore limited in their capacity to yield informative results regarding gene function when the gene plays a crucial role in the organism (Zimmer et al. 2019). However, in this study, complete gene knockout using the CRISPR/Cas9 system was preferable to knockdown with RNA interference (RNAi), as difficulties were experienced in obtaining transgenic RNAi plants with the yel phenotype by reducing COP1 expression. Although several transgenic plants carrying RNAi constructs were generated, the yel phenotype was not observed in T1 or T2 seeds (data not shown). Attempts were also made to generate COP1 loss-of-function plants through anther culture. However, no homozygous recessive plants were obtained from a total of 293 double haploid plants derived from anther culture (data not shown). These experiences indicated that the RNAi approach did not downregulate COP1 expression to a sufficiently low level to induce the yel phenotype, and that recessive deleterious or lethal genes, such as cop1, might cause callus cell death, eliminating haploid plants with homozygous recessive cop1 alleles during anther culture. However, CRISPR/Cas9 editing produced several heterozygous T0 transgenic plants that produced segregating yel phenotype seeds in the T1 generation. Moreover, an egg cell-specific promoter rather than the 35S promoter was used to drive Cas9 expression (Lee et al. 2019; Wang et al. 2015) to overcome potential lethality during tissue culture regeneration of T0 plants and to increase the frequency of the yel phenotype in T1 seeds. Subsequently, homozygous mutant seeds displaying the yel phenotype were observed in the T1 generation (Fig. 4a and b). In this study, targeted gene editing was successfully used to show that COP1 was essential for flavonoid biosynthesis and embryo development, demonstrating the utility of CRISPR/Cas9 editing for examining previously uninducible phenotypes masked by lethality.
Previous studies have shown that the cop1-5 null mutant exhibited seedling lethal and high level of anthocyanin accumulation was successfully restored by full-length AtCOP1 and OsCOP1 (Holm et al. 2001; Ranjan et al. 2014; Stacey et al. 2000) in Arabidopsis. In addition, overexpression of the COP1 (1–282) fragment restored a wild-type phenotype in the cop1-5 mutant seedling in darkness (Stacey et al. 2000). However, normal pericarp color but black colored embryo seeds (partial recovery-type) were produced when the full-length OsCOP1 cDNA was overexpressed in a homozygous recessive yel-hc background seed (Fig. 4a and c). Even though the contents of flavonoids were significantly reduced in grain, embryo, and endosperm, the level of flavonoids in the embryo was still higher than that of wild-type (Fig. 4d–f). Moreover, the embryonic lethality was not rescued in the partially restored seeds. It is known that COP1 interacts with SUPPRESSOR OF PHYA-105 (SPA1) through their coiled-coil regions (Hoecker and Quail 2001), and the COP1/SPA complex targets transcription factors, such as ELONGATED HYPOCOTYL5 (HY5), HY5 HOMOLOG (HYH), LONG HYPOCOTYL IN FAR-RED 1 (HFR1), and PRODUCTION OF ANTHOCYANIN PIGMENT 1 and 2 (PAP1 and PAP2) involved in photomorphogenesis and anthocyanin biosynthesis for ubiquitination and degradation in Arabidopsis (Holm et al. 2002; Jang et al. 2005; Maier et al. 2013; Saijo et al. 2003). In particular, HY5, a bZIP transcription factor, positively regulates anthocyanin biosynthesis related structural genes through direct binding to their promoters (Shin et al. 2007). In the process of restoration, the competition between the mutated form of YEL protein (yel-hc) and normal OsCOP1/YEL to interact with SPA1 may reduce E3 ligase activity of COP1, and it may prevent ubiquitination and degradation of HY5 through the 26S proteasome pathway. Thus, remained HY5 enables to accumulate the flavonoids by inducing the expression of transcription factor in the seed. Therefore, it is assumed that the weakened E3 ligase activity of COP1 caused by the mutated COP1 that interfered the interaction between normal COP1 and SPA1 led to the incomplete COP1-mediated ubiquitination and degradation of HY5, resulting in partial restoration of yel phenotype in rice.
It was previously reported that expression of OsCOP1 was observed in almost all of rice tissues, such as calli, roots, and leaves (Tsuge et al. 2001). In addition, PPS, an ortholog of Arabidopsis COP1 was primarily expressed in leaves than shoot apices and young panicles during the juvenile-adult transition stage in rice; the lowest expression of PPS was shown in second leaves, and the expression was reached a peak in fourth and fifth leaves, but declined in higher leaves (Tanaka et al. 2011). However, unlike previous studies, negligible GUS activity of OsCOP1 was detected in leaf in this study (Fig. 5f). It is probably because the leaf was sampled at the flowering stage, which was after finishing the juvenile-adult transition, or might be sampled at the lower leaf of plant, which had the low expression level of OsCOP1 for GUS staining.
In this study, we identified three novel mutations responsible for the yellowish-pericarp and embryo lethal (yel) phenotype, and showed that COP1, a key repressor of light signaling, played a role in flavonoid biosynthesis and embryogenesis in rice seed. Further examination of the YEL locus will facilitate a better understanding of the molecular mechanisms involved in embryo development, and will provide novel insights into the roles of flavonoid biosynthesis-related genes and their interactions in rice.
References
Abe A, Kosugi S, Yoshida K, Natsume S, Takagi H, Kanzaki H, Matsumura H, Yoshida K, Mitsuoka C, Tamiru M, Innan H, Cano L, Kamoun S, Terauchi R (2012) Genome sequencing reveals agronomically important loci in rice using MutMap. Nat Biotechnol 30:174–178
Ang LH, Chattopadhyay S, Wei N, Oyama T, Okada K, Batschauer A, Deng XW (1998) Molecular interaction between COP1 and HY5 defines a regulatory switch for light control of Arabidopsis development. Mol Cell 1:213–222
Castle LA, Meinke DW (1994) A FUSCA gene of Arabidopsis encodes a novel protein essential for plant development. Plant Cell 6:25–41
Chin J, Kim JH, Jiang W, Chu SH, Mk W, Han L, Brar D, Koh HJ (2007) Identification of subspecies-specific STS markers and their association with segregation distortion in rice (Oryza sativa L.). J Crop Sci Biotechnol 10:137–184
Chory J, Peto C, Feinbaum R, Pratt L, Ausubel F (1989) Arabidopsis Thaliana mutant that develops as a light-grown plant in the absence of light. Cell 58:991–999
Deng XW, Caspar T, Quail PH (1991) cop1: a regulatory locus involved in light-controlled development and gene expression in Arabidopsis. Genes Dev 5:1172–1182
Favory JJ, Stec A, Gruber H, Rizzini L, Oravecz A, Funk M, Albert A, Cloix C, Jenkins GI, Oakeley EJ, Seidlitz HK, Nagy F, Ulm R (2009) Interaction of COP1 and UVR8 regulates UV-B-induced photomorphogenesis and stress acclimation in Arabidopsis. EMBO J 28:591–601
Henikoff S, Comai L (2003) Single-nucleotide mutations for plant functional genomics. Annu Rev Plant Biol 54:375–401
Hoecker U, Quail PH (2001) The phytochrome A-specific signaling intermediate SPA1 interacts directly with COP1, a constitutive repressor of light signaling in Arabidopsis. J Biol Chem 276:38173–38178
Holm M, Hardtke CS, Gaudet R, Deng XW (2001) Identification of a structural motif that confers specific interaction with the WD40 repeat domain of Arabidopsis COP1. EMBO J 20:118–127
Holm M, Ma LG, Qu LJ, Deng XW (2002) Two interacting bZIP proteins are direct targets of COP1-mediated control of light-dependent gene expression in Arabidopsis. Genes Dev 16:1247–1259
Huang X, Yang P, Ouyang X, Chen L, Deng XW (2014) Photoactivated UVR8-COP1 module determines photomorphogenic UV-B signaling output in Arabidopsis. PLoS Genet 10:e1004218
Ilag LL, Kumar AM, Soll D (1994) Light regulation of chlorophyll biosynthesis at the level of 5-aminolevulinate formation in Arabidopsis. Plant Cell 6:265–275
Itoh J, Nonomura K, Ikeda K, Yamaki S, Inukai Y, Yamagishi H, Kitano H, Nagato Y (2005) Rice plant development: from zygote to spikelet. Plant Cell Physiol 46:23–47
Itoh J, Sato Y, Sato Y, Hibara K, Shimizu-Sato S, Kobayashi H, Takehisa H, Sanguinet KA, Namiki N, Nagamura Y (2016) Genome-wide analysis of spatiotemporal gene expression patterns during early embryogenesis in rice. Development 143:1217–1227
Jang IC, Yang JY, Seo HS, Chua NH (2005) HFR1 is targeted by COP1 E3 ligase for post-translational proteolysis during phytochrome A signaling. Gene Dev 19:593–602
Jang K, Lee HG, Jung SJ, Paek NC, Seo PJ (2015) The E3 ubiquitin ligase COP1 regulates thermosensory flowering by triggering GI Degradation in Arabidopsis. Scientific Reports 5
Jefferson RA, Kavanagh TA, Bevan MW (1987) Gus fusions - beta-glucuronidase as a sensitive and versatile gene fusion marker in higher-plants. EMBO J 6:3901–3907
Jiao Y, Lau OS, Deng XW (2007) Light-regulated transcriptional networks in higher plants. Nat Rev Genet 8:217–230
Joazeiro CA, Weissman AM (2000) RING finger proteins: mediators of ubiquitin ligase activity. Cell 102:549–552
Kami C, Lorrain S, Hornitschek P, Fankhauser C (2010) Light-regulated plant growth and development. Curr Top Dev Biol 91:29–66
Kang CY, Lian HL, Wang FF, Huang JR, Yang HQ (2009) Cryptochromes, phytochromes, and COP1 regulate light-controlled stomatal development in Arabidopsis. Plant Cell 21:2624–2641
Khan SH (2019) Genome-editing technologies: concept, pros, and cons of various genome-editing techniques and bioethical concerns for clinical application. Mol Ther Nucl Acids 16:326–334
Kim B, Woo S, Kim MJ, Kwon SW, Lee J, Sung SH, Koh HJ (2018) Identification and quantification of flavonoids in yellow grain mutant of rice (Oryza sativa L.). Food Chem 241:154–162
Kim JY, Jang IC, Seo HS (2016) COP1 controls abiotic stress responses by modulating AtSIZ1 function through Its E3 ubiquitin ligase activity. Front Plant Sci 7:1182
Kosarev P, Mayer KF, Hardtke CS (2002) Evaluation and classification of RING-finger domains encoded by the Arabidopsis genome. Genome Biol 3:RESEARCH0016
Lee BD, Kim MR, Kang MY, Cha JY, Han SH, Nawkar GM, Sakuraba Y, Lee SY, Imaizumi T, McClung CR, Kim WY, Paek NC (2018) Author correction: the F-box protein FKF1 inhibits dimerization of COP1 in the control of photoperiodic flowering. Nat Commun 9:553
Lee JH, Jung JH, Park CM (2017) Light inhibits COP1-mediated degradation of ICE transcription factors to induce stomatal development in arabidopsis. Plant Cell 29:2817–2830
Lee K, Zhang Y, Kleinstiver BP, Guo JA, Aryee MJ, Miller J, Malzahn A, Zarecor S, Lawrence-Dill CJ, Joung JK, Qi Y, Wang K (2019) Activities and specificities of CRISPR/Cas9 and Cas12a nucleases for targeted mutagenesis in maize. Plant Biotechnol J 17:362–372
Li J, Meng XB, Zong Y, Chen KL, Zhang HW, Liu JX, Li JY, Gao CX (2016a) Gene replacements and insertions in rice by intron targeting using CRISPR-Cas9. Nat Plants. https://doi.org/10.1038/nplants.2016.139
Li M, Li X, Zhou Z, Wu P, Fang M, Pan X, Lin Q, Luo W, Wu G, Li H (2016b) Reassessment of the four yield-related genes Gn1a, DEP1, GS3, and IPA1 in rice using a CRISPR/Cas9 system. Front Plant Sci 7:377
Li YY, Mao K, Zhao C, Zhao XY, Zhang HL, Shu HR, Hao YJ (2012) MdCOP1 ubiquitin E3 ligases interact with MdMYB1 to regulate light-induced anthocyanin biosynthesis and red fruit coloration in apple. Plant Physiol 160:1011–1022
Liang D, Zhu T, Deng Q, Lin L, Tang Y, Wang J, Wang X, Luo X, Zhang H, Lv X, Xia H (2020) PacCOP1 negatively regulates anthocyanin biosynthesis in sweet cherry (Prunus avium L.). J Photochem Photobiol B 203:111779
Lowder LG, Zhang D, Baltes NJ, Paul JW 3rd, Tang X, Zheng X, Voytas DF, Hsieh TF, Zhang Y, Qi Y (2015) A CRISPR/Cas9 Toolbox for multiplexed plant genome editing and transcriptional regulation. Plant Physiol 169:971–985
Ma L, Zhao H, Deng XW (2003) Analysis of the mutational effects of the COP/DET/FUS loci on genome expression profiles reveals their overlapping yet not identical roles in regulating Arabidopsis seedling development. Development 130:969–981
Ma LG, Gao Y, Ou LJ, Chen ZL, Li JM, Zhao HY, Deng XW (2002) Genomic evidence for COP1 as a repressor of light-regulated gene expression and development in Arabidopsis. Plant Cell 14:2383–2398
Maier A, Schrader A, Kokkelink L, Falke C, Welter B, Iniesto E, Rubio V, Uhrig JF, Hulskamp M, Hoecker U (2013) Light and the E3 ubiquitin ligase COP1/SPA control the protein stability of the MYB transcription factors PAP1 and PAP2 involved in anthocyanin accumulation in Arabidopsis. Plant J 74:638–651
Matsui M, Stoop CD, von Arnim AG, Wei N, Deng XW (1995) Arabidopsis COP1 protein specifically interacts in vitro with a cytoskeleton-associated protein, CIP1. Proc Natl Acad Sci U S A 92:4239–4243
McNellis TW, von Arnim AG, Araki T, Komeda Y, Misera S, Deng XW (1994) Genetic and molecular analysis of an allelic series of cop1 mutants suggests functional roles for the multiple protein domains. Plant Cell 6:487–500
Michelmore RW, Paran I, Kesseli RV (1991) Identification of markers linked to disease-resistance genes by bulked segregant analysis: a rapid method to detect markers in specific genomic regions by using segregating populations. Proc Natl Acad Sci U S A 88:9828–9832
Misera S, Muller AJ, Weiland-Heidecker U, Jurgens G (1994) The FUSCA genes of Arabidopsis: negative regulators of light responses. Mol Gen Genet 244:242–252
Nagasawa N, Hibara K, Heppard EP, Vander Velden KA, Luck S, Beatty M, Nagato Y, Sakai H (2013) GIANT EMBRYO encodes CYP78A13, required for proper size balance between embryo and endosperm in rice. Plant J 75:592–605
Naito Y, Hino K, Bono H, Ui-Tei K (2015) CRISPRdirect: software for designing CRISPR/Cas guide RNA with reduced off-target sites. Bioinformatics 31:1120–1123
Nishimura A, Aichi I, Matsuoka M (2006) A protocol for Agrobacterium-mediated transformation in rice. Nat Protoc 1:2796–2802
Osterlund MT, Hardtke CS, Wei N, Deng XW (2000) Targeted destabilization of HY5 during light-regulated development of Arabidopsis. Nature 405:462–466
Pacin M, Semmoloni M, Legris M, Finlayson SA, Casal JJ (2016) Convergence of constitutive photomorphogenesis 1 and phytochrome interacting factor signalling during shade avoidance. New Phytol 211:967–979
Park J, Bae S, Kim JS (2015) Cas-designer: a web-based tool for choice of CRISPR-Cas9 target sites. Bioinformatics 31:4014–4016
Pattanayak GK, Biswal AK, Reddy VS, Tripathy BC (2005) Light-dependent regulation of chlorophyll b biosynthesis in chlorophyllide a oxygenase overexpressing tobacco plants. Biochem Biophys Res Commun 326:466–471
Petroni K, Tonelli C (2011) Recent advances on the regulation of anthocyanin synthesis in reproductive organs. Plant Sci 181:219–229
Ponnu J, Riedel T, Penner E, Schrader A, Hoecker U (2019) Cryptochrome 2 competes with COP1 substrates to repress COP1 ubiquitin ligase activity during Arabidopsis photomorphogenesis. Proc Natl Acad Sci U S A
Radoeva T, Vaddepalli P, Zhang Z, Weijers D (2019) Evolution, initiation, and diversity in early plant embryogenesis. Dev Cell 50:533–543
Ranjan A, Dickopf S, Ullrich KK, Rensing SA, Hoecker U (2014) Functional analysis of COP1 and SPA orthologs from Physcomitrella and rice during photomorphogenesis of transgenic Arabidopsis reveals distinct evolutionary conservation. BMC Plant Biol 14:178
Saijo Y, Sullivan JA, Wang H, Yang J, Shen Y, Rubio V, Ma L, Hoecker U, Deng XW (2003) The COP1-SPA1 interaction defines a critical step in phytochrome A-mediated regulation of HY5 activity. Genes Dev 17:2642–2647
Seo HS, Yang JY, Ishikawa M, Bolle C, Ballesteros ML, Chua NH (2003) LAF1 ubiquitination by COP1 controls photomorphogenesis and is stimulated by SPA1. Nature 423:995–999
Shin J, Park E, Choi G (2007) PIF3 regulates anthocyanin biosynthesis in an HY5-dependent manner with both factors directly binding anthocyanin biosynthetic gene promoters in Arabidopsis. Plant J 49:981–994
Spelt C, Quattrocchio F, Mol JN, Koes R (2000) anthocyanin1 of petunia encodes a basic helix-loop-helix protein that directly activates transcription of structural anthocyanin genes. Plant Cell 12:1619–1632
Stacey MG, Hicks SN, von Arnim AG (1999) Discrete domains mediate the light-responsive nuclear and cytoplasmic localization of Arabidopsis COP1. Plant Cell 11:349–364
Stacey MG, Kopp OR, Kim TH, von Arnim AG (2000) Modular domain structure of Arabidopsis COP1. Reconstitution of activity by fragment complementation and mutational analysis of a nuclear localization signal in planta. Plant Physiol 124:979–990
Sun Y, Zhang X, Wu C, He Y, Ma Y, Hou H, Guo X, Du W, Zhao Y, Xia L (2016) Engineering Herbicide-resistant rice plants through CRISPR/Cas9-mediated homologous recombination of acetolactate synthase. Mol Plant 9:628–631
Sun YW, Jiao GA, Liu ZP, Zhang X, Li JY, Guo XP, Du WM, Du JL, Francis F, Zhao YD, Xia LQ (2017) Generation of High-amylose rice through CRISPR/Cas9-mediated targeted mutagenesis of starch branching enzymes. Front Plant Sci. https://doi.org/10.3389/fpls.2017.00298
Takos AM, Jaffe FW, Jacob SR, Bogs J, Robinson SP, Walker AR (2006) Light-induced expression of a MYB gene regulates anthocyanin biosynthesis in red apples. Plant Physiol 142:1216–1232
Tanaka N, Itoh H, Sentoku N, Kojima M, Sakakibara H, Izawa T, Itoh J, Nagato Y (2011) The COP1 ortholog PPS regulates the juvenile-adult and vegetative-reproductive phase changes in rice. Plant Cell 23:2143–2154
Till BJ, Cooper J, Tai TH, Colowit P, Greene EA, Henikoff S, Comai L (2007) Discovery of chemically induced mutations in rice by TILLING. BMC Plant Biol 7:19
Tohge T, de Souza LP, Fernie AR (2017) Current understanding of the pathways of flavonoid biosynthesis in model and crop plants. J Exp Bot 68:4013–4028
Torii KU, McNellis TW, Deng XW (1998) Functional dissection of Arabidopsis COP1 reveals specific roles of its three structural modules in light control of seedling development. EMBO J 17:5577–5587
Tsuge T, Inagaki N, Yoshizumi T, Shimada H, Kawamoto T, Matsuki R, Yamamoto N, Matsui M (2001) Phytochrome-mediated control of COP1 gene expression in rice plants. Mol Genet Genom 265:43–50
Viana VE, Pegoraro C, Busanello C, Costa de Oliveira A (2019) Mutagenesis in rice: the basis for breeding a new super plant. Front Plant Sci 10:1326
von Lintig J, Welsch R, Bonk M, Giuliano G, Batschauer A, Kleinig H (1997) Light-dependent regulation of carotenoid biosynthesis occurs at the level of phytoene synthase expression and is mediated by phytochrome in Sinapis alba and Arabidopsis thaliana seedlings. Plant J 12:625–634
Wang F, Wang C, Liu P, Lei C, Hao W, Gao Y, Liu YG, Zhao K (2016) Enhanced rice blast resistance by CRISPR/Cas9-targeted mutagenesis of the ERF transcription factor gene OsERF922. PLoS ONE 11:e0154027
Wang N, Long T, Yao W, Xiong L, Zhang Q, Wu C (2013) Mutant resources for the functional analysis of the rice genome. Mol Plant 6:596–604
Wang ZP, Xing HL, Dong L, Zhang HY, Han CY, Wang XC, Chen QJ (2015) Egg cell-specific promoter-controlled CRISPR/Cas9 efficiently generates homozygous mutants for multiple target genes in Arabidopsis in a single generation. Genome Biol 16:144
Wei N, Deng XW (1996) The role of the COP/DET/FUS genes in light control of arabidopsis seedling development. Plant Physiol 112:871–878
Xu D, Lin F, Jiang Y, Ling J, Hettiarachchi C, Tellgren-Roth C, Holm M, Wei N, Deng XW (2015) Arabidopsis COP1 suppressor 2 represses COP1 E3 ubiquitin ligase activity through their coiled-coil domains association. PLoS Genet 11:e1005747
Xu D, Zhu D, Deng XW (2016a) The role of COP1 in repression of photoperiodic flowering. F1000Res 5:178. https://doi.org/10.12688/f1000research.7346.1
Xu RF, Yang YC, Qin RY, Li H, Qiu CH, Li L, Wei PC, Yang JB (2016) Rapid improvement of grain weight via highly efficient CRISPR/Cas9-mediated multiplex genome editing in rice. J Genet Genomics 43:529–532
Yang J, Lin R, Sullivan J, Hoecker U, Liu B, Xu L, Deng XW, Wang H (2005) Light regulates COP1-mediated degradation of HFR1, a transcription factor essential for light signaling in Arabidopsis. Plant Cell 17:804–821
Yin R, Arongaus AB, Binkert M, Ulm R (2015) Two distinct domains of the UVR8 photoreceptor interact with COP1 to initiate UV-B signaling in Arabidopsis. Plant Cell 27:202–213
Yu JW, Rubio V, Lee NY, Bai S, Lee SY, Kim SS, Liu L, Zhang Y, Irigoyen ML, Sullivan JA, Zhang Y, Lee I, Xie Q, Paek NC, Deng XW (2008) COP1 and ELF3 control circadian function and photoperiodic flowering by regulating GI stability. Mol Cell 32:617–630
Zeng Y, Wen J, Zhao W, Wang Q, Huang W (2019) Rational improvement of rice yield and cold tolerance by editing the three genes OsPIN5b, GS3, and OsMYB30 with the CRISPR-Cas9 system. Front Plant Sci 10:1663
Zhang A, Liu Y, Wang F, Li T, Chen Z, Kong D, Bi J, Zhang F, Luo X, Wang J, Tang J, Yu X, Liu G, Luo L (2019) Enhanced rice salinity tolerance via CRISPR/Cas9-targeted mutagenesis of the OsRR22 gene. Mol Breed. https://doi.org/10.1007/s11032-019-0954-y
Zhao P, Begcy K, Dresselhaus T, Sun MX (2017) Does early embryogenesis in eudicots and monocots involve the same mechanism and molecular players? Plant Physiol 173:130–142
Zheng Y, Cui X, Su L, Fang S, Chu J, Gong Q, Yang J, Zhu Z (2017) Jasmonate inhibits COP1 activity to suppress hypocotyl elongation and promote cotyledon opening in etiolated Arabidopsis seedlings. Plant J 90:1144–1155
Zhou DX, Kim YJ, Li YF, Carol P, Mache R (1998) COP1b, an isoform of COP1 generated by alternative splicing, has a negative effect on COP1 function in regulating light-dependent seedling development in Arabidopsis. Mol Gen Genet 257:387–391
Zimmer AM, Pan YK, Chandrapalan T, Kwong RWM, Perry SF (2019) Loss-of-function approaches in comparative physiology: is there a future for knockdown experiments in the era of genome editing? J Exp Biol. https://doi.org/10.1242/jeb.175737
Zoratti L, Karppinen K, Escobar AL, Haggman H, Jaakola L (2014) Light-controlled flavonoid biosynthesis in fruits. Front Plant Sci. https://doi.org/10.3389/fpls.2014.00534
Funding
This work was supported by the Next-Generation BioGreen 21 Program (Plant Molecular Breeding Center No. PJ011790 to H-JK) and the Cooperative Research Program for Agriculture Science and Technology Development (Project No. PJ01572901 to H-JK), Rural Development Administration, Republic of Korea; and a grant from the National Research Foundation of Korea (NRF- 2016R1A6A3A03013010 to BK).
Author information
Authors and Affiliations
Contributions
BK designed and performed research, analyzed data, and wrote the manuscript. RP, GL, EK, YL, SW, and R performed research and analyzed data. WJ developed the plant materials. HJK, ES, and MT designed and supervised the experiment and improved the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Additional information
Communicated by Lixi Jiang.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kim, B., Piao, R., Lee, G. et al. OsCOP1 regulates embryo development and flavonoid biosynthesis in rice (Oryza sativa L.). Theor Appl Genet 134, 2587–2601 (2021). https://doi.org/10.1007/s00122-021-03844-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-021-03844-9