
Abstract
Scleractinian corals provide habitats for a broad variety of cryptofauna, which in turn may contribute to the overall functioning of coral symbiomes. Among these invertebrates, hydrozoans belonging to the genus Zanclea represent an increasingly known and ecologically important group of coral symbionts. In this study, we analysed 321 Zanclea colonies associated with 31 coral genera collected from 11 localities across the Indo-Pacific and Caribbean regions, and used a multi-disciplinary approach to shed light on the evolution and biogeography of the group. Overall, we found high genetic diversity of hydrozoans that spans nine clades corresponding to cryptic or pseudo-cryptic species. All but two clades are associated with one or two coral genera belonging to the Complex clade, whereas the remaining ones are generalists associated with both Complex and Robust corals. Despite the observed specificity patterns, no congruence between Zanclea and coral phylogenies was observed, suggesting a lack of coevolutionary events. Most Zanclea clades have a wide distribution across the Indo-Pacific, including a generalist group extending also into the Caribbean, while two host-specific clades are possibly found exclusively in the Red Sea, confirming the importance of this peripheral region as an endemicity hotspot. Ancestral state reconstruction suggests that the most recent common ancestor of all extant coral-associated Zanclea was a specialist species with a perisarc, occurring in what is now known as the Indo-Pacific. Ultimately, a mixture of geography- and host-related diversification processes is likely responsible for the observed enigmatic phylogenetic structure of coral-associated Zanclea.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Much of marine biodiversity remains unknown (Appeltans et al. 2012) and this is especially true for hyper-diverse ecosystems such as coral reefs, in which only a small fraction of species have been discovered and named (Fisher et al. 2015). However, continuous efforts are being made to increase our knowledge on the inhabitants of coral reef ecosystems, leading to the discovery of new species and associations (Plaisance et al. 2011; Leray and Knowlton 2015; Hoeksema 2017; Maggioni et al. 2020a). Moreover, the application of molecular techniques and integrative taxonomy approaches (Dayrat 2005) has allowed the exploration of previously undetectable diversity (Bickford et al. 2007), and the presence of cryptic taxa (i.e. taxa morphologically indistinguishable) in coral reefs seems to be the norm rather than exception (Rocha et al. 2007). Much of the unknown metazoan diversity in coral reefs occurs likely in the cryptofauna, which is composed of highly diverse but understudied invertebrate taxa inhabiting habitat-forming organisms, in particular living, dead, or fragmented (i.e. coral rubble) scleractinian corals (Reaka-Kudla 1997; Takada et al. 2007; Stella et al. 2011; Enochs and Manzello 2012). Indeed, corals provide a multitude of habitats for these organisms (Stella et al. 2011; Hoeksema et al. 2012, 2017) and form multi-species assemblages known as coral symbiomes (sensu Gates and Ainsworth 2011). Coral-associated organisms find shelter, substrate, and resources on their hosts (Stella et al. 2011; Hoeksema 2017), and are believed to contribute to the overall functioning of coral symbiomes, having roles in nutrient cycling, stress tolerance, and defence (Gates and Ainsworth 2011). The study of the taxonomic composition of these multi-species associations, together with the roles of each coral associate, is therefore crucial to improve our knowledge on the functional biology of corals and coral reefs, and consequently on how species interactions may shape the responses of ecological communities to environmental stresses (Gates and Ainsworth 2011; Stella et al. 2011).
Coral-associated hydrozoans have been found to have a possible beneficial role in coral colony protection against predation and diseases (Montano et al. 2017a). Hydrozoans associated with scleractinian corals belong to the genus Zanclea Gegenbaur, 1856 and live as partially endosymbiotic colonies, with their hydrorhiza (i.e. structure that connects different hydranths in the same colony) extending below coral tissues and only polyps extending outwards (Pantos and Bythell 2010). Coral-associated Zanclea hydroids were first reported by Millard and Bouillon (1974) from Mozambique and later by Boero et al. (2000) with the description of Zanclea gilii Boero, Bouillon & Gravili, 2000, associated with an unidentified scleractinian in Papua New Guinea. Three other species have been described so far, namely Z. margaritae Pantos and Bythell 2010 and Z. gallii Montano, Maggioni & Puce, 2015c, associated with Acropora muricata (Linnaeus, 1758) in Australia and the Maldives, respectively (Panthos and Bythell 2010; Montano et al. 2015a), and Z. sango Hirose and Hirose 2011, hosted by Pavona and Psammocora in Japan (Hirose and Hirose 2011). The association is currently thought to have a wide tropical and subtropical distribution, including the Red Sea (Egypt: Montano et al. 2014; Israel: Pica et al. 2017; Saudi Arabia: Maggioni et al. 2017a), Indian Ocean (Mozambique: Millard and Bouillon 1974; Maldives: Montano et al. 2013), Pacific Ocean (Papua New Guinea: Boero et al. 2000; Australia: Pantos and Bythell 2010; Japan: Hirose and Hirose 2011; Taiwan and Indonesia: Fontana et al. 2012; Fiji: Bonito and McInnis 2019) and the Caribbean Sea (Sint Eustatius: Montano et al. 2017b). Since the first reports of Zanclea associated with unidentified corals, our understanding of host ranges has greatly improved, and currently 61 scleractinian species belonging to 31 genera and nine families have been confirmed as hosts (Montano et al. 2015b,c; Bonito and McInnis 2019).
Previous analyses of the genetic diversity of coral-associated Zanclea from the Maldives and Red Sea revealed the presence of multiple clades that could not be fully distinguished using morphology alone, suggesting the presence of cryptic or pseudo-cryptic species (Montano et al. 2015c; Maggioni et al. 2017a). Indeed, the paucity of ‘traditional’ diagnostic morphological characters is common in most zancleid species (Maggioni et al. 2018), as well as in other hydrozoan species (e.g. Cunha et al. 2017; Miglietta et al. 2019). Some coral-associated genetic groups may be recognised according to their host, as they are specifically associated with a single coral genus (Montano et al. 2015c). However, other molecular clades are generalists, and multiple species of Zanclea may occur on the same coral genus from different localities, as observed for Acropora in the Maldives, Red Sea, and Australia which hosts Z. gallii Ia, Z. gallii IIa (sensu Maggioni et al. 2017a), and Z. margaritae, respectively (Pantos and Bythell 2010; Montano et al. 2015c; Maggioni et al. 2017a). Nevertheless, the same coral colony has thus far not been reported to host more than one Zanclea clade.
More recently, the use of novel approaches in hydrozoan taxonomy has shown promising results in discriminating among Zanclea species. For example, Manca et al. (2019) found significant differences in the size of the nematocysts among three coral-associated clades and described the presence of symbiont-related peculiar modifications of the coral skeleton that may have taxonomic value. The latter corresponds to skeletal overgrowths that surround the base of Zanclea polyps, and their size was different among the three investigated clades. Moreover, Maggioni et al. (2020b) found that divergent Zanclea clades associated with Pavona and Goniastrea corals, and with an identical general morphology, show different patterns of green fluorescence in their newly released medusae. Altogether these results suggest that a comprehensive integrative approach may help to shed light on the enigmatic diversity of these symbiotic hydrozoans.
In this study, 321 colonies of Zanclea associated with 31 scleractinian genera from several localities across the entire assumed distributional range of the association were analysed. The integrative study of their diversity, morphology, species boundaries, biogeography, evolution, and relationships with hosts was carried out using genetic information derived from three mitochondrial and four nuclear molecular markers (although nuclear markers appeared to contain little phylogenetic signal in this case).
Materials and methods
Sampling and specimen identification
Sampling was carried out by snorkelling (0–5 m deep) and diving (5–40 m deep) between August 2012 and October 2018 in 11 localities across the Indo-Pacific, Red Sea, and Caribbean Sea (Fig. 1, Table S1). When the presence of Zanclea polyps on scleractinians was recorded in situ (Fig. 2a, b), small fragments of corals and associated hydroids were collected and, when possible, rapidly transferred to bowls filled with seawater, or otherwise directly fixed in 99% ethanol for molecular analyses or 10% formalin for morphological analyses. When laboratory facilities were available, live animals were anesthetised with menthol crystals in order to let them extend and facilitate further manipulation. Hydroids were carefully detached from their hosts using precision forceps, syringe needles, and micropipettes and subsequently fixed in 99% ethanol or 10% formalin. When Zanclea colonies showed developing medusa buds, they were reared with their host corals in constantly oxygenated bowls filled with seawater, at room temperature, under artificial light, and fed with Artemia nauplii until medusa release occurred (Fig. 2c).

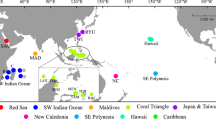
Map showing the updated distribution of coral-associated Zanclea. Sampling localities are indicated by black pins, whereas previously reported localities (not sampled for this study) with yellow pins. Caribbean Sea: EUX, Sint Eustatius. Indian Ocean: MG, Madagascar; MV, Maldives; MZ, Mozambique. Pacific Ocean: AU, Australia; FJ, Fiji; ID, Indonesia; JP, Japan; PG, Papua New Guinea; SG, Singapore; TW, Taiwan; TH, Thailand. Red Sea: EG, Egypt; IL, Israel; SA, Saudi Arabia

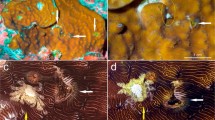
Morphological features of coral-associated Zanclea. a gastrozooids (clade P1) associated with Porites rus, b dactylozooid (clade G2) associated with Echinopora lamellosa, and c newly released medusa from the same colony of b. d–f Nematocysts commonly found in Zanclea polyps, namely d large and e small stenoteles found in polyps associated with Goniastrea (clade G1), and f macrobasic apotrichous euryteles in polyps associated with Gardineroseris (clade G1). g Perisarc covering the distal portion of a gastrozooid associated with Leptoseris (clade G2) and h micro-alteration of the skeleton of a Porites rus caused by the skeletal overgrowth on the distal portion of a Zanclea polyp (clade P1). Scale bars: a–c 200 µm; d–f 5 µm; g–h 50 µm
Scleractinian corals were identified at the genus or species level according to Veron et al. (2016) and the most recent taxonomic classification (e.g. Wallace et al. 2007; Benzoni et al. 2010, 2012; Gittenberger et al. 2011; Arrigoni et al. 2014a, b; Huang et al. 2014; Terraneo et al. 2017, 2019; Arrigoni et al. 2020). Zanclea polyps were identified at the genus or species level according to Boero et al. (2000), Pantos and Bythell (2010), Hirose and Hirose (2011), and Montano et al. (2015a). The morphological characteristics of Zanclea colonies were assessed using a Leica EZ4 D stereomicroscope and a Zeiss Axioskop 40 compound microscope using both fresh and formalin-fixed material. Specifically, we focused our attention on the most distinctive characters of Zanclea hydroids, namely the polymorphism of the colony (Fig. 2a, b), the nematocyst types (Fig. 2d–f), and the presence or absence of a perisarc covering the hydrorhiza (Fig. 2g). Regarding the medusa stage (Fig. 2c), we focused on the general morphology and the nematocyst types.
DNA extraction, amplification, sequencing, and dataset assembling
The total genomic DNA was extracted from ethanol-fixed Zanclea using the following protocol: for each sample, a single polyp was washed with MilliQ water, put in 9 µl of milliQ water, and incubated at 90 °C for 10 min; subsequently 1 µl of proteinase K was added and the sample was incubated at 50 °C for 30 min followed by 10 min at 90 °C; finally, each sample was diluted adding 40 µl of milliQ water. Three mitochondrial DNA marker regions were amplified for all samples, whereas four nuclear regions were amplified for a subset of the samples (two or three specimens per mitochondrial clade recovered as a species hypothesis in downstream analyses), using the primers and protocols as in Maggioni et al. (2020c). The amplified mitochondrial molecular markers were: (1) a ~ 600 bp portion of the 16S ribosomal DNA gene (16S rRNA), (2) a ~ 650 bp portion of the cytochrome c oxidase subunit I gene (COX1), and (3) a ~ 650 bp portion of the cytochrome c oxidase subunit III gene (COX3). The amplified nuclear markers were: (1) a ~ 1700 bp portion of the 18S ribosomal DNA gene (18S rRNA), (2) a ~ 1600 bp portion of the 28S ribosomal DNA gene (28S rRNA), (3) a ~ 650 bp portion of the internal transcribed spacer ribosomal region (ITS), and 4) a ~ 350 bp portion of the Histone 3 gene (H3). The success of polymerase chain reactions (PCRs) was checked through 1.5% agarose electrophoretic runs, and PCR products were purified with Illustra ExoStar (GE Healthcare) and sequenced in both directions with ABI 3730xl DNA Analyzer (Applied Biosystems). The obtained chromatograms were visually checked and corrected with Geneious 6.1.6, primer regions were removed, and protein-coding genes (COX1, COX3, H3) were translated to check for the presence of open reading frames. The sequences were deposited in GenBank and accession numbers (MT356227-MT356588, MT356591-MT356612, MT362091-MT362435, and MT363352-MT363597) are listed in Table S1 and S2. Sequences of each marker were aligned using MAFFT 7.110 (Katoh and Standley 2013) with the E-INS-i option, after adding coral-associated Zanclea sequences generated in previous works (Tables S1, S2). The hydrozoan species Cladocoryne haddoni Kirkpatrick, 1890 and Asyncoryne ryniensis Warren, 1908 were selected as outgroups (Maggioni et al. 2017b). All mitochondrial markers were concatenated using Mesquite 3.2 (Maddison and Maddison 2006). Prior to phylogenetic reconstruction and species delimitation analyses, identical sequences of both single-locus and multi-locus (Table S3) mitochondrial datasets were collapsed into representative sequence types using FaBox (Villesen 2007).
Phylogenetic analyses
General statistics of the Zanclea sequences and the variability of the seven DNA regions were calculated with DnaSP 6 (Rozas et al. 2017) (Table S4). Only mitochondrial datasets were employed in downstream analyses, due to extremely low genetic variability of the nuclear markers (Table S4). Partition schemes and models were determined using jModelTest 2 (Darriba et al. 2012) for single-locus datasets (Table S4), and PartitionFinder 2 (Lanfear et al. 2012) for the multi-locus dataset (Table S5), using the Akaike Information Criterion (AIC), the corrected AIC (AICc), and the Bayesian Information Criterion (BIC) as optimality criteria. Phylogenetic inference was performed using Bayesian inference (BI) and maximum likelihood (ML). BI analyses were run using MrBayes 3.2.6 (Ronquist et al. 2012): two independent runs for four Markov chains were conducted for 50 million generations, with trees sampled every 5000th generation, and burn-in set to 25%. Parameter estimates and convergence were checked using Tracer 1.6 (Rambaut et al. 2014). Maximum likelihood analyses were run with RAxML 8.2.9 (Stamatakis 2014) using 1000 bootstrap replicates. Uncalibrated ultrametric Bayesian trees were also reconstructed in BEAST 1.8.2 (Drummond et al. 2012) for further species delimitation analyses, setting a coalescent tree prior and an uncorrelated lognormal relaxed clock. Three replicate analyses were run for 108 million generations, with trees sampled every 10,000th generation, and were combined using LogCombiner 1.8.2 (Drummond et al. 2012) with a burn-in set to 25%. Maximum clade credibility trees were obtained using TreeAnnotator 1.8.2 (Drummond et al. 2012). All analyses were run on the CIPRES server (Miller et al. 2010).
DNA-based species delimitations, genetic distances, and haplotype networks
Distance- and tree-based species delimitation approaches were used to assess the species boundaries in coral-associated Zanclea. All analyses were run separately on the single-locus mitochondrial datasets, after removing the outgroups.
First, the distance-based Automatic Barcode Gap Discovery (ABGD) method was used. The ABGD delimitations (Puillandre et al. 2012) were run on the website abgd web (https://www.abi.snv.jussieu.fr/public/abgd/abgdweb.html) using matrices of genetic distances (p-distance, Kimura 2-Parameter, Jukes Cantor) as inputs. Parameters were set as follows: Pmin = 0.001, Pmax = 0.1, Steps = 100, X = 1.5, Nb bins = 100.
For tree-based methods, different implementations of the Poisson Tree Process (PTP) and Generalised Mixed Yule Coalescence (GMYC) methods were used. Single-threshold (p = 0.001) and multiple-threshold PTP analyses (PTP: Zhang et al. 2013; mPTP: Kapli et al. 2017) were performed on the website mPTP Webservice (https://mptp.h-its.org), using Bayesian trees as inputs. GMYC analyses were run in the R environment (R Core Team 2019) using ultrametric Bayesian trees as inputs. Single-threshold GMYC analyses (Pons et al. 2006) were run using the packages ‘Ape’ (Paradis et al. 2004) and ‘Splits’ (Ezard et al. 2009). Bayesian GMYC analyses (bGMYC) were performed using the package ‘bGMYC’ (Reid and Carstens 2012) on a subset of 100 trees retrieved from the 10,000 posterior trees obtained with each BI analysis.
MEGA X (Kumar et al. 2018) was used to calculate genetic distances within and among species hypotheses retrieved with species delimitation analyses. Genetic distances were calculated as % uncorrected p-distances with 1000 bootstrap replicates for each mitochondrial region. Additionally, to investigate possible geographic structure within the genetic groups, single-locus mitochondrial datasets were used to generate median-joining haplotype networks in PopART 1.7 (Leigh and Bryant 2015), and the results based on the most complete mitochondrial dataset (i.e. 16S rRNA) are shown.
Ancestral state reconstructions
A set of biogeographical, morphological (perisarc and euryteles), and host specificity characters were mapped onto a reduced Zanclea phylogeny (including one specimen for each clade). For the morphological and host specificity characters, two possible states were coded, namely presence-absence, whereas for the distribution four states were coded, namely Indo-Pacific (including the Red Sea), Pacific only, Red Sea only, and Indo-Pacific (including the Red Sea) + Caribbean Sea. The lack of a time-calibrated phylogeny and the fact that some lineages may be present also in other unsampled localities prevented us from implementing more detailed biogeographic and speciation models. Stochastic mapping (Huelsenbeck et al. 2003) was used to map probable realisations of the evolution of the considered characters on the coral-associated Zanclea tree. The analyses were carried out using the ‘make.simmap’ function available in the R package ‘Phytools’ (Revell 2012). The ‘equal rate’ model was used to evolve the interactions along the phylogenetic trees, and 10,000 stochastic mapping replicates were conducted for each analysis. Simulations were then summarised with density plots (Revell 2013) for morphology and host specificity. The biogeographical results were summarised presenting one of the obtained reconstructions with colours corresponding to the four states and pie charts representing the posterior probability of each internal node being in each state.
Testing for Zanclea-Scleractinia cophylogeny
We tested for cophylogeny between Zanclea and their scleractinian hosts using a reduced multi-locus phylogenetic tree of Zanclea (i.e. including one sample for each clade) and a multi-locus phylogenetic tree including all scleractinian genera known to host Zanclea hydroids. The scleractinian dataset was assembled via mining sequences from GenBank for four loci, namely COX1, ITS, 28S rRNA, and H3 (Table S6), using the corallimorpharian Ricordea florida Duchassaing & Michelotti, 1860 as outgroup. Phylogenetic reconstructions were performed using BEAST 1.8.2 as described above.
Cophylogeny analyses were performed using two methods: the global-fit PACo 1.2 (Balbuena et al. 2013; Hutchinson et al. 2017) and the event-based Jane 4 (Conow et al. 2010). PACo (Procrustes approach to co-phylogenetics) assesses the global-fit of symbiont phylogeny onto the hosts and evaluates the contribution of each individual host-symbiont association to the global-fit. The overall phylogenetic congruence was tested with 104 permutations of the coral-Zanclea association matrix and the contribution of each individual association was assessed by taxon jackknifing. PACo analyses were run in the R environment (R CORE Team 2019). Jane uses an event-based approach that takes into consideration various events, namely cospeciation, intra-host diversification (duplication), host switch, failure to diverge, and loss, to which it assigns different costs. The optimal (minimum) costs found for the host-symbiont dataset are compared against randomised datasets. Jane analysis was run using default cost settings, generations = 100, population size = 500, and sample size = 100. Finally, Zanclea-scleractinians interactions were visualised with TreeMap3 (Charleston and Robertson 2002), using the untangling function to improve the layout.
Results
Updated distribution and host range
A total of 321 Zanclea colonies were examined, 73 of which were already partially analysed in previous studies, and the rest newly examined in this study. Hydroids were obtained from 11 tropical and sub-tropical localities (Fig. 1, Table S1), with most samples collected in the Maldives (n = 128), Red Sea (n = 117), Singapore (n = 30), and Taiwan (n = 23). From other localities we obtained a lower number of samples (total n = 23). Notably, despite the Caribbean Sea being explored in multiple surveys at different localities (Panama, Bonaire, Curaçao, and Sint Eustatius) with a high number of dives and snorkelling activities, only two Zanclea colonies were found in Sint Eustatius. The current distributional range of coral-associated Zanclea covers the Red Sea, Indian Ocean, Western Pacific Ocean, and the Caribbean Sea. In this work we succeeded in obtaining samples from most previously reported localities, with the exception of Mozambique, Papua New Guinea, and Fiji, where we did not perform any field work. Specimens in this work from Singapore, Thailand, and Madagascar represent new geographic records for Zanclea (Fig. 1).
Altogether, the collected Zanclea colonies were associated with 31 coral genera and 10 coral families (Table S1). These records were added to previously reported associations (Table S7), resulting in a total number of at least 90 host-coral species for Zanclea, belonging to 34 genera and 10 families. With this work, we also report new host records, including 32 at the species level, three at the genus level (Acanthastrea, Astrea, Coscinaraea), and one at the family level (Coscinaraeidae) (Table S7).
Overall phylogenetic diversity
Genomic DNA was successfully extracted from all specimens and the seven molecular markers were amplified and sequenced with high success (99%, n = 981), for a total of 972 newly obtained sequences. Nuclear markers were sequenced for a subset of the dataset and showed extremely low levels of variability (e.g. % of variable sites ranging from 0.1 to 2.5%), whereas mitochondrial markers showed higher genetic variability (Table S4). jModelTest and PartitionFinder found different best-fit evolutionary models and partitions according to the information criteria used (Table S4, S5). All downstream analyses were repeated for each model and partition, with no resulting substantial difference in tree topology and support values. Likewise, BI and ML analyses of single- and multi-locus mitochondrial datasets resulted in similar trees (Figs S1, S2), and the multi-locus Bayesian tree is shown in Fig. 3. Specifically, ML and BI trees showed concordant topologies and differences among single-locus trees based on different loci were mostly related to the relationships among clades M1, P1, and G1 and the position of clade P2 (Fig. S1). These uncertainties were also reflected in the low support values in the ML multi-locus tree at these nodes (Fig. S2).

Multi-locus (16S rRNA, COX1, COX3) phylogenetic hypothesis of coral-associated Zanclea. Alpha-numeric codes at terminal nodes indicate the representative sequence types (as shown in Table S3), followed by the number of the collapsed sequences in brackets, and the sampling locality, as coded in the legend. Numbers at nodes represent Bayesian posterior probabilities (> 0.9) and maximum likelihood bootstrap values (> 90), respectively, whereas asterisks denote maximal statistical support (1/100). Each recovered clade is highlighted with a different colour, following Montano et al. (2015c) and with a code, as detailed in the ‘Results’ section. On the right side of the tree, the coral genera associated with each clade are shown, divided by family and with family names coded as in the legend
Coral-associated Zanclea were confirmed as a fully supported monophyletic group showing considerable genetic diversification, with nine highly or fully supported clades identified in all phylogenetic analyses (Fig. 3, Figs. S1, S2). The nine groups were named with alphanumerical codes, with letters indicating host-corals, as follows: A1 (Acropora and Isopora), A2 (Acropora), G1 and G2 (generalists), M1 and M2 (Montipora), P1 and P2 (Porites), and Pa (Pavona). Seven out of the nine clades have already been reported in previous works (Montano et al. 2015c; Maggioni et al. 2017a), whereas the remaining two clades (M2 and P2) were found for the first time in this study. The internal nodes were in some cases well supported, but the relationships among lineages G1, M1, P1 and P2 were not fully resolved, as shown by the low BI posterior probabilities and ML bootstrap values.
General descriptions of the clades
The nine Zanclea clades showed contrasting patterns of host-specificity, as both generalist and specialist (at genus level) groups were found (Fig. 3). The two generalist clades, G1 and G2, are associated with 12 and 24 scleractinian coral genera belonging to six and nine families, respectively. These two clades showed a partial overlap of host-coral genera, as nine out of the 12 scleractinian genera associated with G1 also hosted G2, to the exception of Acanthastrea, Astrea and Gardineroseris. The remaining seven Zanclea clades were coral genus-specific in their associations, given our sampling. Clade A1 corresponds to Zanclea gallii and despite being associated with two genera, Acropora and Isopora, we consider this clade as genus-specific due to the close relationship between the two acroporid genera (Wallace et al. 2007). Australian Acropora-associated specimens were collected relatively close to the type locality of the formally described species Zanclea margaritae, and sequences of these samples also fell in this clade. The sister taxon of clade A1 was A2, which was associated with Acropora and found exclusively in the Red Sea, and previously designated as Z. galli IIa (Maggioni et al. 2017a). The Pavona-associated clade Pa corresponds to Zanclea sango, and this was further supported by our specimens being collected from the type locality (Okinawa, Japan). Zanclea sango was originally described in association with Pavona (= holotype) and Psammocora contigua (Esper, 1794) (= paratypes) (Hirose and Hirose 2011). However, the only specimen collected from Okinawa in this study associated with P. contigua was found to belong to clade G1. Porites corals hosted clade P1, distributed in the Indo-Pacific and Red Sea, and the newly discovered clade P2, collected only from Pacific localities. Finally, Montipora corals hosted a clade found in the Red Sea and Indo-Pacific (M1) and another one exclusive to the Red Sea (M2).
DNA-based species delimitations and genetic distances
Species delimitation analyses consistently suggested that the nine Zanclea phylogenetic clades correspond to nine putative species (Fig. 4a). The distance-based method ABGD identified the nine species hypothesis as most likely in all analyses, whereas the tree-based methods found in some cases a further subdivision, especially concerning clades Pa and G2. However, the vast majority of the analyses agreed in identifying nine putative species. The analysis of intra- and inter-clade genetic distances based on the three mitochondrial markers (Table S8) revealed moderate to high divergence among the Zanclea clades. Specifically, the average inter-clade distances ranged from 3.0 ± 0.6% to 5.6 ± 0.9% for 16S rRNA dataset, from 4.9 ± 0.8% to 9.1 ± 0.9% for the COX1 dataset, and from 7.8 ± 1.0% to 10.8 ± 1.0% for the COX3 dataset. Intra-clade distances were generally low for the 16S rRNA (mean value 0.5 ± 0.1%) and slightly higher for the other, more variable, mitochondrial regions (0.8 ± 0.2% for COX1 and 1.1 ± 0.2% for COX3).

a Summary cladogram showing the DNA-based species delimitation results. Grey cells denote that the clade has been successfully delimited as a putative species, yellow cells indicate a further subdivision in one or more clades. b–g 16S rRNA medium-joining networks of Zanclea clades b G2 (generalist), c Pa (Pavona), d A1 + A2 (Acropora, Isopora; dashed lines delimit the two clades) e P1 (Porites), f M1 (Montipora), g G1 (generalist). Colours denote different provenience of the haplotype, as shown in the legend. n. s. not sequenced
Morphology
The morphology of all polyps included in molecular analyses was investigated in order to detect the presence of perisarc, euryteles, and polymorphic colonies; the morphological characteristics of each clade are summarised in Table 1. Morphology alone was not sufficient to distinguish among most of the clades, according to the morphological characters sampled in this work, although the addition of other characters, such as nematocyst size variation and fluorescence patterns, may reveal fine-scale differences in future works. Specifically, clades G1, G2, and Pa have identical morphologies (perisarc, euryteles, polymorphic colonies), as do clades A1 and A2 (no perisarc, no euryteles), and P1 and P2 (perisarc, no euryteles). Both M1 and M2 clades have morphologies similar to those of G1, G2, and Pa, but euryteles were observed to be very rare. When a perisarc was present, micro-alterations of the host skeleton were generally found (Fig. 2h). Newly released medusae (Fig. 2c) were observed for five clades (Table 1) and were almost identical, with the only difference being the presence of euryteles in medusae belonging to clades in which polyps possessed euryteles. According to ancestral state reconstructions, the most recent common ancestor of all extant coral-associated Zanclea had a perisarc, and this structure was lost only once, in the common ancestor of the two extant Acropora-associated genetic groups (A1 and A2) (Fig. 5a). The latter two clades also lost the presence of euryteles in their cnidome, and the same event also happened independently in the two Porites-associated groups (P1 and P2) (Fig. 5b).

Morphological, host specificity, and biogeographic character mapping on the phylogeny of coral-associated Zanclea. Density plot of 10,000 stochastic character maps of the characters a perisarc, b euryteles, c host specificity. The colour of edges indicates the posterior probability (computed as the relative frequency across stochastic maps) of each character state. d Stochastic character map showing the ancestral distribution reconstruction, with pie charts representing the posterior probability of each node being in each state. The scale bar in a also applies to b–d
Biogeographical patterns
The intra-clade genetic diversity followed geographical patterns only for the Acropora-associated hydroids, showing similar results for each of the mitochondrial region analysed (Fig. 4b–g, Fig. S5). In this clade, different localities never shared identical haplotypes and Maldivian, Pacific, and Red Sea populations could be easily distinguished in clade A1 (Fig. 4d, Fig. S5). Additionally, clade A2, which is found exclusively in the Red Sea, seemed to be more closely related to the A1 Red Sea population rather than to other Indo-Pacific populations. In contrast, all other clades had haplotypes shared by multiple localities and no clear geographic structure was identifiable. Overall, most clades have a wide distribution, including the Indo-Pacific and Red Sea.
According to the biogeographic ancestral state reconstruction analyses (Fig. 5d), only a few terminal branches had different distributions, whereas the most recent common ancestor of coral-associated Zanclea showed uniform and wide distributional ranges across the Indo-Pacific. Specifically, two genetic groups appeared to have originated in the Red Sea (A2 and M2), one is currently found only in Pacific localities (P2), and only the generalist clade, G2, is also found in the Caribbean Sea.
Relationships with the hosts and coevolutionary analyses
Based on the proposed ancestral state reconstruction, the association between Zanclea and scleractinians arose as a host-specific relationship, and generalism emerged independently in the two extant generalist clades (Fig. 5c). The tanglegram (Fig. 6) shows a map of 44 Zanclea-coral associations. All specialist groups were associated with corals belonging to the Complex clade, whereas the generalist clades were associated with both Complex and Robust genera (Fig. 6). The same host coral can be involved in associations with multiple Zanclea clades (Table 1, Fig. 6). However, this pattern was, in most cases, not observed at the same locality, with the exceptions listed in Table 2, and the same coral colony was never observed to host multiple Zanclea clades. The PACo analyses revealed no significant congruence between Zanclea and coral phylogenies (global goodness-of-fit statistic m2 = 1.362, p = 0.147). Despite the absence of significance, different links between generalist clades and their hosts contributed relatively little to the m2 (Fig. S4), and therefore these may represent coevolutionary links. The Jane analysis resulted in 71 putative evolutionary scenarios, namely zero co-speciations, eight duplications, five host switches, 23 losses, and 35 failures to diverge (Fig. S5).

Tanglegram showing the 44 associations between the coral phylogeny (left side) and the Zanclea phylogeny (right side). Associations currently known as host-specific are highlighted in red
Discussion
Molecular phylogenetics of coral-associated Zanclea
The results provided in this study represent the most comprehensive phylogenetic assessment of coral-associated Zanclea to date, including specimens associated with almost all known hosts (31 of 34 reported host genera) and from reported localities. According to the mitochondrial phylogenetic tree, all Zanclea specimens associated with scleractinians clustered together in a fully supported monophyletic group. Moreover, Zanclea is characterised by considerable genetic diversity that it is not reflected in the morphological characters we sampled. Indeed, molecular phylogenetic, species delimitation, and genetic distance analyses revealed the presence of nine well-supported and divergent clades, whereas only three morphotypes were distinguishable: the Z. sango type, with a perisarc and euryteles (Pa, G1, G2, M1, M2); the Z. gallii type, devoid of a perisarc and euryteles (A1, A2); and a third type with a perisarc but no euryteles (P1, P2). Therefore, in all three morphotypes, cryptic or pseudo-cryptic species are present, and except for the Z. gallii type, these morphospecies are not monophyletic. These results are in line with recent DNA-based works showing that the presence of cryptic species is a rather common phenomenon both in Hydrozoa (e.g. Postaire et al. 2016; Vaga et al. 2020) and other cnidarian classes (e.g. Dawson and Jacobs 2001; Bartošová and Fiala 2011; Arrigoni et al. 2019). Hopefully, further detailed morphological assessments of these cryptic groups, including for instance the statistical treatment of morphometric data (Manca et al. 2019) and the observation of green fluorescence patterns (Maggioni et al. 2020b), will eventually allow us to find consistent morphological differences and formally describe the species.
Interestingly, the nuclear region sequences we analysed were extremely conserved. By contrast, mitochondrial sequence divergence was high and comparable to what has been previously observed for taxa that are evolutionarily close to Zanclea, such as Millepora spp. (Arrigoni et al. 2018). Cnidarian mitochondrial DNA has been reported to have slow rates of nucleotide substitution compared to nuclear DNA in Anthozoa (Shearer et al. 2002), but multiple studies have documented faster rates in medusozoan lineages, including hydrozoans (e.g. Govindarajan et al. 2005; Huang et al. 2008), possibly due to mitochondrial DNA linearization and loss of DNA repair genes (Shearer et al. 2002; Hellberg 2006). The observed Zanclea mito-nuclear discordance may be due to incomplete lineage sorting (Toews and Brelsford 2012) as result of a recent origin coupled with higher evolutionary rates of mitochondrial DNA. Alternative possible explanations may be related to phylogenetic inadequacy of nuclear markers, which are nevertheless able to distinguish other closely related cryptic hydrozoans (Maggioni et al. 2016; Montano et al. 2017c), or introgressive hybridisation (Toews and Brelsford 2012). Whatever the cause of the genetic patterns observed in this study may be, the mitochondrial clades show, at least in some cases, morphological variation and peculiar host specificity patterns that suggest the presence of evolutionarily independent units, corresponding to different species.
Translating Zanclea clades into the known morphospecies
With our analyses we could confidently identify the clade Pa as Z. sango, due to the inclusion of specimens associated with Pavona from the locality of the holotype, Okinawa. Hirose and Hirose (2011) referred to hydroids associated with P. contigua in Japan as belonging to Z. sango, due to their completely overlapping morphologies, but here we demonstrate that P. contigua-associated specimens likely actually belong to clade G1 in the same locality. However, we cannot disregard that Pa and G1 are associated with both genera in Japan.
The clades G1, G2, M1, and M2 are morphologically identical to Z. sango, based on the characters analysed in this study, even if genetically divergent. Euryteles were previously not reported in the Montipora-associated clade M1 (Montano et al. 2015c), but they were found in the present work, even if very rare, perhaps the reason why they were not detected in previous works.
Zanclea margaritae and Z. gallii were described from Australia and the Maldives, respectively, and are very similar from a morphological and host-specificity point of view. Both species are associated with Acropora and the only morphological differences are in the cnidome, with Z. margaritae possessing mastigophores and isorhizas (Pantos and Bythell 2010). In this study, we analysed several Acropora-associated Zanclea colonies from many localities, including Australia, but we did not find evidence of these nematocyst types in any investigated polyps, therefore identifying all specimens as Z. gallii. However, in some cases we observed Acropora nematocysts attached to hydranths, including mastigophores and isorhizas similar in shape and size to those characterised by Pantos and Bythell (2010) as part of the Z. margaritae cnidome. Additionally, Pantos and Bythell (2010) found isorhizae in a single hydranth and analysed newly released medusae for three-four hours post-release, increasing the likelihood that mastigophores and isorhizae may come from coral tissues. Therefore, if the mastigophores and isorhizas of Z. margaritae are of coral origin, it is possible that Z. gallii is a junior synonym of Z. margaritae, but further morphological studies are needed to address this issue, since no type material for Z. margaritae was analysed in this work.
The combination of morphological features found in the third morphotype (perisarc, no euryteles) and consisting of clades P1 and P2 in the phylogenetic tree, does not fit with any of the formally described coral-associated Zanclea species. Nevertheless, description of these clades is currently not possible as specimens of this morphotype cluster in two divergent groups that are not sister taxa. Moreover, we currently lack information on their medusa stage.
Zanclea gilii is a polymorphic species with euryteles and no reported perisarc (Boero et al. 2000). Zanclea gilii morphologies were not observed in any of the samples analysed in this work. The presence of a perisarc is sometimes difficult to spot without histological examination due to the high levels of Zanclea-host integration (Manca et al. 2019), and if the perisarc has been overlooked in Z. gilii, this species would then have a morphology identical to Z. sango. However, the lack of genetic material and information on the host-corals of Z. gilii prevents any further conclusions for now.
Biogeography of the association
Overall, the association between Zanclea and scleractinians has a wide distribution and most clades are found across multiple localities in the Indo-Pacific and Red Sea, generally without clear geographic structure. The Zanclea and scleractinian association seems to have originated in the Indo-Pacific, and only a few terminal branches show variations in their distribution, including two groups possibly endemic of the Red Sea (A2 and M2), a clade currently found only in Pacific localities (P2), and a fourth one that is also present in the Atlantic Ocean (G2). For instance, Z. gallii (clade A1) is the only clade that shows a geographic structure, with distinct Indian, Pacific, and Red Sea populations, and the Red Sea-endemic clade A2 may have originated from Z. gallii populations in the Red Sea. Indeed, the Red Sea is considered a biodiversity and endemicity hotspot (Hughes et al. 2002) where new biodiversity is generated and eventually exported (Bowen et al. 2013, 2016), and the high number of endemics in the region is known to have multiple origins, due to many recent and past isolating barriers (DiBattista et al. 2016). The analyses of Zanclea specimens from neighbouring areas together with a temporal characterisation of Zanclea evolution would greatly help in understanding the origin of the Red Sea endemic clades.
The most widespread clade (G2) is found in the Indo-Pacific, Red Sea and Caribbean regions, and is also associated with the highest number of coral genera. Its extensive generalist habits could have promoted its spread into multiple localities, facilitating the establishment of associations with new hosts, as demonstrated by a widespread coral-associated crab that was recently discovered in Hawai´i (Hoeksema et al. 2018). However, the lack of fossil records to time-calibrate the Zanclea radiation and the likely wider Zanclea distribution and diversity prevent us from further inferring historical biogeographic patterns. Therefore, the biogeographic analysis presented here should be interpreted with caution until additional sampling in the Pacific and Atlantic oceans is carried out to achieve a time-calibrated analysis of Zanclea.
Relationships among Zanclea clades and their scleractinian hosts
The updated list of scleractinian hosts reveals that Zanclea is able to establish associations with a diverse array of coral species and genera that show multifaceted morphological and ecological characteristics. Most observations were limited to shallow depths (e.g. Montano et al. 2017a), but coral-associated Zanclea was also reported from depths up to 41 m deep (Montano et al. 2014, 2017b; this study) (Table S1) and further explorations may therefore reveal other host-coral species living in the mesophotic. Similarly, no azooxanthellate scleractinians were reported as hosts of Zanclea, but this could be due to limited observation efforts for such corals.
All but two Zanclea clades are involved in specific associations and this specificity is maintained across distant localities. Contrarily, the generalist Zanclea clades are hosted by a large number of phylogenetically distant coral genera. Zanclea is known to establish symbiotic relationships not only with corals, but also with other sessile invertebrates (Boero et al. 2000), and it has been suggested that ancestral species were generalists without adaptations to their host or substrate, such as the loss of the perisarc (Puce et al. 2002).
However, the present study demonstrates that the association between Zanclea and scleractinians likely arose in combination with host specificity, and generalism has been later achieved twice in the evolution of this taxon. Regarding the hypothesised derived host-related loss of perisarc, this feature is found only in Acropora-associated clades, whereas all the other extant generalist and specialist clades, and also their most recent common ancestors, have their hydrorhiza covered by a perisarc. The loss of the perisarc is therefore better explained as an adaptive modification due to specific association with Acropora (and Isopora) spp. rather than a general derived feature of specialist clades.
An interesting aspect common to all specialist clades is the association with corals belonging only to the Complex clade, whereas generalists are hosted indiscriminately by both Complex and Robust corals. At the moment, it is difficult to suggest possible explanations for this pattern relying on differences between Complex and Robust clades, since the two scleractinian groups are mostly based on molecular and embryogenetic data rather than on morphological, biological, or ecological criteria (Romano and Palumbi 1996; Okubo et al. 2013, 2016; Okubo 2016).
Other than this peculiar pattern, there is limited congruence between the Zanclea and coral phylogenies, refuting the hypothesis of general coevolutionary patterns. This could be due to the very likely fact that the diversification of extant coral genera occurred much earlier than the radiation of extant coral-associated Zanclea. One of the main mechanisms underlying Zanclea radiation, acting both alone and in conjunction with geography-related processes, may be host-switching to exploit new resources and subsequent specialisation of the association and reproductive isolation, as shown in other coral-associated taxa (see review by Potkamp and Fransen 2018). This scenario may apply especially for specialist Zanclea clades. However, a further complication is the presence of host overlap among different clades, which is for instance commonly observed among closely related crustacean symbiont taxa inhabiting the same mushroom coral species (van der Meij et al. 2015; Ivanenko et al. 2018; Rauch et al. 2019). In previous works, no coral genera were found to host more than one Zanclea clade, with the only exception being Acropora spp. (Montano et al. 2015c; Maggioni et al. 2017a), maybe because it is also the most speciose scleractinian genus of reef-dwelling corals (Hoeksema and Cairns 2020). The large-scale sampling conducted in this work indicates that host overlap at genus level is a common phenomenon, both when examining the entire distributional range of clades, and also at the local scale. Therefore, according to the currently available data on the group, the puzzling genetic structure of coral-associated Zanclea seems to be ultimately due to a mixture of relatively recent geography- and host-related processes, in which allopatric, sympatric, and ecological diversifications have played important roles in shaping the current Zanclea diversity.
References
Appeltans W, Ahyong ST, Anderson G, Angel MV, Artois T, Bailly N, Bamber R, Barber A, Bartsch I, Berta A, Blazewicz-Paszkowycz M, Bock P, Boxshall G, Boyko CB, Brandão SN, Bray RA, Bruce NL, Cairns SD, Chan T-Y, Cheng L, Collins AG, Cribb T, Curini-Galletti M, Dahdouh-Guebas F, Davie PJF, Dawson MN, De Clerck O, Decock W, De Grave S, de Voogd NJ, Domning DP, Emig CC, Erséus C, Eschmeyer W, Fauchald K, Fautin DG, Feist SW, Fransen CHJM, Furuya H, Garcia-Alvarez O, Gerken S, Gibson D, Gittenberger A, Gofas S, Gómez-Daglio L, Gordon DP, Guiry MD, Hernandez F, Hoeksema BW, Hopcroft RR, Jaume D, Kirk P, Koedam N, Koenemann S, Kolb JrB, Kristensen RM, Kroh A, Lambert G, Lazarus DB, Lemaitre R, Longshaw M, Lowry J, Macpherson E, Madin LP, Mah C, Mapstone G, McLaughlin PA, Mees J, Meland K, Messing CG, Mills CE, Molodtsova TN, Mooi R, Neuhaus B, Ng PKL, Nielsen C, Norenburg J, Opresko DM, Osawa M, Paulay G, Perrin W, Pilger JF, Poore GCB, Pugh P, Read GB, Reimer JD, Rius M, Rocha RM, Saiz-Salinas JI, Scarabino V, Schierwater B, Schmidt-Rhaesa A, Schnabel KE, Schotte M, Schuchert P, Schwabe E, Segers H, Self-Sullivan C, Shenkar N, Siegel V, Sterrer W, Stohr S, Swalla B, Tasker ML, Thuesen EV, Timm T, Todaro MA, Turon X, Tyler S, Uetz P, van der Land J, Vanhoorne B, van Ofwegen LP, van Soest RWM, Vanaverbeke J, Walker-Smith G, Walter TC, Warren A, Williams GC, Wilson SP, Costello MJ (2012) The magnitude of global marine species diversity. Curr Biol 22:2189–2202
Arrigoni R, Terraneo TI, Galli P, Benzoni F (2014a) Lobophylliidae (Cnidaria, Scleractinia) reshuffled: pervasive non-monophyly at genus level. Mol Phylogenet Evol 73:60–64
Arrigoni R, Kitano YF, Stolarski J, Hoeksema BW, Fukami H, Stefani F, Galli P, Montano S, Castoldi E, Benzoni F (2014b) A phylogeny reconstruction of the Dendrophylliidae (Cnidaria, Scleractinia) based on molecular and micromorphological criteria, and its ecological implications. Zool Scr 43:661–688
Arrigoni R, Maggioni D, Montano S, Hoeksema BW, Seveso D, Shlesinger T, Terraneo TI, Tietbohl MD, Berumen ML (2018) An integrated morpho-molecular approach to delineate species boundaries of Millepora from the Red Sea. Coral Reefs 37:967–984
Arrigoni R, Berumen ML, Stolarski J, Terraneo TI, Benzoni F (2019) Uncovering hidden coral diversity: a new cryptic lobophylliid scleractinian from the Indian Ocean. Cladistics 35:301–328
Arrigoni R, Berumen ML, Mariappan KG, Beck PSA, Hulver AM, Montano S, Pichon M, Strona G, Terraneo TI, Benzoni F (2020) Towards a rigorous species delimitation framework for scleractinian corals based on RAD sequencing: the case study of Leptastrea from the Indo-Pacific. Coral Reefs 39:1001–1025
Balbuena JA, Míguez-Lozano R, Blasco-Costa I (2013) PACo: a novel procrustes application to cophylogenetic analysis. PLoS ONE 8:e61048
Bartošová P, Fiala I (2011) Molecular evidence for the existence of cryptic species assemblages of several myxosporeans (Myxozoa). Parasitol Res 108:573–583
Benzoni F, Stefani F, Pichon M, Galli P (2010) The name game: morpho-molecular species boundaries in the genus Psammocora (Cnidaria, Scleractinia). Zool J Linn Soc Lond 160:421–456
Benzoni F, Arrigoni R, Stefani F, Stolarski J (2012) Systematics of the coral genus Craterastrea (Cnidaria, Anthozoa, Scleractinia) and description of a new family through combined morphological and molecular analyses. Syst Biodivers 10:417–433
Bickford D, Lohman DJ, Sodhi NS, Ng PK, Meier R, Winker K, Ingram KK, Das I (2007) Cryptic species as a window on diversity and conservation. Trends Ecol Evol 22:148–155
Boero F, Bouillon J, Gravili C (2000) A survey of Zanclea, Halocoryne and Zanclella (Cnidaria, Hydrozoa, Anthomedusae, Zancleidae) with description of new species. Ital J Zool 67:93–124
Bonito V, McInnis AJK (2019) New host records of scleractinian-Zanclea symbiosis from Fiji. Mar Biodivers 49:1559–1563
Bowen BW, Rocha LA, Toonen RJ, Karl SA, Laboratory ToBo (2013) The origins of tropical marine biodiversity. Trend Ecol Evol 28:359–366
Bowen BW, Gaither MR, DiBattista JD, Iacchei M, Andrews KR, Grant WS, Toonen RJ, Briggs JC (2016) Comparative phylogeography of the ocean planet. Proc Natl Acad Sci USA 113:7962–7969
Charleston MA, Robertson DL (2002) Preferential host switching by primate lentiviruses can account for phylogenetic similarity with the primate phylogeny. Syst Biol 51:528–535
Conow C, Fielder D, Ovadia Y, Libeskind-Hadas R (2010) Jane: a new tool for the cophylogeny reconstruction problem. Algorithms Mol Biol 5:16
Cunha AF, Collins AG, Marques AC (2017) Phylogenetic relationships of Proboscoida Broch, 1910 (Cnidaria, Hydrozoa): are traditional morphological diagnostic characters relevant for the delimitation of lineages at the species, genus, and family levels? Mol Phylogenet Evol 106:118–135
Darriba D, Taboada GL, Doallo R, Posada D (2012) jModelTest 2: more models, new heuristics and parallel computing. Nat Methods 9:772
Dawson MN, Jacobs DK (2001) Molecular evidence for cryptic species of Aurelia aurita (Cnidaria, Scyphozoa). Biol Bull 200:92–96
Dayrat B (2005) Towards integrative taxonomy. Biol J Linn Soc 85:407–417
DiBattista JD, Howard Choat J, Gaither MR, Hobbs JP, Lozano‐Cortés DF, Myers RF, Paulay G, Rocha LA, Toonen RJ, Westneat MW, Berumen ML (2016) On the origin of endemic species in the Red Sea. J Biogeogr 43:13–30
Drummond AJ, Suchard MA, Xie D, Rambaut A (2012) Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol 29:1969–1973
Enochs IC, Manzello DP (2012) Species richness of motile cryptofauna across a gradient of reef framework erosion. Coral Reefs 31:653–661
Ezard T, Fujisawa T, Barraclough TG (2009) SPLITS: species limits by threshold statistics. R Package Version 1.0–11. https://r-forge.r-project.org/projects/splits
Fisher R, O’Leary RA, Low-Choy S, Mengersen K, Knowlton N, Brainard RE, Caley MJ (2015) Species richness on coral reefs and the pursuit of convergent global estimates. Curr Biol 25:500–505
Fontana S, Keshavmurthy S, Hsieh HJ, Denis V, Kuo CY, Hsu CM, Leung JKL, Tsai WS, Wallace CC, Chen CA (2012) Molecular evidence shows low species diversity of coral-associated hydroids in Acropora corals. PLoS ONE 7:e50130
Gates RD, Ainsworth TD (2011) The nature and taxonomic composition of coral symbiomes as drivers of performance limits in scleractinian corals. J Exp Mar Biol Ecol 408:94–101
Gittenberger A, Reijnen BT, Hoeksema BW (2011) A molecularly based phylogeny reconstruction of mushroom corals (Scleractinia: Fungiidae) with taxonomic consequences and evolutionary implications for life history traits. Contrib Zool 80:107–132
Govindarajan AF, Halanych KM, Cunningham CW (2005) Mitochondrial evolution and phylogeography in the hydrozoan Obelia geniculata (Cnidaria). Mar Biol 146:213–222
Hellberg ME (2006) No variation and low synonymous substitution rates in coral mtDNA despite high nuclear variation. BMC Evol Biol 6:1–8
Hirose M, Hirose E (2011) A new species of Zanclea (Cnidaria: Hydrozoa) associated with scleractinian corals from Okinawa, Japan. J Mar Biol Assoc UK 92:877–884
Hoeksema BW (2017) The hidden biodiversity of tropical coral reefs. Biodiversity 18:8–12
Hoeksema BW, Cairns S (2020) World list of Scleractinia. https://www.marinespecies.org/scleractinia. 18 Apr 2020
Hoeksema BW, van der Meij SET, Fransen CHJM (2012) The mushroom coral as a habitat. J Mar Biol Assoc UK 92:647–663
Hoeksema BW, Butôt R, García-Hernández JE (2018) A new host and range record for the gall crab Fungicola fagei as symbiont of the mushroom coral Lobactis scutaria at Hawai´i. Pac Sci 72:251–261
Hoeksema BW, van Beusekom M, ten Hove HA, Ivanenko VN, van der Meij SET, van Moorsel GWNM (2017) Helioseris cucullata as a host coral at St. Eustatius, Dutch Caribbean. Mar Biodivers 47:71–78
Huang D, Meier R, Todd PA, Chou LM (2008) Slow mitochondrial COI sequence evolution at the base of the metazoan tree and its implications for DNA barcoding. J Mol Evol 66:167–174
Huang D, Benzoni F, Fukami H, Knowlton N, Smith ND, Budd AF (2014) Taxonomic classification of the reef coral families Merulinidae, Montastraeidae, and Diploastraeidae (Cnidaria: Anthozoa: Scleractinia). Zool J Linn Soc Lond 171:277–355
Huelsenbeck JP, Nielsen R, Bollback JP (2003) Stochastic mapping of morphological characters. Syst Biol 52:131–158
Hughes TP, Bellwood DR, Connolly SR (2002) Biodiversity hotspots, centres of endemicity, and the conservation of coral reefs. Ecol Lett 5:775–784
Hutchinson MC, Cagua EF, Balbuena JA, Stouffer DB, Poisot T (2017) paco: implementing procrustean approach to cophylogeny in R. Methods Ecol Evol 8:932–940
Ivanenko VN, Hoeksema BW, Mudrova SV, Nikitin MA, Martínez A, Rimskaya-Korsakova NN, Berumen ML, Fontaneto D (2018) Lack of host specificity of copepod crustaceans associated with mushroom corals in the Red Sea. Mol Phylogenet Evol 127:770–780
Kapli P, Lutteropp S, Zhang J, Kobert K, Pavlidis P, Stamatakis A, Flouri T (2017) Multi-rate Poisson tree processes for single-locus species delimitation under maximum likelihood and Markov chain Monte Carlo. Bioinformatics 33:1630–1638
Katoh K, Standley DM (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30:772–780
Kumar S, Stecher G, Li M, Knyaz C, Tamura K (2018) MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 35:1547–1549
Lanfear R, Calcott B, Ho SY, Guindon S (2012) PartitionFinder: combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol Biol Evol 29:1695–1701
Leigh JW, Bryant D (2015) popart: full-feature software for haplotype network construction. Methods Ecol Evol 6:1110–1116
Leray M, Knowlton N (2015) DNA barcoding and metabarcoding of standardized samples reveal patterns of marine benthic diversity. Proc Natl Acad Sci USA 112:2076–2081
Maddison WP, Maddison DR (2006) Mesquite: a modular system for evolutionary analysis. https://www.mesquiteproject.org
Maggioni D, Montano S, Seveso D, Galli P (2016) Molecular evidence for cryptic species in Pteroclava krempfi (Hydrozoa, Cladocorynidae) living in association with alcyonaceans. Syst Biodivers 14:484–493
Maggioni D, Montano S, Voigt O, Seveso D, Galli P (2020) A mesophotic hotel: the octocoral Bebryce cf. grandicalyx as a host. Ecology 101:e02950
Maggioni D, Galli P, Berumen ML, Arrigoni R, Seveso D, Montano S (2017a) Astrocoryne cabela, gen. nov. et sp. nov. (Hydrozoa: Sphaerocorynidae), a new sponge-associated hydrozoan. Invertebr Syst 31:734–746
Maggioni D, Montano S, Arrigoni R, Galli P, Puce S, Pica D, Berumen ML (2017b) Genetic diversity of the Acropora-associated hydrozoans: new insight from the Red Sea. Mar Biodivers 47:1045–1055
Maggioni D, Arrigoni R, Galli P, Berumen ML, Seveso D, Montano S (2018) Polyphyly of the genus Zanclea and family Zancleidae (Hydrozoa, Capitata) revealed by the integrative analysis of two bryozoan-associated species. Contrib Zool 87:87–104
Maggioni D, Saponari L, Seveso D, Galli P, Schiavo A, Ostrovsky AN, Montano S (2020) Green fluorescence patterns in closely related symbiotic species of Zanclea (Hydrozoa, Capitata). Diversity 12:78
Maggioni D, Schiavo A, Ostrovsky AN, Seveso D, Galli P, Arrigoni R, Berumen ML, Benzoni F, Montano S (2020) Cryptic species and host specificity in the bryozoan-associated hydrozoan Zanclea divergens (Hydrozoa, Zancleidae). Mol Phylogenet Evol 151:106893
Manca F, Puce S, Caragnano A, Maggioni D, Pica D, Seveso D, Galli P, Montano S (2019) Symbiont footprints highlight the diversity of scleractinian-associated Zanclea hydrozoans (Cnidaria, Hydrozoa). Zool Scr 48:399–410
Miglietta MP, Maggioni D, Matsumoto Y (2019) Phylogenetics and species delimitation of two hydrozoa (phylum Cnidaria): Turritopsis (McCrady, 1857) and Pennaria (Goldfuss, 1820). Mar Biodivers 49:1085–1100
Millard NAH, Bouillon J (1974) A collection of hydroids from Mozambique, East Africa. Ann S Afr Mus 65:1–40
Miller MA, Pfeiffer W, Schwartz T (2010) Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In: Proceedings of the gateway computing environments workshop (GCE), New Orleans, pp 1–8
Montano S, Maggioni D, Galli P, Seveso D, Puce S (2013) Zanclea–coral association: new records from Maldives. Coral Reefs 32:701
Montano S, Galli P, Maggioni D, Seveso D, Puce S (2014) First record of coral-associated Zanclea (Hydrozoa, Zancleidae) from the Red Sea. Mar Biodiver 44:581–584
Montano S, Arrigoni R, Pica D, Maggioni D, Puce S (2015a) New insights into the symbiosis between Zanclea (Cnidaria, Hydrozoa) and scleractinians. Zool Scr 44:92–105
Montano S, Seveso D, Galli P, Puce S, Hoeksema BW (2015b) Mushroom corals as newly recorded hosts of the hydrozoan symbiont Zanclea sp. Mar Biol Res 11:773–779
Montano S, Maggioni D, Arrigoni R, Seveso D, Puce S, Galli P (2015c) The hidden diversity of Zanclea associated with scleractinians revealed by molecular data. PLoS ONE 10:e0133084
Montano S, Galli P, Hoeksema BW (2017a) First record from the Atlantic: a Zanclea-scleractinian association at St. Eustatius, Dutch Caribbean. Mar Biodivers 47:81–82
Montano S, Maggioni D, Galli P, Hoeksema BW (2017b) A cryptic species in the Pteroclava krempfi species complex (Hydrozoa, Cladocorynidae) revealed in the Caribbean. Mar Biodivers 47:83–89
Montano S, Fattorini S, Parravicini V, Berumen ML, Galli P, Maggioni D, Arrigoni R, Seveso D, Strona G (2017c) Corals hosting symbiotic hydrozoans are less susceptible to predation and disease. Proc R Soc B 284:20172405
Okubo N (2016) Restructuring the traditional suborders in the order Scleractinia based on embryogenetic morphological characteristics. Zool Sci 33:116–123
Okubo N, Hayward DC, Forêt S, Ball EE (2016) A comparative view of early development in the corals Favia lizardensis, Ctenactis echinata, and Acropora millepora—morphology, transcriptome, and developmental gene expression. BMC Evol Biol 16:48
Okubo N, Mezaki T, Nozawa Y, Nakano Y, Lien Y-T, Fukami H, Hayward DC, Ball EE (2013) Comparative embryology of eleven species of stony corals (Scleractinia). PLoS ONE 8:e84115
Pantos O, Bythell JC (2010) A novel reef coral symbiosis. Coral Reefs 29:761–770
Paradis E, Claude J, Strimmer K (2004) APE: analyses of phylogenetics and evolution in R language. Bioinformatics 20:289–290
Pica D, Bastari A, Vaga CF, Di Camillo CG, Montano S, Puce S (2017) Hydroid diversity of Eilat Bay with the description of a new Zanclea species. Mar Biol Res 13:469–479
Plaisance L, Brainard R, Caley MJ, Knowlton N (2011) Using DNA barcoding and standardized sampling to compare geographic and habitat differentiation of crustaceans: a Hawaiian Islands example. Diversity 3:581–591
Pons J, Barraclough TG, Gomez-Zurita J, Cardoso A, Duran DP, Hazell V, Vogler AP (2006) Sequence based species delimitation for the DNA taxonomy of undescribed insects. Syst Biol 55:595–609
Postaire B, Magalon H, Bourmaud CAF, Bruggemann JH (2016) Molecular species delimitation methods and population genetics data reveal extensive lineage diversity and cryptic species in Aglaopheniidae (Hydrozoa). Mol Phylogenet Evol 105:36–49
Potkamp G, Fransen CHJM (2018) Speciation with gene flow in marine systems. Contrib Zool 88:133–172
Puce S, Cerrano C, Boyer M, Ferretti C, Bavestrello G (2002) Zanclea (Cnidaria: Hydrozoa) species from Bunaken Marine Park (Sulawesi Sea, Indonesia). J Mar Biol Assoc UK 82:943–954
Puillandre N, Lambert A, Brouillet S, Achaz G (2012) ABGD, Automatic Barcode Gap Discovery for primary species delimitation. Mol Ecol 21:1864–1877
R Core Team (2019) R: A language and environment for statistical computing. https://www.R-project.org
Rambaut A, Suchard MA, Xie D, Drummond AJ (2014) Tracer v1.6. https://beast.bio.ed.ac.uk/Tracer/
Rauch C, Hoeksema BW, Hermanto B, Fransen CHJM (2019) Shrimps of the genus Periclimenes (Crustacea, Decapoda, Palaemonidae) associated with mushroom corals (Scleractinia, Fungiidae): linking DNA barcodes to morphology. Contrib Zool 88:201–235
Reaka-Kudla ML (1997) The global biodiversity of coral reefs: a comparison with rain forests. In: Reaka-Kudla ML, Wilson DE, Wilson EO (eds) Biodiversity II: understanding and protecting our biological resources. Joseph Henry Press, Washington, pp 83–108
Reid NM, Carstens BC (2012) Phylogenetic estimation error can decrease the accuracy of species delimitation: a Bayesian implementation of the general mixed Yule-coalescent model. BMC Evol Biol 12:196
Revell LJ (2012) phytools: an R package for phylogenetic comparative biology (and other things). Methods Ecol Evol 3:217–223
Revell LJ (2013) Two new graphical methods for mapping trait evolution on phylogenies. Methods Ecol Evol 4:754–759
Rocha LA, Craig MT, Bowen BW (2007) Phylogeography and the conservation of coral reef fishes. Coral Reefs 26:501–512
Romano SL, Palumbi SR (1996) Evolution of scleractinian corals inferred from molecular systematics. Science 271:640–642
Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S, Huelsenbeck JP (2012) MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol 61:539–542
Rozas J, Ferrer-Mata A, Sánchez-DelBarrio JC, Guirao-Rico S, Librado P, Ramos-Onsins SE, Sánchez-Gracia A (2017) DnaSP 6: DNA sequence polymorphism analysis of large datasets. Mol Biol Evol 34:3299–3302
Shearer TL, Van Oppen MJH, Romano SL, Wörheide G (2002) Slow mitochondrial DNA sequence evolution in the Anthozoa (Cnidaria). Mol Ecol 11:2475–2487
Stamatakis A (2014) RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313
Stella JS, Pratchett MS, Hutchings PA, Jones GP (2011) Coral-associated invertebrates: diversity, ecology importance and vulnerability to disturbance. Oceanogr Mar Biol 49:43–104
Takada Y, Abe O, Shibuno T (2007) Colonization patterns of mobile cryptic animals into interstices of coral rubble. Mar Ecol Prog Ser 343:35–44
Terraneo TI, Arrigoni R, Benzoni F, Tietbohl MD, Berumen ML (2017) Exploring the genetic diversity of shallow-water Agariciidae (Cnidaria: Anthozoa) from the Saudi Arabian Red Sea. Mar Biodivers 47:1065–1078
Terraneo TI, Benzoni F, Baird AH, Arrigoni R, Berumen ML (2019) Morphology and molecules reveal two new species of Porites (Scleractinia, Poritidae) from the Red Sea and the Gulf of Aden. Syst Biodivers 17:491–508
Toews DP, Brelsford A (2012) The biogeography of mitochondrial and nuclear discordance in animals. Mol Ecol 21:3907–3930
Vaga CF, Kitahara MV, Nascimento KB, Migotto AE (2020) Genetic diversity of the Pennaria disticha Goldfuss, 1820 (Cnidaria, Hydrozoa) complex: new insights from Brazil. Mar Biodivers 50:68
Van der Meij SET, Fransen CHJM, Pasman LR, Hoeksema BW (2015) Phylogenetic ecology of gall crabs (Cryptochiridae) as associates of mushroom corals (Fungiidae). Ecol Evol 5:5770–5780
Veron JEN, Stafford-Smith MG, Turak E, DeVantier LM (2016) Corals of the world. https://www.coralsoftheworld.org/page/home/. Accessed 30 Mar 2020
Villesen P (2007) FaBox: an online toolbox for fasta sequences. Mol Ecol Notes 7:965–968
Wallace CC, Chen CA, Fukami H, Muir PR (2007) Recognition of separate genera within Acropora based on new morphological, reproductive and genetic evidence from Acropora togianensis, and elevation of the subgenus Isopora Studer, 1878 to genus (Scleractinia: Astrocoeniidae; Acroporidae). Coral Reefs 26:231–239
Zhang J, Kapli P, Pavlidis P, Stamatakis A (2013) A general species delimitation method with applications to phylogenetic placements. Bioinformatics 29:2869–2876
Acknowledgements
The authors wish to thank all the people involved in collecting material or organising sampling campaigns: Tullia Isotta Terraneo (KAUST, Saudi Arabia), Malek Amr Gusti (KAUST, Saudi Arabia), Timothy Ravasi (OIST, Japan), the captain and crew of the MV Dream-Master (Saudi Arabia), the KAUST Coastal and Marine Resources Core Lab, Inga Dehnert (UNIMIB, Italy), Nicholas WL Yap (NUS, Singapore), Sudhanshi S Jain (NUS, Singapore), Stephen Keable (Australian Museum), Penny Berents (Australian Museum), Anne Hoggett (Australian Museum), Lyle Vail (Australian Museum), Masayuki Tamae (Marine Space, Japan), Hiroki Nakakouji (Marine Space, Japan), Yoshihiro Katsushima (Win Diving, Japan), Yuting V Lin (IONTU, Taiwan), Jian-Wen Chen (IONTU, Taiwan), and staff at the Green Island Marine Research Station (Taiwan) and Academia Sinica (Taiwan). Permissions relevant to undertake the research have been obtained from the applicable governmental agencies. The work was partially funded by PADI Foundation Grants #28634 and #14384 to DM and SM, and baseline research funds of PG and MLB. Samples from Eilat (Israel) were collected during the HyDRa Project funded by the EU FP7 Research Infrastructure Initiative ‘ASSEMBLE’ (Grant #227799) to DP. Samples from Ludao (Taiwan) were collected under the permit #1060174405 issued by Taitung County Government with the support of a grant from the Ministry of Science and Technology of Taiwan (#106-2611-M-002-008) to VD. Financial support to DP for collecting samples at Lizard Island (Australia) was provided by the 2018 John and Laurine Proud Fellowship and the Australian Museum’s Lizard Island Research Station. Fieldwork in Singapore was partially funded by the National Research Foundation, Prime Minister's Office, Singapore under its Marine Science R&D Programme (MSRDP-P03) to DH.
Funding
Open access funding provided by Università degli Studi di Milano - Bicocca within the CRUI-CARE Agreement.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Topic Editor Carly Kenkel
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Maggioni, D., Arrigoni, R., Seveso, D. et al. Evolution and biogeography of the Zanclea-Scleractinia symbiosis. Coral Reefs 41, 779–795 (2022). https://doi.org/10.1007/s00338-020-02010-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00338-020-02010-9