
Abstract
CRISPR and CRISPR-associated (Cas) proteins, which in nature comprise the RNA-based adaptive immune system in bacteria and archaea, have emerged as particularly powerful genome editing tools owing to their unrivaled ease of use and ability to modify genomes across mammalian model systems. As such, the CRISPR–Cas9 system holds promise as a “system of choice” for functional mammalian genetic studies across biological disciplines. Here we briefly review this fast moving field, introduce the CRISPR–Cas9 system and its application to genome editing, with a focus on the basic considerations in designing the targeting guide RNA sequence.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Site-directed DNA endonucleases are powerful tools for genome editing. When introduced into cells, these proteins can bind to a target DNA sequence in the genome and create a DNA double-strand break (DSB), the repair of which leads to varied DNA sequence modifications. The initial efforts on developing these tools were focused on engineering homing endonucleases (Silva et al. 2011) and zinc finger nucleases (ZFN) (Urnov et al. 2005, 2010), and later Transcription Activator-Like Effector Nucleases (TALEN) (Boch et al. 2009; Moscou and Bogdanove 2009; Bogdanove and Voytas 2011). Homing endonucleases use one single domain to perform both DNA recognition and cleavage functions, and as such, are challenging to engineer. For both the ZFN and TALEN systems, the DNA binding domains (DBD) are modular and can be engineered to recognize and bind specific DNA sequences, allowing an attached nuclease domain to generate DSBs at the target site. However, for each genomic target, a unique pair of ZFN or TALEN needs to be designed and generated, which is cumbersome and time-consuming. In 2012, a novel system, Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) and the CRISPR-associated proteins (Cas), emerged from the acquired immune system of bacteria and archaea (Jinek et al. 2012). CRISPR–Cas9 rapidly became the method of choice for genome editing having many advantages over the earlier approaches (Doudna and Charpentier 2014; Hsu et al. 2014). Here we briefly review this fast moving field, introduce the CRISPR–Cas9 system and discuss its application to genome editing, with a focus on the basic considerations in designing the targeting guide RNA sequence.
CRISPR–Cas9-mediated genome editing
The CRISPR–Cas system was first described in the genome of Escherichia coli as a cluster of short palindromic repeats separated by peculiar short spacer sequences (Ishino et al. 1987). Subsequently, it was shown that CRISPR loci are present in the genomes of more than 40 % of bacteria and 90 % of archaea (Horvath and Barrangou 2010) and their function is to serve as an adaptive immune defense mechanism, protecting against phage infection by recognizing and cleaving pathogen DNA (Horvath and Barrangou 2010; Fineran and Charpentier 2012). By 2012, the basic mechanism of CRISPR–Cas9 derived from Streptococcus pyogenes was elucidated (Deltcheva et al. 2011; Jinek et al. 2012). CRISPR–Cas9 is an RNA-guided DNA endonuclease system in which Cas9 endonuclease forms a complex with two naturally occurring RNA species, CRISPR RNA (crRNA) and trans activating CRISPR RNA (tracrRNA). This complex targets specific DNA sequences complementary to the 20 nt (nucleotide) sequence residing at the 5′ end of the crRNA (Jinek et al. 2012). Conveniently, crRNA and tracrRNA can be linked by an arbitrary stem loop sequence to generate a synthetic single-guide RNA (sgRNA). Although naturally evolving as a system in bacteria, upon appropriate codon optimization of the Cas9 coding sequence, CRISPR–Cas9 is highly active in mammalian cells (Cho et al. 2013; Cong et al. 2013; Jinek et al. 2013; Mali et al. 2013b).
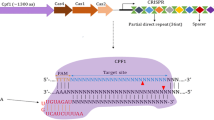
In practice, by simply designing the 5′ 20 nt sequence on the sgRNA to be complementary to the genomic target sequence, the Cas9 nuclease-sgRNA complex can be directed to specific genomic locus generating DNA DSBs. The target defining region of the sgRNA is about 20 nt long, with variations from 17 to 30 nt having been successfully used (Ran et al. 2013; Fu et al. 2014). The other key element in determining target sequence specificity is the Protospacer Adjacent Motif (PAM) that is adjacent to the target site at the genome locus, but is not a part of the guide RNA sequence (see Fig. 1). For Cas9 nuclease from S. pyogenes, the PAM sequence is NGG, while CRISPR–Cas9 systems from other species use different PAM sequences (Cong et al. 2013; Esvelt et al. 2013; Hou et al. 2013). In bacteria, the PAM is thought to effectively distinguish self, with the PAM not being present in the genomic CRISPR loci, from the invading phage, whose genome carries the PAM sequence adjacent to the target sequence (Marraffini and Sontheimer 2010).

CRISPR–Cas9-mediated genome editing. a The structure of Cas9–sgRNA complex binding to target DNA. Cas9 binds to specific DNA sequences via the base-pairing of the guide sequence on sgRNA (pink) with the DNA target (gray). Protospacer adjacent motif (PAM) is downstream of the target sequence. b The CRISPR–Cas9-mediated double-stranded DNA breaks are repaired by endogenous DNA repair machinery: non-homologous end joining (NHEJ) or homology-directed repair (HDR). Various genetic modifications can be generated through these two pathways
CRISPR–Cas9-mediated DNA DSBs are repaired through either the Non-Homologous End Joining (NHEJ) repair process, or the homology-directed repair (HDR) pathway. NHEJ repair often leads to small insertions or deletions (indels) at the targeted site, while HDR pathway leads to perfect repair or precise genetic modification (see Fig. 1) (Doudna and Charpentier 2014; Hsu et al. 2014). Through these two DNA repair pathways, various genetic modifications can be achieved (Fig. 1). The NHEJ-mediated DNA repair pathway can be exploited to generate null mutation alleles. Indel mutations generated at a target site within an exon can lead to frame shift mutations in one or both alleles. One major advantage of the CRISPR–Cas9 system, as compared to conventional gene targeting and other programmable endonucleases, is the ease of multiplexing, where multiple genes can be mutated simultaneously simply by using multiple sgRNAs each targeting a different gene (Wang et al. 2013a, b). In addition, when two sgRNAs are used flanking a genomic region, the intervening region can be deleted or inverted (Blasco et al. 2014; Canver et al. 2014; He et al. 2015). Furthermore, chromosomal translocation can also be achieved by using two sgRNAs targeting two genomic loci located on different chromosomes (Choi and Meyerson 2014).
When a DSB is generated and a donor DNA template is provided, precise genetic modification can be introduced through the HDR pathway (Fig. 1). For small modifications, including incorporation of point mutations, defined indel mutations, as well as insertion of a short sequence such as a loxP site or an epitope tag, single-stranded oligodeoxynucleotide (ssODN) can be used as donor DNA. In this design, donor ssODN is designed to carry homologous sequences flanking the mutation and total size can be up to 200 nt. HDR efficiency does not appear to be directly correlated with donor homology lengths (Yang et al. 2013b), and HDR efficiency variation is likely due to the nature of the target genomic loci, which is still poorly understood. When DNA of larger sizes is to be introduced into a target site, a double-stranded donor plasmid carrying the transgene flanked by homologous arms is used (Yang et al. 2013a).
Because of the ease of use, CRISPR–Cas9 system has swiftly become the most commonly used tool for efficient genome editing of bacteria, plants, cell lines, primary cells, and tissues. Impressively, direct introduction of CRISPR–Cas9 into the zygote leads to efficient genetic modification of the genome in early embryos, which when brought to term develop into genetically modified animals (Hwang et al. 2013; Li et al. 2013a,b; Wang et al. 2013a; Yang et al. 2013a; Hai et al. 2014; Niu et al. 2014). Depending on the experimental setup, different methods can be used to deliver CRISPR–Cas9 system. When used as a genome editing tool in cultured cells, either electroporation or transfection is often used to deliver a plasmid containing a ubiquitous promoter driving Cas9 and sgRNA expression (Cong et al. 2013; Mali et al. 2013b). The genome editing efficiency achieved is highly dependent on a number of variables including the actual transfection efficiency, genomic locus intended to be targeted, and cell types. For genetic engineering in animals, Cas9 mRNA or protein and the sgRNA (with or without donor DNA) are introduced into zygotes by microinjection (Li et al. 2013a,b; Wang et al. 2013a; Yang et al. 2013a; Hwang et al. 2013; Hai et al. 2014; Niu et al. 2014). Germline modification has also been achieved in mice by transfection of plasmids expressing Cas9 and sgRNA into spermatogonial stem cells. After development to spermatids and injection into oocytes (i.e., fertilization), germline transmission of the specific genetic modification was achieved (Wu et al. 2015). Lastly, somatic cell genomic modification in mice has been achieved, by hydrodynamic tail vein injection of plasmids (Xue et al. 2014; Yin et al. 2014), as well as by injecting adeno-associated virus (AAV) expressing CRISPR–Cas9 in brain (Swiech et al. 2015).
The wild-type S. pyogenes-Cas9 (SP-Cas9) endonuclease has two nuclease domains, HNH and RuvC-like, each capable of cleaving one of the double-stranded target DNA when associated with a sgRNA (See Fig. 1). When either one of these domains is mutated, the Cas9-sgRNA complex becomes a sequence and strand-specific nickase (Cas9n). When used with two sgRNAs in close proximity and targeting opposite DNA strands, this “dual” Cas9 nickase generates a DSB with defined overhangs. The more commonly used Cas9n is D10A, where the RuvC domain is mutated and generates 5′ overhang (Mali et al. 2013a; Ran et al. 2013). H840A Cas9n that generates a 3′ overhang has also been successfully applied to mouse model generation (Shen et al. 2014). Furthermore, when both nuclease domains are mutated eliminating all endonuclease activity, Cas9 becomes a programmable DNA binding protein (deadCas9 or dCas9). Guided by sgRNA, dCas9, when fused with different effector domains such as KRAB domain or VP64, can be directed to promoters and directly influence the level of gene transcription (Cheng et al. 2013; Gilbert et al. 2013; Konermann et al. 2014). By using various dCas9-effector fusions, it may be possible to epigenetically modify a specific locus leading to change in gene expression in vitro and in vivo.
In addition to SP-Cas9, several orthologous CRISPR–Cas9 systems from other species have been characterized and applied to genome editing in mammalian cells (Cong et al. 2013; Esvelt et al. 2013; Hou et al. 2013). Compared to SP CRISPR–Cas9 system, most of these orthologous systems have different PAM requirements and crRNA and tracrRNA sequences. Their development and application will greatly expand the sequence space amendable to CRISPR–Cas9 targeting. In addition, by recognizing different sgRNA backbones, Cas9 from different species can be used to perform different functions in the same cells, without interfering with each other (Esvelt et al. 2013). These developments will be useful for applications such as modulation of transcription networks and labeling of multiple genomic sequences for live cell imaging.
Design of CRISPR/Cas9 guide sequence—achieving a high targeting efficiency and specificity
Specificity of the CRISPR–Cas9 system is defined by the 20 nt located at the 5′ end of the sgRNA, which interacts with the target DNA by Watson–Crick RNA–DNA base-pairing. Although highly specific, Cas9-sgRNA binding to the target DNA can tolerate sequence mismatches, leading to mutations in unintended genomic loci (“off-target” effect) (Fu et al. 2013; Hsu et al. 2013; Lin et al. 2014). The principal variables that impact specificity may include target sequence length and composition, concentrations of the Cas9 protein and the sgRNA. Although much needs to be understood to fully define these parameters in a specific targeting experiment, below we attempt to discuss current strategies and available software for the design of the guide RNA.
Rational design of CRISPR guide sequence aims to maximize occurrence of the desired genetic modification at the target site, while minimizing the extent of unintended mutations at off-target sites. To begin defining parameters affecting on-target efficiency, recent work investigated the effect of target sequence composition on targeting efficiency (Wang et al. 2013b; Doench et al. 2014). Both studies concluded that a high or low GC content in guide sequence leads to lower efficiency, while other variables may also impact the efficiency (Wang et al. 2013b; Doench et al. 2014). When a guide sequence capable of mediating efficient on-target cleavage has been identified, it should be assessed for potential off-target activities within the genome of interest. As discussed earlier, CRISPR–Cas9 targeting specificity is determined by a 20 nt guide sequence located at the 5′ end of the sgRNA, plus the PAM sequence adjacent to the target site located at the genomic locus. Mismatches between the guide sequence and target DNA are tolerated to certain extent, especially in the region distal to the PAM sequence (Jinek et al. 2012; Fu et al. 2013; Hsu et al. 2013). Therefore, whenever possible, a guide sequence that matches or is highly similar to multiple genomic loci should be avoided to prevent off-target effects that may lead to unintended and often undetected genetic modification in the genome of the cell or organism. To assist researchers with the design of CRISPR–Cas9 experiments, a growing number of software tools have become available for designing guide RNA and predicting off-target profiles (see Table 1).
Most of the current guide RNA design and off-target prediction tools rely on rules derived from earlier studies based on simple sequence matches/mismatches (Fu et al. 2013; Hsu et al. 2013) and are focused on optimizing computational time, resources, and providing additional features to assist users to design the experiment to meet their specific goals. In general, most of currently available tools allow mismatches of target sequences up to 3 or 4 nucleotides (Sander et al. 2007,2010; Hsu et al. 2013; Heigwer et al. 2014), and in a GPU-based implementation, up to 1–10 mismatches in the online version and up to any number of mismatches in the standalone version (Bae et al. 2014b). Recent studies have begun to collect more experimental data to generate better models for computational predictions. Using experimentally derived models, “sgRNA Designer” predicts on-target efficacy using a logistic regression classifier trained on >1000 sgRNAs targeting multiple genes and scores sgRNAs using position-specific weights for nucleotides and dinucleotides (Doench et al. 2014). “CRISPR Design Tool” incorporates the number of mismatches, position of mismatches, and pairwise distances of mismatches into its off-target scoring scheme, which was derived from a set of systematically designed experiments (Hsu et al. 2013). For guide RNA designs, some tools allow specification of experimental goals by users for different desired modifications (e.g., insertion of tags, disruption of protein domains, etc.) or allow the use of gene architecture annotation to assist guide RNA designs (E-CRISP, CHOPCHOP). While most of the tools are designed for SP-Cas9 with NGG or NAG PAM sequences (ZiFiT, CRISPR Design Tool, E-CRISP, sgRNA design tool), a few provide flexibility for PAM sequences to allow design of guide RNA for orthogonal Cas9 proteins with different PAM requirements (RGEN Tools, CHOP–CHOP). With more experiments investigating how parameters such as target sequence effect on sgRNA expression and folding, as well as epigenetic context of on-target and off-target sites, we foresee in the near future better software packages or updates to existing tools will become available and benefit researchers in the design of more efficient and specific gene editing experiments.
Strategies for mitigating off-target effect
As eluded above, intelligent design of sgRNA guide sequence is still in its infancy. Below, we have listed the main approaches that can be used in conjunction with software systems listed in Table 1.
-
(i)
Choose a guide sequence with minimal potential off-target sites as determined by genome-wide homology searches. Among the guides, choose those with off-targets’ mismatches concentrated at the PAM proximal part of the guide sequence, as these are less tolerated for Cas9 function (Jinek et al. 2012; Fu et al. 2013; Hsu et al. 2013).
-
(ii)
Use a guide sequence of shorter lengths (e.g., 17–19 nt). Fu et al. demonstrated that shorter targeting sequence in the sgRNAs could reduce off-target effect significantly with only a slight reduction of on-target efficiency (Fu et al. 2014).
-
(iii)
Use dual nickase strategy. With a pair of closely positioned sgRNAs, Cas9 nickase (D10A mutant) can introduce two adjacent single-stranded nicks, leading to a DSB with defined overhangs. This approach has been demonstrated to reduce off-target activity by 50- to 1500-fold in cell lines and to achieve gene knockout in mouse zygotes without sacrificing on-target cleavage efficiency (Ran et al. 2013; Shen et al. 2014).
-
(iv)
Use dCas9-FokI strategy. Using a pair of sgRNAs with optimal spacing and orientation, dCas9 fused with Fok1 nuclease domain can form dimer and generate DSB, similar to the design of ZFN and TALEN. The specificity is significantly increased using this strategy (Guilinger et al. 2014; Tsai et al. 2014).
-
(v)
Off-target identification and mitigation. In addition to computational prediction, several strategies have been developed to experimentally identify off-target mutations (Frock et al. 2015; Kim et al. 2015; Tsai et al. 2015; Wang et al. 2015). By genotyping these potential off-target sites, cell lines containing desired genetic modification but free of off-target mutations can be identified. In the case of animal models, breeding can be used to segregate the desired allele from the off-target mutant alleles.
Each of these strategies comes with its own advantages and limitations. Hence, when designing CRISPR–Cas9 experiments, it is important to understand the potential impact of unintentional off-target mutations and the need for mitigating them. For example, if CRISPR–Cas9 is to be used for clinical intervention, it is essential that off-target effect be minimized and its potential impacts understood and/or removed. If, however, the aim is to develop animal models, it is less of a concern, as founder animals will be backcrossed and unintended mutant alleles segregated. A possible simple strategy to avoid misinterpretation of data due to off-target effect is to develop genetically modified models using at least two independent sgRNAs with different guide RNA sequences.
Brief outline of CRISPR–Cas9-meditated genome editing in mouse
To help understand the general process of CRISPR–Cas9-mediated genome editing, here we outline the basic strategy and considerations for generating mouse models using CRISPR–Cas9 system (Table 2).
For generating indel-based null allele, single sgRNA targeting slightly 3′ of ATG or the first coding exon shared by all mRNA isoforms may be a good idea in general. Small indels generated using a single sgRNA can be either in-frame or out-frame mutations. The “RGEN Tools” is designed to analyze sequence surrounding the DSB site for the likelihood of microhomology-mediated repair (MMR) and a guide sequence can be chosen to optimize the occurrence of frameshift mutations (Bae et al. 2014a).
Knock in models can be divided into two categories practically. With “small” alterations, the intended mutation, such as incorporation of a point mutation, tag, loxP site, can be accommodated into a donor ssODN, along with homology sequences flanking the mutation, for a total size of 200 nt which is the limit for current ssODN synthesis. For the larger alterations that could not be accommodated onto a ssODN, a dsDNA plasmid can be synthesized or assembled by molecular techniques, with homology arm lengths from a few hundreds bases to many kb. The timeline for generating these two types of models vary accordingly, as it usually takes only days to synthesize a ssODN, it takes significantly longer to generate dsDNA plasmid.
Genotyping of indel, SNP incorporation and small tag insertions can be accomplished by amplification of the region encompassing the intended mutation (~500 nt) by PCR, followed by sequencing, to identify founder mice. Founders generated by the CRISPR–Cas9 technology often are mosaic, carrying the NHEJ, HDR as well as any remaining wild-type alleles all in one mouse. To identify the successful HDR alleles among the other events, the mixture of PCR product should be cloned into a plasmid and individual clones sequenced to unequivocally confirm the presence of the HDR allele. For transgene insertion alleles generated from use of donor plasmid, long range PCR or Southern blot should be used to examine integrity of the junction regions between donor homology arms and the genomic locus. Of particular notice is the possibility of additional unintended mutations originating from the off-target effect. These may be screened and if positive, mitigated by further breeding.
Current challenges and future development of CRISPR–Cas9
Although CRISPR–Cas9 has been proven powerful and widely applied, it is still a relatively new technology and there is much to be understood and improved.
Improving specificity and efficiency of the CRISPR–Cas9 system
A critical need in CRISPR–Cas9-mediated genome editing is to minimize the risk of off-target damage. As discussed above, various strategies can be used to minimize potential off-target effects, including truncated guide RNA, dual nickase, dCas9-FokI, etc. However, as each guide sequence likely has a variable number of off-target sites, experimental data need to be generated and analyzed to understand the factors related to off-target effect. With the use of multiple recently established methods for detecting off-target mutations and accumulation of data (Frock et al. 2015; Kim et al. 2015; Tsai et al. 2015; Wang et al. 2015), we can expect more comprehensive models for off-target prediction based on all the experimental data.
It is known that efficiency of genome editing using site-specific nucleases varies widely depending on genomic context. This is thought to result from the combined effects of different genetic composition and epigenetic state for each particular locus. For example, DNA accessibility has been correlated with transcription factor binding, and recently with Cas9 protein binding (Kuscu et al. 2014; Wu et al. 2014). Various histone modifications have been correlated with transcription factor binding and chromosome structure (ENCODE Project Consortium et al. 2012; Kundaje et al. 2012; Wang et al. 2012). Due to the complexity of genetic sequences, epigenetic modifications and different genomic loci across different cell types, we currently still lack basic understanding of how the binding, catalytic activity, and ultimately the efficiency of the CRISPR–Cas9 system is affected. A better understanding of these phenomena will significantly enhance our ability to efficiently and accurately target genomic sequences across the genome.
Animal model generation: the challenge of bigger and faster
One major limitation of current method in developing genetically modified animals is that the founder animals are often mosaic, carrying more than two alleles with each appearing at a certain frequency. This is potentially due to CRISPR–Cas9 activity occurring after the first cell division of the zygote. A better understanding of cell division and control of the timing of CRISPR–Cas9 activity, aiming at modifying the genomic locus strictly at the one cell stage, may allow creation of homozygote mutant mice that may be suitable for direct phenotypic analysis without the need for further breeding. Improvement in this aspect will shorten the timeline from model creation to phenotypic analysis. Elimination of mosaicism will be of particular importance for generating genetically modified large animals (e.g., non-human primate), which takes years to breed and often have small liter sizes.
CRISPR–Cas9-mediated genome editing has been used to generate mouse models carrying mutations in a single or multiple genes, as well as reporter and conditional alleles (Wang et al. 2013a; Yang et al. 2013a). However, one of the most impactful uses of a mouse model is genetic humanization that requires replacement of the mouse gene or gene cluster with its human ortholog. This is extremely challenging and time-consuming using gene targeting method (Lee et al. 2014), and has not been achieved using CRISPR–Cas9-mediated genome editing. The average size of a gene is about 50,000 nt and genes from a family can be localized in a specific genomic locus (for example, the immunoglobulin gene cluster resides in a region that occupies a few million nucleotides). Replacement of a few kb long fragment has been demonstrated in human iPS cells (Byrne et al. 2015). More needs to be learnt and explored using the CRISPR–Cas9 system to engineer the genome in larger scale.
Exploiting the sgRNA backbone to maximize efficiency and expand utility of the CRISPR–Cas system
By introducing an A-U flip and extension of the stem loop structure into the trascrRNA portion of the sgRNA backbone, Chen et al. achieved an improved efficiency of gene repression and genomic loci labeling using the CRISPR–Cas9 system (Chen et al. 2013). Moreover, different stem loop structures recognized by RNA binding proteins have been engineered into the sgRNA backbone, which serve as a bait to recruit different effectors, for the purpose of gene activation and repression (Konermann et al. 2014; Zalatan et al. 2015). By using sgRNAs targeting different genomic loci, with different effector recruiting stem loops, the same dCas9 protein could perform different functions at different target sites, therefore allowing for functional multiplexing using one CRISPR–Cas9 system (Zalatan et al. 2015).
In addition to DNA target, Cas9 can also bind with high affinity to single-stranded RNA (ssRNA) targets (O’Connell et al. 2014), although validity of its in vivo application has yet to be demonstrated. There are orthologous CRISPR systems that naturally target RNA molecules (Hale et al. 2009, 2012), therefore would be exciting resources to explore for RNA editing.
Interest in the CRISPR–Cas9 technology as a genome editing tool has increased exponentially in the last three years. Since its debut in 2012, there have already been more than 1000 papers published with CRISPR as the key word. The collective work from the field has culminated in the development of many innovative applications and major breakthroughs have been achieved, including the establishment of whole genome loss of function and gain of function screen (Wang et al. 2013b; Konermann et al. 2014; Shalem et al. 2014), as well as the generation of the first non-human primate knockout model (Niu et al. 2014). It is tantalizing what the future may hold for the CRISPR–Cas9 technology, particularly in the area of gene therapy, but we can be assured that further improvement and development of the technology will deliver even greater achievements.
References
Bae S, Kweon J, Kim HS, Kim J-S (2014a) Microhomology-based choice of Cas9 nuclease target sites. Nat Methods 11:705–706
Bae S, Park J, Kim J-S (2014b) Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics 30(10):1473–1475
Blasco RB, Karaca E, Ambrogio C, Cheong TC, Karayol E et al (2014) Simple and rapid in vivo generation of chromosomal rearrangements using CRISPR/Cas9 technology. Cell Rep 9:1219–1227
Boch J, Scholze H, Schornack S, Landgraf A, Hahn S et al (2009) Breaking the code of DNA binding specificity of TAL-type III effectors. Science 326:1509–1512
Bogdanove AJ, Voytas DF (2011) TAL effectors: customizable proteins for DNA targeting. Science 333:1843–1846
Byrne SM, Ortiz L, Mali P, Aach J, Church GM (2015) Multi-kilobase homozygous targeted gene replacement in human induced pluripotent stem cells. Nucleic Acids Res 43(3):e21
Canver MC, Bauer DE, Dass A, Yien YY, Chung J et al (2014) Characterization of genomic deletion efficiency mediated by clustered regularly interspaced palindromic repeats (CRISPR)/Cas9 nuclease system in mammalian cells. J Biol Chem 289:21312–21324
Chen B, Gilbert LA, Cimini BA, Schnitzbauer J, Zhang W et al (2013) Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell 155:1479–1491
Cheng AW, Wang H, Yang H, Shi L, Katz Y et al (2013) Multiplexed activation of endogenous genes by CRISPR-on, an RNA-guided transcriptional activator system. Cell Res 23:1163–1171
Cho SW, Kim S, Kim JM, Kim JS (2013) Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat Biotechnol 31:230–232
Choi PS, Meyerson M (2014) Targeted genomic rearrangements using CRISPR/Cas technology. Nat Commun 5:3728
Cong L, Ran FA, Cox D, Lin S, Barretto R et al (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339:819–823
Deltcheva E, Chylinski K, Sharma CM, Gonzales K, Chao Y et al (2011) CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 471:602–607
Doench JG, Hartenian E, Graham DB, Tothova Z, Hegde M et al (2014) Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nat Biotechnol 32:1262–1267
Doudna JA, Charpentier E (2014) Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 346:1258096
ENCODE Project Consortium, Bernstein BE, Birney E, Dunham I, Green ED et al (2012) An integrated encyclopedia of DNA elements in the human genome. Nature 489:57–74
Esvelt KM, Mali P, Braff JL, Moosburner M, Yaung SJ et al (2013) Orthogonal Cas9 proteins for RNA-guided gene regulation and editing. Nat Methods 10:1116–1121
Fineran PC, Charpentier E (2012) Memory of viral infections by CRISPR-Cas adaptive immune systems: acquisition of new information. Virology 434:202–209
Frock RL, Hu J, Meyers RM, Ho YJ, Kii E et al (2015) Genome-wide detection of DNA double-stranded breaks induced by engineered nucleases. Nat Biotechnol 33:179–186
Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D et al (2013) High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol 31(9):822–826
Fu Y, Sander JD, Reyon D, Cascio VM, Joung JK (2014) Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat Biotechnol 32:279–284
Gilbert LA, Larson MH, Morsut L, Liu Z, Brar GA et al (2013) CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 154:442–451
Guilinger JP, Thompson DB, Liu DR (2014) Fusion of catalytically inactive Cas9 to FokI nuclease improves the specificity of genome modification. Nat Biotechnol 32:577–582
Hai T, Teng F, Guo R, Li W, Zhou Q (2014) One-step generation of knockout pigs by zygote injection of CRISPR/Cas system. Cell Res 24(3):372–375
Hale CR, Zhao P, Olson S, Duff MO, Graveley BR et al (2009) RNA-guided RNA cleavage by a CRISPR RNA-Cas protein complex. Cell 139:945–956
Hale CR, Majumdar S, Elmore J, Pfister N, Compton M et al (2012) Essential features and rational design of CRISPR RNAs that function with the Cas RAMP module complex to cleave RNAs. Mol Cell 45:292–302
He Z, Proudfoot C, Mileham AJ, McLaren DG, Whitelaw CB et al (2015) Highly efficient targeted chromosome deletions using CRISPR/Cas9. Biotechnol Bioeng 112(5):1060–1064
Heigwer F, Kerr G, Boutros M (2014) E-CRISP: fast CRISPR target site identification. Nat Methods 11:122–123
Horvath P, Barrangou R (2010) CRISPR/Cas, the immune system of bacteria and archaea. Science 327:167–170
Hou Z, Zhang Y, Propson NE, Howden SE, Chu LF et al (2013) Efficient genome engineering in human pluripotent stem cells using Cas9 from Neisseria meningitidis. Proc Natl Acad Sci USA 110:15644–15649
Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S et al (2013) DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol 31(9):827–832
Hsu PD, Lander ES, Zhang F (2014) Development and Applications of CRISPR-Cas9 for Genome Engineering. Cell 157:1262–1278
Hwang WY, Fu Y, Reyon D, Maeder ML, Tsai SQ et al (2013) Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat Biotechnol 31:227–229
Ishino Y, Shinagawa H, Makino K, Amemura M, Nakata A (1987) Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J Bacteriol 169:5429–5433
Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA et al (2012) A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337:816–821
Jinek M, East A, Cheng A, Lin S, Ma E et al (2013) RNA-programmed genome editing in human cells. Elife 2:e00471
Kim D, Bae S, Park J, Kim E, Kim S et al (2015) Digenome-seq: genome-wide profiling of CRISPR-Cas9 off-target effects in human cells. Nat Methods 12(3):237–243
Konermann S, Brigham MD, Trevino AE, Joung J, Abudayyeh OO et al (2014) Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 517(7536):583–588
Kundaje A, Kyriazopoulou-Panagiotopoulou S, Libbrecht M, Smith CL, Raha D et al (2012) Ubiquitous heterogeneity and asymmetry of the chromatin environment at regulatory elements. Genome Res 22:1735–1747
Kuscu C, Arslan S, Singh R, Thorpe J, Adli M (2014) Genome-wide analysis reveals characteristics of off-target sites bound by the Cas9 endonuclease. Nat Biotechnol 32(7):677–683
Lee EC, Liang Q, Ali H, Bayliss L, Beasley A et al (2014) Complete humanization of the mouse immunoglobulin loci enables efficient therapeutic antibody discovery. Nat Biotechnol 32:356–363
Li D, Qiu Z, Shao Y, Chen Y, Guan Y et al (2013a) Heritable gene targeting in the mouse and rat using a CRISPR-Cas system. Nat Biotechnol 31:681–683
Li W, Teng F, Li T, Zhou Q (2013b) Simultaneous generation and germline transmission of multiple gene mutations in rat using CRISPR-Cas systems. Nat Biotechnol 31:684–686
Lin Y, Cradick TJ, Brown MT, Deshmukh H, Ranjan P et al (2014) CRISPR/Cas9 systems have off-target activity with insertions or deletions between target DNA and guide RNA sequences. Nucleic Acids Res 42:7473–7485
Mali P, Aach J, Stranges PB, Esvelt KM, Moosburner M et al (2013a) CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat Biotechnol 31(9):833–838
Mali P, Yang L, Esvelt KM, Aach J, Guell M et al (2013b) RNA-guided human genome engineering via Cas9. Science 339:823–826
Marraffini LA, Sontheimer EJ (2010) Self versus non-self discrimination during CRISPR RNA-directed immunity. Nature 463:568–571
Montague TG, Cruz JM, Gagnon JA, Church GM, Valen E (2014) CHOPCHOP: a CRISPR/Cas9 and TALEN web tool for genome editing. Nucleic Acids Res 42(Web Server issue):W401–W407
Moscou MJ, Bogdanove AJ (2009) A simple cipher governs DNA recognition by TAL effectors. Science 326:1501
Niu Y, Shen B, Cui Y, Chen Y, Wang J et al (2014) Generation of gene-modified cynomolgus monkey via Cas9/RNA-mediated gene targeting in one-cell embryos. Cell 156:836–843
O’Connell MR, Oakes BL, Sternberg SH, East-Seletsky A, Kaplan M et al (2014) Programmable RNA recognition and cleavage by CRISPR/Cas9. Nature 516:263–266
Ran FA, Hsu PD, Lin CY, Gootenberg JS, Konermann S et al (2013) Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 154:1380–1389
Sander JD, Zaback P, Joung JK, Voytas DF, Dobbs D (2007) Zinc Finger Targeter (ZiFiT): an engineered zinc finger/target site design tool. Nucleic Acids Res 35:W599–W605
Sander JD, Maeder ML, Reyon D, Voytas DF, Joung JK et al (2010) ZiFiT (Zinc Finger Targeter): an updated zinc finger engineering tool. Nucleic acids Res 38(1):W462–W468
Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA et al (2014) Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343:84–87
Shen B, Zhang W, Zhang J, Zhou J, Wang J et al (2014) Efficient genome modification by CRISPR-Cas9 nickase with minimal off-target effects. Nat Methods 11:399–402
Silva G, Poirot L, Galetto R, Smith J, Montoya G et al (2011) Meganucleases and other tools for targeted genome engineering: perspectives and challenges for gene therapy. Curr Gene Ther 11:11–27
Swiech L, Heidenreich M, Banerjee A, Habib N, Li Y et al (2015) In vivo interrogation of gene function in the mammalian brain using CRISPR-Cas9. Nat Biotechnol 33:102–106
Tsai SQ, Wyvekens N, Khayter C, Foden JA, Thapar V et al (2014) Dimeric CRISPR RNA-guided FokI nucleases for highly specific genome editing. Nat Biotechnol 32:569–576
Tsai SQ, Zheng Z, Nguyen NT, Liebers M, Topkar VV et al (2015) GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat Biotechnol 33:187–197
Urnov FD, Miller JC, Lee YL, Beausejour CM, Rock JM et al (2005) Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature 435:646–651
Urnov FD, Rebar EJ, Holmes MC, Zhang HS, Gregory PD (2010) Genome editing with engineered zinc finger nucleases. Nat Rev Genet 11:636–646
Wang J, Zhuang J, Iyer S, Lin X, Whitfield TW et al (2012) Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors. Genome Res 22:1798–1812
Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW et al (2013a) One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 153:910–918
Wang T, Wei JJ, Sabatini DM, Lander ES (2013b) Genetic screens in human cells using the CRISPR/Cas9 system. Science 343(6166):80–84
Wang X, Wang Y, Wu X, Wang J, Wang Y et al (2015) Unbiased detection of off-target cleavage by CRISPR-Cas9 and TALENs using integrase-defective lentiviral vectors. Nat Biotechnol 33:175–178
Wu X, Scott DA, Kriz AJ, Chiu AC, Hsu PD et al (2014) Genome-wide binding of the CRISPR endonuclease Cas9 in mammalian cells. Nat Biotechnol 32(7):670–676
Wu Y, Zhou H, Fan X, Zhang Y, Zhang M et al (2015) Correction of a genetic disease by CRISPR-Cas9-mediated gene editing in mouse spermatogonial stem cells. Cell Res 25:67–79
Xue W, Chen S, Yin H, Tammela T, Papagiannakopoulos T et al (2014) CRISPR-mediated direct mutation of cancer genes in the mouse liver. Nature 514:380–384
Yang H, Wang H, Shivalila CS, Cheng AW, Shi L et al (2013a) One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell 154:1370–1379
Yang L, Guell M, Byrne S, Yang JL, De Los Angeles A et al (2013b) Optimization of scarless human stem cell genome editing. Nucleic Acids Res 41:9049–9061
Yin H, Xue W, Chen S, Bogorad RL, Benedetti E et al (2014) Genome editing with Cas9 in adult mice corrects a disease mutation and phenotype. Nat Biotechnol 32(6):551–553
Zalatan JG, Lee ME, Almeida R, Gilbert LA, Whitehead EH et al (2015) Engineering Complex Synthetic Transcriptional Programs with CRISPR RNA Scaffolds. Cell 160:339–350
Acknowledgments
We thank Jesse Hammer for the artwork. This work was in part supported by the National Cancer Institute under award number P30CA034196, award for the The Jackson Laboratory KOMP2 Phenotyping Center under award number 3U54HG006332-04S2, and an institutional grant from the Jackson Laboratory (H.W.). A. W. C. was supported by JAX Postdoctoral Scholar Fellowship. H.W. was supported by “National Natural Science Foundation of China” (31471215), “Strategic Priority Research Program” of the Chinese Academy of Sciences (XDA01010409) and the State 863 Project 2015AA020307. Due to length restrictions, we apologize to all the colleagues whose contribution we could not properly acknowledge in this review.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Wiles, M.V., Qin, W., Cheng, A.W. et al. CRISPR–Cas9-mediated genome editing and guide RNA design. Mamm Genome 26, 501–510 (2015). https://doi.org/10.1007/s00335-015-9565-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00335-015-9565-z