
Abstract
Bone-eating worms of the genus Osedax (Annelida, Siboglinidae) form unique holobionts (functional entity comprising host and associated microbiota), highly adapted to inhabit bone tissue of marine vertebrates. These gutless worms have developed nutritional symbioses housing intracellular, horizontally acquired, heterotrophic bacteria hypothesised to harness nutrients from organic compounds, sequestered within the bone. Despite previous efforts, critical mechanisms mediating activity and acquisition of diverse bacterial assemblages remain unclear. Using 16S rRNA amplicon sequencing, we performed detailed taxonomic and predicted functional analyses shedding light on the microbial communities of two shallow-water Osedax species (Osedax deceptionensis and Osedax ‘mediterranea’) from contrasting habitats (Antarctic and Mediterranean Sea), in two tissue types (roots and palps). Comparative assessments between host species revealed distinct microbial assemblages whilst, within host species and body tissue, relative symbiont frequencies retained high variability. We reported relatively high abundances of microbes previously classified as primary endosymbionts, Ribotype 1 (order Oceanospirillales), and diverse likely secondary epibionts warranting further exploration as recurrent Osedax associates. Surprisingly, O. ‘mediterranea’ exhibited relatively low abundance of Oceanospirillales, but increased abundance of other potentially hydrocarbon degrading bacteria from the family Alteromonadaceae. We hypothesise the presence of functionally similar, non-Oceanospirillales primary endosymbionts within O. ‘mediterranea’. Functional metagenomic profiling (using 16S rRNA sequences) predicted broad metabolic capabilities, encompassing relatively large abundances of genes associated with amino acid metabolism. Comparative analyses between host body tissue communities highlighted several genes potentially providing critical functions to the Osedax host or that confer adaptations for intracellular life, housed within bone embedded host root tissues.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Unique, specialist adaptations are critical for the survival of diverse organisms within nutrient limited, competitive marine zones. The death and fall of large marine mammals can bring temporary and successional oases within such environments, as macrofaunal communities compete to exploit these large influxes of organic matter (Smith and Baco 2003). One such specialist, Osedax (Annelida, Siboglinidae) (Goffredi et al. 2004; Rouse et al. 2004) have successfully colonised these unusual habitats, feeding and living within vertebrate bones from shallow and deep waters of the Pacific, Atlantic and Antarctic Oceans, and even the Mediterranean Sea (see Fujiwara et al. 2019). Commonly known as the ‘bone-eating’ worms, free-swimming Osedax larvae settle upon vertebrate carcasses and develop a distinct network of vascularised ‘roots’ which proliferate into the bone (Goffredi et al. 2005, 2007; Miyamoto et al. 2013). Without a mouth or gut, Osedax rely upon a community of endosymbionts to supply nutrition through the degradation of high energy compounds (Goffredi et al. 2005, 2007; Rouse et al. 2009; Taboada et al. 2015), including collagen and possibly lipids, made available by enzymes secreted from within the roots (Tresguerres et al. 2013; Miyamoto et al. 2017). However, the mechanisms by which endosymbionts acquire such nutrients remain to be fully understood (Goffredi et al. 2014). We now know of 26 Osedax species (Rouse et al. 2004, 2008, 2015; Glover et al. 2005, 2013; Fujikura et al. 2006; Braby et al. 2007; Fujiwara et al. 2007; Goffredi et al. 2007; Vrijenhoek et al. 2008, 2009; Amon et al. 2014; Taboada et al. 2015; Sumida et al. 2016), of which 15 were formally described within the last few years (Rouse et al. 2018; Fujiwara et al. 2019), and a few, as yet remain undescribed, demonstrating the increased knowledge of Osedax prevalence and diversity.
Since their discovery in Monterey Bay, California (Rouse et al. 2004), the unique relationship of Osedax and its symbionts have received much attention. Female Osedax house primary endosymbionts (intracellular symbionts) from the order Oceanospirillales (class Gammaproteobacteria) within specialised bacteriocytes of their interior root tissues, located close to the ovisac (Goffredi et al. 2005). Conversely, with the exception of Osedax priapus (Rouse et al. 2015), paedomorphic dwarf males are developmentally arrested and live as harems within the female’s tube, with no endosymbiotic bacteria ever documented within them (Rouse et al. 2004, 2008). Pre-settling Osedax larvae and gametes are aposymbiotic, strongly indicating horizontal acquisition (Goffredi et al. 2007; Rouse et al. 2009; Verna et al. 2010). Indeed, consistent with horizontally acquired communities (Bright and Bulgheresi 2010), Osedax exhibit diverse endosymbiont assemblages (Goffredi et al. 2005, 2007; Fujikura et al. 2006), particularly within shallow-water hosts (30 m and 125 m; Verna et al. 2010). To date a total of 25 endosymbiont ribospecies have been reported (Goffredi et al. 2004; Rouse et al. 2004; Glover et al. 2005; Fujikura et al. 2006; Braby et al. 2007; Vrijenhoek et al. 2009; Salathé and Vrijenhoek 2012; Taboada et al. 2015), with up to eight distinct endosymbiont clusters (91.4–98.9% similarity between clusters) occurring within a single species (Osedax mucofloris; Verna et al. 2010).
The extent of host Osedax microbial diversity is yet to be fully understood. One popular hypothesis suggests diverse assemblages result from repeated microbial uptake throughout Osedax root development (Verna et al. 2010; Salathé and Vrijenhoek 2012) and their opportunistic, yet selective, mechanisms of acquisition (Salathé and Vrijenhoek 2012; Goffredi et al. 2014). Nonetheless, we are unlikely to fully comprehend such mechanisms without broader assessments encompassing the total Osedax holobiont community (Bosch and McFall-Ngai 2011; Bordenstein and Theis 2015) of which considerably less is known. In addition to primary endosymbionts, several secondary (extracellular) bacterial symbionts belonging to Alphaproteobacteria, Deltaproteobacteria, Epsilonbacteraeota, and Bacteroidetes have also repeatedly been observed from shallow- and deep-water species across a large geographical range (Fujikura et al. 2006; Goffredi et al. 2007; Verna et al. 2010). Despite this consistent association, there are currently no known potential roles of secondary symbionts. Indeed, nine of the above-mentioned studies were conducted ten or more years ago, since which time genomic analysis tools have significantly improved, allowing more detailed assessments. The diverse and complex nature of Osedax microbial relationships have become increasingly apparent. However, key questions regarding Osedax primary and secondary symbiont evolution, functional capacity and mode of acquisition remain unknown and warrant extensive exploration (Verna et al. 2010; Salathé and Vrijenhoek 2012; Goffredi et al. 2014).
Here, we conduct the first holistic assessment of the Osedax holobiont using 16S rRNA amplicon sequencing, to shed light on total microbial community structure and function for two geographically separated shallow-water Osedax species, namely, Osedax deceptionensis from Deception Island, Antarctica and an undescribed species located in the Mediterranean, referred to as Osedax ‘mediterranea’ (Taboada et al. 2015). Unlike previous descriptions, our analyses focus not only on the primary endosymbionts Oceanospirillales but encompass broad scale descriptions of the total holobiont community. We provide detailed microbial community descriptions and predictive functional profiles using PICRUSt2, making comparative analyses between two host species for root samples, and between root and palp body tissues for O. deceptionensis. To the best of our knowledge, Osedax palp tissues are not considered to house primary endosymbionts, and thus provide comparative context for our root tissue analyses. Here we elucidate primary and secondary symbiont structures, detailing microbial diversity at the species and individual level. We aim to provide an initial assessment that may facilitate further investigation into the nature of a microbial partnership, which has likely contributed significantly to the success for members of this genus.
Methods
Sample collection, DNA extraction and amplification
Individual O. deceptionensis and O. ‘mediterranea’ were collected and processed for 16S rRNA amplicon sequencing using DNA extractions from the study by Taboada et al. (2015). Briefly, O. deceptionensis individuals were obtained from the caudal fin of a de-fleshed common minke whale (Balaenoptera acutorostrata), placed at ca. 10 m depth on the seabed of Whalers Bay at Port Foster, Deception Island (South Shetland Islands, Antarctica). All bones were collected using SCUBA in January 2013 after approximately a year of deployment, and there after maintained in filtered sea water (0.22 μm) at ambient temperature (0–5 °C) during a maximum of 5 days whilst bones were observed to investigate the occurrence of Osedax; sea water temperature at the collection site was 0–1 °C. Individuals of O. ‘mediterranea’ were found in 2014 in association with a minke whale bone experimentally deployed at 53 m at the head of the Blanes submarine canyon (NW Mediterranean; 41°40′ 15.5″ N 2°53′ 23.28″ E) after 6 months of deployment, and were kept alive for 5 days in a container with filtered sea water at ambient temperature (20 °C); sea water temperature at the collection site was 17 °C.
For DNA extraction, individuals of O. deceptionensis and O. ‘mediterranea’ were preserved in 96% ethanol and stored at − 20 °C until further processing. Total DNA was extracted using the DNeasy Blood & Tissue Kit (Qiagen) following the manufacturer’s instructions. A total of 7 different individuals of O. deceptionensis (comprising 4 palps and 3 roots) and 2 two different individuals of O. ‘mediterranea’ (all roots) were processed (Table 1). From each individual either palps or roots were sampled, but not both tissue parts, to ensure independent random sampling. About 272 bp amplicon of the 16S rRNA gene was generated using general bacterial primers 515F-Y (Parada et al. 2016) and 806R (Apprill et al. 2015). We used the PCRBIO HiFi Polymerase (PCR Biosystems Ltd, UK) under the following conditions: 95 °C for 3 min, followed by 25 cycles of 95 °C for 20 s, 60 °C for 20 s and 72 °C for 30 s, after which a final elongation step at 72 °C for 5 min was performed. After amplification, PCR products were checked in 1% agarose gel to determine the success of amplification and the relative intensity of bands. PCR products were purified with Agencourt AMPure XP Beads (Beckman Coulter Inc., USA), and libraries prepared with Nextera XT DNA Library Preparation Kit (Illumina Inc., USA). An equimolar pool of DNA was generated by normalising all samples at 4 nM for the sequencing. Next generation, paired-end sequencing was performed at the NHM (https://www.nhm.ac.uk/) on an Illumina MiSeq device.
Analysis of prokaryote 16S rRNA gene amplicon sequences
Raw 16S rRNA amplicon reads were imported for processing into Mothur version 1.41.3 (Schloss et al. 2009). As a quality filter, we removed any read with ambiguous bases, homopolymers longer than 15 nucleotides, or that did not pass the assembled quality score test (average quality score 25). Sequences were pre-clustered with UNOISE3 (Edgar 2016) implemented in Mothur, allowing 1 difference for every 100 bp. This step infers amplicon sequence variants (ASVs), without imposing any arbitrary threshold (Callahan et al. 2017). Remaining singletons were removed at this point. Chimeric sequences were identified with UCHIME (Edgar et al. 2011), referenced to the SILVA database, and discarded. Taxonomies were assigned by classifying sequences with the NR SILVA database v.132 as a reference. Any residual sequences identified as chloroplasts, mitochondria, or eukaryote were excluded from further downstream analysis (1.14% of total derived sequences). Sequences were imported as ASVs into R v.3.5.2 for downstream analysis described below.
We assessed the microbial community structure within our samples using the R package ‘Vegan’ and based on datasets rarefied to the smallest sampling effort. Datasets were filtered, removing any ASV present only in one sample. We visualised variation in community structure amongst samples using both a heat map of the rarefied counts of the 100 most abundant ASVs and a hierarchical clustering dendrogram based on Euclidian distances and average linkage with the R package pheatmap (Kolde 2015). For measures of Alpha diversity, we calculated Chao1, Shannon, and Simpson indices. To evaluate alpha diversity measures between species and body tissues we performed analysis of variance on two separate linear models. Beta diversity was estimated on rarefied counts based on Bray–Curtis dissimilarity and unweighted UniFrac distance separately. Non-metric multidimensional scaling (NMDS) was used to obtain the ordination of community similarity. Samples were compared by performing two separate permutational multivariate analyses of variance (using the adonis function of the R package vegan), comparing roots and palps of O. deceptionensis, and roots of O. ‘mediterranea’ and O. deceptionensis. For analysis of differential abundance, we used the software edgeR (Robinson et al. 2010; McCarthy et al. 2012). Samples were compared by performing two separate exact tests comparing roots and palps of O. deceptionensis, and roots of O. ‘mediterranea’ and O. deceptionensis as before.
Sequences classified as order Oceanospirillales were assessed for phylogenetic analysis. These sequences were imported into Geneious Prime 2019.1 (https://www.geneious.com) and aligned using MAFFT multiple alignment against 168 previously aligned Osedax endosymbiont sequences (Taboada et al. 2015) and some sequences recently published by Taboada et al. (2020). Bayesian inference (BI) analyses were run in MrBayes v.3.2.2 (Ronquist et al. 2012) in the NHMUK cluster, using the most appropriate evolutionary model (GTR + I + G) and consisted of two chains run for 10 million generations, sampled every 1000 generations, and a 25% burn-in. Results were visualised using iTOL 5.5 (Ciccarelli et al. 2006; Letunic and Bork 2019).
Gene content predictions from the 16S rRNA SSU gene
Metagenomic functional profiles of Osedax microbial communities were estimated using PICRUSt2 based on the filtered ASVs (Douglas et al. preprint). PICRUSt2 predicts metagenomic functional abundances based on 16S rRNA data. Briefly, in PICRUSt2 ASV sequences are first aligned (HMMER) and placed within a 16S rRNA reference tree (EPA-NG: Barbera et al. 2019; gappa: Czech and Stamatakis 2019), before implementing the R package, Caster (Louca and Doebeli 2018) to predict gene family abundances (i.e. copy number) for each ASV. Metagenomes are then inferred by dividing the read depth per ASV by predicted 16S rRNA copy numbers and multiplied by predicted gene family copy number for each ASV. Lastly, MetaCyc (Caspi et al. 2014) pathway-level abundances were inferred based on Enzyme Commission number (EC) abundances with MinPath (Ye and Doak 2009). We implemented the script, ‘picrust2_pipeline.py’ following online documentation, https://github.com/picrust/picrust2. ASVs above the nearest-sequenced taxon index (NSTI) cut-off of 2.0 were subsequently removed from further downstream analysis. Predictive functional abundance tables were classified by MetaCyc metabolic pathways (Caspi et al. 2014), EC enzymes, and Kyoto encyclopaedia of genes and genomes (KEGG). Relative abundances down to the sub pathway level were inferred using BURRITO (McNally et al. 2018) from predicted KEGG ortholog (KO) abundances and KEGG BRITE hierarchy (Kanehisa and Goto 2000). Comparative assessments between palp (O. deceptionensis) and root (O. ‘mediterranea’ and O. deceptionensis) tissues were performed on datasets first transformed by relative abundance, before using Welch’s two-sided T-test in the software STAMP (Parks et al. 2014). Differentially abundant pathways, enzymes and KEGG orthologs were visualised with extended error bar plots in STAMP.
Results
Initially we identified 6,381 bacterial ASVs from 465,118 sequences across all samples (ASV sequences, abundances, and taxonomy available online at https://doi.org/10.6084/m9.figshare.12611735). After filtering, we retained a total of 1,551 ASVs from 422,062 sequences across all samples (Table 1). Of these, 1,144 ASVs were identified within O. deceptionensis roots, 923 within O. deceptionensis palps, and 432 within O. ‘mediterranea’ roots (Table 1). Except for individuals, DR21 and MR1, rarefaction curves based on total sequence counts displayed asymptotic behaviour, indicating sequencing depth was sufficient for representative read counts (Online Resource 1). Microbial communities within our samples were distinct between Osedax species and body tissue as displayed by the dendrogram (Fig. 1). In fact, we identified only 3 core ASVs that were present in all individuals (avg: 1.8% of total relative abundance (ra.); Online Resource 2a). A total of 14 core ASVs were present in all root samples from both species (avg: 15% of total root ra.) and 16 core ASVs were present in all root and palp samples from O. deceptionensis (avg: 19% of total O. deceptionensis ra.).

Heatmap of the 100 most abundant ASVs based on rarefied counts. Horizontal orange and blue bars indicate samples belonging to species O. ‘mediterranea’ and O. deceptionensis, respectively. Horizontal light grey and dark grey bars indicate samples from root and palp tissues, respectively. Scale bar indicates log2 counts per million (cpm). Hierarchical clustering dendrogram is based on Euclidian distances and average linkage. DR, O. deceptionensis roots; MR, O. ‘mediterranea’ roots; DP, O. deceptionensis palps
Alpha diversity indices were calculated over rarefied datasets (i.e. 26,947 sequences). The greatest diversity was found amongst O. deceptionensis roots (Shannon H = 4.4); however, there was no statistically significant difference amongst roots of both species (ANOVA; p > 0.05; Online Resource 3). Conversely species diversity was lowest within O. deceptionensis palps (Table 1), but again there was no statistically significant difference between palp and root diversity.
Our assessment of beta diversity based on Bray Curtis dissimilarity and Unifrac distance measures detected distinct microbial community compositions between O. deceptionensis palps and roots only (Fig. 2; adonis: df = 1, 6; p = 0.039; and Online Resource 4; Unifrac p = 0.044). No significant differences were found between the roots of both species (adonis, df = 1, 3; p = 0.1), probably due to the low number of samples available (2 samples from O. ‘mediterranea’, and 3 from O. deceptionensis).

NMDS of beta diversity between O. deceptionensis roots, O. deceptionensis palps and O. ‘mediterranea’ roots. Bray–Curtis measures of dissimilarity estimated based on rarefied library sizes of the smallest sampling size. Osedax deceptionensis palps (DP) and roots (DR) samples exhibited significantly different microbial communities (adonis: p = 0.04)
Total microbial community composition was dominated by Proteobacteria (80% average relative abundance (ara.); Fig. 3a), specifically classes Gammaproteobacteria (54% ara.), followed by Alphaproteobacteria (13.5% ara.), Deltaproteobacteria (10% ara.), and Betaproteobacteria (2% ara.) (Fig. 3b). Other prominent phyla within the total microbial community included Epsilonbacteraeota (8.5% ara.), Bacteroidetes (6.4% ara.), Actinobacteria (1.2% ara.), Kiritimatiellaeota (0.7% ara.), and Firmicutes (0.6% ara.) (Fig. 3a). Within Osedax individuals Gammaproteobacteria was consistently the most abundant. Only in the two most diverse individuals (O. deceptionensis-DR21 and O. mediterranea-MR1), where sequencing depth was the lowest, abundance of Proteobacteria dropped below 80% ara. (with Gammaproteobacteria comprising, 21% and 22% ara., respectively; Fig. 3a), whilst relative abundance of Epsilonbacteraeota was much greater than within other individuals (21% and 22% ara., respectively; Fig. 3a).

Individual O. deceptionensis and O. ‘mediterranea’ taxonomic microbial composition shown by relative abundance. a Phylum composition for phyla of at least 0.5% total relative abundance. b Class composition for members of Proteobacteria and two classes with the highest relative abundance outside Proteobacteria. c Order composition for orders of at least 4% total relative abundance within one of O. deceptionensis roots, O. deceptionensis palps or O. ‘mediterranea’ roots. Letters in brackets indicate phylum (upper case letter) and class (lower case letter)
Comparing total root and palp samples separately, we observed much lower average abundance of classes, such as Deltaproteobacteria and Bacteroidia within palp samples (4.5% and 0.9% ara.) compared to root samples (O. deceptionensis, 16.2% and 12.3% ara.; O. ‘mediterranea’, 12.6% and 8.4% ara.; Fig. 3b). Conversely, abundance of Alphaproteobacteria was lower in O. deceptionensis root samples (2.9% ara.) than O. deceptionensis palp (17.3% ara.) and O. ‘mediterranea’ root samples (22.3% ara.).
We identified 10 dominant ASVs present at relative abundances of 5% or greater in at least one of O. deceptionensis roots, O. deceptionensis palps or O. ‘mediterranea’ roots (Fig. 4; Online Resource 2b). Six of these belonged to three orders of class Gammaproteobacteria, four within the order Oceanospirillales, (ASV1903 and ASV5088 Profundimonas; ASV960 Oleispira; ASV1445 Kangiellaceae uncultured), and the other two within the orders, Alteromonadales (ASV944 Alkalimarinus but see below) and Thiomicrospirales (ASV5513 cluster SUP05 of family Thioglobaceae). The remaining ASVs belonged to the order Campylobacterales (ASV6298 Arcobacter; ASV140 Sulfurospirillum) and unclassified order(s) of Alphaproteobacteria (ASV4230 and ASV4953). Dominant ASVs were largely specific to Osedax species and body region. Additionally, except for ASV1903, none of these 10 dominant ASVs were present in abundances greater than 1% within more than one of the samples types (i.e. O. deceptionensis roots, O. deceptionensis palps or O. ‘mediterranea’ roots), and only three (ASV944, ASV960, and ASV1445) were also present within the identified core root microbiota. However, ASV1903 was abundant within O. deceptionensis roots and palps.

Relative abundance of ASVs above 1% abundance within at least one of O. deceptionensis roots, O. deceptionensis palps or O. ‘mediterranea’ roots. Bubble sizes are proportional to the relative abundance of an ASV within an individual and are coloured according to their order, as assigned using NR SILVA reference database. ASVs displayed on the y-axis are denoted first by their associated family and genus, followed by their unique ASV number. ASV names displayed in blue are the 10 most abundant ASVs (present in abundances of at least 5% within one of O. deceptionensis roots, O. deceptionensis palps or O. ‘mediterranea’ roots). DR, O. deceptionensis roots; MR, O. ‘mediterranea’ Roots; DP, O. deceptionensis palps
Osedax deceptionensis root microbial communities were largely dominated by only two ASVs, belonging to the order Oceanospirillales: ASV1903 comprising 29% ara. and ASV960, comprising 9.1% ara. (Fig. 4). The relative frequencies of these two ASVs were not consistent between individuals (Fig. 4). Remaining O. deceptionensis root taxa composition comprised many less abundant ASVs including ASV3078 (3.3% ara.) and ASV568 (3.0% ara.) within Bacteroidetes, and ASV1442 (3.1% ara.) within class Deltaproteobacteria. These generally less dominant ASVs were observed in much greater relative abundances within the most diverse individual, O. deceptionensis-DR21 (Fig. 4). Conversely, O. deceptionensis palp microbial communities consisted of several dominant ASVs, in which ASV5513 (assigned to the family Thioglobaceae) was most abundant, with 19.8% ara. (Fig. 4). ASV5513 was largely unique to palp samples, only found outside the palps in very low numbers of O. deceptionensis-DR21 (Fig. 4). Also abundant within O. deceptionensis palps were ASV1445 (16.9% ara.) and ASV1903 (12.6% ara.) belonging to the order Oceanospirillales, ASV4230 (13.3% ara.) an unclassified Alphaproteobacterium, and ASV6298 (5% ara.) belonging to Campylobacterales (Fig. 4). Within O. ‘mediterranea’ root microbial communities, order Oceanospirillales was present only at 7.1% ara., of which ASV5088 represented 6.8% ara. (Figs. 3c, 4). Instead, the most dominant ASV was ASV944 (Gammaproteobacteria), which comprised 21.3% ara. We observed high similarity of ASV944 to Alkalimarinus sediminis (99.6% identity), a recently described species within family Alteromonadaceae that clusters separately but shares similarity with Marinobacter (92.8% identity; Zhao et al. 2015). Microbial communities of O. ‘mediterranea’ roots also included high average relative abundances of ASV4953 (9.6% ara.), an unclassified Alphaproteobacterium, and ASV140 (8.9% ara.v), belonging to order Campylobacterales (Fig. 4).
Differential abundance analysis of counts in edgeR, highlighted 19 differentially abundant ASVs between host species (O, deceptionensis roots vs. O. ‘mediterranea’ roots) and 30 between body tissue (palp vs. roots of O. deceptionensis) (p < 0.01; Online Resource 2c). We observed the highest log fold change values between host species. These ASVs included, significantly greater abundance of ASV1903 and ASV960 (order Oceanospirillales) within O. deceptionensis roots, whilst abundance of ASV4953 and ASV518 (class Alphaproteobacteria) were significantly greater within O. ‘mediterranea’ roots (Online Resource 2c).
Metagenomic predictive functional profiling
All 1,551 ASVs were initially included in the PICRUSt2 analysis. After removal of 802 ASVs above the NSTI cut-off 2.0, we had a dataset of 749 ASVs. A total of, 4011 KEGG orthologs (KO), 1,327 enzymes, and 285 metabolic pathways were predicted from these ASVs. There were 226 KO uncharacterized proteins. A comparison of KEGG orthologs predicted from our samples revealed highly comparable functions to those identified within dominant Osedax symbionts, Rs1 and Rs2, as well as the closest described free-living relative Neptumonas japonica (Goffredi et al. 2014; Online Resource 5a). Within our samples, the super pathway amino acid metabolism comprised 25% of total abundance for metabolism classified pathways (Table 2). Specifically, amino acid metabolism was comprised most abundantly of the sub pathway, Phenylalanine, tyrosine and tryptophan biosynthesis (5.4% of total metabolism; Table 2), but also included metabolism of sulphur containing (Cysteine and methionine), and collagen-derived amino acids (Glycine, Proline, Hydroxyproline) (Online Resource 5b). Metagenomic predictions revealed high metabolic flexibility in general, with the potential to metabolise monosaccharides, disaccharides, amino sugars, nucleotide sugars, fatty acids, and organic acids. We identified a total of 252 ABC transporters, including those for oligopeptides (oppABCDF), dipeptides (dppABCDF), nickel (nikABCDE), vitamin B12 (btuCDF), zinc (znuABC), iron (afuABC), and multiple amino acids (aotQ, argT, artIJMPQ, glnHPQ, hisJMPQ, livGFHMK, metINQ, proVWX, tauABC, tcyBCKLM, thiBPQ, aapJMPQ) (Online Resource 5c).
Comparative analysis between root and palp tissue samples highlighted differential abundance of 11 metabolic pathways, 54 enzymes and, 217 KEGG orthologs (Welch’s t test, p < 0.05; Online Resource 6). We identified several functions significantly more abundant within root samples that may shed light on the adaptive traits of root tissue symbionts. These included, urea cycling (urease; EC:3.5.1.5 & nikABCD), vitamin biosynthesis (biotin biosynthesis II; PWY-5005), sulphur detoxification (superpathway of sulphur oxidation; PWY-5304), and intracellular host infection of free-living symbionts (clfAB, fnbAB, sdrCDE) (Online Resource 5d & e).
Phylogenetic assessment of ASV’s belonging to order Oceanospirillales
In total we identified 100 ASVs belonging to 8 families within the order Oceanospirillales. Of these, 82 were identified within O. deceptionensis roots, 16 within O. ‘mediterranea’ roots, and 64 within O. deceptionensis palps (Fig. 5). We identified three core Oceanospirillales ASVs shared across all palp and root samples of O. deceptionensis (ASV960, ASV1445, ASV2484), but none shared between all root samples of O. deceptionensis and O. ‘mediterranea’ (Online Resource 2a).

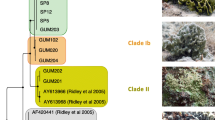
Phylogenetic tree based on Bayesian inference analysis of 100 sequences classified as order Oceanospirillales aligned against 168 previously aligned Osedax endosymbiont sequences (Taboada et al. 2015, 2020) . Branches and text are coloured according to the presence of each sequence within respective samples, Blue: O. deceptionensis palp and root samples, Purple: O. deceptionensis palp samples only, Orange: O. ‘mediterranea’ root samples, Black: Sequences used in Taboada et al. (2015, 2020). Stars indicate the most abundant Oceanospirillales sequences for each sample type, O. deceptionensis roots (blue), O. deceptionensis palps (purple), and O. ‘mediterranea’ roots (orange). Tree based on evolutionary model (GTR + I + G) and two chains run for 10 million generations, sampled every 1000 generations, and a 25% burn-in
Oceanospirillales abundance within O. deceptionensis and O. ‘mediterranea’ roots was dominated by ASV1903 and ASV5088, respectively. Both clustered within the monophyletic lineage, Ribotype 1 (distinguished following Salathé and Vrijenhoek 2012) and most closely associated with Nitrincolaceae Profundimonas (formerly Oceanospirillaceae; Fig. 5). Both host species also harboured many low abundant ASVs closely associated to either Nitrincolaceae Profundimonas or Nitrincolaceae unclassified. The second most abundant within O. deceptionensis roots, ASV960 most closely associated with Saccharospirillaceae Oleispira, but did not cluster significantly within any described Osedax primary endosymbiont ribotypes. Conversely, O. ‘mediterranea’ roots comprised no other dominant Oceanospirillales ASVs. Within O. deceptionensis palps, Oceanospirillales was dominated by ASV1445, belonging to the family Kangiellaceae and did not cluster with any known Osedax endosymbiont Ribotype.
Discussion
Microbial community structure
The shallow-water annelids O. deceptionensis and O. ‘mediterranea’ house diverse primary and secondary microbial assemblages dominated by Proteobacteria (Fig. 3a). Within this bacterial phylum, class Gammaproteobacteria was predominant, distantly followed by Alphaproteobacteria, Deltaproteobacteria, and other non-Proteobacteria groups such as Epsilonbacteraeota and Bacteroidetes (Fig. 3). Such high prevalence of Gammaproteobacteria is unsurprising considering its global dominance within other Osedax and closely related Vestimentiferan tube worms (Thornhill et al. 2008). Conversely, our assessments of species level (i.e. ASVs) diversity revealed highly variable assemblages, both within and between host species (Fig. 4). Individual O. deceptionensis and O. ‘mediterranea’ were characterised by a limited number of dominant ASVs in distinct frequencies, and a large pool of diverse, low abundant ASVs, comprising only a minimal core community (Fig. 4; Online Resource 2a). The taxonomic composition and diversity we observed here is largely consistent with previous Osedax microbial descriptions such as, O. roseus and Osedax sp. ‘yellow collar’ (Goffredi et al. 2007; Osedax lehmani in Rouse et al. 2018). Conversely, other Siboglinidae including Riftia and Tevnia, housing chemotrophic bacteria, display much less diversity, harbouring only a single dominant symbiont phylotype, Candidatus Endoriftia persephone, despite also acquiring their symbionts horizontally within highly disparate environmental extremes (Edwards and Nelson 1991; Di Meo et al. 2000; Gardebrecht et al. 2012; Forget et al. 2015).
Individual Osedax microbial community compositions were diverse, yet distinct between host species and body tissue (Figs. 3, 4). Within O. deceptionensis root communities, consistent with all other described Osedax microbiome assemblages (Goffredi et al 2005; 2007; Verna et al. 2010; Salathé and Vrijenhoek 2012), were predominantly dominated by primary endosymbionts Oceanospirillales, with the exception of the most diverse individual DR21 (Fig. 3c), which may be a reflection of the poorer sampling depth for this individual. The most abundant symbiont, ASV1903 (Profundimonas) lies within family Nitrincolaceae, known to comprise other primary Osedax endosymbionts (Goffredi et al. 2007). Additionally, ASV1903 most closely associated with Ribotype 1, a monophyletic lineage that globally dominates Osedax endosymbionts (Fig. 5; Salathé and Vrijenhoek 2012) and was the most abundant ASV within O. deceptionensis (Fig. 4), indicating it may be a primary endosymbiont involved in nutrient acquisition. The second most abundant symbiont, ASV960 (Oleispira) belongs to the known Osedax endosymbiont family Oceanospirillales, and thus is also a likely functionally significant endosymbiont, though its ribotype placement could not be supported in our phylogenetic tree (Fig. 5). Indeed, we do note the limitations from using relatively short length 16S rRNA markers and the small sample size of this study, when interpreting such results. All root samples of O. deceptionensis harboured both ASV1903 and ASV960; however, their relative abundances of each were distinct (Fig. 4). Despite close spatial and temporal proximity of the O. deceptionensis individuals (they were collected from the same bone and at the same time of the year), relative abundance of ASV960 was much greater (on par with ASV1903) within DR8 than either DR13 or DR21, providing insight into the plasticity of Osedax endosymbiont assemblages.
Little is known regarding the processes by which Osedax horizontally acquire their primary endosymbionts; however, their characteristically diverse assemblages may provide some insight. Unlike other Siboglinidae, Osedax likely acquire their endosymbionts repeatedly during root tissue proliferation, enabling uptake of variable endosymbionts through multiple mechanisms (Goffredi et al. 2007; Verna et al. 2010; Salathe and Vrijenhoek 2012). For instance, the temporally dynamic microbial assemblages within associated whale fall sediments (Goffredi et al. 2008a, b) may facilitate acquisition of distinct lineages through time (Verna et al. 2010; Salathé and Vrijenhoek 2012; Goffredi et al. 2014). That said, here we report distinct frequencies of potential endosymbionts, within individuals of O. deceptionensis and O. ‘mediterranea’ despite collection from a single time point. Alternatively, diverse communities may arise from small-scale variability within the bone-associated microbiota. Verna et al. (2010) hypothesised that a lack of disturbance enables free-living endosymbiont populations within the bone to form structured, clonal growth. Subsequently, because of the large available free-living symbiont populations, any given Osedax individual may acquire a distinct endosymbiont assemblage to that of its neighbour (Verna et al. 2010). Indeed, in their assessment of 20 Osedax mucofloris individuals, Verna et al. (2010) found host individual explained the majority (68%) of observed endosymbiont diversity for this shallow-water Osedax species. Assessment of O. deceptionensis and O. ‘mediterranea’ supports the hypothesis that, at least for shallow-water species, acquisition is not random but selectivity encompasses broad ribospecies diversity through opportunistic acquisition of the best adapted, free-living bacteria accessible within an individuals’ immediate environment (Salathé and Vrijenhoek 2012; Goffredi et al. 2014).
Palp tissues within O. deceptionensis, were less diverse and dominated by unique ASVs, only present in very low numbers within other root tissues. Specifically, these included ASV5513 (Thioglobaceae), most closely related with sulphur oxidising, thioautotrophic endosymbionts of the cold seep mussel Idas sp. (identity 98.8%) which inhabits sunken wood and whale bones (Duperron et al. 2008). This was followed by Oceanospirillales, ASV1445 (Kangiellaceae), and ASV4230 (unclassified Alphaproteobacteria), most closely related with Pseudovibrio sp. (identity 99.2%) (Fig. 3). It is unsurprising to detect such distinct root and palp microbial assemblages given their disparate environment and function (Huusgaard et al. 2012). Embedded within vertebrate bone, root tissues are highly active, exposed to the build-up of toxic sulphide and strong oxygen depletion, whilst palps facilitate ventilation over large diffusive surfaces in the water column (Huusgaard et al. 2012). Osedax palp microbial communities have not previously been explored, and thus the potential functional significance of their ectosymbionts is currently unknown.
Microbial communities were distinct between O. ‘mediterranea’ and O. deceptionensis root tissues (Figs. 3c, 4). Within O. deceptionensis roots, ASVs belonging to the order Oceanospirillales comprised 40% relative abundance, but only 7% (6.8% ASV5088) within O. ‘mediterranea’, none of them being common to both species. Such low abundance of Oceanospirillales in O. ‘mediterranea’ is surprising given its critical role proposed for Osedax nutrient acquisition (Goffredi et al. 2007). If hosts progressively acquire endosymbionts throughout root growth (Verna et al. 2010; Salathé and Vrijenhoek 2012), low Oceanospirillales abundance might be a result of limited root proliferation. Alternatively, in a depth associated comparison of species sampled 1000 m or deeper with those sampled shallower than 500 m, Verna et al. (2010) demonstrated distinct depth-related Osedax microbial communities. Conversely, here depth disparity was not so distinct, with O. deceptionensis and O. ‘mediterranea’ sampled at 10 m and 53 m, respectively. Subsequently instead we suggest that the species’ distinct geographical regions likely exhibit a greater influence on their microbial community composition. Indeed, within warm Mediterranean waters, increased endosymbiont metabolic activity may reduce host minimal endosymbiont requirements for the host to sustain sufficient nutrition (Sarmento et al. 2010). However, primary endosymbiont acquisition and activity remains largely unclear (Goffredi et al. 2014). Furthermore, whilst abundance of Oceanospirillales was relatively low, abundance of family Alteromonadaceae (ASV944 Alkalimarinus and ASV5580 Alteromonas) was relatively high and observed only in limited numbers apart from O. ‘mediterranea’ (Fig. 4). Consequently, we suggest that O. ‘mediterranea’ may house additional primary endosymbionts, other than Oceanospirillales, capable of hydrocarbon degradation, and which may compensate for relatively low Oceanospirillales abundance. As in Oceanospirillales, family Alteromonadaceae, are obligate heterotrophs associated with nutrient rich environments, including marine snow (López-Pérez and Rodriguez-Valera 2014). Additionally, they possess large genomes containing several degradative genes and numerous members of the family, including several species of Marinobacter (96.8% identity to ASV944) and Alteromonas, have been described as hydrocarbon degrading (Jin et al. 2011; López-Pérez and Rodriguez-Valera 2014). Considering such attributes, ASV944 and ASV5580 may feasibly possess the required functionality for Osedax nutrient acquisition, indicating a potential role for non-Oceanospirillales symbionts. Even so, the placing of ASV944 warrants investigation, considering that its closest known relative, Alkalimarinus sediminis shares equally high identity scores with members of both Oceanospirillaceae and Alteromonadaceae (Zhao et al. 2015). Nevertheless, to further explore the functional significance of Alteromonadaceae, we must first determine whether they reside as intracellular endosymbionts. To the best of our knowledge, no free-living bacteria closely related with ASV944 and ASV5580 have previously been detected within associated whale fall sediments (Goffredi and Orphan 2010), or indeed within the microbial mats from which these O. ‘mediterranea’ individuals were sampled (Taboada et al. 2020). Thus, we suggest family Alteromonadaceae might not simply be a casual associate and suggest further exploration as primary endosymbionts within the shallow-water host O. ‘mediterranea’.
Osedax also house a rich community of secondary bacterial symbionts of which current knowledge is severely lacking. Within both root and palp tissues of O. deceptionensis and O. ‘mediterranea’ roots, we detected relatively high abundances of likely secondary symbionts belonging to Alphaproteobacteria, Deltaproteobacteria, Epsilonbacteraeota and Bacteroidetes (root tissues only Fig. 3). Whilst members of these, including families and genera detected here (Desulfobacteraceae, Desulfobulbaceae, Sulfurospirillum, Arcobacter, Sulfurimonas; Fig. 4), are strongly associated to surrounding whale fall sediments (Tringe et al. 2005; Goffredi and Orphan 2010; Taboada et al. 2020), they are notably recurrent within numerous spatially and temporally separated species of Osedax (Goffredi et al. 2005, 2007; Verna et al. 2010). We suspect these bacteria to be epibionts, since several closely related species have been detected across root and trunk surface tissues (Fujikura et al. 2006; Goffredi et al. 2007; Verna et al. 2010). Unusually, Fusobacteria, another recurrent secondary phylum (Goffredi et al. 2005, 2007; Verna et al. 2010), were only detected here within O. deceptionensis roots (Fig. 4). Currently there are no known functional roles of Osedax secondary symbionts. However, considering their consistent association within Osedax microbiota, we suggest investigating whether they are functionally significant as either beneficial, commensal, parasitic or competitive symbionts. Since Osedax are hypothesised to house dominant endosymbionts within core root structures and less abundant assemblages within their peripheral roots (Verna et al. 2010), visualising the spatial distribution of such bacteria may provide an initial proxy for their relative functional significance.
Microbial community functional profile
Metagenomic analysis (predicted from the 16S rRNA genes) of O. deceptionensis and O. ‘mediterranea’ microbial communities using PICRUSt2, revealed predicted functional profiles largely consistent with Osedax’s most dominant symbionts, Rs1 and Rs2 (Goffredi et al. 2014; Online Resource 5a). Similar to Rs1 and Rs2, our analysis indicated broad metabolic flexibility, encompassing metabolism of monosaccharides, disaccharides, amino sugars, nucleotide sugars, fatty acids and organic acids (Online Resource 5b). Additionally, PICRUSt2 analysis predicted large abundances of KEGG orthologs associated with the metabolism of amino acids. Specifically, amino acid metabolism accounted for 25% of the total abundance for metabolism associated pathways (Table 2). These included collagen-derived amino acids (glycine, proline, hydroxyproline), despite observing only limited abundances of collagenase. However, collagenase has been reported from root tissue samples (Goffredi et al. 2007), and indeed Miyamoto et al. (2017) identified proteases (mmp and ctsB genes) capable of collagen degradation, accessing peptides and other nutrients within root epidermal cells. Thus, our data support the hypothesis that Osedax endosymbionts metabolise collagen-derived amino acids or peptides, which are first liberated by the host (Goffredi et al. 2014; Miyamoto et al. 2017). Additionally, oligopeptide ABC transporters (oppABCDF), suggested to facilitate uptake of potentially collagen-derived peptides (Goffredi et al. 2014), were particularly abundant in our samples. The full cassette of dipeptide transporters (dppABCDF) was also present but much less abundant. Expression of SLC genes encoding oligopeptide and amino acid transport have also been reported within Osedax japonicus host root epidermis tissue (Miyamoto et al. 2017). Thus, this supports the hypothesis that host derived, small-peptides and amino acids from the collagenous bone matrix may indeed play a critical role in nutritional acquisition within the Osedax holobiont.
Comparisons of functional abundances between root and palp tissue microbial communities, highlighted several potential adaptations, specific to root tissue symbionts. Within root tissue samples we observed significantly greater prevalence of the urea cycle pathway (PWY-4984; Online Resource 5d). Considering, the urea cycle is a product of amino acid catabolism, and that root tissues house Osedax’s primary endosymbionts, this may provide further indication for amino acid metabolism as a dominant nutritional resource. Additionally, urease (EC:3.5.1.5, subunits ureABC and ureDEFG), a catalyst for the production of ammonia from urea (Varner 1960; Andrews et al. 1984), and transport proteins nikABCDE and nixA, for Nickle, of which urease is reliant on, were significantly more abundant within root tissue samples (Dixon et al. 1976; Blakeley and Zerner 1984; Online Resource 5e). The activity of urease is significant since the assimilation of ammonia (to glutamate and glutamine) facilitates nitrogen recycling, increasing amino acid turnover. PICRUSt2 analysis revealed the potential for de novo microbial synthesis of 6 essential (lysine, valine, leucine, isoleucine, phenylalanine, tryptophan) and three non-essential amino acids within our samples. We hypothesise that Osedax root microbial populations exhibit nitrogen cycling, enabling increased synthesis of amino acids that may be transferred and utilised by their host. Urea recycling has already been suggested in other marine invertebrates hosting microbial endosymbionts, such as in the tropical sponge Xestospongia testudinaria (Su et al. 2013). This mechanism was proposed as a potential source of nitrogen for the host to overcome living in habitats with limited nitrogen availability (Su et al. 2013). Additionally, the production of amino acids through urea recycling has been demonstrated within gut bacterial symbionts of the insect Melolontha hippocastani (Forest Cockchafer; Alonso-Pernas et al. 2017). Similarly, symbiont vitamin biosynthesis may also be critical for the host. For instance, the pathway for Biotin biosynthesis (PWY-5005) was significantly more prevalent within root tissues samples (Online Resource 5d). Biotin is an essential cofactor within cellular metabolism, but which animals cannot produce (Patton et al. 1996; Schneider and Lindqvist 2001) and leads us to speculate whether host Osedax acquire such essential vitamins from their symbionts.
Highly active root tissues are exposed to the build-up of toxic sulphide and strong oxygen depletion (Huusgaard et al. 2012), yet there has been no described mode of detoxification. Within root tissue samples, our analysis predicted significantly greater prevalence of the super pathway for sulphur oxidation (PWY-5304) and five sulphur metabolism classified orthologs, including the sulphite reductase flavoprotein (cysJ) for sulphur assimilation and cystathionine gamma-synthase metB, capable of essential amino acid synthesis for methionine (Ostrowski et al. 1989; Ravanel et al. 1998; Online Resource 5d & e). Consistent with our results, Waits et al. (2016) demonstrated the likely biochemical capacity of O. mucofloris to produce sulphur-binding haemoglobins (Hbs). They also showed that O. mucofloris possess Hb linkers with greater similarity to vestimentiferan siboglinids than sulphide-tolerant polychaetes. Indeed the use of sulphur containing amino acids for detoxification of sulphur has been a suggested mechanism within Riftia pachyptila, a closely related vestimentiferan siboglinid (Pruski et al. 1998). Interestingly, genes involved with metabolism of sulphur derived amino acids, cysteine and methionine, were particularly abundant within our samples and never reported, to our knowledge, in previous studies on Osedax microbes. Thus, investing in microbial mechanisms of sulphur detoxification is likely to be critical for the Osedax holobiont.
We also identified several genes that might be involved in facilitating the infection of free-living microbial symbionts intracellularly within the host (Online Resource 5e). Endosymbiont adaptions for intracellular life likely share analogy with those of intracellular pathogens. Comparative analysis revealed several Staphylococcus aureus associated genes that were significantly more abundant within root tissue samples. These included, serine-aspartic acid repeat proteins sdrCDE, clumping factors clfAB, and fibronectin-binding proteins fnbAB, enabling adhesion of S. aureus to host squamous epithelial and elastin cells, respectively (Corrigan et al. 2009; Roche et al. 2004; Online Resource 5e). Additionally, membrane damaging leukotoxins and haemolysins (hlg, hld and hlyII) associated with pathogenic host infection were also significantly more abundant in root tissue samples (Barrio et al. 2006; Vandenesch et al. 2012; Online Resource 5e). Similarly, within endosymbionts Rs1 and Rs2, Goffredi et al. (2014) also observed haemolysin homologs, encoding large surface exoproteins. Successful mechanisms for host infection are likely to be key for Osedax symbionts, considering their continual horizontal acquisition throughout host root growth (Goffredi et al. 2007; Verna et al. 2010; Salathe and Vrijenhoek 2012).
Conclusion
Broadly, the microbial communities of O. deceptionensis and O. ‘mediterranea’ share some key Osedax microbiome patterns described previously. These include, a dominance of Gammaproteobacteria, high relative abundance of Oceanospirillales within O. deceptionensis root tissues (ASV1903 which closely associated with Ribotype 1, and ASV960), and comparable functional predictions to those observed in Ribotype 1 and Ribotype 2 (Goffredi et al. 2014). Conversely, within O. ‘mediterranea’, abundance of Oceanospirillales was surprisingly low. Thus, we hypothesise that some Osedax species may in fact house other, non-Oceanospirillales functionally significant primary endosymbionts. Within O. ‘mediterranea’, we suggest members belonging to the family Alteromonadaceae as potential candidates.
Deeper assessment, at ASV level revealed a high degree of disparity between individuals, both between and within species, despite individuals of the same species having been sampled from close proximity on the bone matrix. Subsequently, our analyses provide insight to the extent of complexity that comprises the Osedax holobiont and additionally, emphasises the significant gap in the current understanding regarding mode of Osedax symbiont acquisition. Indeed, we document relatively high and consistent abundance of several microbes (including orders Campylobacterales, Desulfobacterales, and Bacteroidales) that we suspect to be more than simply transient, and hence merit further investigation. To date, Osedax microbiome research has largely focussed on primary endosymbionts belonging to order Oceanospirillales. However, the observable holobiont and its functioning is a result of the total nested ecosystem interactions of all microbial members (whether commensal, parasitic, or mutualistic), and thus we stress the importance of such holistic approach in future Osedax holobiont research.
Data availability
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, BioProject Accession Number PRJNA644051, sample accession numbers SAMN15446574 to SAMN15446583, raw 16S rRNA sequences, absolute abundance, relative abundance and taxonomical assignments online at https://doi.org/10.6084/m9.figshare.12611735.
References
Alonso-Pernas P, Bartram S, Arias-Cordero EM, Novoselov AL, Halty-deLeon L, Shao YQ, Boland W (2017) in vivo isotopic labeling of symbiotic bacteria involved in cellulose degradation and nitrogen recycling within the gut of the forest cockchafer (Melolontha hippocastani). Front Microbiol 8:1970. https://doi.org/10.3389/fmicb.2017.01970
Amon DJ, Wiklund H, Dahlgren TG, Copley JT, Smith CR, Jamieson AJ, Glover AG (2014) Molecular taxonomy of Osedax (Annelida: Siboglinidae) in the Southern Ocean. Zoolog Scr 43:1–13. https://doi.org/10.1111/zsc.12057
Andrews RK, Blakeley RL, Zerner B (1984) Urea and urease. In: Eichhorn GL, Marzilli LG (eds) Advances in inorganic biochemistry, vol 6. Elsevier Science Publishing Inc., New York, pp 245–283
Apprill A, McNally S, Parsons R, Weber L (2015) Minor revision to V4 region SSU rRNA 806R gene primer greatly increases detection of SAR11 bacterioplankton. Aquat Microb Ecol 75:129–137. https://doi.org/10.3354/ame01753
Barbera P, Kozlov AM, Czech L, Morel B, Darriba D, Flouri T, Stamatakis A (2019) EPA-ng: massively parallel evolutionary placement of genetic sequences. Syst Biol 68:365–369. https://doi.org/10.1093/sysbio/syy054
Barrio MB, Rainard P, Prevost G (2006) LukM/LukF'-PV is the most active Staphylococcus aureus leukotoxin on bovine neutrophils. Microbes Infect 8:2068–2074
Blakeley RL, Zerner B (1984) Jack bean urease: the first nickel enzyme. J Mol Catal 23:263–292. https://doi.org/10.1016/0304-5102(84)80014-0
Bordenstein SR, Theis KR (2015) Host biology in light of the microbiome: ten principles of holobionts and hologenomes. PLoS Biol 13:e1002226. https://doi.org/10.1371/journal.pbio.1002226
Bosch TCG, McFall-Ngai MJ (2011) Metaorganisms as the new frontier. Zoology 114:185–190. https://doi.org/10.1016/j.zool.2011.04.001
Braby CE, Rouse GW, Johnson SB, Jones WJ, Vrijenhoek RC (2007) Bathymetric and temporal variation among Osedax boneworms and associated megafauna on whale-falls in Monterey Bay, California. Deep-Sea Res Part I 54:1773–1791. https://doi.org/10.1016/j.dsr.2007.05.014
Callahan BJ, McMurdie PJ, Holmes SP (2017) Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME J 11:2639–2643. https://doi.org/10.1038/ismej.2017.119
Caspi R, Altman T, Billington R, Dreher K, Foerster H, Fulcher CA, Holland TA, Keseler IM, Kothari A, Kubo A, Krummenacker M, Latendresse M, Mueller LA, Ong Q, Paley S, Subhraveti P, Weaver DS, Weerasinghe D, Zhang P, Karp PD (2014) The MetaCyc Database of metabolic pathways and enzymes and the BioCyc collection of Pathway/Genome Databases. Nucleic Acids Res 42:D459–D471. https://doi.org/10.1093/nar/gkt1103
Corrigan RM, Miajlovic M, Foster TJ (2009) Surface proteins that promote adherence of Staphylococcus aureus to human desquamated nasal epithelial cells. BMC Evol Biol 9:22. https://doi.org/10.1186/1471-2180-9-22
Ciccarelli FD, Doerks T, von Mering C, Creevey CJ, Snel B, Bork P (2006) Toward automatic reconstruction of a highly resolved tree of life [published correction appears in Science. 5;312:697]. Science 311:1283–1287. https://doi.org/10.1126/science.1123061
Czech L, Stamatakis A (2019) Scalable methods for analyzing and visualizing phylogenetic placement of metagenomic samples. PLoS ONE 14:e0217050. https://doi.org/10.1371/journal.pone.0217050
Di Meo CA, Wilbur AE, Holben WE, Feldman RA, Vrijenhoek RC, Cary SC (2000) Genetic variation among endosymbionts of widely distributed vestimentiferan tubeworms. Appl Environ Microbiol 66:651–658. https://doi.org/10.1128/AEM.66.2.651-658.2000
Dixon NE, Gazzola C, Blakeley RL, Zerner B (1976) Metal ions in enzymes using ammonia or amides. Science 191:1144–1150. https://doi.org/10.1126/science.769157
Douglas GM, Maffei VJ, Zaneveld J, Yurgel SN, Brown JR, Taylor CM, Huttenhower C, Langille MGI (preprint) PICRUSt2: An improved and extensible approach for metagenome inference. bioRxiv:672295. https://doi.org/10.1101/672295
Duperron S, Halary S, Lorion J, Sibuet M, Gaill F (2008) Unexpected co-occurrence of six bacterial symbionts in the gills of the cold seep mussel Idas sp. (Bivalvia: Mytilidae). Environ Microbiol 10:433–445. https://doi.org/10.1111/j.1462-2920.2007.01465.x
Edgar RC (2016) UNOISE2: improved error-correction for Illumina 16S rRNA and ITS amplicon sequencing. https://doi.org/10.1101/081257
Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R (2011) UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27:2194–2200. https://doi.org/10.1093/bioinformatics/btr381
Edwards D, Nelson D (1991) DNA–DNA solution hybridization studies of the bacterial symbionts of hydrothermal vent tube worms (Riftia pachyptila and Tevnia jerichonana). Appl Environ Microbiol 57:1082–1088
Forget NL, Perez M, Juniper SK (2015) Molecular study of bacterial diversity within the trophosome of the vestimentiferan tubeworm Ridgeia piscesae. Mar Ecol 36:35–44. https://doi.org/10.1111/maec.12169
Fujikura K, Fujiwara Y, Kawato M (2006) A new species of Osedax (Annelida: Siboglinidae) associated with whale carcasses off Kyushu, Japan. Zool Sci 23:733–740. https://doi.org/10.2108/zsj.23.733
Fujiwara Y, Kawato M, Yamamoto T, Yamanaka T, Sato-Okoshi W, Noda C, Tsuchida S, Komai T, Cubelio SS, Sasaki T, Jacobsen K, Kubokawa K, Fujikura K, Maruyama T, Furushima Y, Okoshi K, Miyake H, Miyazaki M, Nogi Y, Yatabe A, Okutani T (2007) Three-year investigations into sperm whale-fall ecosystems in Japan. Mar Ecol 28:219–232. https://doi.org/10.1111/j.1439-0485.2007.00150.x
Fujiwara Y, Jimi M, Sumida PYG, Kawato M, Kitazato H (2019) New species of bone-eating worm Osedax from the abyssal South Atlantic Ocean (Annelida, Siboglinidae). ZooKeys 814:53–69. https://doi.org/10.3897/zookeys.814.28869
Gardebrecht A, Markert S, Sievert SM, Felbeck H, Thürmer A, Albrecht D, Wollherr A, Kabisch J, Le Bris N, Lehmann R, Daniel R, Liesegang H, Hecker M, Schweder T (2012) Physiological homogeneity among the endosymbionts of Riftia pachyptila and Tevnia jerichonana revealed by proteogenomics. ISME J 6:766–776. https://doi.org/10.1038/ismej.2011.137
Glover AG, Kallstrom B, Smith CR, Dahlgren TG (2005) World-wide whale worms? A new species of Osedax from the shallow north Atlantic. Proc R Soc B 272:2587–2592. https://doi.org/10.1098/rspb.2005.3275
Glover AG, Wiklund H, Taboada S, Avila C, Cristobo J, Smith CR, Kemp KM, Jamieson AJ, Dahlgren TG (2013) Bone-eating worms from the Antarctic: the contrasting fate of whale and wood remains on the Southern Ocean seafloor. Proc R Soc B 280:20131390. https://doi.org/10.1098/rspb.2013.1390
Goffredi SK, Orphan VJ (2010) Bacterial community shifts in taxa and diversity in response to localized organic loading in the deep sea. Environ Microbiol 12:344–363. https://doi.org/10.1111/j.1462-2920.2009.02072.x
Goffredi SK, Paull CK, Fulton-Bennett K, Hurtado LA, Vrijenhoek RC (2004) Unusual benthic fauna associated with a whale fall in Monterey Canyon, California. Deep Sea Res I 51:1295–1306
Goffredi SK, Orphan VJ, Rouse GW, Jahnke L, Embaye T, Turk K, Lee R, Vrijenhoek RC (2005) Evolutionary innovation: a bone-eating marine symbiosis. Environ Microbiol 7:1369–1378. https://doi.org/10.1111/j.1462-2920.2005.00824.x
Goffredi SK, Johnson SB, Vrijenhoek RC (2007) Genetic diversity and potential function of microbial symbionts associated with newly discovered species of Osedax polychaete worms. J Appl Environ Microbiol 73:2314–2323. https://doi.org/10.1128/AEM.01986-06
Goffredi S, Wilpiszeski R, Lee R, Orphan V (2008a) Temporal evolution of methane cycling and phylogenetic diversity of archaea in sediments from a deep-sea whale-fall in Monterey Canyon, California. ISME J 2:204–220. https://doi.org/10.1038/ismej.2007.103
Goffredi SK, Wilpiszeski R, Lee R, Orphan VJ (2008b) Temporal evolution of methane cycling and phylogenetic diversity of archaea in sediments from a deep-sea whale-fall in Monterey Canyon, California. ISME J 2:204–220. https://doi.org/10.1038/ismej.2007.103
Goffredi SK, Yi H, Zhang Q, Klann JE, Struve IA, Vrijenhoek RC, Brown CT (2014) Genomic versatility and functional variation between two dominant heterotrophic symbionts of deep-sea Osedax worms. ISME J 8:908–924. https://doi.org/10.1038/ismej.2013.201
Huusgaard RS, Vismann B, Kühl M, Macnaugton M, Colmander V, Rouse GW, Glover AG, Dahlgren T, Worsaae K (2012) The potent respiratory system of Osedax mucofloris (Siboglinidae, Annelida): a prerequisite for the origin of bone-eating Osedax? PLoS ONE 7:e35975. https://doi.org/10.1371/journal.pone.0035975
Jin HM, Jeong H, Moon EJ, Math RK, Lee K, Kim HJ, Jeon CO, Oh TK, Kim JF (2011) Complete genome sequence of the polycyclic aromatic hydrocarbon-degrading bacterium Alteromonas sp. strain SN2. J Bacteriol 193:4292–4293. https://doi.org/10.1128/JB.05252-11
Kanehisa M, Goto S (2000) KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28:27–30. https://doi.org/10.1093/nar/28.1.27
Kolde R (2015) pheatmap: pretty heatmaps. https://cran.r-project.org/package=pheatmap
Letunic I, Bork P (2019) Interactive tree of life (iTOL) v4: recent updates and new developments. Nucleic Acids Res 47:W256–W259. https://doi.org/10.1093/nar/gkz239
López-Pérez M, Rodriguez-Valera F (2014) The Family Alteromonadaceae. In: Rosenburg E, DeLong EF, Lory S, Stackebrandt E, Thompson F (eds) The prokaryotes, 4th edn. Springer, Berlin, pp 69–92
Louca S, Doebeli M (2018) Efficient comparative phylogenetics on large trees. Bioinformatics 34:1053–1055. https://doi.org/10.1093/bioinformatics/btx701
McCarthy JD, Chen Y, Smyth KG (2012) Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res 40:4288–4297. https://doi.org/10.1093/nar/gks042
McNally CP, Eng A, Noecker C, Gagne-Maynard WC, Borenstein E (2018) BURRITO: an interactive multi-omic tool for visualizing taxa-function relationships in microbiome data. Front Microbiol. https://doi.org/10.3389/fmicb.2018.00365
Miyamoto N, Yamamoto T, Yusa Y, Fujiwara Y (2013) Postembryonic development of the bone-eating worm Osedax japonicus. Naturwissenschaften 100:285–289. https://doi.org/10.1007/s00114-013-1024-7
Miyamoto N, Yoshida MA, Koga H, Fujiwara Y (2017) Genetic mechanisms of bone digestion and nutrient absorption in the bone-eating worm Osedax japonicus inferred from transcriptome and gene expression analyses. BMC Evol Biol 17:17. https://doi.org/10.1186/s12862-016-0844-4
Ostrowski J, Barber MJ, Rueger DC, Miller BE, Siegel LM, Kredich NM (1989) Characterization of the flavoprotein moieties of NADPH-sulfite reductase from Salmonella typhimurium and Escherichia coli. Physicochemical and catalytic properties, amino acid sequence deduced from DNA sequence of cysJ, and comparison with NADPH-cytochrome P-450 reductase. J Biol Chem 264:15796–15808
Parada AE, Needham DM, Fuhrmam JA (2016) Every base matters: assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ Microbiol 18:1403–1414. https://doi.org/10.1111/1462-2920.13023
Parks DH, Tyson GW, Hugenholtz P, Beiko RG (2014) STAMP: statistical analysis of taxonomic and functional profiles. Bioinformatics 30:3123–3124. https://doi.org/10.1093/bioinformatics/btu494
Patton DA, Volrath S, Ward ER (1996) Complementation of an Arabidopsis thaliana biotin auxotroph with an Escherichia coli biotin biosynthetic gene. Mol Gen Genet 251:261–266
Pruski AM, Fiala Medioni A, Boulegue J, Colomines JC (1998) Sulfur amino acids in symbiotic species from the hydrothermal vents and cold seeps. Cah Biol Mar 39:321–324
Ravanel S, Gakiere B, Job D, Douce R (1998) Cystathionine gamma-synthase from Arabidopsis thaliana: purification and biochemical characterization of the recombinant enzyme overexpressed in Escherichia coli. Biochem J 331:639–648. https://doi.org/10.1042/bj3310639
Robinson MD, McCarthy DJ, Smyth GK (2010) edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26:139–140. https://doi.org/10.1093/bioinformatics/btp616
Roche FM, Downer R, Keane F, Speziale P, Park PW, Foster TJ (2004) The N-terminal A domain of fibronectin-binding proteins A and B promotes adhesion of Staphylococcus aureus to elastin. J Biol Chem 279:38433–38440. https://doi.org/10.1074/jbc.M402122200
Ronquist F, Teslenko M, van der Mark P, Ayres D, Darling A, Höhna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP (2012) MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol 61:539–542. https://doi.org/10.1093/sysbio/sys029
Rouse GW, Goffredi SK, Vrijenhoek RC (2004) Osedax: bone-eating marine worms with dwarf males. Science 305:668–671. https://doi.org/10.1126/science.1098650
Rouse GW, Worsaae K, Johnson SB, Jones WJ, Vrijenhoek RC (2008) Acquisition of dwarf male “Harems” by recently settled females of Osedax roseus n. sp. (Siboglinidae; Annelida). Biol Bull 214:67–82. https://doi.org/10.2307/25066661
Rouse GW, Wilson NG, Goffredi SK, Johnson SB, Smart T, Widmer C, Young CM, Vrijenhoek RC (2009) Spawning and development in Osedax boneworms (Siboglinidae, Annelida). Mar Biol 156:395–405. https://doi.org/10.1007/s00227-008-1091-z
Rouse GW, Wilson NG, Worsaae K, Vrijenhoek RC (2015) A dwarf male reversal in bone-eating worms. Curr Biol 25:236–241. https://doi.org/10.1016/j.cub.2014.11.032
Rouse GW, Goffredi SK, Johnson SB, Vrijenhoek RC (2018) An inordinate fondness for Osedax (Siboglinidae: Annelida): fourteen new species of bone worms from California. Zootaxa 4377:451. https://doi.org/10.11646/zootaxa.4377.4.1
Salathé RM, Vrijenhoek RC (2012) Temporal variation and lack of host specificity among bacterial endosymbionts of Osedax bone worms (Polychaeta: Siboglinidae). BMC Evol Biol 12:189. https://doi.org/10.1186/1471-2148-12-189
Sarmento H, Montoya JM, Domínguez EV, Vaqué D, Josep M, Gasol JM (2010) Warming effects on marine microbial food web processes: how far can we go when it comes to predictions? Phil Trans R Soc B 365:2137–2149. https://doi.org/10.1098/rstb.2010.0045
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF (2009) Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75:7537–7541. https://doi.org/10.1128/AEM.01541-09
Schneider G, Lindqvist Y (2001) Structural enzymology of biotin biosynthesis. FEBS Lett 495:7–11. https://doi.org/10.1016/S0014-5793(01)02325-0
Smith CR, Baco AR (2003) Ecology of whale falls at the deep-sea floor. Oceanogr Mar Biol Ann Rev 41:311–354
Su J, Jin L, Jiang Q, Sun W, Zhang F, Li Z (2013) Phylogenetically diverse ureC genes and their expression suggest the urea utilization by bacterial symbionts in marine sponge Xestospongia testudinaria. PLoS ONE 8:e64848. https://doi.org/10.1371/journal.pone.0064848
Sumida PY, Alfaro-Lucas JM, Shimabukuro M, Kitazato H, Perez JA, Soares-Gomes A, Toyofuku T, Lima AO, Ara K, Fujiwara Y (2016) Deep-sea whale fall fauna from the Atlantic resembles that of the Pacific Ocean. Sci Rep 6:22139. https://doi.org/10.1038/srep22139
Taboada S, Riesgo A, Bas M, Arnedo MA, Cristobo J, Rouse GW, Avila C (2015) Bone-eating worms spread: insights into shallow-water Osedax (Annelida, Siboglinidae) from antarctic, subantarctic, and mediterranean waters. PLoS ONE 10:e0140341. https://doi.org/10.1371/journal.pone.0140341
Taboada S, Bas M, Avila C, Riesgo A (2020) Phylogenetic characterization of marine microbial biofilms associated with mammal bones in temperate and polar areas. Mar Biodiver. https://doi.org/10.1007/s12526-020-01082-8
Thornhill DJ, Fielman KT, Santos SR, Halanych KM (2008) Siboglinid-bacteria endosymbiosis. Commun Integr Biol 1:163–166. https://doi.org/10.4161/cib.1.2.7108
Tresguerres M, Katz S, Rouse GW (2013) How to get into bones: proton pump and carbonic anhydrase in Osedax boneworms. Proc R Soc Lond B: Biol Sci 280:20130625. https://doi.org/10.1098/rspb.2013.0625
Tringe SG, Von Mering C, Kobayashi A, Salamov AA, Chen K, Chang HW, Podar M, Short JM, Mathur EJ, Detter JC, Bork P (2005) Comparative metagenomics of microbial communities. Science 308:554–557. https://doi.org/10.1126/science.1107851
Vandenesch F, Lina G, Henry T (2012) Staphylococcus aureus hemolysins, bi-component leukocidins, and cytolytic peptides: a redundant arsenal of membrane-damaging virulence factors? Front Cell Infect Microbiol 2:12. https://doi.org/10.3389/fcimb.2012.00012
Varner JE (1960) Urease. In: Boyer PD, Lardy H, Myrback K (eds) The enzymes, vol 4, 2nd edn. Academic Press, New York, pp 247–256
Verna C, Ramette A, Wiklund H, Dahlgren T, Glover A, Gaill F, Dubilier N (2010) High symbiont diversity in the bone-eating worm Osedax mucofloris from shallow whale-falls in the North Atlantic. Environ Microbiol 12:2355–2370. https://doi.org/10.1111/j.1462-2920.2010.02299.x
Vrijenhoek RC, Johnson SB, Rouse GC (2008) Bone-eating Osedax females and their ‘harems’ of dwarf males are recruited from a common larval pool. Mol Ecol 17:4535–4544. https://doi.org/10.1111/j.1365-294X.2008.03937.x
Vrijenhoek RC, Johnson SB, Rouse GW (2009) A remarkable diversity of bone-eating worms (Osedax; Siboglinidae; Annelida). BMC Biol 7:74. https://doi.org/10.1186/1741-7007-7-74
Waits DS, Santos SR, Thornhill DJ, Li Y, Halanych KM (2016) Evolution of sulfur binding by hemoglobin in Siboglinidae (Annelida) with special reference to bone-eating worms, Osedax. J Mol Evol 82:219–229. https://doi.org/10.1007/s00239-016-9739-7
Ye Y, Doak TG (2009) A parsimony approach to biological pathway reconstruction/inference for genomes and metagenomes. PLOS Comput Biol 5:e1000465. https://doi.org/10.1371/journal.pcbi.1000465
Zhao JX, Liu QQ, Zhou YX, Chen GJ, Du ZJ (2015) Alkalimarinus sediminis gen. nov., sp. nov., isolated from marine sediment. J Syst Evol Microbiol 65:3511–3516. https://doi.org/10.1099/ijsem.0.000446
Acknowledgements
We are indebted with Dr Ana Riesgo, who provided all the resources needed to conduct all the laboratory analysis. We also would like to acknowledge the project ACTIQUIM-II (CTM2010-17415/ANT) and the environmental association S’Agulla Educació Mediambiental, which contributed with the samples used in this study. Last, but not least, we thank all the members of the Riesgo Lab, Martín Taboada and Otilia Moreno for all the help they provided during the sample processing and writing of the manuscript, and Lorna Page for whose enthusiastic discussions helped inform many of the thoughts presented here.
Funding
This work was supported by the SponGES project (Grant Agreement No. 679848)—European Union Framework Programme for Research and Innovation, H2020. OHH received funding from the European Erasmus + Traineeship programme (2018-1-BE02-KA103-046670). CDV received funding from the EU Horizon 2020 MSCAs DeepSym (grant agreement: 796011). ST received funding from the Juan de la Cierva-Incorporación programme (IJCI-2017-33116), Spanish Government.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hewitt, O.H., Díez-Vives, C. & Taboada, S. Microbial insights from Antarctic and Mediterranean shallow-water bone-eating worms. Polar Biol 43, 1605–1621 (2020). https://doi.org/10.1007/s00300-020-02731-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00300-020-02731-1