
Abstract
Background
Marine animals often exhibit complex symbiotic relationship with gut microbes to attain better use of the available resources. Many animals endemic to deep-sea chemosynthetic ecosystems host chemoautotrophic bacteria endocellularly, and they are thought to rely entirely on these symbionts for energy and nutrition. Numerous investigations have been conducted on the interdependence between these animal hosts and their chemoautotrophic symbionts. The provannid snail Alviniconcha marisindica from the Indian Ocean hydrothermal vent fields hosts a Campylobacterial endosymbiont in its gill. Unlike many other chemosymbiotic animals, the gut of A. marisindica is reduced but remains functional; yet the contribution of gut microbiomes and their interactions with the host remain poorly characterised.
Results
Metagenomic and metatranscriptomic analyses showed that the gut microbiome of A. marisindica plays key nutritional and metabolic roles. The composition and relative abundance of gut microbiota of A. marisindica were different from those of snails that do not depend on endosymbiosis. The relative abundance of microbial taxa was similar amongst three individuals of A. marisindica with significant inter-taxa correlations. These correlations suggest the potential for interactions between taxa that may influence community assembly and stability. Functional profiles of the gut microbiome revealed thousands of additional genes that assist in the use of vent-supplied inorganic compounds (autotrophic energy source), digest host-ingested organics (carbon source), and recycle the metabolic waste of the host. In addition, members of five taxonomic classes have the potential to form slime capsules to protect themselves from the host immune system, thereby contributing to homeostasis. Gut microbial ecology and its interplay with the host thus contribute to the nutritional and metabolic demands of A. marisindica.
Conclusions
The findings advance the understanding of how deep-sea chemosymbiotic animals use available resources through contributions from gut microbiota. Gut microbiota may be critical in the survival of invertebrate hosts with autotrophic endosymbionts in extreme environments.
Similar content being viewed by others
Background
The interaction between eukaryotic animals and microorganisms has resulted in numerous innovative symbiotic adaptations, especially in efficiency of energy utilisation. Such interplay in marine chemosynthetic ecosystems often supports distinctive microbial communities and may involve more than two players: the host animal could be symbiotic with microbes inhabiting different parts of the host body. For instance, gut bacteria contribute to digestive functions of the bone-eating snail Rubyspira [1]; alvinocaridid shrimps rely on epibionts associated with the gill chamber and the gut for both detoxification and nutritional intake [2]; provannid vent snails and bathymodioline mussels rely on gill endosymbionts for energy conversion and nutrition supply [3, 4]. A number of chemosymbiotic invertebrates from deep-sea ecosystems, including siboglinid tubeworms and several families of gastropods and bivalve molluscs, harbour microbes inside the host’s bacteriocytes while the host’s digestive systems is often reduced. These hosts rely mostly or entirely on the autotrophic chemosymbionts for energy and nutrition [5, 6]. Furthermore, laboratory evidence shows bathymodioline mussels are capable of filter-feeding and organic matter contributes to the diet of these mussels, suggesting some chemosymbiotic hosts have the potential to use multiple nutritional resources [7, 8]. Despite numerous investigations conducted on the interdependence between these animal hosts and their chemoautotrophic symbionts over the last decades [3, 4, 9, 10], intestinal functions and associated microbes received little attention [11]. Gut microbes are vital to host survival in mammals and insects, including through maintaining energy homeostasis, conferring metabolic capabilities, enhancing nutrient digestion and absorption and assisting with immunity development and activity [12]. They may also play important roles in chemosymbiotic animals.
Alviniconcha is a genus of chemosymbiotic vent snail in the family Provannidae with six species, five in the Pacific and one in the Indian Ocean [13]. These snails are endemic to hydrothermal vents and harbour chemoautotrophic endosymbionts in the gill epithelium [14]. The aggregated distribution of symbionts near the more exposed, outer surface of the bacteriocytes has been described as ‘semi-endosymbiotic’ condition [15, 16]. The Indian Ocean species A. marisindica hosts a single phylotype of Campylobacterota in its gills that is capable of oxidising sulphur, formate and hydrogen coupled with nitrate or oxygen reduction to produce cellular energy [8, 17]. In symbioses with a single major endosymbiont, the potential contribution of extracellular microbiota in the host gut has not been considered. Unlike many other invertebrate animals that rely on chemosynthetic endosymbionts and have lost most or all of the gut [5], such as siboglinid tubeworms and the awning-clam Solemya reidi, Alviniconcha retains a much reduced yet apparently functional gut and stomach [3, 18]. The stomach of Alviniconcha contains mainly mucus; lots of small white particles with walls and granular contents were found in some specimens, occasionally with mineral particles, sponge spicules, crustacean remains, and similar potential prey [18]. The intestine contains soft biogenic substances and mineral grains [3, 18]. These observations suggest that the stomach and intestine microbiota may contribute to the digestion of these foodstuffs and play nutritional, energetic and other physiological roles.
Integrated metagenomics and metatranscriptomics, which analyse the genomes and gene expression of the host and symbionts simultaneously, are powerful tools for deciphering symbiotic interactions [19, 20]. Here, an integrated metatranscriptomic and metagenomic approach was applied to investigate the gut microbial community and its interplay with the host snail by using A. marisindica from the Wocan hydrothermal vent field on the Carlsberg Ridge (CR), Northern Indian Ocean [21, 22].
Results and discussion
Gut microbial communities of A. marisindica
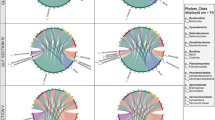
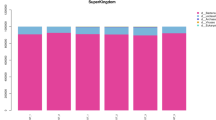
The metagenome assemblage of the intestinal content of A. marisindica using Illumina reads and subsequent taxonomic analyses of prokaryotic sequences was used to describe the intestinal microbial composition (Fig. 1; rarefaction curves for species abundance are shown in Additional file 1: Figure S1). In the intestinal metagenome of A. marisindica, the bacterial assembled contigs accounted for 2.6–5.6% of the total metagenomic sequences, much lower than that in the endosymbionts in the gill at 10.0–7.7% (Fig. 1a, b). Unlike the abundant single dominant campylobacterial endosymbiont in the gill (Fig. 1b), the intestinal tract harboured diverse microbial taxa (Fig. 1c, d) comprising 14 dominant genera (genera constituting > 1% of individual samples) from six phyla in three individuals of A. marisindica (Fig. 1d). The A. marisindica gut microbiome consisted of 8.74–49.30% Proteobacteria, 7.44–11.15% Actinobacteria, 8.98–10.79% Firmicutes, 1.89–9.75% Tenericutes, 3.20–4.42% viruses, and very few other microbes (Fig. 1c).

Abundance and community structure of microbiomes in the intestines of Alviniconcha marisindica from the Wocan vent field. Microbial taxonomic structures are deduced from the intestinal and gill metagenomes. The abundance of bacteria in a intestines and b gills are indicated in red colour. The microbial community compositions of intestines are displayed at the phylum and genus levels on the basis of Kaiju classification. c Phylum totalling > 0.3% and d genus totalling > 1% of the samples are shown
Based on the available metagenomics and 16S rRNA amplicon data, intestines of A. marisindica exhibited a community structure different to the known gut microbiomes of other deep-sea snails from chemosynthesis-based ecosystems, including Rubyspira osteovora, Bathymargarites symplector and Phymorhynchus sp. [1], those of freshwater snail Pomacea canaliculata and land snail Achatina fulica [23, 24] and the environmental compositions of microorganisms in deep-sea whale fall, hydrothermal fluids and sediments [1, 25] (Additional file 2: Table S1). The four snails from deep-sea environments have different feeding modes: Alviniconcha marisindica depends largely on intracellular chemoautotrophic bacteria, Rubyspira osteovora feeds on whalebones, Bathymargarites symplector is a deposit feeder and Phymorhynchus sp. is a predatory and scavenging snail [1, 3, 26]. After the datasets of samples listed above were normalised on the basis of 16S rRNA sequences from Bacteria, Archaea and Fungi (RDP database), similarity analyses of microbial communities among these samples by PCoA (Bray–Curtis dissimilarity) revealed that environmental microbiome of the Crab Spa vent on the East Pacific Rise at 9°N [27, 28], the gut microbiomes of A. marisindica and B. symplector formed a distinct cluster and was separated from other samples (Fig. 2; ANOVA, F = 2.173, P = 0.00371). Environmental and snail gut microbiomes from deep-sea whale fall in Monterey Submarine Canyon were also separated from other samples. These results indicated that the principal environmental factors of different deep-sea habitats, such as vents versus whale falls, might have shaped different structure and function of microbial communities in both the environment and in the gut of animals living there. In particular, the Mollicutes and Campylobacterota in the intestine of A. marisindica made up a greater proportion of the total classified microbial sequences (average ~ 6.06% and 11.51%, respectively) than in the intestines of A. fulica, Phymorhynchus sp., P. canaliculata and hydrothermal fluid of Mariana Trough (~ 3.72%). In addition, Mollicutes and Campylobacterota were commonly identified in guts of the shrimp Rimicaris exoculata from the Mid-Atlantic Ridge hydrothermal vent sites [2], suggesting the broad distribution and importance of Mollicutes and Campylobacterota in guts of vent-endemic invertebrates.

Principal coordinates analysis of three intestinal samples of Alviniconcha marisindica, six intestinal sample of other three deep-sea snails, four intestinal sample of two land snails and five environmental samples of three deep-sea habitats at the level of OTUs (97% sequence similarity). Different shapes represent the location of environmental samples and the snails from marine and land. Different colours represent the species names of all the snails and the types of deep-sea habitats. A. marisindica samples are clustered together with deep-sea scavenger Bathymargarites symplector and Crab Spa hydrothermal vent and well separated from other snails and environmental samples
At the genus level, Sulfurovum accounted for a large proportion of the gut microbes of A. marisindica (Fig. 1d), average ~ 7.87% of the total classified microbial sequences. The endosymbionts of gill epithelial cells in A. marisindica belong to Sulfurovum with one dominant ribotype [17], while two different ribotypes of Sulfurovum were found in the gut of A. marisindica. Based on the similarity of the 16S rRNA gene sequence, one gut ribotype is identical (> 99% similarity) with the gill endosymbiont, and the other gut ribotype shows 97.97% sequence similarity to the gill symbiont in the V6–V9 region of the 16S rRNA gene sequence. A previous study showed that campylobacterial epibionts in the gut and gill chamber of the vent shrimp Rimicaris exoculata were similar and suggested that the ones in the gut could be directly acquired from the environment or ingested from the gill chamber [2]. In A. marisindica, the intestines harboured multiple ribotypes of Sulfurovum compared to Sulfurovum ribotypes of the gills, further suggesting that the Sulfurovum in the gut were likely acquired from the environment, in addition to those from the gills. Sulfurovum is chemolithoautotrophic; it oxidises sulphur and thiosulfate (as electron donors) and uses the produced energy to fix carbon dioxide. A number of novel mesophilic, anaerobic chemosynthetic Sulfurovum have been isolated from hydrothermal sediments, vent plumes, and the tube of siboglinid polychaetes in vents [28,29,30]. In the gut microbiomes of deep-sea snails R. osteovora, B. symplector and Phymorhynchus sp., Sulfurovum is also one of the major components of their microbial communities [1]. Overall, Sulfurovum is abundant in the gut of A. marisindica and could be ingested from the gill and vent environment, the introduction of Sulfurovum may provide detoxification and nutritional intake for the host.
Gut microbial co-occurrence pattern
The gut microbiomes of three A. marisindica individuals exhibited significant non-random co-occurrence networks among component taxa (Fig. 3). The communities could be classified into seven modules differing mainly in the dominance of Campylobacterota, Actinobacteria, Gammaproteobacteria, Bacilli and Mollicutes (Fig. 3). In module A of Campylobacterota, six different genera were positively correlated. Amongst these genera, Sulfurovum, Sulfurimonas and Nitratifractor are chemolithoautotrophic bacteria commonly found in deep-sea hydrothermal environments. They function in sulphur- and/or hydrogen-oxidising respiration [14, 30,31,32], using elemental sulphur, thiosulfate and/or hydrogen as electron donors.

Correlation-based network of intestinal bacteria genera (relative abundance ≥ 0.5%) from three Alviniconcha marisindica individuals. Network analysis displays the intra-associations within each sub-community and inter-associations between sub-communities. Node size is proportional to the number of connections (i.e. degree of connectivity). Connection between nodes represents strong [Spearman correlation efficiency > 0.8 (yellow) or < − 0.8 (blue)] and significant (p-value < 0.01) correlation. The same colour of nodes shows their highly modularised (clustered) property within the network
Module B, which is mainly composed of Actinobacteria, was the second largest group of gut microbes (Fig. 1c), and this module was negatively correlated with Campylobacterota in module A, likely due to their different physiological characteristics. In general, most Actinobacteria were aerobic and chemoheterotrophic [33], which may also apply to the Actinobacteria in the gut of A. marisindica. These Actinobacteria may compete for oxygen with Campylobacterota, which relies on oxygen for sulphur- and hydrogen-oxidising respiration.
In module C of Gammaproteobacteria and Bacilli, three genera of Paenibacillus, Xanthomonas and Haemophilus were positively correlated with one another and negatively correlated with Minicystis. Members of the genera Xanthomonas, Paenibacillus and Haemophilus are known as plant-, human- or insect-associated microorganisms and many of them are pathogenic. Xanthomonas and Paenibacillus species in animals’ guts have the ability to degrade non-digestible carbohydrates such as the cell-wall polysaccharide of plant-like protists and cellulose using xylanases and cellulases [34, 35]. Minicystis species has been identified as omega-3 fatty acid (FA) producer [36]. Omega-3 FA is a predominant polyunsaturated FA, with high concentrations found in marine snails [37]. They are also essential for physiological processes. In addition, putative omega-3 FA-producing bacteria in the gastrointestinal tracts of marine fishes contribute omega-3 FAs to the host in general [38] and have integral roles in regulating membrane fluidity in response to temperature fluctuations [38,39,40]; this may also apply to vent-restricted A. marisindica living in relatively instable environmental conditions.
Module D was mainly composed of Proteus and Mollicutes-related bacteria that are commonly considered as opportunistic pathogens in a wide variety of animals. This module showed a significant negatively correlation with module E, which included Lactobacillus that are typically intestinal probiotics [41] (Fig. 3). In addition, lactic acid bacteria (LAB), which are considered vital for maintaining the gut ecological balance [42], accounted for at least ~ 2.70% of the A. marisindica gut microbiome. Mixed LAB exhibited a mutualistic relationship with the host; they have the potential to prevent pathogens from causing intestinal infections and are vital for food digestion and energy provision to the host [41,42,43]. Various LAB were found to co-exist in the intestine of A. marisindica, including Lactobacillaceae (1.29–1.55%), Streptococcaceae (0.20–0.43%) and Enterococcaceae (0.25–0.34%). This finding further suggested a potential ecological balance in the A. marisindica gut microbiome.
Although microbial sequences obtained from the intestinal metagenome data of A. marisindica were in low abundance (2.61–5.57%), modules of co-occurring microorganisms could be identified from the 3 individuals. Positive and negative associations suggest that their interactions may shape the microenvironment in the intestine of A. marisindica; coexisting microbial species in the gut may have similar, complementary, or competitive ecological functions.
Essential functions of gut microbiome
The gut microbiome of A. marisindica contained ~ 3881–5389 annotated genes exhibiting a wide metabolic spectrum. In this study, the microbial genes were analysed to reveal microbial pathways associated with the metabolism of host-ingested substances, including carbohydrates, proteins, vitamins and minerals. These functions were distinct from, and likely complement the activities of host enzymes in the digestive gland (DG) and intestine, including essential functions of organic matter digestion and nutrient absorption. Accordingly, genes involved in substrates transportation, energy conversion, macromolecular digestion, and absorption were enriched in the intestine (Additional file 1: Figure S2), especially for genes involved in lipid metabolism, indicating the metabolic capacity and activity of intestines. In addition, the gut microbiome of A. marisindica was capable of ammonium assimilation, indicating a role of the guts in recycling the host’s metabolic waste (Fig. 4a) and in producing other metabolites essential for intestinal homeostasis, such as bacteriocins, short-chain FAs (SCFAs), and quorum-sensing autoinducers [44].

Overview of meta-pathways of Alviniconcha marisindica and its gut microbiome. Metabolic pathways of different organisms, including a the gut microbiome and b A. marisindica host, are presented to reveal the functional contribution of gut microbiomes to the host. Important metabolic intermediates and complementary intermediate products are indicated in red colour
Glycoside hydrolases of saccharolytic bacteria in the gut microbiome of A. marisindica (Additional file 2: Table S2) can break down sugars, including host-indigestible carbohydrates, resulting in the production of important metabolites such as vitamins and SCFAs. Most SCFAs are the end products of gut microbial anaerobic metabolism via digestion of the dietary carbohydrates, such as acetate, propionate, and butyrate for nutrition in aquatic animals [45, 46]. In addition, microbial fermentation produces SCFAs together with the gases CO2 and H2, which could be used by chemolithoautotrophic microbiota in the gut of A. marisindica. For example, Sulfurovum and Sulfurimonas could generate energy through hydrogenotrophic energy metabolism and fix CO2 into organics. Evidence showed that meta-pathways of intestinal Campylobacterota contain genes encoding [NiFe]-hydrogenases for hydrogenotrophic metabolism, the Sox system for thiotrophic metabolism, and the reverse tricarboxylic acid cycle for carbon fixation (Fig. 4a). Therefore, these gut microbes of A. marisindica have the potential of chemoautotrophy, consistent with the metagenomic findings described above. In addition, the genera Desulfitobacterium and Desulfovibrio in the gut of A. marisindica are sulphate-reducing microorganisms that could produce H2S which can then be used by thiotrophic gut bacteria, such as Sulfurovum and Sulfurimonas.
Nitrogen is essential for bacterial survival. Genes encoding for dissimilatory nitrate reduction and ammonium assimilation are found in the metagenome of gut microbes (Fig. 4a). Genes encoding for ureases are also found in gut microbiomes, urea produced by the host could be hydrolysed to ammonia by these urease-producing bacteria and the ammonia in turn could be used for protein metabolism (Fig. 4a). The occurrence of urea-nitrogen recycling suggests that the gut microbiomes play an important role in the nitrogen balance of A. marisindica. In addition, genes encoded the sialate O-acetylesterase (SIAE) were highly expressed in the meta-transcriptome of intestinal microbiomes, indicating that active sialic acid (Sia) degradation in the intestine of A. marisindica. Sialic acids are prominent outermost carbohydrates of the intestine; they are important components of the mucus layer [47]. A pathway for the transport and catabolism of Sia is encoded in the metagenome of intestinal microbiomes, indicating Sia could be utilised as a carbon source for heterotrophic microbes in the gut. The intestinal microbiome contains biosynthetic pathways for four vitamins and 10 amino acids, including six amino acids and three vitamins that cannot be synthesised by the host (Table 1), indicating that gut microbiomes could provide essential nutrients for the host.
Co-abundance and meta-pathway analyses indicated that the gut microbiome likely plays critical ecological roles via bacterial interdependencies and mutual cooperation to maintain microbial homeostasis and provide the host with symbiont metabolites that serve as host nutrients. In addition, the gut microbiomes of A. marisindica were found to produce large exo-enzymes for hydrolysing various macromolecules. Thus, they have the potential to digest and supply exogenous nutrients to the host. This finding warranted further investigation on the mutual cooperation of A. marisindica holobiont (that is, involving the A. marisindica host and its gill endosymbiont) in the production of nutrients.
Host–gut microbiome cooperation
In the study of A. marisindica holobiont [17], the gill endosymbionts provide plenty of nutrients to A. marisindica although they still cannot synthesise some nutrients, i.e. two amino acids of taurine and hypotaurine and seven vitamins/cofactors of thiamin, pyridoxine, folate, cobalamin, phylloquinone, menaquinone, and ubiquinone. Neither A. marisindica nor its endosymbionts can synthesise folate, but the host transcriptome contained a complete folate metabolism pathway (including one carbon pool by folate, Fig. 4b), indicating that the host has the ability to use folate and it may be essential to supporting A. marisindica. Furthermore, A. marisindica holobionts cannot synthesise octadecanoic acid and other 10 typical unsaturated FAs [17] but the host harboured a complete pathway of FA metabolism (Fig. 4b). Therefore, the nutrients that cannot be produced by the A. marisindica holobiont are likely supplemented by taking food from the environment [3, 18], followed by intestinal digestion and absorption of substances [48]. This is analogous to the situation in bathymodioline mussels, which also rely on the mixotrophic nutritional strategy of suspension-feeding and endosymbiosis [7, 8], and is in contrast to our understanding of nutrition in other chemosymbiotic animals that entirely rely on endosymbiont nutritional supply [4, 6, 20, 49].
In the present study, the Alviniconcha transcriptome encoded metabolic pathways within lysosomes, endosomes and phagosomes which all served as the main digestive compartment of host cells. Accordingly, functional enzyme-encoding gene distribution in the A. marisindica genome showed that the host contains numerous genes responsible for key hydrolases [17] and the genes for intestinal digestion and absorption of ingested dietary components, such as carbohydrate, fat, protein and vitamin are active (Fig. 4b). For example, genes encoding various large proteases, bile salt-activated lipase, and glycosyl hydrolases were highly expressed in the intestine and genes encoding a gastric intrinsic factor and a bile acid transporter were highly expressed in the digestive gland (Fig. 5a), supporting the active digestion of macromolecular nutrients in the intestine. In addition, genes encoding specialised transport proteins, such as proton-coupled folate transporter (SLC46A1), taurine transporter (SLC6A6), cationic amino acid transporter (SLC7A6), and ferritin (FTH1), were expressed actively in the intestine (Fig. 4b and Fig. 5a). This expression may help the intestinal absorption of folate, amino acids, and minerals that could not be generated by the A. marisindica holobiont. Large numbers of highly expressed genes in the intestine showed that the reduced gut still has functional specialisations responsible for effective and regulated nutrients transport. The number of differentially expressed genes (DEGs) in the intestine, with their biological coefficient of variation (BCV), is shown in the Additional file 1: Figure S3. The annotation of highly expressed genes in the intestine of A. marisindica were shown in Additional file 3: Dataset S1. On the basis of the DEGs, functional enrichments of GO terms/KEGG pathways were also identified in the intestine of A. marisindica. In the intestine, genes involved in vacuolation, hydrolases activity, organic hydroxy compound metabolic processes, and transmembrane transport were enriched (Additional file 1: Figure S2), indicating its activity in nutrients digestion and absorption.

Transcriptional activity of genes participating in the nutrient digestion and absorption of Wocan Alviniconcha marisindica. Heat map of transcriptional activity of genes that a encode specialised transport proteins and various hydrolases, including proteases, glycoside hydrolase, and peptidoglycan recognition proteins (PGRPs) in the foot, neck, mantle, digestive gland (DG), intestine and gill tissues and are involved in b gut microbial exo-hydrolase biosynthesis for intestinal nutrient digestion in A. marisindica
Intestinal microbial enzymes contain 30.6%–34.7% hydrolases, 26.7%–28.2% transferases and 9.6%–17.4% oxidoreductases. The three main types of hydrolases secreted by gut microbiomes are glycoside hydrolases (GHs), which could break down complex carbohydrates (e.g. cellulose and chitin); protease (including aminopeptidases) and esterase (mainly lipases). Genes involved in intestinal microbial hydrolases were extracted from the metagenome and their annotations and expression levels are summarised in Additional file 2: Table S2. In A. marisindica, the digestive exoenzymes produced by gut microbes may help with the enzymatic digestion of intestinal substances. For example, genes encoding α-amylase, which breaks down long-chain saccharides, were abundant and highly expressed in the gut microbiomes (Fig. 5b) that may help degrade the glycogen produced by animal remains found in the gut [18]. Trypsin and zinc-dependent metalloprotease are major exoproteases (Fig. 5b) necessary for protein absorption. GDSL esterases/lipases (Fig. 5b) are the major lipolytic enzymes for carbon source provision. Proteins and lipids are the main components of animal remains and mucus found in the stomach and gut of Alviniconcha snails [18]. In particular, genes encoding GHs were found abundantly in the gut of A. marisindica (Additional file 2: Table S2) and they have important roles in aiding the digestion of dietary carbohydrates. The above results indicated abundant digestive exoenzyme production in the gut of A. marisindica. Complementary to the host metabolic capacities, the intestinal microbiota provides additional enzymes that are not encoded by A. marisindica. For example, comparative analyses among the available lophotrochozoan genomes revealed A. marisindica lacked the enzymes (e.g. cellulases) to degrade the bulk of dietary fibres [17]. The gut microbial metagenome and metatranscriptome data showed that genes encoding cellulases, xylanase/chitin deacetylase, glucanases, phosphorylase and some other GHs were found extensively in Proteobacteria (Dickeya, Xanthomonadales and Cellvibrio), Actinobacteria, Firmicutes, Tenericutes, Chloroflexi and Bacteroidetes (Cytophagia) to form multi‐enzyme complexes that help with the host digestion of ingested carbohydrates such as remains of crustaceans and similar things [18]. Moreover, oligoendopeptidase F (pepF1) and alginate lyase (algL) are not encoded by the host [17] but found encoded by the gut microbiomes and they become complementary for food digestibility in Alviniconcha snail. The results indicated the ability of intestinal microbiomes to assist the host’s intestinal digestion of food for nutrient absorption. All annotated information of gut microbiomes is shown in Additional file 4: Dataset S2.
Host–gut microbiome cooperation in the production and absorption of nutrients is newly revealed here for the vent snail A. marisindica, indicating that the maintenance of the A. marisindica symbiosis is also likely mediated by feeding and the gut microbiome, and not only depends on gill endosymbionts.
Maintenance of gut microbiota
Genes involved in the assembly of bacterial capsular polysaccharides (CPs), surface layer (S-layer) proteins (SLPs), bacterial lipopolysaccharide (LPS) and other surface-associated antigens were found in gut microbiomes such as Campylobacterota, Clostrida, Mollicutes, Bacilli, Erysipelotrichia; they are responsible for the bacterial adhesion to the intestinal epithelium and they activate the complement system [50, 51]. LPS is often the first target of host immune system and it could induce a strong host immune response [50, 52]. Although CPs and SLPs could be recognised by the host as immunodominant antigens [51, 52], they are bacterial physical “cloaks” that protect intracellular bacteria from many of host’s defences [53, 54]. Surface-layer glycoprotein variation in the gut microbiome was evident from the differential expression of S-layer genes, a type of antigenic variation responding to the lytic activity of the host immune system (Fig. 6a). In the gut metagenome of A. marisindica, genes encoding for SIAE were found in Cytophagia (e.g. Leeuwenhoekiella) and Sphingobacteriia in the phylum Bacteroidetes; their high expression levels (Fig. 5b) indicate active sialic acid degradation in the intestine of A. marisindica. Sias are prominent carbohydrates of the intestinal mucus layer [47]. The mucus layer is the interface between the gut microbiome and the host. Thus, Sia breakdown is a mechanism for intestinal bacterial encroachment and survival. Gut-specific bacteria could bind to and degrade mucin glycans as a nutritional source and produce microbial metabolites that facilitate growth of other microbial species [47].

Maintenance of microbiomes in the gut of Alviniconcha marisindica. Heat map of the transcriptional activity of genes that a participate in bacterial surface-associated virulence factors, surface modification, protease synthesis and secretion in the gut microbiome and are b involved in host innate immunity in the foot (F), neck (N), mantle (M), intestine (I) and gill (G) tissues, showing an immune-expression profile of the gut regulated by its inhibited microbiomes. Each grid in the heat map represents an identified gene. The colour represents the gene expression (based on normalised TPM values of the selected tissues). The annotated gene names and their functional classifications are listed on the two sides. c Host–microbiota homeostasis is maintained by the host’s immune compartmentalisation and bacterial counter-defence. All pattern recognition receptors (PRRs) and pathogen-associated molecular patterns (PAMPs) shown here are identified from the transcriptome data. SLPs, surface layer proteins; LPS, lipopolysaccharide; CPS, capsular polysaccharides; SIAE, sialate O-acetylesterase; PGRPs, peptidoglycan recognition proteins; TLR, toll-like receptor; C1q, complement component 1q
The intestine harboured much lower abundance of bacteria than the gill (Fig. 1a, b), but the intestinal immune response was stronger than that of the gill (Fig. 6b). The microbes in animals have the ability to control immune gene transcriptional activity [20, 55]. Host immune responses show the interaction of microbiota with the host. Pattern recognition receptors (PRRs) are crucial to the innate immune system and they could be divided into membrane-bound PRRs and cytoplasmic PRRs. Genes encoding membrane-bound C-type lectin receptors (CLRs) and cytoplasmic RIG-I-like receptors (RLRs) were active in the intestine (Fig. 6b). Toll-like receptors (TLRs) recognise structurally conserved molecules derived from microbes and activate immune responses. Genes encoding membrane-bound TLR2 and TLR6 were active in the intestine (Fig. 6b), indicating a distinct TLR-expression profiles in the intestine that recognise inhabited microbes. Once the host recognised the microbes in the gut, the component cascade is activated, as indicated by the highly expressed complement component 1 complex (C1) and complement C3 (C3, Fig. 6b), which both attack the microbe’s cell membrane and eliminate microbes to control bacterial infections [56] (Fig. 6c). Differentially expressed genes of the gut immune system compartmentalise the intestinal bacteria; these genes are essential for mutual coexistence of A. marisindica and its gut microbiome. The host’s immune compartmentalisation and bacterial counter-defence cooperate to maintain the host–gut microbiome mutualism (Fig. 6c).
Functional analyses of gut microbiomes indicated that the microbiota have a considerable effect on intestinal nutrient digestion and absorption and host immune system stimulation that help defend against pathogens. The digestion processes in the intestine following the action of bile acid and pancreatic and intestinal enzymes are essential for nutritional homeostasis in Alviniconcha holobiont. Thus, even if the gut microbiome has a lower cell abundance than the endosymbionts in the gill, it is important—and likely critical—to the host fitness and survival. Gut microbial ecology and the interplay of this ecology with the host metabolome contribute to meet the nutritional and metabolic demands of Alviniconcha holobiont and they help shape the distinctive microenvironments and physiology of the intestine. These results complement the recent study of the symbiosis between this snail and its gill endosymbiont [17] and enhance our understanding of the contribution of gut microbes to the success of chemosymbiotic snails.
Conclusions
In this study, the microbial community structure and their relative abundance in the gut of the deep-sea vent snail Alviniconcha marisindica were illustrated. More importantly, the metabolite-enabled mutualistic interaction between the host and its gut microbiota was described through gene functional and expression analyses. The energy requirements of A. marisindica were potentially sustained by feeding and through its functionally versatile gut microbiome. The findings advanced general understanding of the mechanisms of animals surviving in extreme chemosynthetic ecosystems. Furthermore, the gut microbiome may have evolved in parallel with host immune systems and has strategies for coping with the host’s immune responses in the gut. Potential interactions of the gut and its microbiome in deep-sea vent invertebrates that primarily rely on endosymbiosis are reported for the first time. These interactions are likely important for the efficient and successful life of these animals at deep-sea vents.
Methods
Sample collection and nucleic acid preparation
Alviniconcha marisindica were collected from the Wocan vent field on the CR of the Northwestern Indian Ocean at a water depth of 2,919 m (60.53°E, 6.36°N). Sampling was conducted using the human-occupied vehicle (HOV) Jiaolong onboard R/V Xiangyanghong 9 on March 19, 2017. Snails were placed into an insulated ‘BioBox’ with a closed lid by using a manipulator to minimise changes in water temperature. Around 2.5 h was needed for HOV Jiaolong to be recovered on deck. Once the snails were on-board the research vessel, all specimens were immediately flash-frozen in liquid nitrogen and then transferred to − 80 °C freezer for storage. The external morphology of a complete Alviniconcha individual preserved in absolute ethanol is shown in Additional file 1: Figure S4. The internal morphology was observed under a Leica MZ9.5 stereozoom microscope and the snails were dissected to isolate their radula. Radular morphologies of two specimens were imaged using scanning electron microscopy (SEM, Additional file 1: Figure S5). Radular sacs were dissected from the body cavities, stored in pure ethanol and then treated with half-strength commercial bleach, leaving the clean radular teeth. Subsequently, the radula for SEM was rinsed in MilliQ water and dehydrated by increasing the concentration of ethanol solution (20%, 40%, 60%, 75% and 100%). The dehydrated radula was air-dried and then SEM observation was undertaken, uncoated at 15 kV using a Hitachi TM-3000 SEM.
Three frozen snails were thawed in RNAlater (Invitrogen, USA) on ice and the intestine was carefully dissected for total DNA and RNA extraction. The total DNA of the intestine was extracted using the E.Z.N.A. Mollusc DNA Kit (Omega Bio-tek, Georgia, USA) and then purified using Genomic DNA Clean & Concentrator-10 Kit (Zymo Research, CA, USA) in accordance with the manufacturer’s protocol. The total RNA was extracted using TRIzol (Invitrogen, USA) following the manufacturer’s protocol and prepared for RNA-Seq. Nucleic acid quality was evaluated using agarose gel electrophoresis and a BioDrop µLITE (BioDrop, Holliston, MA, USA). Nucleic acid concentrations were quantified using a Qubit fluorometer v3.0 (Thermo Fisher Scientific, Singapore).
Library construction and sequencing
The total DNA of the intestine was submitted to Illumina sequencing platform. A library with a 350 bp insert size was constructed following the standard protocol provided by Illumina (San Diego, CA, USA). After paired-end sequencing of the library was conducted at Novogene (Beijing, China), approximately 12 Gb of Illumina Novaseq reads with a read length of 150 bp were generated from each of the three intestine specimens for metagenome analysis.
As the RNA of the intestine includes sequences from the host and microbes, 250–300 bp insert stranded-specific library of each intestine specimen was constructed using Ribo-Zero Magnetic Kit to sequence eukaryotic and microbial RNAs. The meta-transcriptome sequencing of the intestine was conducted on the Illumina Novaseq platform at Novogene to produce 150 bp paired-end reads. Approximately 10 Gb of reads were generated from each intestine specimen.
Microbial metagenome assembly, annotation, and functional analyses
For microbial metagenome assembly of the intestine, Trimmomatic v0.39 [57] and FastUniq [58] were used to trim the Illumina reads and remove duplicates introduced by polymerase chain reaction (PCR) amplification. Four metagenomes were assembled from the eukaryotic reads obtained from each snail individual and the merged dataset of all three individuals. The scaffolds of these metagenomes were individually aligned to the host’s whole genome [17] by using minimap2 [59], which returned the alignment rates of 95–97% and thus confirmed that they originated from Alviniconcha marisindica host. The host’s interference in the analysis of intestinal content was minimised by removing reads that aligned to the host genome by using Bowtie2 [60] before the assembly. The abundance and taxonomic classification of intestinal metagenomic microbial sequences were carried out using Kaiju [61] on the basis of completely assembled and annotated reference genomes of Archaea, Bacteria and Viruses from the NCBI RefSeq database. The clean reads left were assembled using MEGAHIT [62] and metaSPAdes v3.13.1 [63] respectively. Prodigal v2.6.3 [64] was used to predict and translate the coding sequences in the intestinal metagenome. Then, BLASTp was used to align the candidate sequences to the NCBI NR protein database and the taxonomic assignment of each protein was imported to MEGAN v5.7.0 [65] by using the lowest common ancestor (LCA) method with the parameters of Min Score 50, Max Expected 0.01, Top Percent 5 and LCA Percent 100. On the basis of the taxonomic results, the microbial protein sequences were selected for further gene functional analysis, following the gene annotation pipeline described above. Blast2GO [66] and EggNOG mapper [67] were applied to assign GO and COG terms to the intestinal prokaryotic protein sequences, KAAS [68] was used to annotate KEGG numbers of prokaryotic protein sequences by using the single-directional best hit method. Specific metabolic pathways of multiple microorganisms were combined to reconstruct microbial meta-pathways through the KEGG mapper. The associated species annotation of the corresponding protein-encoding genes provides a reference for determining which bacteria contribute to a given enzymatic reaction in the meta-pathway.
Statistical analyses
Data were statistically assessed by using the results from the metagenomics RAST server (MG-RAST) to compare the microbial communities in the gut of Alviniconcha snails with other snails or environments [69]. Whole genome sequencing (WGS) and 16S amplicon samples were mapped against 16S rRNA database RDP, the number of hits of the reads of 16S amplicon and WGS samples mapped against the RDP database were calculated respectively. After automatic normalisation by MG-RAST, the relative abundance of microbial taxonomic groups detected in the WGS and 16S amplicon samples can be compared. The α-diversity of the annotated intestinal metagenomic samples could be estimated from the distribution of the species-level annotations. Annotated species richness is the number of distinct species annotations in the combined MG-RAST data set. Principal coordinates analysis (PCoA) of the microbial communities and Rarefaction curve of annotated species richness were calculated in MG-RAST. Comparison analysis was performed using paired metagenomic samples and gut metagenomics of other snails and microbiomes of deep-sea vent environments from the MG-RAST server were collected for comparison (Additional file 2: Table S1). Differences between the samples were assessed using ANOVA.
Microbial co-occurrence analysis
Network analysis was conducted on the basis of relative abundance of microorganisms in three intestinal samples. The abundance and taxonomic classification of intestinal metagenomic microbial sequences were carried out using Kaiju [61]. To reduce the complexity of the datasets, only relative abundances higher than 0.5% were included. All pairwise Spearman’s rank correlations between genera of three intestinal samples were calculated in the R package ‘picante’. Only robust (r > 0.8 or r < − 0.8) and statistically significant correlations (P < 0.01) were shown in the network. Network visualisation and modular analysis were conducted using Gephi v0.9.2. Nodes in the reconstructed network represent the genera in three intestinal samples, whereas the edges (connections) correspond to a strong (r > 0.8 or r < − 0.8) and significant (P < 0.01) correlation between nodes. The clustering coefficient was 1.0 (the degree to which nodes tend to cluster together) and the modularity index was 0.5 (values > 0.4 suggest that the network has a modular structure [70]).
Gene expression level quantification
For the intestinal meta-transcriptome data, the raw reads were trimmed with Trimmomatic v0.39 [57]. A Salmon index built for the transcripts of the host was obtained and translated from its complete genome data [17] and then the trimmed reads were quantified directly against this index and expressed in transcripts per million (TPM) by Salmon [71]. The read counts for genes were also included in the quantification results. For gut microbiome, the same pipeline was used, with a Salmon index built for the transcripts of microbes obtained and translated from their metagenome data; then, the trimmed reads were quantified directly against this index and expressed in TPM by using Salmon [71]. The gene expression levels of the intestine and its microbiome were produced by using this quantification method and the consistency of gene expression levels for the intestine from three snail individuals indicated the high accuracy of the transcript-level quantification method.
Transcriptome data from the foot, neck, mantle, DG and gills of A. marisindica [17] were compared to show gene differential expression in the intestine. DEGs were determined by DESeq2 using the normalisation method of Loess, a minimum read count of 10 and a paired test (n = 5). Genes considered to be highly expressed in the intestine were overexpressed with over two-fold changes and FDR < 0.05 when compared with other tissue types. WEGO (http://wego.genomics.org.cn/cgi-bin/wego/index.pl) was used to plot GO annotations of highly expressed genes in the intestine. Statistically overrepresented GO terms in the intestine were identified through the topGO package in R session (10.18129/B9.bioc.topGO). The GO enrichment network was visualised using the Cytoscape application [72]. The DEGs of the intestines are shown Additional file 3: Dataset S1.
Availability of data and materials
All raw sequencing data generated in the present study are available from NCBI via the accession numbers SRR11781645, SRR11781642, SRR11781640, SRR11781646, SRR11781639, SRR11781643 and BioSamples via accession numbers SAMN14907815, SAMN14907816, SAMN14907817. The datasets supporting the Conclusions of this article are included within the article (and its Additional files 1–4).
Abbreviations
- BCV:
-
Biological coefficient of variation
- LPS:
-
Lipopolysaccharide
- CLRs:
-
C-type lectin receptors
- CR:
-
Carlsberg Ridge
- CPs:
-
Capsular polysaccharides
- C1q:
-
Complement component 1q
- DG:
-
Digestive gland
- DEGs:
-
Differentially expressed genes
- FA:
-
Fatty acid
- GHs:
-
Glycoside hydrolases
- LAB:
-
Lactic acid bacteria
- PGRPs:
-
Peptidoglycan recognition proteins
- rTCA:
-
Reverse tricarboxylic acid
- RLRs:
-
RIG-I-like receptors
- Sia:
-
Sialic acids
- SLPs:
-
Surface layer proteins
- SIAE:
-
Sialate O-acetylesterase
- TLR:
-
Toll-like receptor
References
Aronson HS, Zellmer AJ, Goffredi SK. The specific and exclusive microbiome of the deep-sea bone-eating snail, Rubyspira osteovora. FEMS Microbiol. Ecol. 2017; 93: fiw250.
Durand L, Roumagnac M, Cueff-Gauchard V, Jan C, Guri M, Tessier C, Haond M, Crassous P, Zbinden M, Arnaud-Haond S, Cambon-Bonavita MA. Biogeographical distribution of Rimicaris exoculata resident gut epibiont communities along the Mid-Atlantic Ridge hydrothermal vent sites. FEMS Microbiol Ecol. 2015; 91: fiv101.
Suzuki Y, Sasaki T, Suzuki M, Nogi Y, Miwa T, Takai K, Nealson KH, Horikoshi K. Novel chemoautotrophic endosymbiosis between a member of the Epsilonproteobacteria and the hydrothermal-vent gastropod Alviniconcha aff. hessleri (Gastropoda: Provannidae) from the Indian Ocean. Appl. Environ. Microbiol. 2005; 71: 5440–5450.
Ponnudurai R, Kleiner M, Sayavedra L, Petersen JM, Moche M, Otto A, Becher D, Takeuchi T, Satoh N, Dubilier N, Schweder T. Metabolic and physiological interdependencies in the Bathymodiolus azoricus symbiosis. ISME J. 2017;11:463–77.
Dubilier N, Bergin C, Lott C. Symbiotic diversity in marine animals: the art of harnessing chemosynthesis. Nat Rev Microbiol. 2008;6:725.
Childress J, Girguis PR. The metabolic demands of endosymbiotic chemoautotrophic metabolism on host physiological capacities. J Exp Biol. 2011;214:312–25.
Riou V, Colaço A, Bouillon S, Khripounoff A, Dando P, Mangion P, Chevalier E, Korntheuer M, Santos RS, Dehairs F. Mixotrophy in the deep sea: a dual endosymbiotic hydrothermal mytilid assimilates dissolved and particulate organic matter. Mar Ecol Prog Ser. 2010;405:187–201.
Page HM, Fiala-Medioni A, Fisher CR, Childress JJ. Experimental evidence for filter-feeding by the hydrothermal vent mussel, Bathymodiolus thermophilus. Deep Sea Res Part I Oceanogr Res Pap. 1991; 38: 1455–1461.
Miyazaki J, Ikuta T, Watsuji TO, Abe M, Yamamoto M, Nakagawa S, Takaki Y, Nakamura K, Takai K. Dual energy metabolism of the Campylobacterota endosymbiont in the chemosynthetic snail Alviniconcha marisindica. ISME J. 2020;14:1273–89.
Sun J, Zhang Y, Xu T, Zhang Y, Mu H, Zhang Y, Lan Y, Fields CJ, Hui JH, Zhang W, Li R, Nong W, Cheung FKM, Qiu JW, Qian PY. Adaptation to deep-sea chemosynthetic environments as revealed by mussel genomes. Nat Ecol Evol. 2017;1:1–7.
Goffredi SK, Warén A, Orphan VJ, Van Dover CL, Vrijenhoek RC. Novel forms of structural integration between microbes and a hydrothermal vent gastropod from the Indian Ocean. Appl Environ Microbiol. 2004;70:3082–90.
Barko P, McMichael M, Swanson K, Williams D. The gastrointestinal microbiome: a review. J Vet Intern Med. 2018;32:9–25.
Johnson SB, Warén A, Tunnicliffe V, Dover CV, Wheat CG, Schultz TF, Vrijenhoek RC. Molecular taxonomy and naming of five cryptic species of Alviniconcha snails (Gastropoda: Abyssochrysoidea) from hydrothermal vents. Syst Biodivers. 2015;13:278–95.
Beinart RA, Luo C, Konstantinidis KT, Stewart FJ, Girguis PR. The bacterial symbionts of closely related hydrothermal vent snails with distinct geochemical habitats show broad similarity in chemoautotrophic gene content. Front Microbiol. 2019;10:1818.
Endow K, Ohta S. The symbiotic relationship between bacteria and a mesogastropod snail, Alviniconcha hessleri, collected from hydrothermal vents of the Mariana Back-Arc Basin. Bull Japan Soc Microbial Ecol. 1989;3:73–82.
Windoffer R, Giere O. Symbiosis of the hydrothermal vent gastropod Ifremeria nautilei (Provannidae) with endobacteria-structural analyses and ecological considerations. Biol Bull. 1997;193:381–92.
Yang Y, Sun J, Chen C, Zhou Y, Lan Y, Van Dover CL, Wang C, Qiu JW, Qian PY. Tripartite holobiont system in a vent snail broadens the concept of chemosymbiosis. bioRxiv 2020.
Warèn A, Bouchet P. New records, species, genera, and a new family of gastropods from hydrothermal vents and hydrocarbon seeps. Zool Scr. 1993;22:1–90.
Hansen AK, Moran NA. Aphid genome expression reveals host–symbiont cooperation in the production of amino acids. Proc Natl Acad Sci USA. 2011;108:2849–54.
Yang Y, Sun J, Sun Y, Kwan YH, Wong WC, Zhang Y, Xu T, Feng D, Zhang Y, Qiu JW, Qian PY. Genomic, transcriptomic, and proteomic insights into the symbiosis of deep-sea tubeworm holobionts. ISME J. 2020;14:135–50.
Zhou Y, Chen C, Sun Y, Watanabe HK, Zhang R, Wang C. Amphisamytha (Annelida: Ampharetidae) from Indian Ocean hydrothermal vents: Biogeographic implications. Deep Sea Res Part I Oceanogr Res Pap. 2019; 154: 103148.
Wang Y, Han X, Petersen S, Frische M, Qiu Z, Li H, Li H, Wu Z, Cui R. Mineralogy and trace element geochemistry of sulfide minerals from the Wocan Hydrothermal Field on the slow-spreading Carlsberg Ridge, Indian Ocean. Ore Geol Rev. 2017;84:1–9.
Li LH, Lv S, Lu Y, Bi DQ, Guo YH, Wu JT, Yue ZY, Mao GY, Guo ZX, Zhang Y, Tang YF. Spatial structure of the microbiome in the gut of Pomacea canaliculata. BMC Microbiol. 2019;19:273.
Cardoso AM, Cavalcante JJ, Cantão ME, Thompson CE, Flatschart RB, Glogauer A, Scapin SM, Sade YB, Beltrão PJ, Gerber AL, Martins OB. Metagenomic analysis of the microbiota from the crop of an invasive snail reveals a rich reservoir of novel genes. PLoS One 2012; 7: e48505.
Nakai R, Abe T, Takeyama H, Naganuma T. Metagenomic analysis of 0.2-μm-passable microorganisms in deep-sea hydrothermal fluid. Mar. Biotechnol. 2011; 13: 900–908.
Warén A, Bouchet P, von Cosel R, Desbruyères D, Segonzac M, Bright M. Gastropoda: Neomphalina. Handbook of deep-sea hydrothermal vent fauna (second completely revised edition.). Denisia 2006; 18: 104–120.
Labonté JM, Pachiadaki M, Fergusson E, McNichol J, Grosche A, Gulmann LK, Vetriani C, Sievert SM, Stepanauskas R. Single cell genomics-based analysis of gene content and expression of prophages in a diffuse-flow deep-sea hydrothermal system. Front Microbiol. 2019;10:1262.
Inagaki F, Takai K, Nealson KH, Horikoshi K. Sulfurovum lithotrophicum gen. nov., sp. nov., a novel sulfur-oxidizing chemolithoautotroph within the epsilon-proteobacteria isolated from Okinawa trough Hydrothermal sediments. Int J Syst Evol Microbiol. 2004; 54:1477–1482.
Mino S, Kudo H, Arai T, Sawabe T, Takai K, Nakagawa S. Sulfurovum aggregans sp. nov., a hydrogen-oxidizing, thiosulfate-reducing chemolithoautotroph within the Epsilonproteobacteria isolated from a deep-sea hydrothermal vent chimney, and an emended description of the genus sulfurovum. Int J Syst Evol Microbiol. 2014; 64:3195–3201.
Giovannelli D, Chung M, Staley J, Starovoytov V, Le Bris N, Vetriani C. Sulfurovum riftiae sp. nov., a mesophilic, thiosulfate-oxidizing, nitrate-reducing chemolithoautotrophic epsilonproteobacterium isolated from the tube of the deep-sea hydrothermal vent polychaete Riftia pachyptila. Int J Syst Evol Microbiol. 2016; 66: 2697–2701.
Takai K, Campbell BJ, Cary SC, Suzuki M, Oida H, Nunoura T, Hirayama H, Nakagawa S, Suzuki Y, Inagaki F, Horikoshi K. Enzymatic and genetic characterization of carbon and energy metabolisms by deep-sea hydrothermal chemolithoautotrophic isolates of Epsilonproteobacteria. Appl Environ Microbiol. 2005;71:7310–20.
Nakagawa S, Takai K, Inagaki F, Horikoshi K, Sako Y. Nitratiruptor tergarcus gen. nov., sp. nov. and Nitratifractor salsuginis gen. nov., sp. nov., nitrate-reducing chemolithoautotrophs of the ε-Proteobacteria isolated from a deep-sea hydrothermal system in the Mid-Okinawa Trough. Int J Syst Evol. 2005; 55: 925–33.
Barka EA, Vatsa P, Sanchez L, Gaveau-Vaillant N, Jacquard C, Klenk HP, Clément C, Ouhdouch Y, van Wezel GP. Taxonomy, physiology, and natural products of Actinobacteria. Microbiol Mol Biol Rev. 2016;80:1–43.
Déjean G, Blanvillain-Baufumé S, Boulanger A, Darrasse A, Dugé de Bernonville T, Girard AL, Carrére S, Jamet S, Zischek C, Lautier M, Solé M, Büttner D, Jacques MA, Lauber E, Arlat M. The xylan utilization system of the plant pathogen Xanthomonas campestris pv campestris controls epiphytic life and reveals common features with oligotrophic bacteria and animal gut symbionts. New Phytol. 2013; 198: 899–915.
de Castro AL, Vollú RE, Peixoto RS, Grigorevski-Lima AL, Coelho RR, Bon EP, Rosado AS, Seldin L. Cellulolytic potential of a novel strain of Paenibacillus sp. isolated from the armored catfish Parotocinclus maculicauda gut. Braz J Microbiol. 2011; 42: 1608–15.
Garcia R, Gemperlein K, Müller R. Minicystis rosea gen. nov., sp. nov., a polyunsaturated fatty acid-rich and steroid-producing soil myxobacterium. Int. J. Syst. Evol. 2014; 64: 3733–42.
D’Armas H, Yáñez D, Reyes D, Salazar G. Fatty acids composition of the marine snails Phyllonotus pomum and Chicoreus brevifrons (Muricidae). Rev Biol Trop. 2010;58:645–54.
Dailey FE, McGraw JE, Jensen BJ, Bishop SS, Lokken JP, Dorff KJ, Ripley MP, Munro JB. The microbiota of freshwater fish and freshwater niches contain omega-3 fatty acid-producing Shewanella species. Appl Environ Microbiol. 2016;82:218–31.
Tocher DR. Metabolism and functions of lipids and fatty acids in teleost fish. Rev Fish Sci. 2003;11:107–84.
Budge SM, Devred E, Forget MH, Stuart V, Trzcinski MK, Sathyendranath S, Platt T. Estimating concentrations of essential omega-3 fatty acids in the ocean: supply and demand. ICES J Mar Sci. 2014;71:1885–93.
Koleva Z, Dedov I, Kizheva J, Lipovanska R, Moncheva P, Hristova P. Lactic acid microflora of the gut of snail Cornu aspersum. Biotechnol Equip. 2014;28:627–34.
Pessione E. Lactic acid bacteria contribution to gut microbiota complexity: lights and shadows. Front Cell Infect Microbiol. 2012;2:86.
Ren D, Gong S, Shu J, Zhu J, Liu H, Chen P. Effects of mixed lactic acid bacteria on intestinal microbiota of mice infected with Staphylococcus aureus. BMC Microbiol. 2018;18:109.
Li Z, Quan G, Jiang X, Yang Y, Ding X, Zhang D, Wang X, Hardwidge PR, Ren W, Zhu G. Effects of metabolites derived from gut microbiota and hosts on pathogens. Front Cell Infect Microbiol. 2018;8:314–314.
den Besten G, Van Eunen K, Groen AK, Venema K, Reijngoud DJ, Bakker BM. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J Lipid Res. 2013;54:2325–40.
Tran NT, Li Z, Wang S, Zheng H, Aweya JJ, Wen X, Li S. Progress and perspectives of short-chain fatty acids in aquaculture. Rev Aquac. 2020;12:283–98.
Schroeder BO. Fight them or feed them: how the intestinal mucus layer manages the gut microbiota. Gastroenterol Rep. 2019;7:3–12.
Dar MA, Pawar KD, Pandit RS. Gut microbiome analysis of snails: a biotechnological approach. Organ Mol Malacology Intech. 2017;16:189–217.
Newton IL, Woyke T, Auchtung TA, Dilly GF, Dutton RJ, Fisher MC, Fontanez KM, Lau E, Stewart FJ, Richardson PM, Barry KW. The Calyptogena magnifica chemoautotrophic symbiont genome. Science. 2007;315:998–1000.
Futoma-Koloch B. Immune response against bacterial lipopolysaccharide. J Mol Immunol. 2016;1:2.
Sára M, Sleytr UB. S-layer proteins. J Bacteriol Res. 2000;182:859–68.
Zamze S, Martinez-Pomares L, Jones H, Taylor PR, Stillion RJ, Gordon S, Wong SY. Recognition of bacterial capsular polysaccharides and lipopolysaccharides by the macrophage mannose receptor. J Biol Chem. 2002;277:41613–23.
Spinosa MR, Progida C, Tala A, Cogli L, Alifano P, Bucci C. The Neisseria meningitidis capsule is important for intracellular survival in human cells. Infect Immun. 2007;75:3594–603.
Taverniti V, Stuknyte M, Minuzzo M, Arioli S, De Noni I, Scabiosi C, Cordova ZM, Junttila I, Hämäläinen S, Turpeinen H, Mora D. S-layer protein mediates the stimulatory effect of Lactobacillus helveticus MIMLh5 on innate immunity. Appl Environ Microbiol. 2013;79:1221–31.
Detree C, Haddad I, Demey-Thomas E, Vinh J, Lallier FH, Tanguy A, Mary J. Global host molecular perturbations upon in situ loss of bacterial endosymbionts in the deep-sea mussel Bathymodiolus azoricus assessed using proteomics and transcriptomics. BMC Genom. 2019;20:109.
Janeway Jr CA, Travers P, Walport M, Shlomchik MJ. The complement system and innate immunity. Immunobiology: The Immune System in Health and Disease. 5th edition 2001. Garland Science.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20.
Xu H, Luo X, Qian J, Pang X, Song J, Qian G, Chen J, Chen S. FastUniq: a fast de novo duplicates removal tool for paired short reads. PLoS One 2012; 7: e52249.
Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018;34:3094–100.
Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357.
Menzel P, Ng KL, Krogh A. Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nat Commun. 2016;7:11257.
Li D, Liu CM, Luo R, Sadakane K, Lam TW. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics. 2015;31:1674–6.
Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19:455–77.
Hyatt D, Chen GL, LoCascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010;11:119.
Huson DH, Mitra S, Ruscheweyh HJ, Weber N, Schuster SC. Integrative analysis of environmental sequences using MEGAN4. Genome Res. 2011;21:1552–60.
Götz S, García-Gómez JM, Terol J, Williams TD, Nagaraj SH, Nueda MJ, Robles M, Talón M, Dopazo J, Conesa A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008;36:3420–35.
Huerta-Cepas J, Forslund K, Coelho LP, Szklarczyk D, Jensen LJ, Von Mering C, Bork P. Fast genome-wide functional annotation through orthology assignment by eggNOG-mapper. Mol Biol Evol. 2017;34:2115–22.
Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30.
Meyer F, Paarmann D, D’Souza M, Olson R, Glass EM, Kubal M, Paczian T, Rodriguez A, Stevens R, Wilke A, Wilkening J. The metagenomics RAST server: a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinform. 2008;9:386.
Newman MEJ. Modularity and community structure in networks. Proc Natl Acad Sci USA. 2006; 103: 8577.
Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods. 2017;14:417–9.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–504.
Acknowledgements
The authors wish to thank the operations team of the HOV Jiaolong, the captain and crew of vehicle R/V Xiangyanghong 9 for collecting the samples and Mr. Yanhong Lu for his assistance on microbial co-occurrence analysis.
Funding
This work was supported by grants from the China Ocean Mineral Resources Research and Development Association (DY135-E2-1-03); the Key Special Project for Introduced Talents Team of Southern Marine Science and Engineering Guangdong Laboratory (Guangzhou) (GML2019ZD0409); the Major Basic and Applied Basic Research Projects of Guangdong Province (2019B030302004-04); and the Hong Kong Branch of Southern Marine Science and Engineering Guangdong Laboratory (Guangzhou) (SMSEGL20SC01), which was awarded to P.Y.Q.; and the National Natural Science Foundation of China (grant No. 91951201).
Author information
Authors and Affiliations
Contributions
PYQ conceived the project. YY designed the experiments. YZ and CW collected the Alviniconcha snails. CC and JS identified the snails, carried out morphological investigations, and dissected the specimens. YY performed DNA extraction, RNA extraction, and data analyses. YY prepared the figures and tables and drafted the manuscript. CC, JS, JWQ, CVD and PYQ contributed to manuscript editing. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
. Figure S1. Rarefaction curves for gut microbial samples of three Alviniconcha marisindica individuals from the Wocan vent field. Figure S2. Gene Ontology (GO) enrichment network of differentially expressed genes (DEGs). The significantly (p-value < 0.01) enriched GO terms of selected highly expressed genes in the intestine of Alviniconcha marisindica are clustered in accordance with their functional category. The connecting pairs of nodes showing the intra-cluster and inter-cluster similarities of enriched terms. The colour code represents different cluster annotations. Each node represents an enriched term. Figure S3. Differentially expressed genes (DEGs) in the intestine and gill of Alviniconcha marisindica. Volcano plot and biological coefficient of variation (BCV) plot of DEGs in the intestine are identified by DESeq2 analysis. The log10 (FDR corrected p-values) are plotted against the log2 (FC) in gene expression. Upregulated genes by twofold or more and with a FDR corrected p-value < 0.05 are marked as blue dots, whilst down-regulated genes (FRD ≤ 2 with P < 0.05) are marked in red colour. Figure S4. Photograph of snail Alviniconcha marisindica collected from the Wocan hydrothermal field (WHF) and stored in absolute ethanol. Figure S5. SEM images of radula. Overview: (a) Alviniconcha marisindica (individual 01); scale bar = 300 μm (b) A. marisindica (individual 01); scale bars = 200 μm. Central and lateral teeth close-up: (c) A. marisindica (individual 01); scale bars = 200 μm (d) A. marisindica (individual 02); scale bars = 200 μm. Marginal teeth close-up: (e) A. marisindica (individual 02); scale bars = 30 μm.
Additional file 2
. Table S1. Metagenomic and 16S rRNA amplicon data of intestines of Alviniconcha marisindica from the Wocan field, intestines of giant land snails, intestines of deep-sea bone-eating, scavenger and predatory snails and environmental samples from deep-sea habitats. Table S2. Annotation and expression levels of genes involved in encoding representative exo-hydrolases in intestinal microbiomes of three A. marisindica individuals from the Wocan site.
Additional file 3
. Dataset S1 The annotation of highly expressed genes in the intestine of A. marisindica.
Additional file 4
. Dataset S2 The annotation of genes predicted from the metagenome of intestinal flora from three snail individuals.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yang, Y., Sun, J., Chen, C. et al. Metagenomic and metatranscriptomic analyses reveal minor-yet-crucial roles of gut microbiome in deep-sea hydrothermal vent snail. anim microbiome 4, 3 (2022). https://doi.org/10.1186/s42523-021-00150-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s42523-021-00150-z