
Abstract
There is only one species representing Magnoliopsida which is considered as native to the Antarctic, i.e., Antarctic pearlwort (Colobanthus quitensis). Although it was intensively studied toward the morphophysiological adaptation to extreme environmental conditions of that area, there is still a lack of sufficient data on its genetic variability. Nine C. quitensis populations from Chile and the Maritime Antarctic were sampled to estimate the pattern of genetic variation in relation to the geographic distribution of analyzed populations and postglacial history of the species. The retrotransposon-based DNA marker system used in our studies appeared to be effective in revealing genetic polymorphism between individuals and genetic differentiation among populations. Although the level of polymorphism was low (9.57%), the Analysis of Molecular Variance showed that overall population differentiation was high (FST = 0.6241) and revealed significant differentiation between the Northern and Southern Group of populations as well as the population from Conguillio Park. The observed genetic subdivision of C. quitensis populations was confirmed by Bayesian clustering and results of Principal Coordinates Analysis. The Southern Group of populations was characterized by generally higher genetic diversity, which was expressed by the values of the effective number of alleles, expected heterozygosity and by the distribution of private alleles. Our results suggest that the species may have survived the Last Glacial Maximum in refugia located both on the South American continent and in geographically isolated islands of the Maritime Antarctic, i.e., they support the concept of the multiregional origin of C. quitensis in Antarctica.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Patterns of population genetic structure revealed in the widely distributed plant species are shaped by the interaction of many factors, including natural processes such as climate fluctuations, natural barriers, life history (e.g., breeding system and life-form) as well as human impacts (Convey et al. 2014). Also, demographic events occurring during range contractions and expansions are responsible for modifications of the genome-wide patterns of population genetic diversity (Hewitt 2000; Excoffier et al. 2009). Genetic drift, founder, and bottleneck effects are expected to be especially intense during range expansion, causing gradients or even abrupt modulations in allele frequencies, genetic diversity and population structure (Klopfstein et al. 2006; Excoffier and Ray 2008; François et al. 2008). Expansions can affect the geographic distribution of both neutral and selectively important genetic variation and thus complicate the inferences drawn from clines in alleles or phenotypic traits (Vasemagi 2006; Keller et al. 2009) or genetic association mapping (Pritchard et al. 2000; Price et al. 2006). Therefore, a proper understanding of how expansion can shape diversity and population structure is crucial for both inferring from the demographic history of the species but also for studying the molecular basis and evolution of the locally adaptive traits in natural populations (Nielsen 2005; Wright and Gaut 2005).
There are a number of molecular phyleographic studies in which maternally inherited organellar DNA markers were used for determining the patterns of plant population genetic structure and inferring processes such as the number and locations of glacial refugia, past migration routes, and colonization dynamics (Comes and Kadereit 1998). The reason for this approach is that the organellar genomes are usually characterized by uniparental inheritance and generally does not undergo recombination, and therefore haplotypes remain mostly unchanged when passed to the next generation. Nevertheless, the organellar genome reveals only a single gene genealogy and since the genealogical process is highly variable among genes and is affected by demographic events or unexpected loss of lineages (Knowles and Maddison 2002) such analyses may reveal genealogical processes associated with single genes only, which are not necessarily characteristic for the whole species’ history.
On the contrary, nuclear DNA markers which are bi-parentally inherited allow for recombination, integrating many genealogical processes. Currently SSRs (Simple Sequence Repeats) or SNPs (Single Nucleotide Polymorphisms) are the markers of choice for many phyleographic studies (e.g., DeFaveri et al. 2013; Gao et al. 2013; Faivre-Rampant et al. 2016; Foote and Morin 2016), however due to the high cost and labor intensity involved in their development, they are available generally for model species and those which are economically important. For genetic studies on underutilized crops or plants for which genomic data is limited, several PCR (Polimerase Chain Reaction) marker systems are available, varying in complexity, reliability, and information generating capacity.
Recently, a versatile method of organism genotyping based on the use of transposon sequences has been developed. The iPBS method (inter Primer Binding Sites) is based on the virtually universal presence of a tRNA complement as a reverse transcriptase primer binding site (PBS) in LTR (Long Terminal Repeat) retrotransposons (Kalendar et al. 2010). The iPBS technique has been introduced as a powerful DNA fingerprinting technology which can be applied without the need of prior sequence knowledge (Kalendar et al. 2010). Moreover, iPBS markers are highly reproducible due to the length of primers and the high stringency achieved by the annealing temperature, and they have already found application in clone identification, genetic diversity analyses and phylogenetic studies (e.g., Smýkal et al. 2011; Mehmood et al. 2016; Özer et al. 2016). Colobanthus is often described as a genus showing southern circum-temperate distribution (e.g., West 1991). 26 species are recognized at present in the genus, with the major diversity, occurring in New Zealand—14 species (Sneddon 1999). It exhibits a Gondwanan distribution, occurring in South America, the sub-Antarctic islands, the Antarctic Peninsula, and the Australasian area including New Zealand, Australia, and Tasmania (West 1991). The most recent taxonomic studies in Colobanthus are scatter a descriptions of two new species from Australia: Colobanthus nivicola (Gray 1976) and Colobanthus curtisiae (West 1991) and a revision of Colobanthus quitensis (Kunth) Bartl., which is the most wide-ranging species in the genus, occurring from the Antarctic Peninsula (Greene and Greene 1963) north along the southern Andes mountains up to scattered locations northward to Mexico (Moore 1970; Parnikoza et al. 2011). Antarctic pearlwort—C. quitensis. and Antarctic hairgrass—Deschampsia antarctica Desv. are the two flowering plant species considered as native to the Maritime Antarctic. Due to the broad distribution range, both species undergo various selection forces which shape both their morphological and genetic variability. However, information on the genetic variation of these species is limited and devoted almost exclusively to D. antarctica (e.g., Chwedorzewska and Bednarek 2008; van de Wouw et al. 2008; Volkov et al. 2010; Chwedorzewska and Bednarek 2011; González et al. 2016). As regards C. quitensis, results of the genetic variation studies can be found so far in only a few papers, which suffer from limited geographic range or the low number of populations studied (Lee and Postle 1975; Gianoli et al. 2004; Acuña-Rodríguez et al. 2014). More recently, two more papers concerning C. quitensis genetics appeared, but they were focused on selected areas of the species genomics like the sequence of chloroplast genome (Kang et al. 2016) or genome size estimated by flow cytometry (Cuba-Díaz et al. 2017b).
Our previous studies on the genetic variation of C. quitensis (Androsiuk et al. 2015) encompassed the highest number of populations of the species analyzed so far in one study, i.e., eight populations from King George Island (Southern Shetland Islands). They represented diversified habitats of the Maritime Antarctic, which vary considerably when microclimatic conditions as well as soil moisture and its nutrient content are considered. The applied DNA markers (iPBS) revealed low genetic variability and moderate population differentiation. Moreover, the pattern of population differentiation corresponds well with their geographic location. This study demonstrated the usefulness of iPBS markers for genetic diversity studies in non-model plant species and confirmed our hypothesis that genetic variation revealed by this technique is influenced by the abiotic stress and thus shaped in the response to local environment conditions.
In this study, we used the iPBS markers to assess the range-wide genetic variation of C. quitensis in order to infer about the population history of the vicariants of C. quitensis in South America and the Maritime Antarctic. We aimed to address the following questions: (1) Do the iPBS markers reveal cryptic genetic structure in C. quitensis? (2) Do the levels of genetic diversity in small, isolated marginal populations differ from those in other populations?
Materials and methods
DNA extraction and genotyping
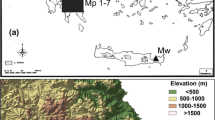
The research material consisted of 174 individuals of C. quitensis representing its natural stands (hereinafter referred to as populations) from South America and the Maritime Antarctic (South Shetland Islands) (Table 1; Fig. 1). C. quitensis from South America was collected from wet environments of both fresh and brackish water, in river deposits flooded by high tides and less frequent on coastal rocks. In the high mountain ranges, it was found mainly in bogs flooded by meltwater runoff. It is usually associated with other herbaceous species. The pPar and pV populations manifest a high degree of deterioration due to human activity: the presence of pitches for winter sports in pPar and heavy grazing in pV. In case of pPA and pL, the ample influence of human activity is observed: both populations are close to roads, especially the pL population is located very nearby to the sites with high industrial activity. In the Antarctic samples were taken from coastal zones, from areas with minor human influence. Each population was represented by 6–30 individuals. The number of sampled individuals was limited by the size of a given population. The plant material was dried after collection and stored at − 20 °C until DNA extraction in presence of silica gel. The DNA from each individual was extracted using Syngen Plant DNA Mini Kit. The quality of DNA was verified on 1% agarose gel and visualized by staining with 0.5 μg* mL−1 ethidium bromide, while the amount and purity of DNA samples were assessed spectrophotometrically.

Map depicting the location of the studied sampling sites of Colobanthus quitensis: a Geographic location of the populations from South America, b contour map of the Drake passage with c the enlargement area of South Shetland Archipelago with Elephant Island, King George Island and Livingston Island. pPar La Parva; pC Conguillio Park; pV La Vega; pL Laredo; pPA La Marisma; pOm Omora Park; pEl Elephant Is.; pA Arctowski; pBy Byers
Initially, we screened 20 iPBS primers and their combinations for C. quitensis (Kalendar et al. 2010). Six of the 20 iPBS primers which gave polymorphic, clearly identifiable and repeatable bands were selected for further analyses. The PCR was performed with the 6 iPBS primers applied individually or in combination with two primers (Table 2), according to the procedure described in Androsiuk et al. (2015). The amplification products were analyzed by gel electrophoresis in 1.5% agarose gels stained with 0.5 μg* mL−1 ethidium bromide.
Data analysis
All amplification products (bands), that could be reliably read were treated as single dominant loci and scored either present (1) or absent (0) across genotypes (Online Resources 1). On the basis of the obtained binary matrix of bands the following genetic parameters were estimated: effective number of alleles (NB), the percentage of polymorphic bands (P), Shannon’s Information Index (I), and expected heterozygosity (He). Pairwise FST values among all analyzed C. quitensis populations were also estimated. The abovementioned calculations were performed with GenAlEx 6.5 software (Peakall and Smouse 2006, 2012), except pairwise FST values which were estimated using Arlequin 3.5 software (Excoffier et al. 2005).
In order to infer about the geographic pattern of genetic variation among C. quitensis populations, two methods were used. The first approach was a model-based clustering method using the Bayesian analysis with the STRUCTURE ver. 2.3.4 software (Pritchard et al. 2000). The model assigns individual multilocus genotypes probabilistically to a user-defined number of clusters (K), achieving linkage equilibrium within clusters (Pritchard et al. 2000). We conducted 10 replicate runs for each K, ranging from 1 to 9. Each run consisted of a burn-in of 500,000 iterations, followed by data collection over 500,000 iterations. The analysis, with the implemented admixture model, was conducted without any prior information on the original population of each sampled individual. In order to determine the optimal number of clusters (K), an ad hoc statistic ΔK (Evanno et al. 2005) was used. The ΔK was evaluated in Structure Harvester ver. 0.6.94 (Earl and Vonholdt 2012). The second method was a Principal Coordinates Analysis (PCoA), based on the matrix of Euclidean distances between individuals from all analyzed populations, performed in PAST software (Hammer et al. 2001).
Hierarchical Analysis of Molecular Variance (AMOVA; Excoffier et al. 1992) was carried out using Arlequin 3.5. For that analysis, the iPBS data were treated as haplotypic, comprising a combination of alleles at one or several loci (Excoffier et al. 2005). The significance of the fixation indices was tested using a non-parametric permutation approach (Excoffier et al. 1992).
The spatial genetic structure was investigated by testing the significance of isolation by distance (IBD) using a Mantel test with 9999 permutations of the relationship between the matrix of pairwise FST/(1 − FST) and that of the logarithm of geographical distance between populations (Rousset 1997), using GenAlEx 6.5.
Results
Efficiency of applied PCR primers
Our analysis of C. quitensis populations, using six iPBS primers/primers combination, yielded 137 clearly distinguished bands (Table 2). The highest number, 27 bands, was revealed by iPBS2231, whereas the lowest number of bands (20) was scored for iPBS2228 and iPBS2076×2085 primers combination. The average number of bands per primer was 22.83. Out of all identified loci, 53 (38.68%) were polymorphic.
Of a total of 137 scored bands, eight (5.84%) were represented as private bands—i.e., observed only in one population and absent in the others. Three private bands were revealed by each of the following primers: iPBS2085 and iPBS2231; whereas primer iPBS2378 revealed two (Table 3). In this context, pEl (Elephant Island) population appeared as the most abundant in private bands—six of them were scored in individuals representing that population. The remaining two private bands were characteristic for pPA (La Marisma) population. Moreover, one of the bands revealed for pEl population (iPBS2378-12), was characteristic for all individuals representing that sampling site, and thus may be considered as a potentially diagnostic marker for that population. In the case of the other private bands, they were scored only for a few (1–5) individuals from a given population.
Genetic diversity and differentiation
The iPBS markers revealed both the presence of genetic polymorphism between individuals within populations and genetic variation between populations (Table 4). The number of iPBS bands ranged from 122, for pPar, pC, and pV populations, to 131 for pPA and pEl populations. The highest number of polymorphic bands was scored for pPA population (26.28%) whereas the lowest polymorphism was observed for pV and pBy populations (1.46%). The genetic variation was assessed with two parameters: Shannon`s Information Index and expected heterozygosity, and in both cases the highest values were observed for pPA population from La Marisma, whereas the lowest—for pV population (La Vega).
The Bayesian clustering revealed that ΔK, the second-order rate of change of the likelihood function with respect to K, has a maximum at K = 3 (Fig. 2). Consequently, we chose K = 3 as the optimal number of clusters of the uppermost hierarchical level of population structure. The first cluster consists of pPA, pOm populations from the southern edges of the South American continent and pA, pEl, pBy populations from the South Shetland Islands, the pPar, pV, and pL populations were gathered in the second cluster, whereas pC population form solitary, third cluster.

The uppermost hierarchical level of genetic structure of Colobanthus quitensis from Chile and Maritime Antarctic using STRUCTURE (Pritchard et al. 2000). The values of second-order rate of change of L (K), ΔK, of data between successive K values
The AMOVA results revealed that most of the described genetic variation occurred between populations (59.48%), whereas the remaining 40.52% of variation was attributed to the variation among individuals within populations (Table 5). With the purpose of investigating how the region of origin of each population (affiliation to one of the two clusters revealed by STRUCTURE) influences the partition of revealed genetic variation of C. quitensis, further analysis of variance was performed using the region of origin as an additional factor. The AMOVA revealed that while the most genetic variation still occurs between populations (35.65%), and among individuals within populations (37.59%), there is a significant variation (26.76%) among geographical regions (Table 5).
PCoA indicated that 50.89% of variation was explained by the first three components (20.86, 19.75, and 10.28%, respectively). Figure 3 illustrates the projection of the analyzed populations on the first two axes. The grouping revealed by PCoA pointed on the close relationship between three populations from Arctowski (pA), Elephant Island (pEl), and Byers (pBy) representing the South Shetland Islands. The individuals representing Populations La Marisma (pPA) and Omora Park (pOm) from South America form two the most dispersed clouds, which overlap with each other and also with populations from South Shetlands Islands. The high dispersion of individuals from pPA and pOm point on the high level of genetic variation found within that populations. Individuals from populations La Parva (pPar) and Laredo (pL) form two overlapping clouds which departed from the others along the Coordinate 1. The most distinct character is shown by Conguillio Park (pC) which individuals departed from the others along Coordinate 2. The individuals representing La Vega population form the smallest and the densest cloud of individuals in the center of Fig. 3.

Plot of Coordinate 1 versus Coordinate 2 obtained by Principal Coordinates Analysis (PCoA) based on Euclidean distances between all individuals from nine Colobanthus quitensis populations. pPar La Parva (plus sign); pC Conguillio Park (open square); pV La Vega (filled square); pL Laredo (open circle); pPA La Marisma (filled circle); pOm Omora Park (asterisk); pEl Elephant Is.(times); pA Arctowski (open triangle); pBy Byers (open diamond)
A Mantel test showed the highly significant correlation between genetic divergence, denoted by pairwise FST/(1 − FST), and the logarithm of the geographical distances between populations (R2 = 0.1299, p < 0.016) (Fig. 4). Pairwise FST/(1 − FST) values plotted against the logarithm of geographical distances between populations indicated that the values increased with the distance between them (IBD).

Analysis of isolation by distance. The pairwise FST/(1 − FST) values are plotted against the logarithm of the geographical distances (km) between studied populations of Colobnathus quitensis
Discussion
Genetic diversity and differentiation of Colobanthus quitensis
Colobanthus quitensis geographic range spreads over thousands of kilometers from Mexico (17°N) to the Antarctic Peninsula (68°S) and from sea level to 4200 m a.s.l. (Moore 1970). Our studies covered the area of Chile and the Maritime Antarctic from 33°S to 62°S and spread over 3 300 km, which is the distance between the most distant pPar and pBy populations. In Chile, C. quitensis can be found along the Andes, usually in bogs at high altitudes in the north, but close to sea level in the south (Moore 1970; Hoffmann et al. 1998). In the Antarctic, the sites of C. quitensis occurrence are disjunct, situated mainly on the Antarctic islands and the Antarctic Peninsula coastal ice-free areas, often separated from one another by natural barriers (open sea, glacial, or mountains) limiting the exchange of gene pool (Lewis-Smith 2003).
Colobanthus. quitensis became an interesting subject of morphophysiological investigations aiming to identify ecotypic variation (Gianoli et al. 2004), to analyze its reproduction performance (Giełwanowska et al. 2011; Sanhueza et al. 2017) or identify the characteristic endophytic fungal communities (Santiago et al. 2012, 2017). Moreover, as the only representative of Magnoliopsida that grows in the extreme environmental conditions of the Antarctic, C. quitensis was extensively studied in terms of the morphological, physiological, and biochemical basis of adaptation to extremely cold climate, ample thermal oscillations, short vegetation season, high UV-B radiation, and salinity (Bravo et al. 2007; Bascunan-Godoy et al. 2012; Navarrete-Gallegos et al. 2012; Cuba-Díaz et al. 2017a). In contrast, the genetic diversity of this species is still poorly studied and deserves more attention.
Our data on the genetic variation of C. quitensis showed that on average 9.57% of the bands revealed by iPBS primers were polymorphic. However, the uneven distribution of the revealed polymorphism between the analyzed population needs to be emphasized here—the value of this parameter ranged from 26.28% in pPA to 1.46% in pV and pBy. The level of polymorphism seems to be associated here with a sample size for individual populations: the highest polymorphism was found in pPA population, which was represented by 30 individuals, whereas the lowest number of polymorphic loci was characteristic for C. quitensis stands represented by 10 and 6 individuals, pV and pBy respectively. However, our data show also that even populations represented by a relatively low number of individuals, e.g., pEl (6 plants) or pOm (7 plants), can reveal higher polymorphism than populations which consist of 4-5 times higher number of individuals, i.e., pC and pL. Similarly, uneven distribution of polymorphism can be found in our previous studies on C. quitensis from King George Island, where the level of revealed polymorphism was on average 14.25% (Androsiuk et al. 2015). In the studies of other authors, the average level of polymorphism for iPBS markers was much higher and reached 85.7% for guava accessions (Mehmood et al. 2013), 86.3% for grape varieties (Guo et al. 2014) or even 97.4% for Myrica rubra (Chen and Liu 2014). So far the lowest level of polymorphism of iPBS markers (4.88%) was reported by Baránek et al. (2012), but their analyses aimed at genetic identification of clones of the apricot cultivar.
The AMOVA revealed that the genetic variation was almost equally partitioned between populations (35.65%) and among individuals within studied populations (37.59%); remaining 26.76% was observed between geographical regions with a high fixation index (FST) pointing at a low historical gene flow between them. Previously high genetic variability between populations (71%) was revealed by AMOVA in Acuña-Rodríguez et al. (2014), who describe the results of the genetic study of C. quitensis from populations separated by the Drake Passage; the remaining 29% of revealed genetic variation was portioned between individuals. The results obtained by van de Wouw et al. (2008) for another flowering plant from the Antarctic region, D. antarctica, reveal the opposite situation: 74.6% of detected genetic variation was portioned among studied regions and only 15.1% among populations. It needs to be emphasized that the study area covered an extremely large geographic range which included the Indian Ocean, South Georgia, the Falklands, and Antarctic Zone. However, when only the regions within the Antarctic Zone were considered, the situation changed significantly: 45.6% of the variation was found among populations within Antarctic sites and 33.9% among Antarctic sites, the rest of variation was portioned among individuals within populations. High values of the fixation index also indicated low historical gene flow (van de Wouw et al. 2008).
The results of model-based clustering analysis showed that there was a clear gene pool subdivision within the nine populations of C. quitensis. According to STRUCTURE, pPA, pOm, pBy, pA, and pEl populations form one cluster (hereinafter called the Southern Group), C. quitensis from pL, pV, and pPar form another one (further called the Norther Group), whereas population pC departed from the others and form solitary third cluster PCoA revealed a similar pattern of population subdivision, where an individual character of population pC was also observed. The unique character of pC population was previously revealed by Cuba-Díaz et al. (2017b). The authors, using flow cytometry, estimated the genome size of three C. quitensis populations and found that mountainous populations from pC present a smaller genome size (0.84 pg), almost half of that found in the other studied pA and pPA populations originating from lower altitudes (1.95 pg), which may suggest that pA and pPA are tetraploids. This variation in genome size could be attributed to ecographic, adaptive variation to site-specific local environmental conditions, and interpreted as a sign of incipient speciation (Murray 2005). The hypothesis on adaptive character of the genetic variation pattern revealed here can be supported by the results of our previous study on C. quitensis with application of iPBS markers, which proved to be a very helpful tool in assessment of genome rearrangements, associated with activation of transposable elements arising in a response to various abiotic stress characteristic for a given population site (Androsiuk et al. 2015). Undoubtedly, C. quitensis populations analyzed in the present study also represent a very wide range of environmental conditions and therefore undergo various selecting forces which independently shape their genetic variation.
The presence of genetic structure within the analyzed C. quitensis population was accompanied by an interesting geographic pattern of genetic diversity distribution. Although low values of the effective number of alleles and expected heterozygosity were found among the populations studied, populations from the Southern Group had generally higher genetic diversity (on average NB = 1.0939 and He = 0.053) when compared to those from the Northern Group and population pC all together (on average NB = 1.024 and He = 0.014) (data not shown). Moreover, the Southern Group was characterized by a higher number of bands scored for each population (on average 127.4 bands per population for the Southern Group and 122.25 for the Northern Group + pC population), among which eight private bands can be found which were absent in C. quitensis from pC population as well as in populations from the Northern Group. Six of the revealed private bands were scored in pEl, one of the three populations representing a geographically isolated area of the South Shetland Islands. Population pEl was also characterized by a higher than average value of He. The unique character of pEl was not an exception since another population (pA) from this remote geographic area has He which was higher than the average value of that parameter for the whole Northern Group of C. quitensis populations. Only C. quitensis from pBy represents a very low level of genetic diversity, which can be attributed to a limited number of individuals available for that site. Significantly higher levels of allelic richness and higher expected heterozygosity for the Maritime Antarctic populations of C. quitensis in comparison to the population from Punta Arenas (South America) was also observed by Acuña-Rodríguez et al. (2014).
Phytogeography
Postglacial migration and gene flow associated with this process had major consequences for the population genetics of both animal and plant species. On the one hand, species repeatedly encountered founder events and experienced more or less drastic bottlenecks (Newton et al. 1999; Hewitt 2000). This could be observed both during retreat and re-immigration, which may lead to a reduction in the number of genotypes with increasing distance from glacial refugium (Hewitt 1996). On the other hand, the merging and expanding population from different glacial refugia, which had evolved genetically isolated, could increase genetic variation in zones of secondary contact (Taberlet et al. 1998).
In case of the Antarctic flora, it is difficult to draw conclusions about its evolutionary history since data on contemporary species distributions are not sufficient and this question requires an approach combining knowledge of Antarctic plant species distribution with studies concerning their genetic diversity and rates of evolution. Unfortunately, in case of the C. quitensis studies which can provide insights into previous population fragmentation and bottlenecks, historical patterns of dispersal and gene flow, relatedness between the Antarctic and South American populations are still at a very early stage (Gianoli et al. 2004; Acuña-Rodríguez et al. 2014).
Currently available data for C. quitensis point at two possible scenarios describing its history and dispersal in the Antarctic. According to one of them, C. quitensis is, most likely, a recent postglacial immigrant in the Antarctic and its presence in this region is estimated on Holocene, which was proved by identification of pollen grains and macro-remains of the species in peat deposits dated on 6000 years BP on King George Island, South Shetlands Islands (Birkenmajer et al. 1985). In this context, the Antarctic pearlwort is not an exception—the recent origin for much of the flowering plants is supported by species richness patterns with the highest number of species occurring in localities closest to neighboring continents and areas where there are more favorable climatic conditions for establishment. It is certainly clear that Antarctica experiences a continuous, but at the low level, the input of propagules from the other Southern Hemisphere continents (Marshall 1996). Some species may have potentially overcome natural colonization barriers of the Antarctic, those transported in a natural manner like anemochory (Lewis-Smith 1984, 1991, 1993, 2014; Bargagli et al. 1996; Marshall 1996; Muñoz et al. 2004), hydrochory (Coulson et al. 2002), zoochory (Barnes et al. 2004), or marine debris (Barnes et al. 2004; Lewis et al. 2005), but recent evidence of this kind of transportation is rather limited (Hughes et al. 2006).
According to the second scenario, C. quitensis could have survived in highly isolated refugia south of Drake Passage and inhabited this region from a Mid-Early Cenozoic or even Late Cretaceous (Convey and Stevens 2007; Parnikoza et al. 2007, 2011). Moreover, although during Last Glacial Maximum (LGM) in Pleistocene, all low altitude, coastal regions in the Maritime Antarctic are believed to have been obliterated by expanding glaciers and ice sheets (Larter and Vanneste 1995) and only re-exposed as ice has retreated over, at most, the last 10,000 years (Maslen and Convey 2006; Pugh and Convey 2008), there are palaeolimnological data for coastal continental Antarctic according to which some oases have hosted terrestrial life for up to 150,000 years (Hodgson et al. 2001; Squier et al. 2005), and hence that at least some of them have a considerably longer continuous history of exposure extending beyond the LGM (Peat et al. 2007). Recently even, when propagules from lower latitudes cross geographical barrier isolating the Antarctic the most important obstacle are harsh abiotic conditions, diminishing the probability of their establishment. There are a lot of species which have ecophysiological features required for survival in the polar environment including those from the Colobanthus genus like e.g., C. lycopodioides, C. subulatus and, C. lechleri, that partly cover C. quitensis range and tolerate similar edaphic conditions. That support partly fact that only records of successful introduction and establishment of species in Maritime Antarctic form lower latitudes (e.g., Chwedorzewska et al. 2015; Pertierra et al. 2017a) are via human vectors (Lityńska-Zając et al. 2012).
Our molecular data which revealed the presence of evident genetic structure within analyzed C. quitensis populations seems to support the concept of the multiregional origin of C. quitensis in the Antarctic. The gene pool subdivision as well as relatively high genetic diversity found in the Southern Group of analyzed C. quitensis populations suggest that the species may have survived the LGM in refugia located both on the South American continent and also in geographically isolated islands of the Maritime Antarctic. Moreover, high values of fixation index as well as significant results of IBD point at limited gene flow between populations.
However, the connectivity between populations from the Maritime Antarctic and south edges of the South American continent revealed within the Southern Group of C. quitensis populations cannot categorically reject the thesis that the current pattern of genetic variation of the species could have been also modified more recently (i.e., during the Holocene) due to low intense, random dispersal events (Muñoz et al. 2004; Parnikoza et al. 2012). Solving of this complex subject needs further, more detailed investigations using more extensive sampling of additional population and application of DNA markers which can provide a better resolution of molecular study, like SNPs or microsatellites.
In face of expanding the human footprint and climate change, the largest redistribution of species since the Last Glacial Maximum (Pecl et al. 2017) have been observed even in remote Antarctic terrestrial ecosystems (Pertierra et al. 2017b). Expansion of the distribution ranges of two native Antarctic flowering plant species has been the most studied example of a biological response to the recent environmental change in maritime the Antarctic (e.g., Gerighausen et al. 2003; Znój et al. 2017; Wódkiewicz et al. 2018). Thus, with the growing risk of homogenization of Antarctic biota (Terauds et al. 2012) the distribution, population size and diversity of Antarctic native plants became an important ecological bioindicators of environmental changes.
References
Acuña-Rodríguez IS, Oses R, Cortés-Vasquez J, Torres-Díaz C, Molina-Montenegro MM (2014) Genetic diversity of Colobanthus quitensis across the Drake Passage. Plant Genet Resour 12:147–150. https://doi.org/10.1017/S1479262113000270
Androsiuk P, Chwedorzewska K, Szandar K, Giełwanowska I (2015) Genetic variability of Colobanthus quitensis from King George Island (Antarctica). Pol Polar Res 36:281–295. https://doi.org/10.1515/popore-2015-0017
Baránek M, Meszáros M, Sochorová J, Čechová J, Raddová J (2012) Utility of retrotransposon-derived marker systems for differentiation of presumed clones of the apricot cultivar Velkopavlovická. Sci Hortic 143:1–6. https://doi.org/10.1016/j.scienta.2012.05.022
Bargagli R, Broady PA, Walton DWH (1996) Preliminary investigation of the thermal biosystem of Mount Rittmann fumaroles (northern Victoria Land, Antarctica). Antarct Sci 8:121–126. https://doi.org/10.1017/S0954102096000181
Barnes DKA, Warren N, Webb K, Phalan B, Reid K (2004) Polar pedunculate barnacles piggy-back on pycnogona, penguins, pinniped seals and plastics. Mar Ecol Prog Ser 284:305–310. https://doi.org/10.3354/meps284305
Bascunan-Godoy L, Sanhueza C, Cuba M, Zuniga GE, Corcuera LJ, Bravo LA (2012) Cold-acclimation limits low temperature induced photoinhibition by promoting a higher photochemical quantum yield and a more effective PSII restoration in darkness in the Antarctic rather than the Andean ecotype of Colobanthus quitensis (Kunt) Bartl. (Cariophyllaceae). BMC Plant Biol 12:114. https://doi.org/10.1186/1471-2229-12-114
Birkenmajer K, Ochyra R, Olsson IU, Stuchlik L (1985) Mid-Holocene radiocarbon-dated peat at Admirality Bay King George Island (South Shetlands, West Antarctica). Bull Pol Acad Sci Earth-Sci 33:7–12
Bravo LA, Saavedra-Mella FA, Vera F, Guerra A, Cavieres LA, Ivanov AG, Huner NPA, Corcuera LJ (2007) Effect of cold acclimation on the photosynthetic performance of two ecotypes of Colobanthus quitensis (Kunth) Bartl. J Exp Bot 58:3581–3590. https://doi.org/10.1093/jxb/erm206
Chen FY, Liu JH (2014) Germplasm genetic diversity of Myrica rubra in Zhejiang Province studied using inter−primer binding site and start codon−targeted polymorphism markers. Sci Hortic 170:169–175. https://doi.org/10.1016/j.scienta.2014.03.010
Chwedorzewska KJ, Bednarek PT (2008) Genetic variability in the Antarctic hairgrass Deschampsia antarctica Desv. from maritime Antarctic and sub-Antarctic sites. Pol J Ecol 56:209–216
Chwedorzewska KJ, Bednarek PT (2011) Genetic and epigenetic studies on populations of Deschampsia antarctica Desv. from contrasting environments at King George Island (Antarctic). Pol Polar Res 32:15–26. https://doi.org/10.2478/v10183-011-0005-9
Chwedorzewska KJ, Giełwanowska I, Olech M, Molina-Montenegro MA, Wódkiewicz M, Galera H (2015) Poa annua L. in the maritime-Antarctic—an overview. Polar Rec 51:637–643. https://doi.org/10.1007/s00300-007-0353-4
Comes HP, Kadereit JW (1998) The effect of Quaternary climatic changes on plant distribution and evolution. Trends Plant Sci 3:432–438. https://doi.org/10.1016/S1360-1385(98)01327-2
Convey P, Stevens MI (2007) Antarctic biodiversity. Science 317:1877–1878. https://doi.org/10.1126/science.1147261
Convey P, Chown SL, Clarke A, Barnes DKA, Bokhorst S, Cummings V, Ducklow HW, Frati F, Green TGA, Gordon S, Griffiths HJ, Howard-Williams C, Huiskes ADHL, Laybourn-Parry J, Lyons WB, Mcminn A, Morley SA, Peck LS, Quesada A, Robinson SA, Schiaparelli S, Wall DH (2014) The spatial structure of Antarctic biodiversity. Ecol Monogr 84:203–244. https://doi.org/10.1890/12-2216.1
Coulson SJ, Hodkinson ID, Webb NR, Harrison JA (2002) Survival of terrestrial soil-dwelling arthropods on and in seawater: implications for trans-oceanic dispersal. Funct Ecol 16:353–356. https://doi.org/10.1046/j.1365-2435.2002.00636.x
Cuba-Díaz M, Castel K, Acuña D, Machuca Á, Cid I (2017a) Sodium chloride effect on Colobanthus quitensis seedling survival and in vitro propagation. Antarct Sci 29:45–46. https://doi.org/10.1017/S0954102016000432
Cuba-Díaz M, Cerda G, Rivera C, Gómez A (2017b) Genome size comparison in Colobanthus quitensis populations show differences in species ploidy. Polar Biol 40:1475–1480. https://doi.org/10.1007/s00300-016-2058-z
DeFaveri J, Viitaniemi H, Leder E, Merila J (2013) Characterizing genic and nongenic molecular markers: comparison of microsatellites and SNPs. Mol Ecol Resour 13:377–392. https://doi.org/10.1111/1755-0998.12071
Earl DA, Vonholdt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361. https://doi.org/10.1007/s12686-011-9548-7
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620. https://doi.org/10.1111/j.1365-294X.2005.02553.x
Excoffier L, Ray N (2008) Surfing during population expansions promotes genetic revolutions and structuration. Trends Ecol Evol 23:347–351. https://doi.org/10.1016/j.tree.2008.04.004
Excoffier L, Smouse P, Quattro JM (1992) Analysis of molecular variance inferred from metric distances among DNA haplotypes: Application to human mitochondrial DNA restriction sites. Genetics 131:479–491
Excoffier L, Laval G, Schneider S (2005) Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evol Bioinform Online 1:47–50
Excoffier L, Foll M, Petit RJ (2009) Genetic consequences of range expansions. Annu Rev Ecol Evol Syst 40:481–501. https://doi.org/10.1146/annurev.ecolsys.39.110707.173414
Faivre-Rampant P, Zaina G, Jorge V, Giacomello S, Segura V, Scalabrin S, Guerin V, De Paoli E, Aluome C, Viger M, Cattonaro F, Payne A, Paulstephenraj P, Le Paslier MC, Berard A, Allwright MR, Villar M, Taylor G, Bastien C, Morgante M (2016) New resources for genetic studies in Populus nigra: genome-wide SNP discovery and development of a 12 k Infinium array. Mol Ecol Resour 16:1023–1036. https://doi.org/10.1111/1755-0998.12513
Foote AD, Morin PA (2016) Genome-wide SNP data suggest complex ancestry of sympatric North Pacific killer whale ecotypes. Heredity 117:316–325. https://doi.org/10.1038/hdy.2016.54
François O, Blum MGB, Jakobsson M, Rosenberg NA (2008) Demographic history of European populations of Arabidopsis thaliana. PLoS Genet 4:e1000075
Gao CH, Ren XD, Mason AS, Li JN, Wang W, Xiao ML, Fu DH (2013) Revisiting an important component of plant genomes: microsatellites. Funct Plant Biol 40:645–661. https://doi.org/10.1071/FP12325
Gerighausen U, Brautigam K, Mustafa O, Peter HU (2003) Expansion of vascular plants on an Antarctic island—a consequence of climate change? In: Huiskes AHL, Gieskes WWC, Rozema J, Schorno RML, van der Vies SM, Wolff WJ (eds) Antarctic biology in a global context. Backhuys, Leiden, pp 79–83
Gianoli E, Inostroza P, Zuniga-Feest A, Reyes-Diaz M, Cavieres LA, Bravo LA, Corcuera LJ (2004) Ecotypic differentiation in morphology and cold resistance in populations of Colobanthus quitensis (Caryophyllaceae) from the Andes of Central Chile and the Maritime Antarctic. Arct Antarct Alp Res 36:484–489
Giełwanowska I, Bochenek A, Gojło E, Gorecki R, Kellmann W, Pastorczyk M, Szczuka E (2011) Biology of generative reproduction of Colobanthus quitensis (Kunth) Bartl. from King George Island, South Shetland Islands. Pol Polar Res 32:139–155. https://doi.org/10.2478/v10183-011-0008-6
González M, Urdampilleta JD, Fasanella M, Premoli AC, Chiapella JO (2016) Distribution of rDNA and polyploidy in Descampsia antarctica Desv. in Antarctic and Patagonic populations. Polar Biol 39:1663–1677. https://doi.org/10.1007/s00300-016-1890-5
Greene SW, Greene DM (1963) Check list of the sub-Antarctic and Antarctic Vascular Flora. Polar Rec 73:411–418
Guo DL, Guo MX, Hou XG, Hang GH (2014) Molecular diversity analysis of grape varieties based on iPBS markers. Biochem Syst Ecol 52:27–32. https://doi.org/10.1016/j.bse.2013.10.008
Hammer Ø, Harper DAT, Ryan PD (2001) PAST: paleontological statistics software package for education and data analysis. Palaeontol Electronica 4:1–9
Hewitt GM (1996) Some genetic consequences of ice ages, and their role in divergence and speciation. Biol J Linn Soc Lond 58:247–276. https://doi.org/10.1111/j.1095-8312.1996.tb01434.x
Hewitt GM (2000) The genetic legacy of the Quaternary ice ages. Nature 405:907–913. https://doi.org/10.1038/35016000
Hodgson DA, Noon PE, Vyverman W, Bryant CL, Gore DB, Appleby P, Gilmour M, Verleyen E, Sabbe K, Jones VJ, Ellis-Evans JC, Wood PB (2001) Were the Larsemann Hills ice-free through the last glacial maximum? Antarct Sci 13:440–454. https://doi.org/10.1017/S0954102001000608
Hoffmann A, Arroyo MTK, Liberona F, Munoz M, Watson J (1998) Plantas Altoandinas en la Flora Silvestre de Chile. Edicione Fundacion Claudio Gay, Santiago
Hughes KA, Ott S, Bölter M, Convey P (2006) Colonization Processes. In: Bergstrom DM, Convey P, Huiskes AHL (eds) Trends in Antarctic terrestrial and limnetic ecosystems: Antarctica as a global indicator. Springer, Dordrecht, pp 35–54
Kalendar R, Antonius K, Smýkal P, Schulman AH (2010) iPBS: a universal method for DNA fingerprinting and retrotransposon isolation. Theor Appl Genet 121:1419–1430. https://doi.org/10.1007/s00122-010-1398-2
Kang Y, Lee H, Kim MK, Shin SC, Park H, Lee J (2016) The complete chloroplast genome of Antarctic pearlwort, Colobanthus quitensis (Kunth) Bartl. (Caryophyllaceae). Mitochondrial DNA 27:4677–4678. https://doi.org/10.3109/19401736.2015.1106498
Keller SR, Sowell DR, Neiman M, Wolfe LM, Taylor DR (2009) Adaptation and colonization history affect the evolution of clines in two introduced species. New Phytol 183:678–690. https://doi.org/10.1111/j.1469-8137.2009.02892.x
Klopfstein S, Currat M, Excoffier L (2006) The fate of mutations surfing on the wave of a range expansion. Mol Biol Evol 23:482–490. https://doi.org/10.1093/molbev/msj057
Knowles LL, Maddison WP (2002) Statistical phylogeography. Mol Ecol 11:2623–2635
Larter RD, Vanneste LE (1995) Relict subglacial deltas on the Antarctic Peninsula outer shelf. Geology 23:33–36
Lee DW, Postle RL (1975) Isozyme variation in Colobanthus quitensis (Kunth) Bartl.: Methods and preliminary analysis. Br Antarct Surv B 41:133–137
Lewis PN, Riddle M, Smith SDA (2005) Assisted passage or passive drift: a comparison of alternative transport mechanisms for non − indigenous coastal species into the Southern Ocean. Antarct Sci 17:183–191. https://doi.org/10.1017/S0954102005002580
Lewis-Smith RI (1984) Colonization and recovery by cryptogams following recent volcanic activity on Deception Island, South Shetland I. Br Antarct Surv B 62:25–51
Lewis-Smith RI (1991) Exotic sporomorpha as indicators of potential immigrant colonists in Antarctica. Grana 30:313–324. https://doi.org/10.1080/00173139109431986
Lewis-Smith RI (1993) Dry costal ecosystems of Antarctica. In: Maarel E (ed) Ecosystems of the world, 2A; dry coastal ecosystems, polar regions and Europe. Elsevier, Amsterdam, pp 51–57
Lewis-Smith RI (2003) The enigma of Colobanthus quithensis and Deschampsia antarctica in Antarctica. In: Huiskes AHL, Gieskes WWC, Rozema J, Schorno RML, van der Vies SM, Wolff WJ (eds) Antarctic biology in a global context. Backhuys, Leiden, pp 234–239
Lewis-Smith RI (2014) A fern cultured from Antarctic glacier detritus. Antarct Sci 26:341–344. https://doi.org/10.1017/S0954102013000606
Lityńska-Zając M, Chwedorzewska KJ, Olech M, Korczak-Abshire M, Augustyniuk-Kram A (2012) Diaspores and phyto-remains accidentally transported to the Antarctic Station during three expeditions. Biodivers Conserv 21:3411–3421. https://doi.org/10.1007/s10531-012-0371-6
Marshall WA (1996) Biological particles over Antarctica. Nature 383:680. https://doi.org/10.1038/383680a0
Maslen NR, Convey P (2006) Nematode diversity and distribution in the southern maritime Antarctic–clues to history? Soil Biol Biochem 38:3141–3151. https://doi.org/10.1016/j.soilbio.2005.12.007
Mehmood A, Jaskani MJ, Ahmad S, Ahmad R (2013) Evaluation of genetic diversity In open pollinated guava by iPBS primers. Pak J Agric Sci 50:591–597
Mehmood A, Luo S, Ahmad NM, Dong C, Mahmood T, Sajjad Y, Jaskani MJ, Sharp P (2016) Molecular variability and phylogenetic relationships of guava (Psidium guajava L.) cultivars using inter-primer binding site (iPBS) and microsatellite (SSR) markers. Genet Resour Crop Evol 63:1345–1361. https://doi.org/10.1007/s10722-015-0322-7
Moore DM (1970) Studies in Colobanthus quitensis (Kunth) Bartl. and Deschampsia antarctica Desv.: II. Taxonomy, distribution and relationships. Br Antarct Surv B 23:63–80
Muñoz J, Felicísimo AM, Cabezas F, Burgaz AR, Martínez I (2004) Wind as a long-distance dispersal vehicle in the southern hemisphere. Science 304:1144–1147. https://doi.org/10.1126/science.1095210
Murray BG (2005) When does intraspecific C-value variation become taxonomically significant? Ann Bot 95:119–125. https://doi.org/10.1093/aob/mci007
Navarrete-Gallegos AA, Bravo LA, Molina-Montenegro MA, Corcuera LJ (2012) Antioxidant responses in two Colobanthus quitensis (Caryophyllaceae) ecotypes exposed to high UV-B radiation and low temperature. Rev Chil Hist Nat 85:419–433. https://doi.org/10.4067/S0716-078X2012000400005
Newton AC, Allnutt TR, Gillies ACM, Lowe AJ, Ennos RA (1999) Molecular phylogeography, intraspecific variation and the conservation of tree species. Trends Ecol Evol 14:140–145. https://doi.org/10.1016/S0169-5347(98)01555-9
Nielsen R (2005) Molecular signatures of natural selection. Annu Rev Genet 39:197–218. https://doi.org/10.1146/annurev.genet.39.073003.112420
Özer G, Bayraktar H, Baloch FS (2016) iPBS retrotransposons ‘A Universal Retrotransposons’ now in molecular phylogeny of fungal pathogens. Biochem Syst Ecol 68:142–147. https://doi.org/10.1016/j.bse.2016.07.006
Parnikoza IY, Maidanuk DN, Kozeretska IA (2007) Are Deschampsia antarctica Desv. and Colobanthus quitensis (Kunth) Bartl. migratory relicts? Cytol Genet 41:226–229
Parnikoza I, Kozeretska I, Kunakh V (2011) Vascular plants of the maritime antarctic: origin and adaptation. Am J Plant Sci 2:381–395. https://doi.org/10.4236/ajps.2011.23044
Parnikoza I, Dykyy I, Ivanets V, Kozeretska I, Kunakh V, Rozhok A, Ochyra R, Convey P (2012) Use of Deschampsia antarctica for nest building by the kelp gull in the Argentine Islands area (maritime Antarctica) and its possible role in plant dispersal. Polar Biol 35:1753–1758. https://doi.org/10.1007/s00300-012-1212-5
Peakall R, Smouse PE (2006) GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes 6:288–295. https://doi.org/10.1111/j.1471-8286.2005.01155.x
Peakall R, Smouse PE (2012) GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics 28:2537–2539. https://doi.org/10.1093/bioinformatics/bts460
Peat HJ, Clarke A, Convey P (2007) Diversity and biogeography of the Antarctic flora. J Biogeogr 34:132–146. https://doi.org/10.1111/j.1365-2699.2006.01565.x
Pecl GT, Araújo MB, Bell JD, Blanchard J, Bonebrake TC, Chen I-C, Clark TD, Colwell RK, Danielsen F, Evengård B, Falconi L, Ferrier S, Frusher S, Garcia RA, Griffis RB, Hobday AJ, Janion-Scheepers C, Jarzyna MA, Jennings S, Lenoir J, Linnetved HI, Martin VY, McCormack PC, McDonald J, Mitchell NJ, Mustonen T Pandolfi JM, Pettorelli N, Popova E, Robinson SA, Scheffers BR, Shaw JD, Sorte CJB, Strugnell JM, Sunday JM, Tuanmu M-N, Vergés A, Villanueva C, Wernberg T, Wapstra E, Williams SE (2017) Biodiversity redistribution under climate change: impacts on ecosystems and human well-being. Science 355:eaai9214. https://doi.org/10.1126/science.aai9214
Pertierra LR, Aragón P, Shaw JD, Bergstrom DM, Terauds A, Olalla-Tárraga MÁ (2017a) Global thermal niche models of two European grasses show high invasion risks in Antarctica. Glob Chang Biol 23:2863–2873. https://doi.org/10.1111/gcb.13596
Pertierra LR, Hughes KA, Vega GC, Olalla-Tárraga MÁ (2017b) Correction: high resolution spatial mapping of human footprint across Antarctica and its implications for the strategic conservation of avifauna. PLoS ONE 12:e0173649. https://doi.org/10.1371/journal.pone.0173649
Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D (2006) Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 38:904–909. https://doi.org/10.1038/ng1847
Pritchard JK, Stephens P, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Pugh PJA, Convey P (2008) Surviving out in the cold: Antarctic endemic invertebrates and their refugia. J Biogeogr 35:2176–2186. https://doi.org/10.1111/j.1365-2699.2008.01953.x
Rousset F (1997) Genetic differentiation and estimation of gene flow from F-statistics under isolation by distance. Genetics 145:1219–1228
Sanhueza C, Vallejos V, Cavieres LA, Saez P, Bravo LA, Corcuera LJ (2017) Growing temperature affects seed germination of the antarctic plant Colobanthus quitensis (Kunth) Bartl. (Caryophyllaceae). Polar Biol 40:449–455. https://doi.org/10.1007/s00300-016-1972-4
Santiago IF, Alves TMA, Rabello A, Sales PA, Romanha AJ, Zani CL, Rosa CA, Rosa LH (2012) Leishmanicidal and antitumoral activities of endophytic fungi associated with the Antarctic angiosperms Deschampsia antarctica Desv. and Colobanthus quitensis (Kunth) Bartl. Extremophiles 16:95–103. https://doi.org/10.1007/s00792-011-0409-9
Santiago IF, Rosa CA, Rosa LH (2017) Endophytic symbiont yeasts associated with the Antarctic angiosperms Deschampsia antarctica and Colobanthus quitensis. Polar Biol 40:177–183. https://doi.org/10.1007/s00300-016-1940-z
Smýkal P, Bačová-Kerteszová N, Kalendar R, Corander J, Schulman AH, Pavelek M (2011) Genetic diversity of cultivated flax (Linum usitatissimum L.) germplasm assessed by retrotransposon-based markers. Theor Appl Genet 122:1385–1397. https://doi.org/10.1007/s00122-011-1539-2
Sneddon BV (1999) The Taxonomy and Breeding System of Colobanthus squarrosus (Caryophyllaceae). New Zeal J Bot 37:195–204. https://doi.org/10.1080/0028825X.1999.9512627
Squier AH, Hodgson DA, Keely BJ (2005) Evidence of late Quaternary environmental change in a continental East Antarctic lake for lacustrine sedimentary pigment distributions. Antarct Sci 17:361–376. https://doi.org/10.1017/S0954102005002804
Taberlet P, Fumagalli L, Wust-Saucy AG, Cosson JF (1998) Comparative phylogeography and postglacial colonization routes in Europe. Mol Ecol 7:453–464. https://doi.org/10.1046/j.1365-294x.1998.00289.x
Terauds A, Chown SL, Morgan F, Peat HJ, Watts D, Keys H, Convey P, Bergstrom DM (2012) Conservation biogeography of the Antarctic. Divers Distrib 18:726–741. https://doi.org/10.1111/j.1472-4642.2012.00925.x
van de Wouw M, van Dijk P, Huiskes AHL (2008) Regional genetic diversity patterns in Antarctic hairgrass (Deschampsia antarctica Desv.). J Biogeogr 35:365–376. https://doi.org/10.1111/j.1365-2699.2007.01784.x
Vasemagi A (2006) The adaptive hypothesis of clinal variation revisited: single-locus clines as a result of spatially restricted gene flow. Genetics 173:2411–2414. https://doi.org/10.1534/genetics.106.059881
Volkov RA, Kozeretska IA, Kyryachenko SS, Andreev IO, Maidanyuk DN, Parnikoza IY, Kunakh VA (2010) Molecular evolution and variability of ITS1–ITS2 in populations of Deschampsia antarctica from two regions of the maritime An tarctic. Polar Sci 4:469–478. https://doi.org/10.1016/j.polar.2010.04.011
West JG (1991) Colobanthus curtisiae (Caryophyllaceae), a new species from Eastern Australia. In: Banks MR, Smith SJ, Orchard AE, Kantvilas G (eds) Aspects of Tasmanian botany: a tribute to Winifred Curtis. Royal Society of Tasmania, Hobart, pp 75–77
Wódkiewicz M, Chwedorzewska KJ, Bednarek PT, Znój A, Androsiuk P, Galera H (2018) How much of the invader’s genetic variabilsity can slip between our fingers? A case study of secondary dispersal of Poa annua on King George Island (Antarctica). Ecol Evol 8:592–600. https://doi.org/10.1002/ece3.3675
Wright SI, Gaut BS (2005) Molecular population genetics and the search for adaptive evolution in plants. Mol Biol Evol 22:506–519. https://doi.org/10.1093/molbev/msi035
Znój A, Chwedorzewska KJ, Androsiuk P, Cuba-Diaz M, Giełwanowska I, Koc J, Korczak-Abshire M, Grzesiak J, Zmarz A (2017) Rapid environmental changes in the Western Antarctic Peninsula region due to climate change and human activity. Appl Ecol Env Res 15:525–539. https://doi.org/10.15666/aeer/1504_525539
Acknowledgements
This research was partly supported by the Polish Ministry of Scientific Research and Higher Education Grant NN303796240. Samples were also collected on Henryk Arctowski Polish Antarctic Station. We would like to thank Editor and three anonymous Reviewers for constructive advice that has improved our paper.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
Authors declare no conflicts of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Online Resources 1
Binary matrix of genotyping data for studied Colobanthus quitensis populations. Supplementary material 1 (XLSX 81 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Koc, J., Androsiuk, P., Chwedorzewska, K.J. et al. Range-wide pattern of genetic variation in Colobanthus quitensis. Polar Biol 41, 2467–2479 (2018). https://doi.org/10.1007/s00300-018-2383-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00300-018-2383-5