
Abstract
Key message
Here, we show that Au SINE elements have strong associations with protein-coding genes in wheat. Most importantly Au SINE insertion within introns causes allelic variation and might induce intron retention.
Abstract
The impact of transposable elements (TEs) on genome structure and function is intensively studied in eukaryotes, especially in plants where TEs can reach up to 90% of the genome in some cases, such as in wheat. Here, we have performed a genome-wide in-silico analysis using the updated publicly available genome draft of bread wheat (T. aestivum), in addition to the updated genome drafts of the diploid donor species, T. urartu and Ae. tauschii, to retrieve and analyze a non-LTR retrotransposon family, termed Au SINE, which was found to be widespread in plant species. Then, we have performed site-specific PCR and realtime RT-PCR analyses to assess the possible impact of Au SINE on gene structure and function. To this end, we retrieved 133, 180 and 1886 intact Au SINE insertions from T. urartu, Ae. tauschii and T. aestivum genome drafts, respectively. The 1886 Au SINE insertions were distributed in the seven homoeologous chromosomes of T. aestivum, while ~ 67% of the insertions were associated with genes. Detailed analysis of 40 genes harboring Au SINE revealed allelic variation of those genes in the Triticum–Aegilops genus. In addition, expression analysis revealed that both regular transcripts and alternative Au SINE-containing transcripts were simultaneously amplified in the same tissue, indicating retention of Au SINE-containing introns. Analysis of the wheat transcriptome revealed that hundreds of protein-coding genes harbor Au SINE in at least one of their mature splice variants. Au SINE might play a prominent role in speciation by creating transcriptome variation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Transposable elements (TEs) make up a large fraction of plant genomes (Kidwell 2002), as they can reach up to 90% of the wheat genome (Charles et al. 2008). Retrotransposons are the most abundant class of TEs in plants (Kejnovsky et al. 2012; Kumar and Bennetzen 1999); they are divided into LTR retrotransposons and non-LTR retrotransposons, the latter of which include Long INterspersed Nuclear Elements (LINEs) and Short INterspersed Nuclear Elements (SINEs). SINEs are miniature elements (80–500 bp) that probably originated from an accidental retroposition of polymerase III-derived (pol III, e.g. tRNAs) transcripts (Wicker et al. 2007). Their 5′ region harbors an internal pol III promoter [composed of A and B boxes that are recognized by RNA polymerase III (Arnaud et al. 2001)], a family unique internal region (sized 50–200 bp), and a 3′ region. Their 3′ region can be either AT or A rich and it contains short tandem repeats (3–5 bp) or a poly(A) tail. SINE superfamilies (tRNA, 7SL RNA and 5S RNA) are defined by conserved pol III promoters. SINEs are non-autonomous, as they are only capable of transposition using proteins encoded by LINEs elements, while creating TSDs (5–15 bp) (Wicker et al. 2007). Several SINE families have been discovered in plants, such as in Brassica napus (Deragon et al. 1994) Oryza sativa (Hirano et al. 1994), Nicotiana tabacum (Yoshioka et al. 1993), Myotis daubentonii (Borodulina and Kramerov 1999) and others (Deragon and Zhang 2006; Wenke et al. 2011). While SINEs are less abundant in grasses compared to LTR retrotransposons (Kumar and Bennetzen 1999; Sabot et al. 2004), a SINE family termed Au SINE, discovered in high copy numbers in wheat (Ben-David et al. 2013; Yasui et al. 2001) was found to be widely distributed in higher plants (Fawcett et al. 2006; Yagi et al. 2011). The impact of SINEs on plant genomes is poorly studied, while it has been well studied in mammalians, e.g., MIR and Alu elements (Deininger and Batzer 1999; Lev-Maor et al. 2003; Makalowski 2003; Schmid 1998; Schmitz and Brosius 2011; Smit 1996, 1999).
Both MIR and Alu elements were found to play a role in the exonization process of protein-coding genes (Schmitz and Brosius 2011). In the exonization process, non-protein-coding sequences, primarily introns, become part of the mature RNA, creating alternative splice variants (Clavijo et al. 2017). It has been reported that fragments of Alu sequences, which exist in ~ 1.4 million copies in the human genome, may appear in the protein-coding region of mature RNAs (Makałowski et al. 1994; Nekrutenko and Li 2001). Nearly 1800 retrotransposon-derived exons were found in humans, mostly Alu-containing transcripts (Schmitz and Brosius 2011). However, the frequency of Alu-containing transcripts was found to be much lower than the alternatively spliced exons that do not contain an Alu sequence (Schmitz and Brosius 2011). In most cases, the insertion of Alu into the coding regions of mRNAs creates frame-shifts or premature termination codons, but sometimes it also creates new protein functions or had modified existing ones (Hilgard et al. 2002).
In a previous report, we showed that Au SINE retains retrotranspositional activity following allopolyploidization events in wheat (Ben-David et al. 2013). In this study, the availability of updated genome drafts for several wheat species, especially the genome draft and the RNA-seq database of bread wheat (T. aestivum) facilitated a genome-wide analysis of Au SINE in the wheat genome and transcriptome. We have retrieved Au SINE-containing sequences distributed among the seven homoeologous chromosomes of T. aestivum, and found strong association with hundreds of protein-coding genes; in most of our cases, Au SINE was found to be inserted within the introns of a gene. We then analyzed the impact of Au SINE on the structure of genes and found allelic variations of many genes, based on insertional polymorphism of Au SINE in various wheat species. Expression analysis of several genes by real-time RT-PCR, revealed that Au SINE might undergo exonization in T. aestivum. Genome-wide, in-silico analysis of the T. aestivum transcriptome revealed that tens of protein-coding genes harbor Au SINE in their coding sequence. Detailed analysis of 83 genes showed that at least 50 of them showed splice variants including or excluding an Au SINE. The possible impact of Au SINE on gene structure and function is discussed.
Results and discussion
Genome-wide analysis of Au SINE in genome drafts of T. urartu, Ae. tauschii and T. aestivum
The publicly available sequence drafts for T. urartu, Ae. tauschii and T. aestivum facilitated a genome-wide analysis, including: copy numbers, insertion sites and distribution of Au SINE elements in bread wheat and its diploid ancestors. The relatively short sequence (181 bp) (Deragon and Zhang 2006; Yasui et al. 2001) of Au SINE allowed us to identify and characterize intact elements together with their insertion sites. In addition, the updated genome draft sequence of T. aestivum was published for each chromosome separately, which allowed the analysis of Au SINE content in each one of the three subgenomes (A, B and D), and analysis of the distribution of Au SINE in the seven homoeologous chromosomes. To this end, using the MITE analysis kit (MAK) (Yang and Hall 2003b), we have retrieved 133, 180 and 1886 intact Au SINE insertions from T. urartu, Ae. tauschii and T. aestivum genome drafts, respectively. The copy number of Au SINE in the allohexaploid T. aestivum genome was ~ tenfold its copy number in the diploid genomes, T. urartu and Ae. tauschii, indicating the massive retrotransposition burst of Au SINE following allopolyploidization events; most probably, the retrotransposition burst occurred following allotetraploidization because a similar content of Au SINE was found in the genome draft of Triticum turgidum ssp. dicoccoides (data not shown). This finding provides additional evidence for our previous report, wherein, using real-time quantitative PCR analysis, we found that the content of Au SINE was up to tenfold higher in allopolyploid wheat species compared to diploid species (Yaakov et al. 2013b).
Of the 1886 retrieved Au SINE insertions from the T. aestivum genome, 1849 were mapped to the seven homoeologous chromosomes (Fig. 1), distributed among the seven chromosomes of the three subgenomes: AA, BB and DD. The copy number of Au SINE in the AA subgenome was ~ fivefold higher than its copy number in the diploid AA genome (753 vs. 133, respectively), indicating proliferation of the element in the AA genome following allopolyploidization. In addition, the copy number of Au SINE in the DD subgenome was nearly similar to its copy number in the DD diploid genome (221 vs. 180, respectively), indicating a lack of proliferation in the DD subgenome following allohexaploidization, and thus the retrotransposition burst of Au SINE might occurred at the allotetraploid level, around 0.5 million years ago (Feldman and Levy 2005) Our data strongly indicate that although Au SINE is an ancient retrotransposon family (arising prior to the divergence of monocots and eudicots), found in many groups of higher plants (Fawcett et al. 2006; Yagi et al. 2011), it retained retrotranspositional activity in the Triticum–Aegilops genus.

Copy number and distribution of Au SINE in T. aestivum (genome composition AABBDD) genome. Each chromosome (1–7) is defined by its genome composition (AA, BB and DD subgenomes). A total of 1886 Au SINE insertions were retrieved from the T. aestivum genome draft, while 753, 875, and 221 insertions were retrieved from A, B and D subgenomes, respectively. Note that 37 insertions were not mapped in the seven homeologous chromosomes and they are indicated as “unknown”
Analysis of the common insertions of Au SINE among the three genome drafts: T. urartu, Ae. tauschii and T. aestivum revealed that only 24 of the 2199 total insertions are common (monomorphic insertions in the three species), indicating the massive proliferation of Au SINE after the divergence of the diploid species, around 4 million years ago (Feldman and Levy 2005). Note that the analysis was done based on 100% identity (e value = 0) of the insertions sites (Au SINE-flanking sequences), as such the results might be underestimated. In addition, 124 (53.1%) of the 221 insertions in the DD subgenome of T. aestivum are in common with Ae. tauschii, while only 77 (10.2%) of the 753 insertions in the AA subgenome of T. aestivum are in common with T. urartu (the diploid donor of genome AA). This finding supports our above conclusion that the retrotransposition burst of Au SINE might have occurred at the allotetraploidization level in the AA and BB subgenomes.
In a previous study (Ben-David et al. 2013), we reported on ~ 38% of the 3706 retrieved Au SINE insertions from the publicly available unassembled 454 pyrosequencing of T. aestivum were in transcribed regions. Here, we report on nearly half of the number of Au SINE insertions (1886) in the assembled and sorted genome draft of T. aestivum [(Clavijo et al. 2017) (http://www.ebi.ac.uk/ena/data/view/GCA_900067645.1, plants.ensembl.org/Triticum_aestivum/)], indicating the sequence redundancy in the 454 pyrosequencing data (Brenchley et al. 2012), as we have noted in our previous study (Ben-David et al. 2013). Annotation of the 1886 Au SINE insertion sites revealed that 1268 (67.2%) insertions were located within or near (up to 500 bp upstream or downstream) the DNA sequence of predicted protein-coding genes, 213 (11.2%) insertions within non-coding RNA (ncRNA) sequences, 253 (13.4%) insertions within other class I (173 insertions) and class II (80 insertions) TEs, and the remaining 152 Au SINE insertions were in non-coding DNA sequences. The data demonstrate that ~ 78.5% of the Au SINE insertions are in transcribed sequences (excluding insertions in other TEs), which might indicate a strong association of Au SINE with genes. Protein-coding genes that harbor Au SINE include: Transcription factors, Zinc finger-containing proteins, Homeobox genes, Methyltransferase, RNA-directed DNA polymerase, DNA-damage–repair, Ethylene-forming-enzyme-like dioxygenase, chromatin-associated protein, WRKY transcription factor, and others (Table S1).
Allelic variation in protein-coding genes caused by Au SINE
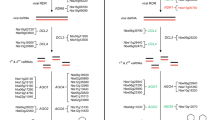
To examine whether Au SINE insertions into protein-coding genes might cause allelic variation (based on presence/absence of the element) among species in the Aegilops–Triticum genus, we have performed site-specific PCR analysis to amplify Au SINE elements within genes in Aegilops and Triticum species (see plant material), including diploid (AA, BB and DD genome species), tetraploid (wild emmer and durum, AABB genome), and hexaploid (AABBDD genome) species. Primers were designed from Au SINE-flanking sequences; so in each case, a larger PCR product represents a full site (presence of an Au SINE in the gene), while a smaller PCR product represents an empty site (absence of an Au SINE in the gene) (see Fig. 2). Note that in most cases PCR products were sequenced for validation of the presence/absence of Au SINE. To this end, 40 arbitrarily selected genes harboring Au SINE in T. aestivum were analyzed by site-specific PCR for presence/absence of the element in the genome of other wheat species (Table 1, supplementary Figure S1). Monomorphic Au SINE insertions in all the tested Aegilops and Triticum species were seen in 6 of the 40 analyzed genes (cases 1–6 in Table 1, see an example in Figure S1a), indicating old insertions of Au SINE, most probably before the divergence of the Aegilops and Triticum species. For the remaining 34 genes, polymorphic Au SINE insertions in Aegilops and Triticum species were seen (Fig. 2 and Figure S1). We have classified the insertion patterns into two main classes: (1) Au SINE has inserted into a gene only in the allopolyploid species (Fig. 2a–c), either in the T. aestivum only (cases 7–8 in Table 1) or in both T. turgidum and T. aestivum (cases 9–24 in Table 1), indicating that these insertions occurred following allopolyploidization; (2) Au SINE insertion occurred in the diploid species and was further inherited to the derived polyploid species, either from the DD genome (Ae. tauschii) donor (cases 25–29 in Table 1; Fig. 2d, e), the AA genome (T. urartu) donor (cases 30–33 in Table 1; Fig. 2f), the BB genome (Ae. speltoides and/or Ae. searsii) donors (cases 34–36 in Table 1, Figure S1w), or the insertion was seen in different diploid donors (cases 37–40 in Table 1, Fig. 2g, h). Note that in some cases polymorphic insertions were seen among different accessions of the same species, creating genetic variation within the same species; for example, insertions in some accessions of T. aestivum (Fig. 2a) or in some accessions of Ae. tauschii (Fig. 2d, e). Additionally, we have used accessions of Ae. speltoides and Ae. searsii because they are considered the potential donors of BB genome to wheat (Feldman and Levy 2005; Yaakov et al. 2012). To this end, our data indicate the dynamic nature of Au SINE throughout wheat evolution and its strong association with genes that might impact gene structure and function.

Site-specific PCR analysis using primers from Au SINE-flanking sequences. In each panel, the upper arrow represents a “full site” and the lower arrow represents an “empty site”. “M” represents the size marker in all the gels, “NC” represents for negative control, ddH20 was used as template in PCR reactions. The PCR analysis was performed in accessions of: BB1 = Ae. searsii, BB2 = Ae. speltoides, AA = T. urartu, DD = Ae. tauschii, AABB1 = T. durum, AABB2 = T. dicoccoides, AABBDD = T. aestivum. Note that for all polymorphic Au SINE insertions the difference between the “full site” fragment and the “empty site” is ~ 181 bp, the size of Au SINE. Numbers above each lane represent the genomic replicates. a Au SINE insertion in Putative Serine/threonine-protein kinase (case 8 in Table 1). The “full site” is 399 bp and the “empty site” is 218 bp. The insertion is unique to T. aestivum (amplified in two accessions 1 and 3). Note that the rest of the upper bands are non-specific PCR products as seen by sequence validation. b Au SINE insertion in an Predicted protein (case 22 in Table 1). The “full site” is 395 bp and the “empty site” is 214 bp. The insertion was seen in accessions of T. durum, T. dicoccoides and T. aestivum. c Au SINE insertion in Putative ATP-dependent RNA helicase DHX36 (cases 13 in Table 1). The “full site” is 387 bp and the “empty site” is 208 bp. The insertion was seen in T. durum, T. dicoccoides and T. aestivum. d Au SINE insertion in Inositol hexakisphosphate and diphosphoinositol-pentakisphosphate kinase (cases 27 in Table 1). The “full site” is 377 bp and the “empty site” is 196 bp. The insertion was seen in Ae. tauschii and T. aestivum. e Au SINE insertion in Chloride channel protein CLC-c (cases 25 in Table 1). The “full site” is 323 bp, and the “empty site” is140bp. The insertion was seen in Ae. tauschii and T. aestivum. f Au SINE insertion in an Predicted protein (case 31 in Table 1). The “full site” is 274 bp and the “empty site” is 97 bp. The insertion was seen in T. urartu, T. dicoccoides and T. aestivum. g Au SINE insertion in SIN3 transcription regulator family member B (cases 40 in Table 1). The “full site” is 342 bp and the “empty site” is 165. The insertion was seen in all tested species except for Ae. searsii accessions. h Au SINE insertion in Calcineurin-like metallo-phosphoesterase (cases 36 in Table 1). The “full site” is 352 bp and the “empty site” is 180 bp. The insertion was seen in Ae. speltoides, T. durum, T. dicoccoides and T. aestivum
Expression analysis of protein-coding genes harboring Au SINE
Sequence analysis of the 40 genes (Table 1) revealed that in most cases (35 of the 40) Au SINE had inserted in the intron region of the gene, indicating that Au SINE might be spliced out in the mature transcripts. In 4 cases (cases 3, 7, 18 and 33), the Au SINE was found to be part of an exon region, indicating that it might have underwent exonization throughout wheat evolution. In one case (case 4), the insertion was found 100 bp downstream to the gene. The phenomenon of SINE exonization has been reported in several studies in humans and other primates (Lev-Maor et al. 2003; Makałowski et al. 1994; Nekrutenko and Li 2001; Sorek et al. 2002), but has not been reported previously in plants. Here, we have analyzed the expression of several genes harboring Au SINE insertions in T. aestivum, using realtime RT-PCR, and found that most of those genes are expressed in bread wheat. Note that we have used two sets of primers (Table S2) for the expression analysis; the first set was designed to amplify exon–exon junction rescripts, and the second set of primers was designed to amplify chimeric (Au-SINE/flanking) transcripts, if they exist (see schemes on top of each one of the four panels in Fig. 3). While for most genes the exon–exon junction transcripts were amplified, no chimeric transcripts were seen, indicating that the intron harboring Au SINE was spliced out in the mature RNA. However, the expression analysis of four genes revealed that both the regular transcript (based on exon–exon junction amplification) and the chimeric (Au SINE/flanking) were simultaneously amplified in the same tissue, indicating retention of Au SINE-containing intron (Fig. 3). Note that the purity of each cDNA sample was tested using site-specific PCR reaction with primers from two exons of Actin gene, giving different amplification products for cDNA and genomic DNA. No DNA contamination was detected (Figure S2). In addition, the melting curves of the 4 cases are presented in Figure S3.

Relative expression levels of genes harboring Au SINE in two accessions of T. aestivum, as seen by realtime RT-PCR analysis. In each accession, the “regular transcript” compared to the Au SINE-containing transcript were analyzed. On top of each panel, a scheme of the analyzed gene, while the numbered boxes the exons and the black boxes note the Au SINE insertion. White arrows note the primers used to amplify the “regular transcript”, while the black arrows note the primers used to amplify the Au SINE-containing transcript. Expression levels (Y axis) were measured relative to ACTIN, and the exact relative expression (fold) is indicated by on top of each bar. Standard error on top of each bar was measured using three biological replicates. All the analyzed genes display the same trend of significantly higher expression levels of the regular transcript compared to the Au SINE-containing transcript, in the two tested T. aestivum accessions. The 4 analyzed genes: a DnaJ homolog (case 1 in Table 1), b Calcineurin-like metallo-phosphoesterase (case 36 in Table 1), c Putative Serine/threonine-protein kinase (case 8 in Table 1), and d Superoxide dismutase (case 6 in Table 1)
The expression analysis of a gene (case 1 in Table 1) that codes for DnaJ homolog [considered as chaperones in eukaryotes (Westermann et al. 1996)], in two accessions of T. aestivum revealed that both the regular transcript (based on primers designed from exon 1-exon two junction), and the chimeric (Au SINE/flanking) were simultaneously amplified at different levels (Fig. 3a). The regular transcript level was ~ 22-fold higher compared to the chimeric transcript in one accession, and ~ 11-fold higher in the second accession. Note that the transcript levels were relative to ACTIN transcription and three biological replicates were used in each accession. The expression analysis of a gene (case 36 in Table 1) that codes for Calcineurin-like metallo-phosphoesterase [involved in phosphorylated proteins substrates, nucleic acids or phospholipids (KEPPETIPOLA and SHUMAN 2006)] revealed that both the regular and the chimeric transcripts were simultaneously amplified, while the level of the regular transcript was over 160 fold higher compared to the chimeric transcript in both T. aestivum accessions (Fig. 3b). The Au SINE inserted into the intron located between exon 5 and exon 6 of this gene. The expression analysis of a gene (case 8, Table 1) that codes for Putative Serine/threonine-protein kinase [belong to the family of transferases (Huala et al. 1997)] revealed that both the regular and the chimeric transcripts were simultaneously amplified, while the level of the regular transcript was over 250 fold higher compared to the chimeric transcript in both T. aestivum accessions (Fig. 3c). The Au SINE inserted into the intron located between exon 8 and exon 9 of this gene. Finally, the expression analysis of a gene (case 6 in Table 1) that codes for Superoxide dismutase [catalyzes the dismutation of the superoxide (Kliebenstein et al. 1998)] revealed that both the regular and the chimeric transcripts were simultaneously amplified, while the level of the regular transcript was over ~ 130 fold higher compared to the chimeric transcript in both T. aestivum accessions. In this case, the Au SINE had inserted into the intron located between exon 5 and exon 6 of this gene. The relatively very low expression of the Au SINE-containing transcripts might indicate that these alternative transcripts do not have a major impact on the normal function of the proteins, but they might lead to the creation of modified proteins with new functions, similarly as was reported in animal and human systems (Lev-Maor et al. 2003; Makałowski et al. 1994; Nekrutenko and Li 2001; Schmitz and Brosius 2011; Schwartz et al. 2009; Sorek et al. 2002).
Genome-wide analysis of Au SINE-containing transcripts in T. aestivum
We have performed a genome-wide analysis of Au SINE-containing transcripts from the updated RNA-seq database of bread wheat (plants.ensembl.org/Triticum_aestivum/Info/Annotation/) to reveal exonization events of Au SINE. Using the MAK software, 113 Au SINE-containing transcripts (Au SINE and flanking sequences 500 bp upstream and downstream of the element) were retrieved from the T. aestivum transcriptome database. Detailed analysis of the 113 transcripts revealed that they belong to 83 protein-coding genes. Of the 83 genes that harbor Au SINE in their mature transcript, 76 were mapped in the seven homoeologous chromosomes, while 27, 47 and 2 were found in AA, BB and DD subgenomes, respectively (Fig. 4). Detailed analysis using the EnsemblPlants scripts revealed that 50 of the 83 genes showed different splice variants (Table 2), while many of those transcripts harbor Au SINE (Table 2). The number of splice variants for the 50 genes ranged between 2 and 9 transcripts, while at least 1 splice variant was an Au SINE-containing transcript (Table 2). For example: a gene that codes for putative methyltransferase (Table 2) showed 5 splice variants in chromosome 1A of T. aestivum, while 3 variants harbor Au SINE within their transcript (Fig. 5a); a gene that codes for Zinc finger CCCH domain-containing protein (Table 2) showed 4 splice variants in chromosome 3B, while one variant contained Au SINE within its transcript (Fig. 5b); a Putative WRKY transcription factor (Table 2) showed 2 splice variants in chromosome 5B, one of them harbored Au SINE within its transcript (Fig. 5c); and a Transcription initiation factor (Table 2) showed three splice variants in chromosome 7B, one of them contained Au SINE within its transcript (Fig. 5d). Figure S4 shows genes whereas some of their variants contain an Au SINE insertion within an exon. For example, Figure S4a presents a gene coding for 3-deoxy-manno-octulosonate cytidylyltransferase, mitochondrial protein that two of its variants (1, 2) contain Au SINE insertion in their last exon. Detailed analysis of the coding regions (CDS) in each splice variant revealed that in most cases the CDS of Au SINE-containing transcripts is shorter than the regular transcript leading, if translated, to a shorter protein (Fig. 5). For example: the CDS of the three Au SINE-containing splice variants of putative methyltransferase (Fig. 5a) lead to predicted protein sizes of 433 aa, while the CDS of the regular transcript (Fig. 5a) leads to a predicted protein size of 600 aa; the size of the predicted protein of Zinc finger CCCH domain-containing protein can reach up to 435 aa in the regular transcript, while it is 386 aa in the Au SINE-containing transcript (Fig. 5b); and the size of the predicted WRKY transcription factor is 495, while 355 aa in the Au SINE-containing transcript (Fig. 5c). In some cases, Au SINE or part of it became part of the coding sequence. For example, in TRIAE_CS42_2BL_TGACv1_131783_AA0432150 gene (Table 2, Figure S4f), the coding sequence of variant 1 does not contain Au SINE, but the coding sequence of variant 2 starts in a start codon located within an Au SINE insertion. Another example is TRIAE_CS42_3B_TGACv1_224095_AA0792250 gene (Table 2, Figure S4i) in which the coding sequence of variants 4 + 5 do not contain Au SINE, while the coding sequence of variants 1 + 2 + 3 + 6 contain different parts of the Au SINE insertion in their coding sequence. In variants 3 + 6, the coding sequence starts inside the Au SINE insertion. These data clearly indicate that Au SINE-containing introns underwent retention/exonizaion and became part of the mature transcript of many protein-coding genes.

Distribution of 84 Au SINE-containing genes in the seven homoeologous chromosomes of T. aestivum. Each chromosome (1–7) is defined by its genome composition (AA, BB and DD subgenomes). A total of 28, 47 and 2 Au SINE-containing genes were retrieved from AA, BB and DD subgenomes, respectively. Note that 7 Au SINE-containing genes were not mapped in the seven homoeologous chromosomes and they are indicated as “unknown”

Splice variants (transcripts) of four Au SINE-harboring genes (a–d). The name of the gene and EnsemblPlants accessions number are indicated on top. Gray boxes represent exons and lines represent introns. White boxes represent CDS (coding sequences) regions. Note that the mature transcripts consist of exons only; thus, we kept here the intron regions to indicate the exact location of Au SINE (black boxes) in the mature transcript. The predicted protein for each splice variant is indicated on right. a Transcripts 2, 4 and 5 contain Au SINE. b Transcript 2 contains Au SINE. c Transcript 2 contains Au SINE. d Transcript 3 contains Au SINE
Conclusions
An updated genome sequence draft for T. aestivum revealed that bread wheat consists of ~ 100,000 genes (Clavijo et al. 2017) and that over 80% of its genome consist of TEs. Our estimation based on the current study and our previous reports (Ben-David et al. 2013; Yaakov et al. 2013a; Yaakov and Kashkush 2012) is that many wheat genes harbor at least one TE insertion, while most of the insertions are in intron regions. Plant TEs are considered one of the main components of the genome that are implicated in creating genetic variation among species. The insertion of TEs within genes might create allelic variation, and by such might impact gene expression. In this study, we provide data which led us to conclude that transposable elements, in this case a non-LTR retrotransposon termed Au SINE in wheat, might considerably impact gene structure and function by creating allelic variation and exonization in protein-coding genes. We have used very stringent parameters in the MAK software to retrieve Au SINE insertions from the updated RNA-seq database of T. aestivum (Clavijo et al. 2017); thus, the number of Au SINE-containing transcripts that were retrieved here (113 transcripts) might be an underestimate. We estimate that the intron retention of Au SINE might occur in hundreds of wheat genes. To this end, transcriptional interference induced by intronic retrotransposons might impact the transcription of large number of genes. Alternative splicing generates transcriptome variation that could lead to sub-functionalization of genes and speciation. Finally, because Au SINE is found in the entire plant kingdom (Deragon and Zhang 2006; Fawcett et al. 2006), we hypothesize based on our data that intron retention of Au SINE might be a general phenomenon in plants.
Materials and methods
Genomic data
In this study, three publicly available genome drafts were analyzed: (1) T. urartu, the donor of AA genome that was paired-end sequenced using whole-genome shotgun by Illumina [plants.ensembl.org/Aegilops_tauschii/Info/Index, (Ling et al. 2013)]. (2) Ae. tauschii, the donor of DD genome that was sequenced and assembled in the same way as T. urartu and the assembled scaffolds cover 83.4% of its genome with 90-fold depth reads. These reads combined with Roche-454 sequenced reads represent 97% of Ae. tauschii genome [plants.ensembl.org/Triticum_urartu/Info/Index, (Jia et al. 2013)]. (3) T. aestivum, the hexaploid bread wheat, which was published on June 2016 in EnsemblPlants [(Clavijo et al. 2017) pre.plants.ensembl.org/Triticum_aestivum/Info/Index]. This updated T. aestivum assembly was generated by The Genome Analysis Center in Norwich (TGACv1).
Transcriptomic data
Here, we used the updated publicly available RNA-seq database of T. aestivum found in Ensemblplants [(Clavijo et al. 2017), plants.ensembl.org/info/website/ftp/index.html]. The library includes cDNA, CDS and ncRNA sequences that were used for annotation analysis in our study.
Retrieval of Au SINE insertions
The sequences of Au SINE were retrieved from these genome drafts and transcriptome, using the MITE analysis kit (MAK) software [a standalone version was kindly provided by Guojun Yang, University of Toronto, (Janicki et al. 2011; Yang and Hall 2003a)]. The publicly available consensus sequence of the Au SINE family (GIRI database at http://www.girinst.org/repbase/update/browse.php) was used as an input (query sequence) in the MAK software and BLASTN was performed against the genomic drafts. For the retrieval of Au SINE-containing sequences from the genome drafts, we have used the MAK function “Member”, an e value of 10−3 and an end mismatch tolerance of 20 nucleotides. In addition, flanking sequences (500 bp from each end) were retrieved together with each one of the insertions, to characterize the insertion sites. A rice-specific MITE, called mPing, was used as a negative control in this analysis and no mPing-related sequences were retrieved in wheat. Redundant sequences were detected by BLAST + software (Camacho et al. 2009) using BLASTN function. We have compared sequences against themselves and excluded the paired element from each couple of sequences that were found to have a 100% identity (100% coverage with an e value of 0 and no gaps). The final output files were then edited using Textpad 7.4 ‘Regular Expression’ functions for cleaning excess data. It is important to mention that we have considered in this analysis truncated elements (at one of the terminal sequences) as being nearly intact elements.
Insertion sites annotation
Annotation of Au SINE-flanking sequences was performed using the complementary-DNA (cDNA), coding sequences (CDS) and non-coding RNA (ncRNA) databases of T. aestivum (taken from EnsemblPlants at plants.ensembl.org/index.html). In addition, Transposable element consensus sequences from different plant genomes were also used as database in this annotation analysis (taken from ITMI at botserv2.uzh.ch/kelldata/trep-db/index.html). Annotation was performed using BLAST + standalone version 2.2.3 with an e value of 10−10. The merged 5′ and 3′ flanking sequences were used as query against the mentioned databases.
Plant material, DNA and RNA extraction
In this study, we have used 21 accessions of seven Triticum and Aegilops species including the possible donors of AA (T. urartu, three accessions), BB (Ae. speltoides, and Ae. searsii, three accessions from each species), DD (Ae. tauschii, three accessions) genomes, and the allopolyploid species, T. turgidum (wild emmer and durum wheat, three accessions from each species) and T. aestivum (bread wheat, three accessions) seeds were kindly provided by Moshe Feldman, the Weizmann Institute of Science, Israel and the US Department of Agriculture (npgsweb.ars-grin.gov/gringlobal/search.aspx). Young leaves of ~ 4 weeks post germination plants were used for DNA (using GeneJET plant genomic DNA Purification Mini Kit, Thermo scientific) and RNA (using TRI reagent, Sigma) extractions. First strand cDNA was created using 5X All-In-One RT MasterMix (Applied Biological Material).
Site-Specific PCR analysis
Insertional polymorphism of Au SINE was analyzed based on primers designed from flanking sequences (both sides) of Au SINE insertion. Primers were designed using PRIMER3 version 4.0.0 (bioinfo.ut.ee/primer3/) (see Table 1 for primer sequences). A full site includes a PCR product containing an Au SINE and flanking sequences, while an empty site lacks Au SINE (amplification of flanking sequences only). The reaction consisted of 12 µl ultrapure water (Biological Industries), 2 µl of 10× Taq DNA polymerase buffer (EURX), 2 µl of 25 mM MgCl2 (EURX), 0.8 µl of 2.5 mM dNTPs, 0.2 µl Taq DNA polymerase (5 U µl-1, EURX), 1 µl of each site-specific primer (50 ng µl−1) and 1 µl of template genomic DNA (approximately 50 ng µl−1). The PCR conditions for these reactions were 94 °C for 3 min, 30 cycles of 94 °C for 1 min, 58 °C for 1 min and 72 °C for 1 min, then 72 °C for 3 min. For sequence validation, PCR products were extracted from agarose gels using the QIAquick PCR Purification Kit (QIAGEN). Next, products were ligated into the pGEM-T easy vector (Promega, Madison, WI, USA) which was used for transformation into E. coli DH5α. Finally, for sequence validation, DNA products were sequenced by 3730 DNA Analyzer (Applied Biosystems, Foster City, CA, USA) at Ben-Gurion University, Israel.
Expression analysis
Real-time quantitative RT-PCR (using 7500 Fast Real-Time PCR system, Applied Biosystems) was used to analyze the expression of genes harboring Au SINE in leaves of bread wheat (T. aestivum). For each gene, two primer pairs were designed; the first to amplify a regular transcript based on exon–exon junction amplification, and the second primer pair designed to amplify a chimeric transcript, if produced, consisting of Au SINE and flanking intron sequence. Primers were designed using the Primer Express v2.0 software and the PRIMER3 version 4.0.0 software (bioinfo.ut.ee/primer3/). Each reaction contained: 7.5 µl KAPA SYBR FAST qPCR Master Mix, 0.3 µl ROX Low 509—a reference dye for fluorescence normalization (KAPA BIOSYSTEMS), 1 µl forward primer (10 µM), 1 µl reverse primer (10 µM), 0.2 µl H2O (nuclease free water, Hylabs) and 5 µl or of cDNA template (50X dilution). The data were analyzed using the 7500 version 2.0.5 software (Applied Biosystems). The reaction conditions were 20 s at 95 °C, followed by 40 cycles of 3 s at 95 °C and 30 s at 60 °C. To differentiate specific PCR products from nonspecific ones, a melting curve was generated right after amplification by employing a 15 s incubation at 95 °C and a 1 min incubation at 60 °C, after which the temperature was raised by increments of 0.1 °C per sec until reaching 95 °C.
Data of each sample were received as Ct, threshold cycle of the PCR amplification reaching a certain level of fluorescence (Livak and Schmittgen 2001), normalized to the Ct of ACTIN, a known single copy gene used as an endogenous control. A comparative \({{\text{2}}^{ - \Delta \Delta {{\text{C}}_t}}}\) method was then used to determine the relative expression level of the two targets in each sample. First, each one of the normalized target expression levels in each sample was compared to the normalized target expression level of the reference sample, based on the following equation:
Therefore, \({\text{RQ }}({\text{the relative expression level}}){\text{ }}={\text{ }}{({\text{2}}\, \times \,{\text{primer efficiency}})^{^{{ - \Delta \Delta {C_t}}}}}.\) Second, the two targets in each sample were compared to find their relative expression levels. Three technical replicates were used for each reaction to evaluate reproducibility. Standard deviations (SD) were calculated based on these three replications. Note that total RNA (not treated with reverse transcriptase) was used in RT-PCR reaction as a negative control for DNA contamination.
In-silico analysis of Au SINE-containing transcripts
Au SINE-containing transcripts were further examined for all predicted variants of the same gene, as found in Ensemblplants. To validate these sequences are real transcripts and not genomic DNA sequences (due to contamination of the transcriptome), we compared these transcripts with the T. aestivum genome using BLASTN analysis to check whether we find full hits (meaning, genomic DNA sequence), or multiple partial hits for each sequence (meaning, mature RNA transcript). This was done by BLASTN algorithm with comparison of transcripts (query) to genomic database with an e value < 1e−100 (Table S3). This analysis showed whether each transcript had a full match (100% coverage) to sequence in the genomic database, or multiple partial matches to sequences in different locations of the genomic database. Using this analysis, we can eliminate transcripts that had full match and suspected to be genomic DNA or precursor RNA. To this end, all transcripts used here are mature RNA transcripts. The translated region of each transcript was determined by the CDS (coding sequence) as found in Ensemblplants. Each transcript containing an Au SINE insertion was traced back to its gene by transcript accession. All predicted variants of the same gene were examined in BLASTN analysis vs. Au SINE sequence and the specific location of insertion was determined in each variant.
Author contribution statement
DK: Generated the in-silico analysis data, analyzed results and manuscript preparation. CD: Generated the PCR and RT-PCR data, analyzed results. KK: (corresponding author). analyzed results, manuscript preparation and submission.
References
Arnaud P, Yukawa Y, Lavie L, Pélissier T, Sugiura M, Deragon JM (2001) Analysis of the SINE S1 Pol III promoter from Brassica; impact of methylation and influence of external sequences. Plant J 26:295–305
Ben-David S, Yaakov B, Kashkush K (2013) Genome-wide analysis of short interspersed nuclear elements SINES revealed high sequence conservation, gene association and retrotranspositional activity in wheat. Plant J 76:201–210
Borodulina OR, Kramerov DA (1999) Wide distribution of short interspersed elements among eukaryotic genomes. Febs Lett 457:409–413
Brenchley R et al (2012) Analysis of the bread wheat genome using whole-genome shotgun sequencing. Nature 491:705–710
Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL (2009) BLAST+: architecture and applications BMC. bioinformatics 10:421
Charles M et al (2008) Dynamics and differential proliferation of transposable elements during the evolution of the B and A genomes of wheat. Genetics 180:1071–1086
Clavijo BJ et al (2017) An improved assembly and annotation of the allohexaploid wheat genome identifies complete families of agronomic genes and provides genomic evidence for chromosomal translocations. Genome Res 27:885–896
Deininger PL, Batzer MA (1999) Alu repeats and human disease. Mol Genet Metab 67:183–193
Deragon J-M, Zhang X (2006) Short interspersed elements (SINEs) in plants: origin, classification, and use as phylogenetic markers. Syst Biol 55:949–956
Deragon J, Landry B, Pelissier T, Tutois S, Tourmente S, Picard G (1994) An analysis of retroposition in plants based on a family of SINEs from Brassica napus. J Mol Evol 39:378–386
Fawcett JA, Kawahara T, Watanabe H, Yasui Y (2006) A SINE family widely distributed in the plant kingdom and its evolutionary history. Plant Mol Biol 61:505–514
Feldman M, Levy A (2005) Allopolyploidy—a shaping force in the evolution of wheat genomes. Cytogenet Genome Res 109:250–258
Hilgard P, Huang T, Wolkoff AW, Stockert RJ (2002) Translated Alu sequence determines nuclear localization of a novel catalytic subunit of casein kinase 2. Am J Physiol Cell Physiol 283:C472-C483
Hirano H-Y, Mochizuki K, Umeda M, Ohtsubo H, Ohtsubo E, Sano Y (1994) Retrotransposition of a plant SINE into the wx locus during evolution of rice. J Mol Evol 38:132–137
Huala E, Oeller PW, Liscum E, Han I-S, Larsen E, Briggs WR (1997) Arabidopsis NPH1: a protein kinase with a putative redox-sensing domain. Science 278:2120–2123
Janicki M, Rooke R, Yang G (2011) Bioinformatics and genomic analysis of transposable elements in eukaryotic genomes. Chromosome Res 19:787–808
Jia J et al (2013) Aegilops tauschii draft genome sequence reveals a gene repertoire for wheat adaptation. Nature 496:91–95
Kejnovsky E, Hawkins JS, Feschotte C (2012) Plant transposable elements: biology and evolution. In: Plant genome diversity, vol 1. Springer, New York, pp 17–34
Keppetipola N, Shuman S (2006) Mechanism of the phosphatase component of Clostridium thermocellum polynucleotide kinase-phosphatase. Rna 12:73–82
Kidwell MG (2002) Transposable elements and the evolution of genome size in eukaryotes. Genetica 115:49–63
Kliebenstein DJ, Monde R-A, Last RL (1998) Superoxide dismutase in Arabidopsis: an eclectic enzyme family with disparate regulation and protein localization. Plant Physiol 118:637–650
Kumar A, Bennetzen JL (1999) Plant retrotransposons. Ann Rev Genet 33:479–532
Lev-Maor G, Sorek R, Shomron N, Ast G (2003) The birth of an alternatively spliced exon: 3’splice-site selection in Alu exons. Science 300:1288–1291
Ling H-Q et al (2013) Draft genome of the wheat A-genome progenitor Triticum urartu. Nature 496:87–90
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)) method. Methods 25:402–408 doi:10.1006/meth.2001.1262
Makalowski W (2003) Not junk after all. Science 300:1246–1247
Makałowski W, Mitchell GA, Labuda D (1994) Alu sequences in the coding regions of mRNA: a source of protein variability. Trends Genet 10:188–193
Nekrutenko A, Li W-H (2001) Transposable elements are found in a large number of human protein-coding genes. TRENDS Genet 17:619–621
Sabot F, Simon D, Bernard M (2004) Plant transposable elements with an emphasis on grass species. Euphytica 139:227–247
Schmid CW (1998) Does SINE evolution preclude Alu function? Nucleic Acids Res 26:4541–4550
Schmitz J, Brosius J (2011) Exonization of transposed elements: a challenge and opportunity for evolution. Biochimie 93:1928–1934
Schwartz S, Gal-Mark N, Kfir N, Oren R, Kim E, Ast G (2009) Alu exonization events reveal features required for precise recognition of exons by the splicing machinery. PLoS Comput Biol 5:e1000300
Smit AF (1996) The origin of interspersed repeats in the human genome. Curr Opin Genet Dev 6:743–748
Smit AF (1999) Interspersed repeats and other mementos of transposable elements in mammalian genomes. Curr Opin Genet Dev 9:657–663
Sorek R, Ast G, Graur D (2002) Alu-containing exons are alternatively spliced. Genome Res 12:1060–1067
Wenke T, Döbel T, Sörensen TR, Junghans H, Weisshaar B, Schmidt T (2011) Targeted identification of short interspersed nuclear element families shows their widespread existence and extreme heterogeneity in plant genomes. Plant Cell 23:3117–3128
Westermann B, Gaume B, Herrmann JM, Neupert W, Schwarz E (1996) Role of the mitochondrial DnaJ homolog Mdj1p as a chaperone for mitochondrially synthesized and imported proteins. Mol Cell Biol 16:7063–7071
Wicker T et al (2007) A unified classification system for eukaryotic transposable elements. Nat Rev Genet 8:973–982
Yaakov B, Kashkush K (2012) Mobilization of stowaway-like MITEs in newly formed allohexaploid wheat species. Plant Mol Biol 80:419–427
Yaakov B, Ceylan E, Domb K, Kashkush K (2012) Marker utility of miniature inverted-repeat transposable elements for wheat biodiversity and evolution. Theor Appl Genet 124:1365–1373
Yaakov B, Ben-David S, Kashkush K (2013a) Genome-wide analysis of stowaway-like MITEs in wheat reveals high sequence conservation, gene association, and genomic diversification. Plant Physiol 161:486–496
Yaakov B, Meyer K, Ben-David S, Kashkush K (2013b) Copy number variation of transposable elements in Triticum–Aegilops genus suggests evolutionary and revolutionary dynamics following allopolyploidization. Plant Cell Rep 32:1615–1624
Yagi E, Akita T, Kawahara T (2011) A novel Au SINE sequence found in a gymnosperm. Genes Genet Syst 86:19–25
Yang G, Hall TC (2003a) MAK, a computational tool kit for automated MITE analysis. Nucleic Acids Res 31:3659–3665
Yang GJ, Hall TC (2003b) MAK, a computational tool kit for automated MITE analysis. Nucleic Acids Res 31:3659–3665 doi. Doi 10.1093/Nar/Gkg531
Yasui Y, Nasuda S, Matsuoka Y, Kawahara T (2001) The Au family, a novel short interspersed element (SINE) from Aegilops umbellulata. Theor Appl Genet 102:463–470
Yoshioka Y, Matsumoto S, Kojima S, Ohshima K, Okada N, Machida Y (1993) Molecular characterization of a short interspersed repetitive element from tobacco that exhibits sequence homology to specific tRNAs. Proc Natl Acad Sci 90:6562–6566
Acknowledgements
We would like to thank Dr. Guojun Yang, Toronto University for providing the updated stand-alone MAK software, Moshe Feldman and Hakan Ozkan for providing the seed material. We thank Vadim Khasdan for his assistance with the wet-bench experiments and Beery Yaakov for his critical reading of the manuscript. This work was supported by a grant from the Israel Science Foundation (grant # 322/15) to K. K.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Communicated by Mark C. Jordan.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Keidar, D., Doron, C. & Kashkush, K. Genome-wide analysis of a recently active retrotransposon, Au SINE, in wheat: content, distribution within subgenomes and chromosomes, and gene associations. Plant Cell Rep 37, 193–208 (2018). https://doi.org/10.1007/s00299-017-2213-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00299-017-2213-1