
Abstract
Background
Treating patients with inflammatory joint diseases (rheumatoid arthritis, psoriatic arthritis) according to established treatment algorithms often requires the simultaneous use of three or more medications to relieve symptoms and prevent long-term joint damage as well as disability.
Objective
To assess and give an overview on drug-drug interactions in the pharmacotherapy of inflammatory joint diseases with regards to their clinical relevance.
Methods
All possible drug combinations were evaluated using three commercially available drug interaction programs. In those cases where only limited/no data were found, a comprehensive hand search of Pubmed was carried out. Finally, the drug–drug interactions of all possible combinations were classified according to evidence-based medicine and a specifically generated relevance-based system.
Results
All three interaction software programs showed consistent results. All detected interactions were combined in clearly structured tables.
Conclusion
A concise overview on drug-drug interactions is given. Especially in more sophisticated cases extensive knowledge of drug interactions supports optimisation of therapy and results in improved patient safety.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
When treating inflammatory joint diseases [e.g., rheumatoid arthritis (RA), psoriatic arthritis (PsA)] one therapeutic goal is to reach and maintain remission or low disease activity. Over time a reduction in functional loss as well as radiographic progression and an improved social integration for patients should be achieved. Meanwhile broad evidence supports the early use of aggressive therapy for patients with active inflammatory arthritis. According to the EULAR treat-to-target recommendations antirheumatic therapy contains conventional (c), biologic (b) and targeted synthetic (ts) disease-modifying antirheumatic drugs (DMARDs) as well as corticosteroids used to attenuate the inflammatory process. In addition, non-steroidal-antirheumatic drugs (NSAIDs) in combination with proton-pump inhibitors (PPIs) as well as analgesics are part of the therapeutic armamentarium [1, 2]. This mandatory long-term polypharmacy can result in potentially harmful drug–drug interactions (DDIs). A considerable amount of literature is available on the risk of potential interactions of single drug combinations [3,4,5,6,7,8] but to our knowledge there is no detailed review on the whole range of drugs used for managing inflammatory joint diseases.
RA and PsA were singled out on the fact that in these two inflammatory joint diseases there is an especially large variety of medication involved in the treatment. The aim of this paper is to provide a current and detailed overview on DDIs and their clinical relevance to support safety and consequently also optimise pharmacotherapy.
Methods
Search strategy
To overcome limitations of separate online sources and comprehensively cover scientific information, three different databases as well as Pubmed and SciFinder were applied. Based on the three drug interaction screening programs (Apotronik, mediQ, and Micromedex Solutions [9,10,11]), a comprehensive literature research (PubMed, SciFinder) was performed to evaluate possible interactions especially for drugs new to the market (up to Feb. 27th 2019). Apotronik contains drugs available in Austria, mediQ is a Swiss software and Micromedex includes drugs available at European level. Apotronik and mediQ classify the severity of DDIs with a colour code and Micromedex with a coloured symbol code. The severity of interactions is graded in three categories in the Apotronic and mediQ database: (1) minor risk/unlikely to be clinically significant; (2) moderate risk/clinically relevant/monitoring recommended; (3) major risk/potentially severe or life-threatening interaction. The Micromedex program uses the four categories mild, moderate, major and contraindicated. It should be noted that this paper only focuses on major risks and contraindications. All programs provide information about DDIs on 1:1 interaction.
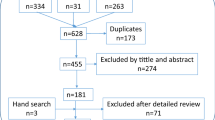
The following search strategy was applied and included English and German literature: If appropriate data for a drug were available from the three interaction databases (Apotronik, mediQ, Micromedex), PubMed was used for searching clinical trials, reviews and case reports. In case of no or limited data available for a drug, an extensive search regarding general interactions was performed via PubMed and SciFinder. To identify all suitable articles, the following keywords were applied: drug AND drug (e.g., MTX AND pantoprazole) OR drug AND group of drugs (e.g., MTX AND PPIs) AND interaction OR combination AND rheumatoid (Fig. 1). If further little/no data were obtained with this procedure the terms were omitted stepwise from behind. Finally, if no results were found with this strategy, a specific search with regard to CYP-enzymes and transport systems was carried out to ascertain theoretically possible interactions. This procedure was mainly applied with regards to tsDMARDs.

Flow chart of search procedure strategy
Main outcome variable
Evaluation and classification of the results were performed with the system of evidence-based medicine (EBM) and a specifically generated relevance-based system (RBS). RBS was developed as a decision-making tool for helping identify potential DDIs. In a first step, the RBS differentiates between clinically relevant and non-relevant interactions based on data from interaction studies. Furthermore, in case of lack of study data, a third category of interactions which might occur due to pharmacological mechanisms was introduced [12]. This approach is based on the chemical structure of drugs and their corresponding mechanisms found in interaction programs and literature. Therefore, the RBS can also be used as a theoretical framework flagging DDIs especially for newly registered pharmaceuticals in case there are no head-to-head data available. In Table 1 the classification criteria of both systems are summarised.
Results
NSAIDs, PPIs, analgesics and glucocorticoids
NSAIDs are a commonly prescribed medication for treating inflammatory arthritis patients. NSAIDs such as diclofenac, ibuprofen, naproxen, and piroxicam are mainly metabolised via CYP2C9 [13]. Co-prescribing of PPIs (pantoprazole, omeprazole, esomeprazole, and lansoprazole) with non-selective NSAIDs is strongly recommended. In vitro studies showed that almost all PPIs are metabolised via CYP2C19 and CYP3A4 into inactive metabolites. Pantoprazole additionally showed inhibitory effects on CYP2C9 [14]. DDIs between NSAIDs and PPIs, especially pantoprazole, might be possible. However, all three interaction programs and the literature research did not show any clinically relevant interactions between NSAIDs and PPIs.
A pharmacodynamic interaction is given between NSAIDs and glucocorticoids. Their concomitant use increases the risk for gastrointestinal bleeding [9,10,11] and even short term NSAIDs intake can cause ulcers. Normally, body tissue regenerates by producing prostaglandin E. Concomitantly administered steroids inhibit the production of cytokines as well as the secretion of gastric mucosa and protective growth factors. As a result, the autoregulatory mechanisms of ulcer repair are inhibited [15]. With regards to severity the interaction is graded as major risk/monitoring recommended.
COX-2 inhibitors and opioids (tramadol, oxycodone and buprenorphine) are alternatives for mitigating pain. These two classes of drugs are metabolised by CYP2D6, resulting in a pharmacokinetic interaction [9, 10]. Tramadol is changed into its active form o-desmethyltramadol by CYP2D6. Its analgesic efficacy can be reduced by inhibition of its enzymatic activation [16]. When coxibes and oxycodone or buprenorphine are administered together, the plasma concentration of the two opioids may rise, leading to an increased risk for side effects [17]. However, no clinical studies documenting a direct interaction between coxibes and opioids could be found. A summary of the above mentioned interactions is shown in Table 2.
Therapy with conventional disease-modifying antirheumatic drugs (cDMARDs)
cDMARDs combined with NSAIDs
Between MTX and NSAIDs a pharmacokinetic interaction was found [9,10,11]. Approximately 80% of MTX is excreted unchanged through glomerular filtration. Furthermore, it is eliminated by active transporters such as organic anionic transporters (OAT) 1 and 3 and multidrug-resistance-proteins (MRP) 2 and 4 in the kidney [18]. NSAIDs inhibit glomerular filtration through impairment of renal blood flow following inhibition of prostaglandin synthesis. Subsequently MTX is filtrated more slowly [19]. NSAIDs are also inhibitors of the active transporters mentioned above in vitro [19, 20]. In terms of severity the interaction is graded as major risk. However, the interaction between MTX and NSAIDs seems to have little relevance in low-dose-MTX therapy, equivalent to a dose of up to 30 mg once weekly.
With regards to leflunomide (LEF) and NSAIDs there is a possible pharmacokinetic interaction [9,10,11]. NSAIDs are metabolized by CYP2C9 in the liver. The active metabolite of LEF, teriflunomide, inhibits CYP2C9. It was shown that LEF inhibits NSAID metabolism via CYP2C9 in vitro. There seems to be no relevant interaction in vivo [21].
The interaction between sulfasalazine (SSZ) and NSAIDs is a pharmacodynamic one. The interaction programs state an interaction between NSAIDs and salicylates because SSZ is partly a salicylate. This can potentially lead to an increased risk for gastrointestinal bleeding. The concomitant use of NSAIDs and aspirin causes increased gastrotoxicity, but it is not supported by any data if the same interaction also applies to SSZ [11, 22, 23]. Furthermore, there is an increased risk for hepatotoxicity when SSZ and diclofenac are administered together [10].
The pharmacodynamic interaction between hydroxychloroquine (HCQ) and diclofenac leads to hepatotoxicity, as both substances can cause elevated liver enzymes. Diclofenac is the NSAID with the greatest potential for hepatotoxicity [10], whereas HCQ normally has a small risk for hepatotoxic effects. No studies could be found where these two drugs were administered and investigated together. DDIs between cDMARDs and NSAIDs are summarised in Table 3.
cDMARDs combined with other analgesics
The pharmacodynamic interaction between MTX and the analgesic drug metamizole can trigger haematological side effects [10, 11]. However, only one case report describing bone marrow suppression due to the concomitant use of MTX and metamizole could be found [24]. In terms of severity the interaction is rated as major risk.
Based on a pharmacodynamic interaction, combining SSZ and metamizole increases the risk of haematological side effects, especially agranulocytosis, although no studies regarding the concomitant use could be identified [10, 25, 26].
Between the antimalarials (CQ, HCQ) and paracetamol a theoretical pharmacokinetic interaction is described as paracetamol is changed partly by CYP2D6 into one of its toxic metabolites, N-acetyl-p-benzoquinone-imine (NAPQI) [10, 27]. Chloroquine (CQ) and HCQ both have the ability to inhibit CYP2D6, so the toxification of paracetamol might be inhibited [28]. However, the toxicity of already formed NAPQI can be enhanced by CQ and HCQ inhibiting lysosomal digestion of injured organelles. This results in cell death and subsequent liver necrosis [29].
The antimalarials may also interact with opioids through pharmacokinetic mechanisms [10]. As mentioned before, some opioids are metabolised by CYP2D6 [16]. The ability of CQ and HCQ to inhibit CYP2D6 [28] might reduce the analgesic efficacy of tramadol and lead to higher plasma concentrations of oxycodone and buprenorphine. No studies regarding the concomitant use of these two classes of drugs could be identified, so the clinical relevance remains unclear. Possible interactions of cDMARDs and analgesics are shown in Table 3.
cDMARDs combined with glucocorticoids
The interaction between MTX and glucocorticoids can be characterized as a pharmacokinetic one. Referring to mediQ, glucocorticoids inhibit cellular uptake of MTX and therefore the efficacy of MTX might be decreased [10]. Literature research in PubMed showed that hepatotoxicity is increased when combining MTX and long-term glucocorticoids. This is probably due to the steroids influencing various active transporters involved in the excretion of MTX, particularly multidrug-resistance protein (MRP) and breast cancer resistance protein (BCRP) [30].
Between LEF and glucocorticoids a pharmacodynamic interaction is given. Both drugs can cause hypertension and hyperlipidaemia [10]. Hypertension is a common side effect of both drugs so the combination is likely to increase the risk of high blood pressure [31]. Regarding hyperlipidaemia, it remains unclear if their combination increases the risk [32].
SSZ and glucocorticoids can trigger hematotoxicity due to a pharmacodynamic interaction [10]. However, few studies regarding the concomitant use of the two drugs could be found [33, 34].
The interaction between the antimalarials and glucocorticoids is a pharmacodynamic one. As both drugs carry a risk for myopathy and cardiomyopathy [10], the combination of two myotoxic agents might increase the risk of developing such severe side effects [35, 36]. An overview on these DDIs is given in Table 3.
cDMARDs combined with PPIs
The interaction between MTX and PPIs is a pharmacokinetic interaction [9,10,11]. PPIs are able to inhibit the excretion of MTX through various transport systems like OAT3 [37] and BRCP [38]. The interaction seems to be less relevant in low-dose-MTX therapy [7, 39], nevertheless it might be important to pay attention in individual cases.
The interaction of PPIs with the antimalarials is a pharmacokinetic one [10]. PPIs increase the lysosomal pH-value. Theoretically, the lysosomal resorption and therefore efficacy of CQ and HCQ might be decreased subsequently [40]. However, it remains unclear if this mechanism is relevant in clinical practice (Table 3).
No interactions were found for the combination of PPIs, LEF, and SSZ.
Methotrexate combined with folic acid
In clinical practice, it is mandatory to add folic acid to a therapy with MTX to reduce the side effects caused by folate deficiency. It was suggested by the interaction programs that folic acid might reduce the efficacy of MTX therapy [10, 11]. It could be figured out that supplementation with folic acid significantly reduces side effects without a noteworthy reduction of its efficacy [41] (Table 3).
Therapy with biological DMARDs (bDMARDs)
bDMARDs often used in rheumatology are TNFα-inhibitors (adalimumab, etanercept, certolizumab pegol, golimumab, infliximab), various IL-antagonists (tocilizumab, sarilumab, secukinumab, ustekinumab, ixekizumab) and antibodies against B-cells and T-cell activation (rituximab, abatacept). As they are not metabolised by CYP-isoenzymes or excreted by active transporters, there are no pharmacokinetic interactions with other drugs involving liver enzymes and transport proteins [42]. However, some of them influence the expression of CYP-enzymes [10].
There may be a pharmacodynamic interaction when MTX and TNFα-inhibitors are administered together. Elevation of liver enzymes is a frequent side effect of MTX, but this can also occur during therapy with TNFα-inhibitors. Due to the fact that there are only few case reports available, it could not be fully elucidated if the combination results in an increased risk for hepatotoxicity [43]. Between MTX and TNFα-inhibitors there is a potential for pharmacokinetic interaction, too. The occurrence of anti-drug-antibodies seems to be reduced when MTX is applied concomitantly. Other DMARDs don’t seem to have such “protective” effects on the immunogenicity of biologicals [42, 44, 45].
The combination of the TNFα-inhibitors with LEF leads to a potentially increased risk for hepatotoxicity, because both agents have hepatotoxic effects. However, no studies could be found investigating the combination of both [45].
Tocilizumab, an inhibitor of the IL-6 receptor causes pharmacokinetic interactions with various CYP-substrates. IL-6, which is formed during inflammatory processes, suppresses the expression of CYP and therefore the metabolism of the respective substrates is impaired [10, 11]. By inhibiting the IL-6-mediated signalling pathway the activity of CYP normalises and the metabolism of CYP-substrates is increased subsequently. This makes dose adjustments necessary [46]. The same type of interaction was suggested for the IL-17A-antagonist secukinumab and IL-17-antagonist ixekizumab [47] in the interaction program mediQ, but no studies could be identified [10]. Other cytokines inhibiting IL-12/IL-23 have no effect on microsomal CYP-enzymes in vitro [48]. Due to lack of data no information regarding severity is available.
A summary of possible interactions is given in Table 4.
Therapy with targeted synthetic DMARDs (tsDMARDs)
Novel drugs used in the therapy of inflammatory arthritis are the two Janus kinase inhibitors (JAKinibs) tofacitinib and baricitinib and the PDE-inhibitor apremilast.
Tofacitinib is metabolised through various CYP-enzymes, especially CYP3A4, but CYP1A2, 2C9, 2C19 as well as 2D6 seem to be involved to a smaller extent. Therefore interactions with CYP3A4-inhibitors are mostly relevant [49] (Tables 5, 6). Among PPIs, lansoprazole seems to have the greatest potential for inhibiting CYP2C19, whereas pantoprazole has the lowest potential for causing interactions [50]. Tofacitinib is often combined with MTX in patients with inflammatory joint diseases. No change in the pharmacokinetics of MTX and tofacitinib was found [51]. In another study, the incidence of side effects was similar in the monotherapy and the combination therapy [51, 52] (Table 5).
Baricitinib is metabolised mainly via CYP3A4, but only to the extent of approximately 10%. As the drug is a substrate for OAT3, there could be interactions with NSAIDs and PPIs, which are inhibitors of OAT3 [53], however, no study was found. Baricitinib is also an inhibitor of OAT3 in vitro which could lead to possible interactions with MTX [53], but again no study was found.
Apremilast is metabolised mainly via CYP3A4, with participation of CYP1A2 and 2A6. Interactions with CYP-inhibitors do not seem to be critical, but the drug should not be combined with potent CYP-inducers. The combination with MTX did not show any changes in pharmacokinetics for these two drugs [54] (Table 6).
Due to lack of data no information regarding severity is available.
Discussion
A systematic database supported analysis of DDIs in the therapy of inflammatory joint diseases was carried out. Drug-related problems in RA patients like drug-choice, dosing, drug use and drug interaction problems were generally discussed by Ma et al. [3]. A further study was found focusing on the quantification of polypharmacy in RA patients and investigating the interaction risk of these patients [4]. The majority of previous reviews, however, is focused on the risk of potential interactions of single drug combinations with methotrexate (MTX) [5,6,7,8], but so far there is no detailed review on the whole range of drugs used for managing inflammatory joint diseases.
Interactions between drugs used for the pharmacotherapy of inflammatory arthritis were assessed using a software detection approach based on three different computerised interaction programs and classified according to their clinical relevance by applying EBM and the specifically developed RBS. Drug combinations were selected according to the EULAR treat-to-target recommendations for treating rheumatoid arthritis and psoriatic arthritis patients [1, 2]. Nowadays a considerable amount of computer programs based on pharmacological studies is available for checking drug interactions. We selected three programs offering clinically useful information about DDIs: Apotronik was decided upon as this is a frequently applied program in retail pharmacies in Austria. MediQ is a highly approved Swiss program and Micromedex is a leading international program widely used in hospital pharmacies. This selection allows the concise evaluation of DDIs from a national as well as international point of view. The theoretical background of relevant effects is described in greater detail in the Micromedex program and is additionally supported by an included description of the underlying DDI mechanisms and references [55].
Generally, and for the most part DDIs are attributed to metabolising enzymes including phase I and phase II reactions as well as CYP genes. Depending on the molecular structure and based on comparable functional chemical moieties, CYP3A4 is the key enzyme for metabolising drugs used in the therapy of inflammatory joint diseases. Therefore, it has to be kept in mind that all inhibitors of CYP3A4 can lead to increased plasma levels of the drug and consequently to an increased risk of side effects. We obtained similar results with regards to type and severity of interactions, mentioning that no contraindication was found. Based on the results of these programs, the screened combinations seem to generally be quite safe. Noteworthy is the fact that clinically relevant drug interactions within the group of bDMARDs seem to be limited to IL-6 receptor blockers, IL-17A- and IL-17-antagonists, substances normalising down-regulated CYP activity caused by uncontrolled inflammation. A subsequent increase of the metabolism of CYP-substrates makes dose adjustments necessary.
Knowledge of CYP enzyme metabolism and transport systems is crucial especially in case of polypharmacy in elderly patients suffering from treatable comorbidities (e.g., hypertension, heart failure, renal failure, depression and osteoporosis). The risk for interactions increases with the number of drugs taken. According to Rottenkolber et al. medication side effects account for 3.25% of all non-elective admissions to internal medical departments [56]. Whereas this is already an unexpectedly high number, drug-related hospitalisation rises up to 11% in the group of elderly patients [57]. Hence, alertness with regards to potential interactions saves patients wellbeing. Closer interdisciplinary cooperation between drug prescribing physicians and pharmacists is recommended as a very important safety enhancing tool.
As all possible DDIs evaluated in this paper refer to standard dosage schemes, thus the interaction(s) may be significantly more distinct or only present in higher therapeutic drug concentrations. For the anchor drug MTX the interaction potential may well be overestimated due to a reduced dosage in rheumatology as opposed to high dose administration in oncology. Interactions with active transporters for drugs new to the market such as tofacitinib and baricitinib are possible and therefore more studies investigating pharmacokinetic interactions with JAK inhibitors are required. Furthermore, every additional single drug as well as ethanol and dietary supplements can influence drugs’ kinetics. Hence these factors as well as comorbidity, aspects related to patients, and medication administration including dosages, time, and sequences, were not reviewed and therefore are the limitations in this paper and warrant further investigation.
Summarising these investigations, it can be emphasised that pharmacotherapy in the treatment of inflammatory arthritis is quite safe and well evidence-based. Interactions between drugs used for this treatment were screened by comparing three different computerised interaction programs supported with extensive literature research, especially for drugs new to the market. The applied screening software in this paper is limited to a 1:1 analysis so you only get one tessera of the mosaic for inflammatory joint disease patients. To obtain a more comprehensive picture, the results were combined in clearly structured tables which allows a quick overview on potential interactions. They can be used on an individual basis to identify patients at risk, e.g., patients with (multiple) comorbidities and therefore under polypharmacy. Furthermore, it allows simple adjustment to new drugs and thus maybe contributes to a reshaping of future treatment algorithm.
References
Smolen JS, Aletaha D, Bijlsma JWJ et al (2010) Treating rheumatoid arthritis to target: recommendations of an international task force. Ann Rheum Dis 69:631–637. https://doi.org/10.1136/ard.2009.123919
Smolen JS, Schöls M, Braun J et al (2018) Treating axial spondyloarthritis and peripheral spondyloarthritis, especially psoriatic arthritis, to target: 2017 update of recommendations by an international task force. Ann Rheum Dis 77:3–17. https://doi.org/10.1136/annrheumdis-2017-211734
Ma SN, Zaman Huri H, Yahya F (2019) Drug-related problems in patients with rheumatoid arthritis. Ther Clin Risk Manag 15:505–524. https://doi.org/10.2147/TCRM.S194921
Bagatini F, Blatt CR, Maliska G et al (2011) Potential drug interactions in patients with rheumatoid arthritis. Rev Bras Reumatol 51:20–39. https://doi.org/10.1590/S0482-50042011000100003
Colebatch AN, Marks JL, van der Heijde DM, Edwards CJ (2012) Safety of nonsteroidal antiinflammatory drugs and/or paracetamol in people receiving methotrexate for inflammatory arthritis: a cochrane systematic review. J Rheumatol Suppl 90:62–73. https://doi.org/10.3899/jrheum.120345
Patanè M, Ciriaco M, Chimirri S et al (2013) Interactions among low dose of methotrexate and drugs used in the treatment of rheumatoid arthritis. Adv Pharmacol Sci 2013:313858. https://doi.org/10.1155/2013/313858
Hall JJ, Bolina M, Chatterley T, Jamali F (2017) Interaction between low-dose methotrexate and nonsteroidal anti-inflammatory drugs, penicillins, and proton pump inhibitors. Ann Pharmacother 51:163–178. https://doi.org/10.1177/1060028016672035
Bourré-Tessier J, Haraoui B (2010) Methotrexate drug interactions in the treatment of rheumatoid arthritis: a systematic review. J Rheumatol 37:1416–1421. https://doi.org/10.3899/jrheum.090153
Apotronik Datenservice GmbH ApotronikXP. Apotheken-Managementsystem, www.apotronik.at. Accessed 26 Feb 2019.
Psychiatrische Dienste Aargau AG, mediQ-Interaktionsprogramm, www.mediq.com. Accessed 27 Feb 2019.
Truven Health Analytics LLC. Micromedex® Solutions, www.micromedexsolutions.com. Accessed 27 Feb 2019.
Espinosa-Bosch M, Santos-Ramos B, Gil-Navarro MV, Santos-Rubio MD, Marín-Gil R, Villacorta-Linaza P (2012) Prevalence of drug interactions in hospital healthcare. Int J Clin Pharm 34:807–817. https://doi.org/10.1007/s11096-012-9697-0
Miners JO, Coulter S, Tukey RH, Veronese ME, Birkett DJ (1996) Cytochromes P450, 1A2, and 2C9 are responsible for the human hepatic O-demethylation of R- and S-naproxen. Biochem Pharmacol 51:1003–1008. https://doi.org/10.1016/0006-2952(96)85085-4
Li X-Q, Andersson TB, Ahlström M, Weidolf L (2004) Comparison of inhibitory effects of the proton pump-inhibiting drugs omeprazole, esomeprazole, lansoprazole, pantoprazole, and rabeprazole on human cytochrome P450 activities. Drug Metab Dispos 32:821–827. https://doi.org/10.1124/dmd.32.8.821
Chi C-W (2009) Inhibition of the healing of gastric ulcer by glucocorticoid and its relation to proinflammatory cytokines. J Chin Med Assoc 72:559–560. https://doi.org/10.1016/S1726-4901(09)70429-0
Leppert W (2011) CYP2D6 in the metabolism of opioids for mild to moderate pain. Pharmacology 87:274–285. https://doi.org/10.1159/000326085
Samer CF, Daali Y, Wagner M et al (2010) Genetic polymorphisms and drug interactions modulating CYP2D6 and CYP3A activities have a major effect on oxycodone analgesic efficacy and safety. Br J Pharmacol 160:919–930. https://doi.org/10.1111/j.1476-5381.2010.00709.x
El-Sheikh AAK, van den Heuvel JJMW, Koenderink JB, Russel FGM (2007) Interaction of nonsteroidal anti-inflammatory drugs with multidrug resistance protein (MRP) 2/ABCC2- and MRP4/ABCC4-mediated methotrexate transport. J Pharmacol Exp Ther 320:229–235. https://doi.org/10.1124/jpet.106.110379
Maeda A, Tsuruoka S, Kanai Y et al (2008) Evaluation of the interaction between nonsteroidal anti-inflammatory drugs and methotrexate using human organic anion transporter 3-transfected cells. Eur J Pharmacol 596:166–172. https://doi.org/10.1016/j.ejphar.2008.08.023
Kawase A, Yamamoto T, Egashira S, Iwaki M (2016) Stereoselective inhibition of methotrexate excretion by glucuronides of nonsteroidal anti-inflammatory drugs via multidrug resistance proteins 2 and 4. J Pharmacol Exp Ther 356:366–374. https://doi.org/10.1124/jpet.115.229104
Rozman B (2002) Clinical pharmacokinetics of leflunomide. Clin Pharmacokinet 41:421–430. https://doi.org/10.2165/00003088-200241060-00003
Lanas A, García-Rodríguez LA, Arroyo MT et al (2006) Risk of upper gastrointestinal ulcer bleeding associated with selective cyclo-oxygenase-2 inhibitors, traditional non-aspirin non-steroidal anti-inflammatory drugs, aspirin and combinations. Gut 55:1731–1738. https://doi.org/10.1136/gut.2005.080754
Sostres C, Gargallo CJ, Lanas A (2013) Nonsteroidal anti-inflammatory drugs and upper and lower gastrointestinal mucosal damage. Arthritis Res Ther 15(Suppl 3):S3. https://doi.org/10.1186/ar4175
Blanchet E, Beau P, Frat J-P (2004) Aplasie médullaire après prise de noramidopyrine chez une malade traitée au long cours par méthotrexate pour maladie de Crohn. Gastroenterol Clin Biol 28:502–503
Huber M, Andersohn F, Bronder E et al (2014) Drug-induced agranulocytosis in the Berlin case-control surveillance study. Eur J Clin Pharmacol 70:339–345. https://doi.org/10.1007/s00228-013-1618-1
Navarro-Martínez R, Chover-Sierra E, Cauli O (2016) Non-chemotherapy drug-induced agranulocytosis in a tertiary hospital. Hum Exp Toxicol 35:244–250. https://doi.org/10.1177/0960327115580603
Dong H, Haining RL, Thummel KE, Rettie AE, Nelson SD (2000) Involvement of human cytochrome P450 2D6 in the bioactivation of acetaminophen. Drug Metab Dispos 28:1397–1400
Projean D, Baune B, Farinotti R et al (2003) In vitro metabolism of chloroquine: identification of CYP2C8, CYP3A4, and CYP2D6 as the main isoforms catalyzing N-desethylchloroquine formation. Drug Metab Dispos 31:748–754. https://doi.org/10.1124/dmd.31.6.748
Ni H-M, Bockus A, Boggess N, Jaeschke H, Ding W-X (2012) Activation of autophagy protects against acetaminophen-induced hepatotoxicity. Hepatology 55:222–232. https://doi.org/10.1002/hep.24690
Fuksa L, Brcakova E, Kolouchova G et al (2010) Dexamethasone reduces methotrexate biliary elimination and potentiates its hepatotoxicity in rats. Toxicology 267:165–171. https://doi.org/10.1016/j.tox.2009.11.010
Panoulas VF, Metsios GS, Pace AV et al (2008) Hypertension in rheumatoid arthritis. Rheumatology 47:1286–1298. https://doi.org/10.1093/rheumatology/ken159
Schroeder LL, Tang X, Wasko MCM, Bili A (2015) Glucocorticoid use is associated with increase in HDL and no change in other lipids in rheumatoid arthritis patients. Rheumatol Int 35:1059–1067. https://doi.org/10.1007/s00296-014-3194-9
Marabani M, Madhok R, Capell HA, Hunter JA (1989) Leucopenia during sulphasalazine treatment for rheumatoid arthritis. Ann Rheum Dis 48:505–507. https://doi.org/10.1136/ard.48.6.505
Farr M, Symmons DP, Blake BPA (1986) Neutropenia in patients with inflammatory arthritis treated with sulphasalazine. Ann Rheum Dis 45:761–764
Katzberg HD, Kassardjian CD (2016) Toxic and Endocrine Myopathies. Continuum (Minneap Minn) 22:1815–1828. https://doi.org/10.1212/CON.0000000000000407
Tselios K, Gladman DD, Su J, Urowitz MB (2016) Antimalarials as a risk factor for elevated muscle enzymes in systemic lupus erythematosus. Lupus 25:532–535. https://doi.org/10.1177/0961203315617845
Chioukh R, Noel-Hudson M-S, Ribes S, Fournier N, Becquemont L, Verstuyft C (2014) Proton pump inhibitors inhibit methotrexate transport by renal basolateral organic anion transporter hOAT3. Drug Metab Dispos 42:2041–2048. https://doi.org/10.1124/dmd.114.058529
Suzuki K, Doki K, Homma M et al (2009) Co-administration of proton pump inhibitors delays elimination of plasma methotrexate in high-dose methotrexate therapy. Br J Clin Pharmacol 67:44–49. https://doi.org/10.1111/j.1365-2125.2008.03303.x
Vakily M, Amer F, Kukulka MJ, Andhivarothai N (2005) Coadministration of lansoprazole and naproxen does not affect the pharmacokinetic profile of methotrexate in adult patients with rheumatoid arthritis. J Clin Pharmacol 45:1179–1186. https://doi.org/10.1177/0091270005280100
Namazi MR (2009) The potential negative impact of proton pump inhibitors on the immunopharmacologic effects of chloroquine and hydroxychloroquine. Lupus 18:104–105. https://doi.org/10.1177/0961203308097574
Khanna D, Park GS, Paulus HE et al (2005) Reduction of the efficacy of methotrexate by the use of folic acid: post hoc analysis from two randomized controlled studies. Arthritis Rheum 52:3030–3038. https://doi.org/10.1002/art.21295
Xu Z, Davis HM, Zhou H (2015) Clinical impact of concomitant immunomodulators on biologic therapy: pharmacokinetics, immunogenicity, efficacy and safety. J Clin Pharmacol 55(Suppl 3):S60–74. https://doi.org/10.1002/jcph.380
Tobon GJ, Cañas C, Jaller J-J, Restrepo J-C, Anaya J-M (2007) Serious liver disease induced by infliximab. Clin Rheumatol 26:578–581. https://doi.org/10.1007/s10067-005-0169-y
Schaeverbeke T, Truchetet M-E, Kostine M, Barnetche T, Bannwarth B, Richez C (2016) Immunogenicity of biologic agents in rheumatoid arthritis patients: lessons for clinical practice. Rheumatology 55:210–220. https://doi.org/10.1093/rheumatology/kev277
Jani M, Barton A, Warren RB, Griffiths CEM, Chinoy H (2014) The role of DMARDs in reducing the immunogenicity of TNF inhibitors in chronic inflammatory diseases. Rheumatology 53:213–222. https://doi.org/10.1093/rheumatology/ket260
Kim S, Östör AJK, Nisar MK (2012) Interleukin-6 and cytochrome-P450, reason for concern? Rheumatol Int 32:2601–2604. https://doi.org/10.1007/s00296-012-2423-3
Liu L, Lu J, Allan BW et al (2016) Generation and characterization of ixekizumab, a humanized monoclonal antibody that neutralizes interleukin-17A. J Inflamm Res 9:39–50. https://doi.org/10.2147/JIR.S100940
Dallas S, Chattopadhyay S, Sensenhauser C, Batheja A, Singer M, Silva J (2013) Interleukins-12 and -23 do not alter expression or activity of multiple cytochrome P450 enzymes in cryopreserved human hepatocytes. Drug Metab Dispos 41:689–693. https://doi.org/10.1124/dmd.112.048884
Dowty ME, Lin J, Ryder TF et al (2014) The pharmacokinetics, metabolism, and clearance mechanisms of tofacitinib, a janus kinase inhibitor, in humans. Drug Metab Dispos 42:759–773. https://doi.org/10.1124/dmd.113.054940
Ogawa R, Echizen H (2010) Drug-drug interaction profiles of proton pump inhibitors. Clin Pharmacokinet 49:509–533. https://doi.org/10.2165/11531320-000000000-00000
Cohen S, Zwillich SH, Chow V, Labadie RR, Wilkinson B (2010) Co-administration of the JAK inhibitor CP-690,550 and methotrexate is well tolerated in patients with rheumatoid arthritis without need for dose adjustment. Br J Clin Pharmacol 69:143–151. https://doi.org/10.1111/j.1365-2125.2009.03570.x
Tanaka Y, Suzuki M, Nakamura H, Toyoizumi S, Zwillich SH (2011) Phase II study of tofacitinib (CP-690,550) combined with methotrexate in patients with rheumatoid arthritis and an inadequate response to methotrexate. Arthritis Care Res 63:1150–1158. https://doi.org/10.1002/acr.20494
Payne C, Zhang X, Shahri N, Williams W, Cannady E (2015) AB0492 Evaluation of potential drug-drug interactions with baricitinib. Ann Rheum Dis 74:1063. https://doi.org/10.1136/annrheumdis-2015-eular.1627
Liu Y, Zhou S, Nissel J, Wu A, Lau H, Palmisano M (2014) The pharmacokinetic effect of coadministration of apremilast and methotrexate in individuals with rheumatoid arthritis and psoriatic arthritis. Clin Pharmacol Drug Dev 3:456–465. https://doi.org/10.1002/cpdd.109
Roblek T, Vaupotic T, Mrhar A, Lainscak M (2015) Drug-drug interaction software in clinical practice: a systematic review. Eur J Clin Pharmacol 71:131–142. https://doi.org/10.1007/s00228-014-1786-7
Rottenkolber D, Schmiedl S, Rottenkolber M et al (2011) Adverse drug reactions in Germany: direct costs of internal medicine hospitalizations. Pharmacoepidemiol Drug Saf 20:626–634. https://doi.org/10.1002/pds.2118
Farker K (2017) Arzneimittelwechselwirkungen bei Polypharmakotherapie. Arzneiverordnung in der Praxis 44:58–62
Laine JE, Auriola S, Pasanen M, Juvonen RO (2009) Acetaminophen bioactivation by human cytochrome P450 enzymes and animal microsomes. Xenobiotica 39:11–21. https://doi.org/10.1080/00498250802512830
Mazaleuskaya LL, Sangkuhl K, Thorn CF, FitzGerald GA, Altman RB, Klein TE (2015) PharmGKB summary: pathways of acetaminophen metabolism at the therapeutic versus toxic doses. Pharmacogenet Genom 25:416–426. https://doi.org/10.1097/FPC.0000000000000150
Czock D, Keller F, Rasche FM, Häussler U (2005) Pharmacokinetics and pharmacodynamics of systemically administered glucocorticoids. Clin Pharmacokinet 44:61–98. https://doi.org/10.2165/00003088-200544010-00003
Takeda M, Khamdang S, Narikawa S et al (2002) Characterization of methotrexate transport and its drug interactions with human organic anion transporters. J Pharmacol Exp Ther 302:666–671. https://doi.org/10.1124/jpet.102.034330
Kis E, Nagy T, Jani M et al (2009) Leflunomide and its metabolite A771726 are high affinity substrates of BCRP: implications for drug resistance. Ann Rheum Dis 68:1201–1207. https://doi.org/10.1136/ard.2007.086264
Kalgutkar AS, Nguyen HT, Vaz ADN et al (2003) In vitro metabolism studies on the isoxazole ring scission in the anti-inflammatory agent lefluonomide to its active alpha-cyanoenol metabolite A771726: mechanistic similarities with the cytochrome P450-catalyzed dehydration of aldoximes. Drug Metab Dispos 31:1240–1250. https://doi.org/10.1124/dmd.31.10.1240
Ma LL, Wu ZT, Wang L et al (2016) Inhibition of hepatic cytochrome P450 enzymes and sodium/bile acid cotransporter exacerbates leflunomide-induced hepatotoxicity. Acta Pharmacol Sin 37:415–424. https://doi.org/10.1038/aps.2015.157
Wiese MD, Schnabl M, O'Doherty C et al (2012) Polymorphisms in cytochrome P450 2C19 enzyme and cessation of leflunomide in patients with rheumatoid arthritis. Arthritis Res Ther 14:R163. https://doi.org/10.1186/ar3911
Elsby R, Fox L, Stresser D et al (2011) In vitro risk assessment of AZD9056 perpetrating a transporter-mediated drug-drug interaction with methotrexate. Eur J Pharm Sci 43:41–49. https://doi.org/10.1016/j.ejps.2011.03.006
Posada MM, Cannady EA, Payne CD et al (2017) Prediction of transporter-mediated drug-drug interactions for baricitinib. Clin Transl Sci 10:509–519. https://doi.org/10.1111/cts.12486
Keating GM (2017) Apremilast: a review in psoriasis and psoriatic arthritis. Drugs 77:459–472. https://doi.org/10.1007/s40265-017-0709-1
Acknowledgements
Open access funding provided by University of Graz. All authors would like to thank Dr. Schäffer and Mag. Knoll for their helpful technical assistance.
Funding
No funding was received.
Author information
Authors and Affiliations
Contributions
All authors take full responsibility for the integrity of the manuscript. SP and AO conceived, designed and coordinated the search. KB and ML extracted the data. RP and WG revised the data and provided critical feedback. All authors approved the revised version manuscript and agreed to be accountable for all aspects of the current review.
Corresponding author
Ethics declarations
Conflict of interest
All authors declare that they have no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pflugbeil, S., Böckl, K., Pongratz, R. et al. Drug interactions in the treatment of rheumatoid arthritis and psoriatic arthritis. Rheumatol Int 40, 511–521 (2020). https://doi.org/10.1007/s00296-020-04526-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00296-020-04526-3