
Abstract
Sporadic inclusion body myositis (s-IBM) is a progressive, skeletal muscle disease with poor prognosis. However, establishing the final diagnosis is difficult because of the lack of clear biomarkers in the blood serum and very slow development of clinical symptoms. Moreover, most other organs function normally without any disturbance. Here, in patients with this untreatable disease, we have underlined the importance of immunohistochemical and ultrastructural assessment of skeletal muscle in patients diagnosed with s-IBM. The goal of this study was to identify the distribution of specific antigens and to determine morphological features in order to localize pathological protein aggregates, rimmed vacuoles, and loss of myofibrils, which are key elements in the diagnosis of s-IBM. All studied patients were between 48 and 83 years of age and were hospitalized in the Department of Rheumatology and Internal Medicine between 2011 and 2016. Anamneses revealed an accelerated progression of muscle atrophy, weakness of limb muscles, and difficulties with climbing stairs. Based on histopathology and transmission electron microscopy examination, inflammatory infiltrations consisting of mononuclear cells, severe atrophy and focal necrosis of myofibers, splitting of myofilaments, myelinoid bodies and rimmed vacuoles were observed. Primary antibodies directed against CD3, CD8, CD68, cN1A, beta-amyloid, Tau protein and apolipoprotein B made it possible to identify types of cells within infiltrations as well as the protein deposits within myofibers. Using a combination of immunohistochemistry and electron microscopy methods, we were able to establish the correct final diagnosis and to implement a specific treatment to inhibit disease progression.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
In adult patients both sporadic inclusion body myositis (s-IBM) and hereditary inclusion body myopathy (h-IBM) are classified as progressive muscular diseases [1, 2]. They are characterized by similar pathological features, including abnormal inclusions and vacuoles in myofibers, however, h-IBM shows no signs of inflammation in the muscle biopsy [3, 4].
Sporadic IBM belongs to a group of idiopathic inflammatory myopathies of unknown cause with inflammatory and degenerative features resulting from autoimmunity [5,6,7,8,9]. However, many scientists feel that it is a primary degenerative disorder that triggers inflammation [10, 11]. It is a progressive disease and is the most common disease of skeletal muscle in patients over the age of 50 [6, 12], leading to serious disability [3, 7, 13]. The onset of symptoms in patients younger than 60 years of age is typical for 18–20% of patients [6]. The occurrence of IBM varies between 4.3 and 9.3 per a million births, with an increase to 35.3 among people over 50 years of age [5]. Clinically this disease entity is characterized by slow and progressive weakening of distal muscles of the arms (emaciated forearms) and proximal muscles of the legs. A biopsy reveals inflammatory cell infiltrates in the endomysium, vacuoles with granularities, the so-called rimmed vacuoles (RVs), as well as necrosis of muscle fibers and deposits of abnormal proteins [2, 3, 6, 7, 9, 14, 15]. Many authors distinguish four basic subgroups of idiopathic inflammatory myopathies: dermatomyositis (DM), polymyositis (PM), immune-mediated necrotizing myopathy (NM) and IBM [6, 7, 15, 16]. However, from a clinical standpoint s-IBM, is distinguishable from the aforementioned diseases by asymmetric weakness of the finger flexor and knee extensor muscles, atrophy of the quadriceps muscles of the thighs with an inability to fully straighten the knees and resistance to immunosuppressive therapy [3, 6, 7, 17]. It should be emphasized that patients demonstrating clinical symptoms typical for IBM with few inflammatory cells or with the presence of RVs, as seen in a muscle biopsy examination, could be difficult to diagnose in terms of histopathology [6]. Moreover, patients suffering from PM may have several RVs [6]. Although RVs are a hallmark of IBM, they can also be found in 30 other muscular or neurogenic disorders, e.g., in spinal muscular atrophy or muscular dystrophies [18, 19]. Therefore, morphological determinants of s-IBM are not specific and appear in various neuromuscular disorders [20]. Similarly, electromyography (EMG) examination makes it possible to include this disease in a group of muscle damage of the myogenic type, without the possibility of precisely determining its cause [21]. Hence, the initial diagnosis is often incorrect, and it is likely to be assessed as PM or as amyotrophic lateral sclerosis (ALS) [7, 9].
The most reliable way of identifying this disease is a muscle biopsy followed by histopathological and immunohistochemical assessment [5, 14]. Immunohistochemistry (IHC) is a method which enables the detection of various proteins that accumulate in muscle fibers of patients suffering from s-IBM and that are associated with inflammation processes, autophagy or endoplasmic reticulum stress [7, 17, 22]. Accumulated proteins have the immunoreactivity of beta-amyloid, Tau protein and apolipoproteins [20, 23,24,25]. For the final diagnosis of this disease it is necessary to carry out research with a transmission electron microscope (TEM), in order to demonstrate the presence of RVs and disorganization of myofibrils [7, 23, 26, 27].
The aim of this study was to identify the distribution of specific antigens as well as to determine morphological features in the skeletal muscles of patients diagnosed with s-IBM.
It is important to emphasize that the very slow progress of clinical symptoms makes the diagnosis difficult. Another problem was a study in transmission electron microscope (TEM) which is currently pivotal in the diagnostic scheme. Not much research has focused exclusively on the pathologically defined IBM (according to modified IBM diagnostic criteria proposed by Hilton-Jones et al. [28]). For this reason, we would like to emphasize the morphological aspect during the diagnosis of this disease and underline the role of IHC reaction and transmission electron microscopy that are crucial for pathologically defined IBM. Therefore, we have tried to accentuate the practical aspects of our study which include unique muscle images resulting from an innovative combination of IHC and TEM techniques. To the best of our knowledge there is no literature in this area of research.
Materials and methods
Patients
All patients were hospitalized and treated between 2011 and 2016 in the Department of Rheumatology and Internal Medicine of the Wroclaw Medical University. They were between 48 and 83 years old and on the average had felt symptoms for the previous 5 years. The research material was constituted of small skin samples (13 mm wide and 4 mm deep) with fragments of subcutaneous tissue and skeletal muscles (12 mm wide and 4 mm deep) of the lower limbs: quadriceps femoris, rectus femoris, gastrocnemius and of the upper limbs: biceps brachii and deltoid muscle collected from six patients diagnosed with s-IBM.
All patients signed an informed consent for muscle biopsy and histopathological study, based on local regulations and approved by the Wroclaw Medical Hospital (part of Wroclaw Medical University). Diagnostic criteria for patients are concomitant with definite IBM according to Griggs et al. [29] and Dalakas [5] (without tubulofilaments in TEM), together with pathologically defined IBM after the modified criteria of Hilton-Jones et al. [28]. In our study, we could definitely exclude possible IBM by using anti-beta-amyloid antibody which detected the presence of amyloid deposits. Exclusion criteria were as follows: toxic and drug-induced (including statins, glucocorticoids) myopathies, rhabdomyolysis, and injury; polymyositis was eliminated on the basis of clinical features and histopathological examination. Clinical characteristics of patients are given in Table 1.
Light microscopy
Tissue samples were embedded in paraffin blocks (Merck, Darmstadt, Germany) which were cut on fully automated Rotary Microtome (Leica RM 2255, Nussloch, Germany) into 4-µm-thick paraffin sections. Subsequently, sections were stained with Mayer’s hematoxylin and eosin (H&E) (Bio-Optica Milano, Italy) and were analyzed with the use of a light microscope BX41 (Olympus, Tokyo, Japan), connected to a digital camera Colorview IIIu (Olympus).
Immunohistochemistry (IHC)
Immunohistochemical reactions were performed on 4-µm-thick paraffin sections using Autostainer Link48 (Dako, Glostrup, Denmark) and the following antibodies: anti-beta-amyloid (Dako), anti-Tau protein (Invitrogen, Carlsbad, USA), anti-apolipoprotein B (apo-B; Santa Cruz Biotechnology, Dallas, USA), anti-CD3 (T lymphocytes marker; Dako), anti-CD8 (cytotoxic T lymphocytes marker; Dako), anti-CD68 (macrophages marker; Dako) and anti-cN1A (anti-NT5C1A; Abcam). Briefly, deparaffinization, rehydration and the antigens unmasking were performed with EnVision FLEX Target Retrieval Solution (pH 9, 20 min, 97 °C; Dako) using PTLink platform (Dako). The activity of endogenous peroxidase was blocked by 5-min incubation with EnVision FLEX Peroxidase-Blocking Reagent (Dako). Afterwards, primary antibodies directed against beta-amyloid (1:300), Tau protein (1:3000), apolipoprotein B (1:100), cN1A (1:200), CD3 (RTU), CD8 (RTU), and CD68 (RTU) were applied for 20 min. For visualization EnVision FLEX/HRP (Dako, 20 min) was used. As a peroxidase substrate 3,3′-diaminobenzidine (DAB; Dako) was used for 10 min of incubation. Finally, all sections were counterstained with EnVision FLEX Hematoxylin (Dako) for 5 min. After dehydration in gradually increasing ethanol concentrations (70%, 96% and 99.8%), followed by incubation in xylene, slides were closed with coverslips in Dako Mounting Medium (Dako). The primary antibodies were diluted in EnVision FLEX Antibody Diluent (Dako).
Transmission electron microscope (TEM)
For TEM examination skeletal muscle samples from six patients were fixed in 2.5% glutaraldehyde (Serva Electrophoresis, Heidelberg, Germany). After 24 h, the specimens were washed in cacodylate buffer (0.1 M, pH 7.4, Serva) and postfixed for 1 h in 1% osmium tetroxide (Serva). Subsequently, they were rinsed with the cacodylate buffer. Afterwards, the specimens were dehydrated in highly concentrated ethyl alcohol and twice in pure acetone (Chempur, Piekary Slaskie, Poland). Samples were embedded in epoxy resin (Epon 812, Serva). To select the examined area for TEM Epon blocks were cut into semithin, 600-nm-thick sections, stained with toluidine blue (Serva) and closed with the use of Euparal mounting agent (Roth, Mannheim, Germany). Finally, ultrathin, 50-nm-thick sections were prepared using an ultramicrotome Power Tome XL (RMC, Tucson, USA). To improve the contrast, ultrathin sections placed on the copper grids were counterstained with uranyl acetate and lead citrate (Serva), and examined in the TEM JEM-1011 (JEOL, Tokyo, Japan). Digital micrographs were prepared with the use of TEM imaging platform iTEM1233 equipped with a Morada Camera (Olympus, Münster, Germany) at magnifications ranging from 5 to 20 K.
Results
Clinical features of patients
In the physical examination the following characteristics were found: muscular atrophy, particularly in the proximal muscles of the lower limbs and distal muscles of the upper limbs, with a predominance on the left side compared to the right. Furthermore, flexor muscles were affected more than the extensor muscles. The widening of circuit calves was also observed. Furthermore, all patients experienced decreased muscle strength in the lower limbs, especially weakness in the quadriceps muscles, deterioration of the locomotion function, inability to climb stairs and get up from a sedentary position without the use of hands. One patient suffered from dysphagia (a case is described in detail by Misterska-Skóra et al. [8]). At the onset of weakness, the patients were over the age of 45 and had had symptoms for at least a year or more. These clinical findings together with muscle biopsies confirmed diagnosis of definite or pathologically defined IBM according to criteria established by Griggs et al. [29] and Hilton-Jones et al. [28], respectively. The EMG test confirmed that the muscle damage was of the myogenic type. The patients received immunosuppressive agents, such as methotrexate (MTX, orally), glucocorticosteroids (GKS, orally), mycophenolate mofetil (MMF, orally), azathioprine (AZA, orally) and rituximab (RTX, intravenously) (Table 1). For all patients, normal blood cell morphology and acceptable values of inflammatory parameters (i.e., ESR and CRP) were recorded. The serum level of the creatine kinase enzyme (CK) was normal or mildly elevated, but as a rule it was not greater than 15 times the upper normal limit. However, a temporary increase in the serum level of the lactate dehydrogenase enzyme (LDH) was observed (the maximum value: 1011 IU/L; upper normal limit: 248). In addition, a periodic increase in the serum level of CK was found (the maximum value: 6956 IU/L; upper normal limit: 145). The highest value of 6956 IU/L was recorded only once. Among the studied patients, the average value of LDH was 344 IU/L and of CK—1068 IU/L.
Histopathologic, immunohistochemical and ultrastructural studies of muscle biopsies
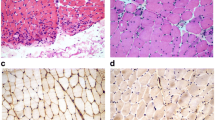
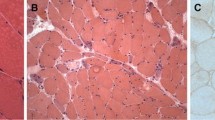
Skin and subcutaneous tissues did not show significant pathological changes or features of connective tissue hyperplasia. Only small clusters of lymphocyte infiltration were present around the blood vessels, however, no vessel wall infiltration nor blood clots in the lumen were observed. The biopsies of skeletal muscle revealed advanced and acute changes (Figs. 1, 2, 3, 4, 5). Histopathological examination found that myofibers had different diameters. Moreover, there was moderately severe atrophy (Fig. 1a). Hyperplasia of connective (Fig. 1a) and adipose (Fig. 1b, c) tissues, splitting of myofibrils (Fig. 1b) and blurring of cross striations (Fig. 1d) were seen, as well as some necrotic myofibers (Fig. 2a, b, d). Only a few myofibers with centrally located nuclei were observed (Figs. 1d, 2c). In the sarcoplasm of individual muscle fibers (usually less than 10 per sample) there were centrally or paracentrally located electron-lucent vacuoles or vacuoles with heterogeneous contents (Fig. 2a–d). At the ultrastructural level the vacuoles were surrounded by granularities (RVs) (Fig. 4a, b). Further observations revealed mononuclear cell infiltration of moderate intensity into the endomysium of both, normal and changed myofibers as well as around blood vessels (Fig. 1a, b). Inflammatory cells invaded nonnecrotic fibers (Fig. 1a). Immunohistochemical analysis confirmed the presence of T lymphocytes (CD3+ and CD8+ cells) and macrophages (CD68+ cells) in the endomysial infiltrate (Fig. 3a–c, respectively). Positive immunoreactivity for cN1A was observed around the vacuoles and in the vicinity of myonuclei (Fig. 3d). Numerous aggregates of pathological protein deposits of Tau, beta-amyloid (Fig. 3e) and apolipoprotein B (apo-B, Fig. 3f) were observed in the sarcoplasm around the vacuoles or unrelated to them. Additionally, deposits of apo-B protein were found in endomysium. Focal and significant destruction of myofibrils and dispersion of myofilaments in many areas of the sarcoplasm were also detected by TEM (Fig. 4c, d). Whorled osmiophilic membrane debris, myelinoid bodies and abnormalities in the triad structure (Fig. 5a, b) or lipid-like inclusion bodies (Fig. 5c, d) were seen close to the nucleus or between bundles of myofibrils.

Infiltrations in the endomysium and degenerative changes in skeletal muscles. Longitudinal (a, b) and transverse (c, d) section of myofibers. Inflammatory infiltrations of mononuclear cells (a, b, arrows), atrophic muscle fibers with connective tissue hyperplasia (a arrowheads), splitting of myofibrils (b arrowhead) and blurring of cross striations (d asterisk arrow). Adipose tissue hyperplasia (b, c, asterisks). A centrally located nucleus (d). N nucleus. Paraffin sections. H&E staining. a–c Patient 6; d patient 4

Degenerative changes in skeletal muscles. Longitudinal (a) and transverse (b–d) sections of muscle fibers. Necrosis of myofibers (a, b, d, arrows) and distinct, numerous vacuoles (a–d, arrowheads). A centrally located nucleus (c). N nucleus. Paraffin sections. H&E staining (a–c). Semithin section. Toluidine blue staining (d). a, d Patient 6; b, c patient 4

Specific antigen distribution in skeletal muscles after resorting to immunohistochemical reactions. Longitudinal (a–c, e, f) and transverse (d) sections of muscle fibers immunohistochemical stained. T lymphocytes (CD3+, a, CD8+, b, arrows) and macrophages (CD68+, c, arrows) in infiltrations. cN1A antigen was identified around the myonuclei (d, arrowheads) or vacuole (d, arrow) which were delineated by it. Deposits of beta-amyloid (e, arrowheads) and apolipoprotein B (f, arrowhead) proteins. Paraffin sections. a, c Patient 3; b, d, e patient 6, f patient 2

Localization of rimmed vacuoles and ultrastructural changes in myofibrils of skeletal muscles. The ultrastructure of myofibers at the transverse (a, b, d) and longitudinal (c) sections. Rimmed vacuoles (a, b). Significant loss and destruction of myofibrils (c). The total disorganization of myofibrils (d, arrows). TEM. a, b Patient 4; c, d patient 6

Localization of inclusion bodies in the sarcoplasm. The ultrastructure of myofibers at the longitudinal (a–d) sections. Osmiophilic membranes debris and myelinoid bodies are conspicuous (a, b). Triad abnormalities (a, inserts). Lipid-like inclusion bodies under sarcolemma (c, arrow) or in the region close to the nucleus (d, arrow). TEM. a–c Patient 6; d patient 3
Discussion
Early clinical symptoms which should raise suspicion of s-IBM include difficulty in climbing stairs and rising from a sedentary position, aching of thighs and knees and difficulties in using different hand tools [30]. In addition, weakness of the thigh’s quadriceps muscles, increased serum CK level, the presence of RVs and atrophic myofibers in muscle biopsies could help to diagnose patients suffering from s-IBM [9, 31]. There are some reports showing that physical effort may improve skeletal muscle strength and function, which would encourage its implementation immediately after s-IBM has been diagnosed [6, 32]. Since s-IBM is an untreatable disease establishing the final diagnosis is critical for patients in order to avoid an ineffective immunosuppressive therapy with its many side effects [9, 15], and to start physiotherapy [33].
In our study the clinical features, including weakness of upper and lower extremities, difficulty in climbing stairs as well as patients’ ages were typical and similar to other reports [12, 27, 34]. Moreover, similarly to Hermanns et al. [4] and Sakai et al. [17] we observed periodical increases in the levels of CK and LDH enzymes in the studied patients’ serum. In patients from a study conducted in China the average value of CK was 397.38 IU/L, whereas for LDH it was 218.21 IU/L [12]. In our study, the average values of CK and LDH were much higher, 1068 IU/L and 344 IU/L, respectively. As is well known, serum CK and LDH levels increase during skeletal muscle damage and degeneration. Therefore, a muscle biopsy in every case where IBM is suspected is essential for a definitive diagnosis even if there are no features of muscle weakness according to the MRC scale [13, 26, 35, 36].
We found that skeletal muscle biopsies of IBM patients presented typical changes which have been well described by others, including hypertrophy of connective and adipose tissues, focal occurrence of necrotic fibers, RVs, a pattern of inflammatory cell infiltrations or pathological protein aggregates [3, 4, 9, 12, 17, 37]. In most cases we saw myofiber atrophy with subsequent invasion of connective and adipose tissue. The results are in accordance with observations made by others [12, 17, 27, 36, 37]. In a few cases hypertrophic myofibers in IBM patients were also reported [12, 27]. However, muscle fiber hypertrophy seems to be more characteristic of skeletal muscle dystrophies than inflammatory myopathy [38]. We have also demonstrated the presence of necrotic myofibers which could occur in muscles of IBM patients, as was described by Kierdaszuk et al. [37]. The necrotic process is probably caused by the action of perforin and granzymes, enzymes secreted by T lymphocytes [15]. The necrosis of single myofibers is very often associated with muscle fiber regeneration. Indeed, our biopsies found myofibers with centrally located myonuclei which are typical for muscle regeneration. Moreover, in our biopsies we saw moderately intense infiltration of mononuclear cells. To identify the type of inflammatory cell we used an immunohistochemical method, which had been previously suggested by others [17, 37, 39]. Our results indicated that cytotoxic CD8 + T lymphocytes [40] as well as macrophages in infiltrates are present. Current reports suggest that cN1A is a reliable marker for s-IBM, although information on this subject is still insufficient. To our knowledge, only two research teams have presented the diagnostic utility of cN1A antibody on paraffin sections [41, 42]. Our results are the third and so far, they confirm the accumulation of cN1A protein around the vacuoles and myonuclei. Moreover, aggregates of Tau protein, beta-amyloid and apolipoprotein B were detected in the examined fibers. These results are in accordance with some other researchers who suggest that they are typical for most IBM cases [9, 37, 39]. However, Sakai et al. [17] found that in muscle biopsies taken from IBM patients beta-amyloid aggregates were not always present, especially in those patients with granuloma formation inside myofibers. We also found in the sarcoplasm of individual muscle fibers the central or paracentral RVs which are key elements in diagnosing s-IBM in muscles. Here, we presented unique images from the transmission electron microscope showing the real ultrastructure of RVs. To the best of our knowledge, the actual ultrastructure of vacuoles with granules at the margins has not been described. It has been suggested that the formation of RVs could be associated with the process of autophagy or myonuclei degeneration in myofibers. Recently, typical autophagic proteins and nuclear membrane proteins in these vacuoles were seen [10, 13, 30, 43].
In some cases, tubulofilamentous inclusions within myofibers have been seen at the ultrastructural level [3, 4]. In our study such structures were not found, implying that they do not always exist in this disease, which is consistent with diagnostic criteria of definite IBM according to Griggs (1995) [29]. However, as others, we found typical, for IBM, whorled osmiophilic membrane debris, myelinoid bodies and lipid-like inclusion bodies which were predominant in the sarcoplasm. A few authors have reported the presence of some nuclear and cytoplasmic inclusions in IBM muscle biopsies which were located adjacent to the tubulofilamentous structures [4] or between the myofilaments [44]. However, in contrast to others, we did not detect inclusion bodies in the myonuclei [2, 37]. According to other researchers [6], at least three vacuolated fibers should be examined by electron microscopy in order to find inclusions. Interestingly, we noticed abnormalities in the triad ultrastructure, which are characteristic of centronuclear myopathy [45, 46].
As was described above, the morphological features of the disease and detection of typical proteins are not homogenous in each case. We are aware of the main limitation of this work, namely the small number of patients, and the lack of some laboratory tests such as antibodies or magnetic resonance imaging which are not available in our university. Furthermore, they are not routinely used in regular practice. Diagnostic imaging can primarily support the differential diagnosis of inflammatory muscle disease, due to characteristic patterns of muscle involvement [47]. This article summarizes the practical aspects of the s-IBM-diagnosis by using a combination of immunohistochemical and ultrastructural assessment. Current studies on early recognition of IBM are focused on searching for new molecular biomarkers which could be detected in skeletal muscle biopsies [7, 9, 10, 24, 48,49,50].
Conclusions
To date, there is no effective therapy for IBM patients. Therapy only provides temporary health improvement. Therefore, establishing the final diagnosis is so important and necessary for patients having IBM. Our results confirmed most previous observations, and we strongly recommend employing IHC and TEM techniques in the diagnostics of skeletal muscle biopsies. Early recognition of IBM even without fully developed clinical symptoms might help to slow the progression of the disease. This will encourage implementation of exercises, which will enable functional efficiency as long as possible. Even if there is muscle damage, through rehabilitation patients could learn how to compensate by using other muscle groups.
Abbreviations
- s-IBM:
-
Sporadic inclusion body myositis
- h-IBM:
-
Hereditary inclusion body myopathy
- RVs:
-
Rimmed vacuoles
- TEM:
-
Transmission electron microscope
- DM:
-
Dermatomyositis
- PM:
-
Polymyositis
- NM:
-
Immune-mediated necrotizing myopathy
- EMG:
-
Electromyography
- ALS:
-
Amyotrophic lateral sclerosis
- IHC:
-
Immunohistochemistry
- apo-B:
-
Apolipoprotein B
- MTX:
-
Methotrexate
- GCS:
-
Glucocorticosteroids
- MMF:
-
Mycophenolate mofetil
- AZA:
-
Azathioprine
- RTX:
-
Rituximab
- LDH:
-
Lactate dehydrogenase
- CK:
-
Creatine kinase
- MRC scale:
-
Medical Research Council scale
References
Kazamel M, Sorenson EJ, Milone M (2016) Clinical and electrophysiological findings in hereditary inclusion body myopathy compared with sporadic inclusion body myositis. J Clin Neuromuscul Dis 17:190–196
Fidziańska A, Glinka Z (2006) Rimmed vacuoles with beta-amyloid and tau protein deposits in the muscle of children with hereditary myopathy. Acta Neuropathol 112:185–193
Askanas V, Engel WK, Bilak M, Alvarez RB, Selkoe DJ (1994) Twisted tubulofilaments of inclusion body myositis muscle resemble paired helical filaments of Alzheimer brain and contain hyperphosphorylated Tau. Am J Pathol 144:177–187
Hermanns B, Molnar M, Schröder JM (2000) Peripheral neuropathy associated with hereditary and sporadic inclusion body myositis: confirmation by electron microscopy and morphometry. J Neurol Sci 179:92–102
Dalakas MC (2006) Sporadic inclusion body myositis—diagnosis, pathogenesis and therapeutic strategies. Nat Clin Pract Neurol 2:437–447
Dimachkie MM, Barohn RJ (2012) Inclusion body myositis. Semin Neurol 32:237–245
Machado P, Brady S, Hanna MG (2013) Update in inclusion body myositis. Curr Opin Rheumatol 25:763–771
Misterska-Skóra M, Sebastian A, Dzięgiel P, Sebastian M, Wiland P (2013) Inclusion body myositis associated with Sjögren’s syndrome. Rheumatol Int 33:3083–3086
Chilingaryan A, Rison RA, Beydoun SR (2015) Misdiagnosis of inclusion body myositis: two case reports and a retrospective chart review. J Med Case Rep 9:169
Weihl CC, Mammen AL (2017) Sporadic inclusion body myositis—a myodegenerative disease or an inflammatory myopathy. Neuropathol Appl Neurobiol 43:82–91
Jabari D, Vedanarayanan VV, Barohn RJ, Dimachkie MM (2018) Update on inclusion body myositis. Curr Rheumatol Rep 20(8):52
Li K, Pu Ch, Huang X, Liu J, Mao Y, Lu X (2015) Clinicopathologic features of sporadic inclusion body myositis in China. Neurol Neurochir Pol 49:245–250
Askanas V, Engel WK, Nogalska A (2015) Sporadic inclusion-body myositis: a degenerative muscle disease associated with aging, impaired muscle protein homeostasis and abnormal mitophagy. Biochim Biophys Acta 1852:633–643
Vattemi G, Mirabella M, Guglielmi V, Lucchini M, Tomelleri G, Ghirardello A, Doria A (2014) Muscle biopsy features of idiopathic inflammatory myopathies and differential diagnosis. Autoimmun Highlights 5:77–85
De Paepe B, Zschüntzsch J (2015) Scanning for therapeutic targets within the cytokine network of idiopathic inflammatory myopathies. Int J Mol Sci 16:18683–18713
Lundberg IE, Miller FW, Tjärnlund A, Bottai M (2016) Diagnosis and classification of idiopathic inflammatory myopathies. J Intern Med 280:39–51
Sakai K, Ikeda Y, Ishida Ch, Matsumoto Y, Ono K, Iwasa K, Yamada M (2015) Inclusion body myositis with granuloma formation in muscle tissue. Neuromuscul Disord 25:706–712
Neudecker S, Krasnianski M, Bahn E, Zierz S (2004) Rimmed vacuoles in facioscapulohumeral muscular dystrophy: a unique ultrastructural feature. Acta Neuropathol 108:257–259
Momma K, Noguchi S, Malicdan MC, Hayashi YK, Minami N, Kamakura K, Nonaka I, Nishino I (2012) Rimmed vacuoles in Becker muscular dystrophy have similar features with inclusion myopathies. PLoS One 7:e52002
Gang Q, Bettencourt C, Machado P, Hanna MG, Houlden H (2014) Sporadic inclusion body myositis: the genetic contributions to the pathogenesis. Orphanet J Rare Dis 9:88
Dalakas MC (2008) Therapeutic advances and future prospects in immune-mediated inflammatory myopathies. Ther Adv Neurol Disord 1:157–166
Amici DR, Pinal-Fernandez I, Mázala DA, Lloyd TE, Corse AM, Christopher-Stine L, Mammen AL, Chin ER (2017) Calcium dysregulation, functional calpainopathy, and endoplasmic reticulum stress in sporadic inclusion body myositis. Acta Neuropathol Commun 5:24
Dalakas MC (2011) Review: an update on inflammatory and autoimmune myopathies. Neuropathol Appl Neurobiol 37:226–242
Weihl CC, Pestronk A (2010) Sporadic inclusion body myositis: possible pathogenesis inferred from biomarkers. Curr Opin Neurol 23:482–488
Murata-Shinozaki Y, Takahashi T, Matsubara T, Maruyama H, Izumi Y, Matsumoto M (2017) The origins of rimmed vacuoles and granulovacuolar degeneration bodies are associated with the Wnt signaling pathway. Neurosci Lett 638:55–59
Nadaj-Pakleza A, Kierdaszuk B, Kamińska A (2010) The role of skeletal muscle biopsy in the diagnosis of neuromuscular disorders. Neurol Neurochir Pol 44:481–491
Hamann PD, Roux BT, Heward JA, Love S, McHugh NJ, Jones SW, Lindsay MA (2017) Transcriptional profiling identifies differential expression of long non-coding RNAs in Jo-1 associated and inclusion body myositis. Sci Rep 7:8024
Hilton-Jones D, Miller A, Parton M, Holton J, Sewry C, Hanna MG (2010) Inclusion body myositis: MRC Centre for Neuromuscular Diseases, IBM workshop, London, 13 June 2008. Neuromuscul Disord 20:142–147
Griggs RC, Askanas V, DiMauro S, Engel A, Karpati G, Mendell JR, Rowland LP (1995) Inclusion body myositis and myopathies. Ann Neurol 38:705–713
Mastaglia FL, Needham M (2015) Inclusion body myositis: a review of clinical and genetic aspects, diagnostic criteria and therapeutic approaches. J Clin Neurosci 22:6–13
Badrising UA, Maat-Schieman ML, van Houwelingen JC, van Doorn PA, van Duinen SG, van Engelen BG, Faber CG, Hoogendijk JE, de Jager AE, Koehler PJ, de Visser M, Verschuuren JJ, Wintzen AR (2005) Inclusion body myositis. Clinical features and clinical course of the disease in 64 patients. J Neurol 252:1448–1454
Jørgensen AN, Aagaard P, Nielsen JL, Frandsen U, Diederichsen LP (2016) Effects of blood-flow-restricted resistance training on musclefunction in a 74-year-old male with sporadic inclusion body myositis: a case report. Clin Physiol Funct Imaging 36:504–509
Alexanderson H (2016) Physical exercise as a treatment for adult and juvenile myositis. J Intern Med 280:75–96
Munshi SK, Thanvi B, Jonnalagadda SJ, Da Forno P, Patel A, Sharma S (2006) Inclusion body myositis: an underdiagnosed myopathy of older people. Age Ageing 35:91–94
Paternostro-Sluga T, Grim-Stieger M, Posch M, Schuhfried O, Vacariu G, Mittermaier C, Bittner C, Fialka-Moser V (2008) Reliability and validity of the Medical Research Council (MRC) scale and a modified scale for testing muscle strength in patients with radial palsy. J Rehabil Med 40:665–671
Bello R, Bertorini T, Ganta K, Mays W (2017) A case of asymptomatic inclusion body myositis. J Clin Neuromuscul Dis 18:132–134
Kierdaszuk B, Berdynski M, Palczewski P, Golebiowski M, Zekanowski C, Kaminska AM (2015) Sporadic inclusion body myositis: clinical, pathological, and genetic analysis of eight Polish patients. Folia Neuropathol 53:355–366
Kornegay JN, Childers MK, Bogan DJ, Bogan JR, Nghiem P, Wang J, Fan Z, Howard JF Jr, Schatzberg SJ, Dow JL, Grange RW, Styner MA, Hoffman EP, Wagner KR (2012) The paradox of muscle hypertrophy in muscular dystrophy. Phys Med Rehabil Clin N Am 23:149 – 172
Malik A, Hayat G, Kalia JS, Guzman MA (2016) Idiopathic inflammatory myopathies: clinical approach and management. Front Neurol 7:64
Schmidt J (2018) Current classification and management of inflammatory myopathies. J Neuromuscul Dis 5:109–129
Larman HB, Salajegheh M, Nazareno R, Lam T, Sauld J, Steen H, Kong SW, Pinkus JL, Amato AA, Elledge SJ, Greenberg SA (2013) Cytosolic 5′-nucleotidase 1A autoimmunity in sporadic inclusion body myositis. Ann Neurol 73:408–418
Eura N, Sugie K, Kinugawa K, Nanaura H, Ohara H, Iwasa N, Shobatake R, Kiriyama T, Izumi T, Kataoka H, Ueno S (2016) Anti-cytosolic 5′-nucleotidase 1A (cn1a) positivity in muscle is helpful in the diagnosis of sporadic inclusion body myositis: a study of 35 Japanese patients. J Neurol Neurosci 7:155
Greenberg SA, Pinkus JL, Amato AA (2006) Nuclear membrane proteins are present within rimmed vacuoles in inclusion-body myositis. Muscle Nerve 34:406–416
Askanas V, Engel WK, Nogalska A (2009) Inclusion body myositis: a degenerative muscle disease associated with intra-muscle fiber multi-protein aggregates, proteasome inhibition, endoplasmic reticulum stress and decreased lysosomal degradation. Brain Pathol 19:493–506
Dowling JJ, Vreede AP, Low SE, Gibbs EM, Kuwada JY, Bonnemann CG, Feldman EL (2009) Loss of myotubularin function results in T-tubule disorganization in zebrafish and human myotubular myopathy. PLoS Genet 5:e1000372
Gibbs EM, Davidson AE, Telfer WR, Feldman EL, Dowling JJ (2014) The myopathy-causing mutation DNM2-S619L leads to defective tubulation in vitro and in developing zebrafish. Dis Model Mech 7:157–161
Lilleker JB (2018) Advances in the early diagnosis and therapy of inclusion body myositis. Curr Opin Rheumatol. https://doi.org/10.1097/BOR.0000000000000537
Lu X, Peng Q, Wang G (2015) Discovery of new biomarkers of idiopathic inflammatory myopathy. Clin Chim Acta 444:117–125
Schmidt K, Kleinschnitz K, Rakocevic G, Dalakas MC, Schmidt J (2016) Molecular treatment effects of alemtuzumab in skeletal muscles of patients with IBM. BMC Neurol 16:48
Schmidt K, Schmidt J (2017) Inclusion body myositis: advancements in diagnosis, pathomechanisms, and treatment. Curr Opin Rheumatol 29:632–638
Funding
No financial support was received to carry out the work described in this manuscript.
Author information
Authors and Affiliations
Contributions
KH wrote a first draft of the manuscript, carried out the study in transmission electron microscope and performed the analysis and interpretation of images, prepared the figures and was responsible for literature searching. AS diagnosed patients, contributed to the literature search and to drafting the manuscript. AP performed immunohistochemistry reactions and analyzed the results. MMS diagnosed patients and contributed to drafting the manuscript. AH performed histopathologic description. MSk participated in sample collection and data analysis. MSe participated in sample collection and data analysis. PW and PD revised the manuscript critically. MPO performed the analysis and interpretation of images from transmission electron microscope, supervised and coordinated the entire study. All authors contributed to the interpretation of the results, gave advice while carrying out the experiments, improved and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
Katarzyna Haczkiewicz, Agata Sebastian, Aleksandra Piotrowska, Maria Misterska-Skóra, Agnieszka Hałoń, Marta Skoczyńska, Maciej Sebastian, Piotr Wiland, Piotr Dzięgiel and Marzenna Podhorska-Okołów declare that they have no conflict of interest.
Ethical approval
Obeying the principles of Good Clinical Practice and the principles of the Declaration of Helsinki the authors obtained the ethical approval from The Bioethics Committee of the Wroclaw Medical University (ethics approval date: 27 September 2018, protocol number: 501/2018), where the work was carried out.
Research involving human participants and/or animals
All procedures involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Haczkiewicz, K., Sebastian, A., Piotrowska, A. et al. Immunohistochemical and ultrastructural analysis of sporadic inclusion body myositis: a case series. Rheumatol Int 39, 1291–1301 (2019). https://doi.org/10.1007/s00296-018-4221-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00296-018-4221-z