
Abstract
Enterobacter cloacae exhibits strong adhesion and invasion properties that contribute its ability to infect the host; it is considered an important opportunistic pathogen throughout the world. To control the spread of E. cloacae, simple, rapid, and accurate detection methods are required. Current methods suffer from various shortcomings and do not meet the demand for on-site quickly detection. Using recombinase polymerase amplification combined with lateral flow strip (RPA-LFS), an isothermal detection method was developed to target the outer membrane protein X (ompX) gene of E. cloacae. This reaction can be performed in 30 min at 37 °C. Limit of detection of 10 CFU/reaction was equivalent to that of the qPCR method. The detection accuracy of clinical samples was also equal to that of the qPCR method. In this study, we developed the RPA-LFS assay, which is simple, rapid, accurate, and does not require a laboratory facility. This assay may prove useful for detecting E. cloacae on-site.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Enterobacter cloacae (E. cloacae) belongs to the Enterobacter genus and is a kind of Gram-negative bacterium [1]. It has been widely found in soil, plants, and various aquatic environments [2]. It has strong adhesion and invasion properties that contribute to its ability to infect the host [3]. E. cloacae is an important opportunistic pathogen and the leading cause of nosocomial infections worldwide [4, 5]. It can cause septicemia, meningitis, endocarditis, septic arthritis, and bone marrow. It is also possible for inflammation to cause infections of the lower respiratory tract, skin, soft tissues, urinary tract, abdominal cavity, and eyes [4, 6]. Recently, there have been many reports of E. cloacae infections in veterinary clinics. E. cloacae can also cause diseases in aquatic animals and cause a large number of deaths [7]. In remote areas, it can result in the deaths of many aquatic animals on farms, due to a failure to detect and treat in time.
Now, with the overuse of antibiotics, E. cloacae has a high prevalence of multi-drug resistance [8]. This brings several challenges to clinical treatment and infection control [9, 10]. To deal with the global epidemic of E. cloacae, monitoring and early identification is important for human life, farm animals, and aquaculture.
The diagnosis of pathogens is essential for the prevention and treatment of diseases for both patients and hospitals [11]. Traditional culture methods can identify pathogen accurately, but are a lengthy process. Recently, many functional enzymes have been used to establish conventional methods of biochemical identification of clinical pathogens, such as polymerase chain reaction (PCR)-based, quantitative PCR (qPCR)-based, biochemical analysis, conventional culture procedure, and immunology-based diagnosis tests [12, 13,14,15]. Although many useful detection methods have been developed, they all suffer from a variety of drawbacks. It is possible to perform most of these methods in laboratories with the proper equipment, but it is often necessary to detect pathogens in peoples’ houses, poorly equipped hospitals, and farms; in these cases, it can be difficult to detect pathogens in a short period of time [16]. It is particularly important to establish a relatively simple and straightforward method as there is often a lack of trained personnel. For this reason, isothermal amplification methods have been established (such as loop-mediated isothermal amplification (LAMP) and recombinase polymerase amplification (RPA)) [17, 18].
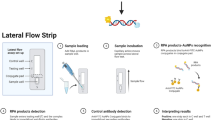
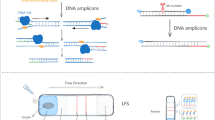
Recombinase polymerase amplification (RPA), first reported in 2006 [19], it amplifies nucleic acids by using recombinases (UvsX and UvsY), single-stranded binding proteins (gp32), and strand-displacing DNA polymerases (BSU). RPA does not require a stringent incubation temperature; exponential amplification can be performed at 37 °C rather than thermocycling between 55 °C and 95 °C. It also only requires 30 min or less to complete the reaction. RPA amplification products can be detected by gel electrophoresis, real-time fluorescence, and lateral flow strips (LFS), and other methods [17, 20, 21]. Of these detection methods, as detection results can be analyzed with the naked eye, the lateral flow strip is suitable for simple testing and do not require complex instruments and trained personnel.
Materials and Methods
Collection of Samples and DNA Extraction
Reference strains of Enterobacter cloacae, Candida parapsilosis, Candida tropicalis, Candida albicans, and Acinetobacter baumannii were obtained from American Type Culture Collection (Manassas, VA, USA). In addition, sputum isolated strains of E. cloacae and other infectious pathogens were provided by The Second People’s Hospital of Lianyungang (Lianyungang, China). Clinical samples were collected from patients. Strain information is provided in Table 1. All strains have been confirmed by 16S rRNA sequencing [22]. The strains used in this study were cultured in Luria–Bertani (LB) broth at 37 °C for 200 rpm. Cultures of 107 colony-forming units per microliter (CFU/μL) were tenfold diluted from 106–100 CFU/μL as templates. All templates were boiled at 100 °C for 10 min, and RPA-LFS was detected using a template of 1 μL.
Design of Primers and Probes
An RPA-LFS assay was developed using primers and probes targeted at the sequence of the ompX gene (GenBank accession number CP009756.1). The FASTA sequence of the fragment was input into the NCBI Primer-BLAST (https://www.ncbi.nlm.nih.gov/tools/primer-blast) and primer BLAST was performed with the following criteria: product size minimum was 100 and maximum 300; primer size minimum was 30 and maximum 33; primer GC content minimum was 20 and maximum 80.
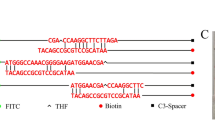
In the RPA reaction, a probe can increase amplification specificity and reduce primer-dependent artifacts [19]. A spacer C3 group (SpC3) was located at the 3 end of the probe, preventing strand extension, and a tetrahydrofuran group (THF) at the middle (position 32) made the probe function. For the 3 end of the probe to be released from SpC3 blocking, it was necessary for the bases flanking the THF site to pair well with the template. For probe design, the forward primer previously screened was used, and new forward primers for the 5ʹ end of the probe. The probe was designed using Primer Premier 5 software. Key parameters were as follows: size of probe, 45–48 bp; melting temperature (Tm), 50–100 ˚C; GC content, 20–70%. Other parameters were set as default. Additionally, if the probe and primers matched three times consecutively, to avoid false-positive results, primers and probes were mutated. The primers and probes used in this study are listed in Table 2.
RPA-LFS Procedure
For RPA-LFS detection, the primers and templates mentioned above were used. The Twist Amp® DNA Amplification Kit (TwistDx Ltd., Maidenhead, UK) should be used according to the manufacturer's instructions. In each 50 μL reaction mixture, the following components were included: 2.1 μL of each primer (10 μM), 0.6 μL of probe, and 1 μL of template. Following a few minutes of centrifugation, the reaction was conducted at 37 °C for 30 min. Ten microliters of the RPA product were then spotted on the LFS (Ustar Biotechnologies Ltd., Hangzhou, China). The LFS was composed of a sample pad, conjugate pad (soaked with mouse-originated AuNP-tagged anti-FITC antibody), test line (coated with streptavidin), control line (coated with anti-mouse antibody), and absorbent pad that lined up through the solvent migration route. Test and control lines were visualized after 5 min of immersion in 100 μL of the solvent with the LFS stick containing the RPA product.
The qPCR Procedure
The qPCR detection of E. cloacae was performed as reported previously [23]. A pair of specific primers (qPCR-F and qPCR-R) targeting the ompX gene of E. cloacae was used. The reaction program was set as follows: denaturation at 95 °C for 30 s, 40 cycles of denaturation at 95 °C for 3 s, and annealing and extension at 60 °C for 20 s on the StepOne™ real-time PCR System from Applied Biosystems (CA, USA).
Results
Design and Screening of RPA Primers and Probe for E. cloacae Detection
The gene ompX was selected as the target gene for E. cloacae detection. RPA detection indicated no nonspecific bands, and the size of the products was consistent with the expected size. The two sets of primer pairs demonstrated good ability to detect E. cloacae (Fig. 1a). To avoid primer dimers, the primer–probe set EC-F1/R1 was selected for further screening. RPA-LFS system must be able to incorporate the probe without excessive amplification, the forward primer (EC-F1) was changed to the probe, and possible mismatches with the reverse primer (EC-R1B) was analyzed (Fig. 1b). There can be a few mismatches between primers and probes when using RPA-LFS (Wu et al., 2020). To reduce the possibility of nonspecific amplification, match bases of the reverse primer and probe were changed and marked in red (Table 2). F4/P/R1B, F5/P/R1B, F6/P/R1B, and F7/P/R1B showed false-positive signal from the no template control (NTC); only F3/P/R1B was used for further detection (Fig. 1c).

Primer and probe amplification. a Agarose gel showing primer pairs that target the ompX gene and their recombinase polymerase amplification (RPA) results. On each lane, primer pairs are indicated. Three independently conducted experiments are represented in the image. b Primer Premier 5 software was used to analyze and modify reverse primer–probe sets for the detection of EF. DNA bases that are relevant to the probe and reverse primer have been excluded. Horizontal lines represent DNA strands, vertical lines represent matching DNA bases. Under the figure, you will find a list of molecular markers. c Screening of forward primers. Image shows RPA-LFS results of different forward primers. On each lane, primer–probe pairs are indicated. Three independently conducted experiments are represented in the image. NTC no template control
Specificity of the RPA-LFS Method for Detection
To confirm the RPA-LFS method could effectively detect different E. cloacae, 4 reference strains and 16 sputum isolated strains were detected with the primer–probe set F3/P/R1B; this primer–probe effectively detected the pathogen (Fig. 2). To evaluate specificity of the method, 28 common pathogens (including E. cloacae) were detected using RPA-LFS. Only the E. cloacae sample was positive; the other strains were negative (Fig. 3). Thus, the primer–probe set of F3/P/R1B was used for future reactions.

Compatibility confirmation. Image shows RPA-LFS results of different reference and isolated strains of E. cloacae. In each lane, bacterial templates are identified by their names. Three independently conducted experiments are represented in the image. NTC no template control

Specificity confirmation. Image shows RPA-LFS results of different bacterial templates. In each lane, bacterial templates are identified by their names. Three independently conducted experiments are represented in the image. NTC no template control
Detection Limit of RPA-LFS
Using RPA-LFS, we evaluated its limit of detection. First, E. cloacae at 106–100 CFU per microliter was tested (1 μL for each reaction). A red weak band appeared at the test line of 101 CFU. A higher CFU resulted in a stronger signal on the test line (Fig. 4)a. Second, E. cloacae was tenfold diluted from 106–100 CFU/µL spiked with 106 CFU/µL of E. coli O157. A signal at 101 CFU was observed (Fig. 4b). Thus, detection limit of RPA-LFS was 101 CFU/reaction.

Limit of detection of RPA-LFS. a Image shows detection results with different amounts (106 to 100 CFU/µL) of E. cloacae. b Image shows detection results with different amounts (106 to 100 CFU/µL) of E. cloacae, and 106 CFU/µL of E. coli O157. Reactions were performed at 37 °C for 30 min. Three independently conducted experiments are represented in the image. NTC no template control
Application of the RPA-LFS Method for E. cloacae Detection
A total of 217 clinical samples collected from different patients were tested for E. cloacae with RPA-LFS and qPCR. The detection results of RPA-LFS were consistent with those of qPCR (Table 3). Thus, RPA-LFS is a reasonable alternative for diagnosing E. cloacae on-site with qPCR.
Discussion
Enterobacter cloacae has emerged as a serious threat to hospitals, farms, and aquaculture worldwide. E. cloacae infections can cause septicemia, meningitis, endocarditis, septic arthritis, and bone marrow [4], [6]. In addition, E. cloacae can also cause disease in aquatic animals, resulting in a large number of deaths [7]. These situations also occur in resource-poor regions and countries. Current methods (e.g., PCR, qPCR) cannot meet the demand in remote areas. Although LAMP is an isothermal amplification technology, it requires a temperature control instrument. Fortunately, compared to existing detection methods, RPA-LFS can compensate for their shortcomings and can be tolerated for crude templates. Human body heat can even be used to complete the reaction, so no special equipment is required [19], it is useful for the detection of E. cloacae in hospitals, farms, and aquaculture.
A good amplification target for effective detection of a particular species is very important; the outer membrane protein X (ompX) gene has been used as an amplification target in other methods [23]. Our data also indicated that the ompX gene had a good specificity. In addition, the primer–probe set F3/P/R1B selected in this study could effectively distinguish E. cloacae from other common pathogens. In the RPA-LFS method, E. cloacae was detected with excellent accuracy. The limitation of detection was 101 CFU, which is consistent with the qPCR method. This method could specifically detect E. cloacae in the presence of E. coli O157; the sensitivity was not affected in the presence of E. coli O157, infected patients can be identified by this method in order to determine which microorganisms are pathogenic. Importantly, the RPA-LFS method can be performed within 35 min (30 min of amplification and 5 min of LFS analysis), while the PCR and qPCR methods require more than 50 min to complete. Some areas without adequate equipment may spend more time transporting clinical samples to specialized laboratories for detection, this is not conducive to clinical treatment.
It was found that the RPA-LFS assay was just as accurate as qPCR when used on clinical samples, but did not require extraction of DNA, and it was only necessary to use bacterial liquid after high temperature lysis as a template, which means this method can be easily performed by untrained personnel.
In conclusion, its simplicity, rapidity, and accuracy made it easy to perform without the need for laboratory facilities. In combination with a simple, fast method for extracting DNA, the method may be useful for detection of E. cloacae on-site as well as in hospitals, farms, and aquaculture, saving both time and resources.
Conclusion
A reliable RPA-LFS method for E. cloacae was established after screening several primers-probe pairs. Reactions could be completed within 30 min at 37 °C, and the results could be observed on a LFS in 5 min. This method had the same limit of detection (101 CFU/reaction) as the qPCR method when the results of 217 clinical samples were compared. Thus, the method established in this study may be useful in remote areas or farms with poor resources.
Statement
The study protocol was approved by the Medical Ethics Committee of the Second People's Hospital of Lianyungang City (Lianyungang, Jiangsu, China) and informed consent was obtained from patients prior to collection of clinical samples. All methods were carried out in accordance with relevant guidelines and regulations.
Data Availability
The data presented in this study are included in the article. Further inquiries should be directed to the corresponding authors.
References
Lockhart SR, Abramson MA, Beekmann SE, Gallagher G, Riedel S, Diekema DJ, Quinn JP, Doern GV (2007) Antimicrobial resistance among Gram-negative bacilli causing infections in intensive care unit patients in the United States between 1993 and 2004. J Clin Microbiol 45(10):3352–3359. https://doi.org/10.1128/JCM.01284-07
Liu F, Wang FT, Du LH, Zhao T, Doyle MP, Wang DY, Zhang XX, Sun ZL, Xu WM (2017) Antibacterial and antibiofilm activity of phenyllactic acid against enterobacter cloacae. Food Control 84:442–448. https://doi.org/10.1016/j.foodcont.2017.09.004
Krzymińska S, Koczura R, Mokracka J, Puton T, Kaznowski A (2010) Isolates of the Enterobacter cloacae complex induce apoptosis of human intestinal epithelial cells. Microb Pathog 49(3):83–89. https://doi.org/10.1016/j.micpath.2010.04.003
Davin-Regli A, Pagès JM (2015) Enterobacter aerogenes and Enterobacter cloacae; versatile bacterial pathogens confronting antibiotic treatment. Front Microbiol 18(6):392. https://doi.org/10.3389/fmicb.2015.00392
Cabral AB, Maciel M, Barros JF, Antunes MM, Barbosa de Castro C, Lopes A (2017) Clonal spread and accumulation of β-lactam resistance determinants in Enterobacter aerogenes and Enterobacter cloacae complex isolates from infection and colonization in patients at a public hospital in Recife, Pernambuco. Brazil J Medi Microbiol 66(1):70–77. https://doi.org/10.1099/jmm.0.000398
Cisse H, Vernet-Garnier V, Hentzien M, Bajolet O, Lebrun D, Bonnet M, Ohl X, Diallo S, Bani-Sadr F (2019) Treatment of bone and joint infections caused by Enterobacter cloacae with a fluoroquinolone-cotrimoxazole combination. Int J Antimicrob Agents 54(2):245–248. https://doi.org/10.1016/j.ijantimicag.2019.05.010
Sekar VT, Santiago TC, Vijayan KK, Alavandi SV, Raj VS, Rajan JJ, Sanjuktha M, Kalaimani N (2018) Involvement of Enterobacter cloacae in the mortality of the fish. Mugil cephalus Letters in applied microbiology 46(6):667–672. https://doi.org/10.1111/j.1472-765X.2008.02365.x
Nordmann P, Naas T, Poirel L (2011) Global spread of Carbapenemase-producing Enterobacteriaceae. Emerg Infect Dis 17(10):1791–1798. https://doi.org/10.3201/eid1710.110655
Castanheira M, Deshpande LM, Mathai D, Bell JM, Jones RN, Mendes RE (2011) Early dissemination of NDM-1- and OXA-181-producing Enterobacteriaceae in Indian hospitals: Report from the SENTRY antimicrobial surveillance program, 2006–2007. Antimicrob Agents Chemother 55(3):1274–1278. https://doi.org/10.1128/AAC.01497-10
Ho HJ, Toh CY, Ang B, Krishnan P, Lin RT, La MV, Chow A (2016) Outbreak of New Delhi metallo-β-lactamase-1-producing Enterobacter cloacae in an acute care hospital general ward in Singapore. Am J Infect Control 44(2):177–182. https://doi.org/10.1016/j.ajic.2015.08.028
Agashe V, Shenai S, Mohrir G, Deshmukh M, Bhaduri A, Deshpande R, Mehta A, Rodrigues C (2009) Osteoarticular tuberculosis–diagnostic solutions in a disease endemic region. J Infect Dev Ctries 3(7):511–516. https://doi.org/10.3855/jidc.469
Yen PW, Hwang CH, Lin MY, Lu YP, Lin CT, Huang KS, Yeh JA (2014) Emerging electrical biosensors for detecting pathogens and antimicrobial susceptibility tests. Curr Org Chem 18(2):165–172. https://doi.org/10.2174/13852728113176660140
Haugland RA, Siefring SC, Wymer LJ, Brenner KP, Dufour AP (2005) Comparison of Enterococcus measurements in freshwater at two recreational beaches by quantitative polymerase chain reaction and membrane filter culture analysis. Water Res 39(4):559–568. https://doi.org/10.1016/j.watres.2004.11.011
He JW, Jiang S (2005) Quantification of enterococci and human adenoviruses in environmental samples by real-time PCR. Appl Environ Microbiol 71(5):2250–2255. https://doi.org/10.1128/AEM.71.5.2250-2255.2005
Kim YB, Seo HJ, Seo KW, Jeon HY, Kim DK, Kim SW, Lim SK, Lee YJ (2018) Characteristics of high-level ciprofloxacin-resistant Enterococcus faecalis and Enterococcus faecium from retail chicken meat in Korea. J Food Prot 81(8):1357–1363. https://doi.org/10.4315/0362-028X.JFP-18-046
Fukuta S, Iida T, Mizukami Y, Ishida A, Ueda J, Kanbe M, Ishimoto Y (2003) Detection of Japanese yam mosaic virus by RT-LAMP. Adv Virol 148(9):1713–1720. https://doi.org/10.1007/s00705-003-0134-5
Wang L, Wang Y, Wang F, Zhao MD, Gao XZ, Chen HM, Li N, Zhu Q, Liu LP, Zhu WJ, Liu X, Chen YJ, Zhou P, Lu YZ, Wang K, Zhao WG, Liang W (2022) Development and application of rapid clinical visualization molecular diagnostic technology for Cryptococcus neoformans/C. gattii based on recombinase polymerase amplification combined with a lateral flow strip. Front Cell Infec Microbiol 11:803798. https://doi.org/10.3389/fcimb.2021.803798
Wu D, Kang J, Li B, Sun D (2018) Evaluation of the RT-LAMP and LAMP methods for detection of Mycobacterium tuberculosis. J Clinic Lab Anal 32(4):e22326. https://doi.org/10.1002/jcla.22326
Piepenburg O, Williams CH, Stemple DL, Armes NA (2006) DNA detection using recombination proteins. PLoS biology 4(7):e204. https://doi.org/10.1371/journal.pbio.0040204
Khater M, Escosura-Muñiz A, Altet L, Merkoçi A (2019) In Situ plant virus nucleic acid isothermal amplification detection on gold nanoparticle-modified electrodes. Anal Chem 91(7):4790–4796. https://doi.org/10.1021/acs.analchem.9b00340
Ma C, Fan SH, Wang Y, Yang HT, Qiao Y, Jiang, G, Lyu MS, Dong JQ, Shen H, Gao S (2021) Rapid detection of Enterocytozoon hepatopenaei infection in shrimp with a real-time isothermal recombinase polymerase amplification assay. Front Cell Infec Microbiol 11:631960. https://doi.org/10.3389/fcimb.2021.631960
Hiergeist A, Gläsner J, Reischl U, Gessner A (2015) Analyses of intestinal microbiota: Culture versus sequencing. ILAR J 56(2):228–240. https://doi.org/10.1093/ilar/ilv017
Wang YQ, Liu K, Pei-Jing LI, Xiong Y, Chang-Yun YE (2017) Genetic diversity of Enterobacter cloacae strains isolated from hospital in china and taqman probe real-time polymerase chain reaction for rapid detection of it. Chinese J Zoonoses 33(1):1–8. https://doi.org/10.3969/j.issn.1002-2694.2017.01.001
Acknowledgements
We thank International Science Editing (http://www.internationalscienceediting.com) for editing this manuscript.
Funding
This study was supported by grants from the Lianyungang Health Science and Technology Project Youth Project (grant no. QN202208), the Lianyungang General Health Science and Technology Project in 2022 (grant no. 202224), the 2022 Medical Research Guidance Project of Jiangsu Provincial Health Commission(grant no. Z2022070), the Clinical Medical Science and Technology Development Fund Project of Jiangsu University (grant no. JLY2021087), the Lianyungang Second People's Hospital Youth Medical Talent Growth Funding(grant no. TQ202006).
Author information
Authors and Affiliations
Contributions
WJZ, YW, and CC designed the experiments and wrote the manuscript. JH and YW collected the clinical samples. JH, JX, and YZL performed the experiments. LW analyzed the data. All authors reviewed and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Conflict of Interest
The authors have no conflicts of interest to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Hu, J., Xu, J., Lu, Y. et al. Rapid Detection of Enterobacter cloacae With a Visualized Isothermal Recombinase Polymerase Amplification Assay. Curr Microbiol 80, 233 (2023). https://doi.org/10.1007/s00284-023-03269-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00284-023-03269-1