
Abstract
Acinetobacter baumannii is a multidrug-resistant bacteria responsible for nosocomial infections with significant fatality rates globally. Therapeutic failure and relapse of infection has been associated with persister cells formation which can also lead to resistance in A. baumannii. In the present study, we observed that A. baumannii ATCC 17978 in exponential phase survived lethal concentrations of amikacin, rifampicin and ciprofloxacin by generating persister cells but was unable to survive tobramycin treatment. The transcriptome of A. baumannii ATCC 17978 was analyzed following exposure to a high concentration of tobramycin (10 × MIC) for a short period of time to study the possible mechanisms responsible for lethality. Tobramycin reduced the expression of genes involved in energy production (nuoH, nuoN, nuoM, cydA, sucC), oxidative stress protection (tauD, cysD), and nutrition uptake (ompW) significantly. In addition, hemerythrin (non-heme di-iron oxygen-binding protein) was found to be the most downregulated gene in response to tobramycin which needs to be further studied for its role in susceptibility to antibiotics. Tobramycin upregulated the expression of genes that are mainly involved in stress response (leucine catabolism, DNA repair and HicAB toxin-antitoxin system). The differentially expressed genes highlighted in the study provided insight into the probable molecular mechanism of tobramycin-induced cell death and revealed some novel targets that can be explored further for their potential to control A. baumannii.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Acinetobacter baumannii is a Gram-negative coccobacillus that causes ventilator-associated pneumonia (VAP), bacteraemia, meningitis and infections related to skin and soft tissue, central nervous system, and urinary tract in patients admitted to intensive care units [1]. It is one of the six superbugs in the “ESKAPE” group that are responsible for nosocomial infections [2]. A. baumannii has also been associated with secondary infections of lower respiratory tract in COVID-19 patients [3]. It has been designated as a “red alert” human pathogen due to its high level of antibiotic resistance [4]. The World Health Organization (WHO) has listed A. baumannii in the “critical” category of priority pathogens against which new antibiotics are urgently needed [5].
The formation of persister cells is a key factor in the development of recalcitrant A. baumannii infections and antibiotic treatment failure [6, 7]. Persisters are a subset of antibiotic-sensitive bacterial cells that enter a slow or non-growing transient state to survive lethal dose of antibiotics. They can be distinguished from resistant cells as they do not divide actively in the presence of antibiotics and their MIC does not change after regrowth [8, 9]. A. baumannii has been shown to form persister cells in exponential phase against amikacin, carbenicillin, colistin, rifampicin, polymixin B, meropenem, ceftazidime, tetracycline, and imipenem [10,11,12,13]. However, tobramycin has been found to make A. baumannii cultures sterile [12].
In the present study, A. baumannii ATCC 17978 culture in exponential phase survived against amikacin, rifampicin, and ciprofloxacin by forming persister cells but could not survive tobramycin treatment as reported earlier. To gain insight into the reasons for eradication shown by tobramycin, transcriptome of A. baumannii ATCC 17978 cells in exponential phase exposed, for short duration, to lethal concentration of tobramycin was analyzed. Our results highlight the relevance of genes involved in energy production, sulphur metabolism, OmpW, and hemerythrin for tobramycin-induced cell death. These findings provide new target genes necessitating their further investigation as potential therapeutic targets.
Materials and Methods
Determination of Minimum Inhibitory Concentration (MIC)
A. baumannii ATCC 17978 was grown and maintained in Lysogeny Broth (LB) at 37 °C. MIC of ciprofloxacin, rifampicin, amikacin and tobramycin was determined by broth micro-dilution method as per the Clinical and Laboratory Standards Institute guidelines [14]. Overnight grown culture was diluted in Mueller Hinton (MH) broth to a turbidity equivalent to 0.5 McFarland standard. The culture was mixed with serial two-fold dilutions of antibiotics and incubated for 24 h at 37 °C, 180 rpm. The lowest antibiotic concentration that inhibited visible growth was defined as the MIC.
Persister Assays
A single colony of A. baumannii was inoculated in LB medium and cultured at 37 °C, 180 rpm for 16 h. 100 µl of overnight grown culture was transferred to 10 ml of LB and grown to exponential phase (OD600 ~ 0.5). Concentration-dependent persister assay was done using different antibiotic concentrations (×MIC) for 3 h. The time-dependent persister assay was performed with constant concentrations of antibiotics as selected from the concentration-dependent assay for different (3–24 h) time intervals. After the addition of antibiotics, cells were separated by centrifugation at 8000× g for 10 min at different time points, washed twice with phosphate buffer saline (PBS), and diluted tenfold serially in PBS. 10 µl aliquot of each dilution was spot-plated on LB agar plates to determine colony-forming units per milliliter (CFU/mL) [15].
RNA Isolation, Sequencing and Transcriptome Analysis
Exponential phase A. baumannii cells were treated with 10 × MIC of tobramycin for 60 min and total RNA was extracted using TRI Reagent (Sigma) and RNeasy Mini Kit (Qiagen, USA). Untreated cells were used as control. The quality and quantity of isolated RNA was assessed on 1% agarose gels and NanoDrop (Eppendorf) respectively. DNA was removed from the samples using Turbo DNase I (Invitrogen) according to the manufacturer’s instructions. RNA samples were evaluated for integrity using Agilent 2100 Bioanalyzer (Agilent Technologies), and samples with RNA integrity number (RIN) greater than 7 were used to build cDNA libraries. RNA sequencing was outsourced to SciGenom Labs Pvt. Ltd. (Kochi), India where Illumina HiSeq 2500 platform with paired-end sequencing (2 × 100 bp) was used. RNA-Seq reads were aligned with A. baumannii ATCC 17978 genome using Hisat2 [16], and differential gene expression analysis was done with Cuffdiff [17]. A gene was considered significantly differentially expressed when the log|FC|≥ 2 with an adjusted P value ≤ 0.05. RNA-Sequencing raw data have been deposited in NCBI BioProject database under Accession no. PRJNA787919.
Functional Annotation of Differentially Expressed Genes (DEGs)
To analyze the functional roles of the DEGs, OmicsBox (version 3.2) was used for Gene Ontology (GO) annotation [18]. The Kyoto Encyclopedia of Genes and Genomes (KEGG) metabolic pathways database and Cluster of Orthologous Groups (COGs) protein database were used to further classify the genes.
Protein–Protein Interaction (PPI) Network Analysis
The PPI network of DEGs was constructed using the Search Tool for the Retrieval of Interacting Genes (STRING; http://string-db.org) and a combined score > 0.7 (high confidence) was set as the cut-off criterion. Nodes in the network that were not connected were excluded. Based on the results from STRING, the open source platform Cytoscape (version 3.8.2, https://cytoscape.org/) was used to visualize molecular interaction networks, and the degree centrality was determined using the plug-in CytoHubba.
Quantitative Reverse Transcription Polymerase Chain Reaction (qRT-PCR) Validation
To verify transcriptome results, RNA was extracted as described above. The cDNA was synthesized by the Verso cDNA synthesis kit (Thermo Fisher, Co., USA) following the manufacturer’s manual. Primer3 software was used to design specific primers for qRT-PCR (Supplementary Table 3). qRT-PCR was performed using iTaq Universal SYBR® Green Supermix (Biorad, USA). The 16S rRNA gene was used as the reference gene, and the gene expression was calculated using the 2−∆∆Ct method.
Results
Persister Cell Subpopulation of A. baumannii ATCC 17978 on Treatment with Different Classes of Antibiotics
A. baumannii ATCC 17978 culture in exponential phase formed varying percentage of persister cells in response to ciprofloxacin (MIC, 0.25 µg/ml), rifampicin (MIC, 4 µg/ml) and amikacin (MIC, 2 µg/ml) (Supplementary Fig. 1). The time-dependent assay revealed maximum persisters against 10 × rifampicin (1.71%) followed by 50 × ciprofloxacin (0.022%) and 10 × amikacin (0.0065%) at 24 h (Fig. 1). However, increasing concentration of tobramycin led to a rapid decline in viable cells. Tobramycin at 1 × MIC in a time-dependent assay did not affect the viability of cells, while treatment with 3 × MIC led to an effective reduction in the number of viable cells, with only 0.005% cells surviving after 5 h. Treatment with 5 × MIC resulted in 0.03% cell survival after 2 h but after 3 h, the viable cell count fell below detection limit. Treatment with 10 × MIC for 1 h had 0.04% cell survival which dropped below the detection limit in 2 h (Supplementary Fig. 2). Hence to study the effect of tobramycin in A. baumannii, cells in exponential phase were treated with 10 × MIC for 1 h.

Exponential phase cells of A. baumannii ATCC 17978 on treatment with ciprofloxacin (50×), rifampicin (10×), amikacin (10×) and tobramycin (10×) for different time intervals. Untreated cells at 0 h (3.2 ± 0.36 × 108 CFU/ml) were taken as 100% and percent survival was calculated accordingly. Data are represented as mean log percent survival ± SD of three independent experiments. The asterisk indicates cell count below the detection limit
Identification of Differentially Expressed Genes in A. baumannii After Exposure to Lethal Concentration of Tobramycin
The transcriptome profiles of A. baumannii ATCC 17978 cells with (10 × MIC for 1 h) and without tobramycin treatment were analyzed by high-throughput sequencing. The differential gene expression analysis showed that 97 genes were differentially expressed, including 62 genes that were upregulated and 35-genes that were downregulated significantly (Fig. 2, Supplementary Table 1). Among these, 25 genes (14 upregulated and 11 downregulated) encoded hypothetical proteins.

A Plot showing log2 fold change values of all significantly (P ≤ 0.05) differentially expressed genes identified from transcriptome analysis of A. baumannii ATCC 17978 in response to tobramycin treatment. Log2 fold change values were plotted with respect to gene locus tag number. Genes with log2 fold change ≥ 4 and ≤ 4 but ≥ 2 are shown in red and blue, respectively. B Depiction of genes that are differentially expressed in response to tobramycin treatment. Downregulated genes are highlighted with a red downward pointing arrows, and upregulated genes with green upward pointing arrows. (Color figure online)
Functional Annotation of Differentially Expressed Genes
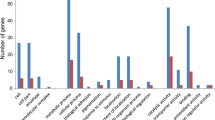
In GO classification, a total of 81 differentially expressed genes (DEGs) having annotated biological functions were assigned 188 GO terms based on their location in the cell, their biological and molecular functions. Two main enriched terms in biological process (BP) category were metabolic process and cellular process. In cellular component (CC) category, catalytic activity and binding were the most enriched GO terms. In molecular function (MF) category, the most enriched term was catalytic activity, followed by binding (Fig. 3A). In COG classification, DEGs were functionally classified into 17 COG categories (Fig. 3B). Among all COG categories, Transcription (K) occupied the largest proportion, followed by the Energy production and conversion (C), and Lipid transport and metabolism (I). Further, the KEGG pathway analysis revealed that the upregulated DEGs were linked to 40 pathways, with valine, leucine and isoleucine degradation and fatty acid degradation being the most dominant. The downregulated DEGs were linked to 22 KEGG pathways, with oxidative phosphorylation and sulphur metabolism being the most enriched (Fig. 3C).

A Gene ontology (GO) classification of DEGs grouped into three categories: biological process (BP), molecular function (MF), and cellular component (CC). Blue and red bars indicate downregulated and upregulated genes, respectively. The Y-axis indicates number of genes. B Cluster of orthologous genes (COG) classification of DEGs. Yellow bars represent upregulated genes, and orange bars, downregulated genes in each COG category. C KEGG pathway classification of DEGs. Top 10 KEGG terms of downregulated and upregulated DEGs represented by green and purple bars, respectively. (Color figure online)
PPI Network Analysis of Differentially Expressed Genes
PPI network analysis was performed to gain insight into the interaction among DEGs. The PPI network of DEGs contained 33 nodes (including 26 upregulated and 7 downregulated genes) and 27 interactions (Supplementary Fig. 3). Genes identified with the highest degree of connections (Supplementary Table 2) included putative ferredoxin (A1S_0945), putative vanillate o-demethylase oxygenase subunit (A1S_0947), NADH dehydrogenase I chain N membrane subunit (nuoN, A1S_0764), hypothetical protein (A1S_0953), putative dioxygenase (A1S_0949), acyl-CoA dehydrogenase (ivd, A1S_1376), regulatory protein (repA, A1S_0663), type IV secretion system protein TrbJ (A1S_0665), type IV secretion system protein TrbL (A1S_0666), and NADH dehydrogenase I chain H membrane subunit (nuoH, A1S_0758).
Genes DownRegulated by Tobramycin Treatment
Genes encoding hemerythrin (A1S_0891), outer membrane protein ompW (A1S_0292), putative membrane protein (A1S_1665), transcriptional regulator Cro/CI family (A1S_1582) and six genes involved in metabolism (cydA, A1S_1924; dadA, A1S_1075; sucC, A1S_2718; tauD, A1S_1445; cysD, A1S_1000; cysP, A1S_2531) were among the top 10 downregulated genes. Other significantly downregulated genes were involved in oxidative phosphorylation (nuoH, A1S_0758; nuoN, A1S_0764; nuoM, A1S_0763), methylation (A1S_1188), translation (efp, A1S_2419) and transportation (bamD, A1S_0840).
Genes UpRegulated by Tobramycin Treatment
Two genes involved in leucine catabolism pathway (mccB, A1S_1375; ivd, A1S_1376), two genes encoding type II toxin-antitoxin system (hicB, A1S_2019; hicA, A1S_2020), four genes encoding transcriptional regulators (A1S_2699; A1S_0094; A1S_1377; A1S_3294), ddrR (A1S_1388) and putative NADPH:quinone reductase and related Zn-dependent oxidoreductase (A1S_3293) were among the top ten upregulated genes. Apart from these, genes involved in DNA repair (umuD, A1S_1389), type IV secretion system (trbJ, A1S_0665; trbL, A1S_0666), iron homeostasis (A1S_0040; A1S_0945; iscU, A1S_1631) and transporters (arsB, A1S_1454; A1S_1503; A1S_1504; A1S_2302; A1S_0161; A1S_3240; A1S_1046; A1S_1333; A1S_1390; A1S_2472; A1S_3914) were also among the significantly overexpressed genes.
Validation of DEGs by qRT-PCR
Highly upregulated and the most downregulated genes from the RNA-Seq data were validated by qRT-PCR. ddrR (A1S_1388) and hicB (A1S_2019) genes upregulated with 5.86- and 4.92 log2 fold change, respectively in RNA-Seq data, showed 2.97- and 3.2 log2 fold upregulation respectively by qRT-PCR. RNA-Seq data showed hemerythrin (A1S_0891), OmpW family protein (A1S_0292) and cytochrome d ubiquinol oxidase subunit 1, cydA (A1S_1924) to be downregulated with −8.0, −5.02 and −4.97 log2 fold change, respectively, were also found to be −7.5, −4.91 and −5.62 log2 fold downregulated by qRT-PCR (Fig. 4). Hence, the expression pattern of five genes determined by qRT-PCR was found to be consistent with the transcriptome data.

Relative expression of five DEGs (ddrR, hicB, hemerythrin, ompW and cydA) in exponential phase cells of A. baumannii ATCC 17978 upon treatment with tobramycin (10 × MIC for 1 h). Data are representative of three independent experiments. Bars represent the mean ± SD. **P ≤ 0.01; ***P ≤ 0.001
Discussion
A. baumannii has become a major global public health concern [19]. Bacterial persistence allows antibiotic susceptible bacteria to survive lethal concentrations of antibiotics. Persistence is responsible for the recurrence of infections and is also considered an important contributor to the emergence of resistance [20]. The present study showed that A. baumannii ATCC 17978 in exponential phase formed persister cells against rifampicin (rifamycin), ciprofloxacin (fluoroquinolone) and amikacin (aminoglycoside) but could not survive lethal concentrations of another aminoglycoside tobramycin. Therefore, to explore genes and pathways that might be important in eradicating A. baumannii, the transcriptome of exponential phase A. baumannii cells after exposure to lethal concentration of tobramycin (10 × MIC for 1 h) was evaluated.
Transcriptome analysis revealed that upregulated genes outnumbered the downregulated genes in response to tobramycin stress (Fig. 2). Aminoglycosides have been shown to inhibit protein synthesis in bacteria [21]. In accordance 23S rRNA methyltransferase and translation elongation factor (efp) was found to be downregulated in the present study. In A. baumannii ATCC 17978, there was significant downregulation of genes involved in energy production and conversion (cydA, nuoH, nuoN, nuoM, sucC) after tobramycin treatment. cydA encodes subunit I of cytochrome bd oxidase, which generates proton motive force across the membrane for ATP production [22]. Lack of a functional cytochrome severely compromised virulence and intracellular viability of Staphylococcus aureus [23], Salmonella enterica [24], E. coli [25], Mycobacterium tuberculosis [26] and Mycobacterium smegmatis [27]. The cydA deletion mutant of M. tuberculosis was found to be more susceptible to the FDA-approved drug bedaquiline and resulted in rapid killing of cells, underscoring the role of this respiratory protein in survival [28]. nuoH, nuoM and nuoN genes encode subunits of NADH-quinone oxidoreductase (complex I) that catalyzes the transfer of electrons from NADH to quinone in cytoplasmic membrane, forming a proton gradient required for energy production [29]. sucC encodes β subunit of succinyl-CoA synthetase that converts succinyl-CoA into succinate accompanied by the production of ATP. In E. coli, ubiF and sucB deletion mutants have been shown to reduce persister survival against ampicillin and gentamicin [30]. PPI network analysis of DEGs also highlighted nuoN and nuoH as the hub genes. The downregulation of these genes reflected decreased respiration, which probably lowered PMF and subsequently ATP production contributing to tobramycin-induced cell death.
Expression of genes involved in sulphur metabolism (tauD and cysD) was found to be significantly downregulated under tobramycin stress. tauD encodes taurine dioxygenase that is required for the utilization of taurine as a sulphur source. In A. oleivorans DR1, deletion of tauD exhibited increased sensitivity to H2O2, highlighting its protective role in oxidative stress [31]. cysD encodes a subunit of sulfate adenylyltransferase that catalyzes the first step in sulfate assimilation pathway. Upregulation of cysD has been shown in M. tuberculosis in response to nutritional deprivation, hypoxia, hydrogen peroxide, and vancomycin treatment [32]. cysD deletion mutant of Aeromonas hydrophila showed decreased survival rate against furazolidone treatment [33]. Since aminoglycosides produce reactive oxygen species [34], the downregulation of tauD and cysD genes in A. baumannii might have compromised the management of oxidative stress related to tobramycin.
Hemerythrin was also found to be downregulated by tobramycin. In A. baumannii, downregulation of hemerythrin has been reported under iron-limiting conditions [35]. Hemerythrin is non-heme, di-iron and O2-binding protein involved in iron and oxygen detoxification, transport, and storage [36]. Tobramycin may have disarmed the protective effect by reducing the expression of hemerythrin in A. baumannii. Downregulation of hemerythrin expression in A. baumannii was found to be concentration-dependent as 10 × MIC showed higher downregulation than 3 × MIC (Supplementary Fig. 4). Hemerythrin-like proteins affect oxidation–reduction regulation and antibiotic resistance [37]. The number of hemerythrin-like proteins varies in different organisms. This multiplicity makes it difficult to understand their role. However, A. baumannii ATCC 17978 has only one gene (A1S_0891) coding for hemerythrin. Since not much is known about hemerythrin-like proteins in A. baumannii, knockout of hemerythrin may help in providing insight into its function.
Expression of ompW was found to be significantly downregulated under tobramycin stress. OmpW is a β-barrel shaped outer membrane protein with a hydrophobic channel that is involved in the transport of hydrophobic molecules [38, 39]. In A. baumannii, OmpW is implicated in uptake of iron and its homeostasis, and binding of colistin to the membrane [40]. Downregulation of ompW might have reduced the uptake of iron by A. baumannii under tobramycin stress.
mccB, ivd, and liuC genes involved in leucine catabolism were upregulated on exposure to tobramycin. Leucine catabolism produces important intermediates of tricarboxylic acid (TCA) cycle (acetyl-CoA and acetoacetate) [41, 42], which are probably required to meet energy requirements under stress conditions. Genes related to leucine degradation and transport were found to be induced in Acinetobacter harbinensis HITLi7T under stress of low temperatures [43]. Tobramycin treatment upregulated the expression of ddrD and umuD genes involved in DNA repair [44]. Kanamycin, tobramycin, and gentamicin have been shown to induce the SOS response in Vibrio cholera as a result of drug-induced oxidative DNA damage [45]. Increased expression of these genes may be due to DNA damage at high concentrations of tobramycin.
Tobramycin also increased the expression of the hicA and hicB genes of the HicAB toxin-antitoxin system. Earlier, we had shown increased expression of hicA and hicB genes in A. baumannii in response to ciprofloxacin which may be related to persistence against it [46]. Since expression of these genes was significantly upregulated even under tobramycin stress, further studies are required to understand their role in persistence. In Burkholderia pseudomallaei, the deletion of hicAB was found to reduce persistence against ciprofloxacin but not ceftazidime [47].
Conclusion
The present study shows that A. baumannii ATCC 17978 cannot survive lethal concentrations of tobramycin. Various genes and pathways were differentially expressed in A. baumannii as a response to tobramycin treatment. Significant downregulation of genes involved in energy production might have contributed to the lethality of tobramycin to A. baumannii cells. Downregulation of outer membrane protein W might have reduced the uptake of nutrients required for survival. Furthermore, decreased expression of hemerythrin and genes involved in sulphur metabolism (tauD and cysD) may have disarmed the protective response against tobramycin. Further work involving knockouts of these DEGs are required to confirm their involvement in tobramycin induced cell death.
Data Availability
The data used to support the findings of this study are available from the corresponding author upon request.
Code Availability
Not applicable.
Material Availability
The data used to support the findings of this study are available from the corresponding author upon request.
References
Garnacho-Montero J, Timsit JF (2019) Managing Acinetobacter baumannii infections. Curr Opin Infect Dis 32:69–76. https://doi.org/10.1097/qco.0000000000000518
De Oliveira DM, Forde BM, Kidd TJ, Harris PN, Schembri MA, Beatson SA, Paterson DL, Walker MJ (2020) Antimicrobial resistance in ESKAPE pathogens. Clin Microbiol Rev 33:e00181-e219. https://doi.org/10.1128/CMR.00181-19
Sharifipour E, Shams S, Esmkhani M, Khodadadi J, Fotouhi-Ardakani R, Koohpaei A, Doosti Z, Golzari SE (2020) Evaluation of bacterial co-infections of the respiratory tract in COVID-19 patients admitted to ICU. BMC Infect Dis 20:1–7. https://doi.org/10.1186/s12879-020-05374-z
Cerqueira GM, Peleg AY (2011) Insights into Acinetobacter baumannii pathogenicity. IUBMB Life 63:1055–1060. https://doi.org/10.1002/iub.533
Tacconelli E, Carrara E, Savoldi A, Harbarth S, Mendelson M, Monnet DL et al (2018) Discovery, research, and development of new antibiotics: the WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect Dis 18:318–327. https://doi.org/10.1016/s1473-3099(17)30753-3
Defraine V, Fauvart M, Michiels J (2018) Fighting bacterial persistence: current and emerging anti-persister strategies and therapeutics. Drug Resist Updates 38:12–26. https://doi.org/10.1016/j.drup.2018.03.002
Wong FH, Cai Y, Leck H, Lim TP, Teo JQ, Lee W, Koh TH, Tan TT, Tan KW, Kwa AL (2020) Determining the development of persisters in extensively drug-resistant Acinetobacter baumannii upon exposure to polymyxin B-based antibiotic combinations using flow cytometry. Antimicrob Agents Chemother 64:e01712-e1719. https://doi.org/10.1128/aac.01712-19
Van den Bergh B, Fauvart M, Michiels J (2017) Formation, physiology, ecology, evolution and clinical importance of bacterial persisters. FEMS Microbiol Rev 41:219–251. https://doi.org/10.1093/femsre/fux001
Balaban NQ, Helaine S, Lewis K, Ackermann M, Aldridge B, Andersson DI, Brynildsen MP, Bumann D, Camilli A, Collins JJ, Dehio C et al (2019) Definitions and guidelines for research on antibiotic persistence. Nat Rev Microbiol 17:441–448. https://doi.org/10.1038/s41579-019-0196-3
Bhargava N, Sharma P, Capalash N (2014) Pyocyanin stimulates quorum sensing-mediated tolerance to oxidative stress and increases persister cell populations in Acinetobacter baumannii. Infect Immun 82:3417–3425. https://doi.org/10.1128/iai.01600-14
Gallo SW, Ferreira CA, de Oliveira SD (2017) Combination of polymyxin B and meropenem eradicates persister cells from Acinetobacter baumannii strains in exponential growth. J Med Microbiol 66:1257–1260. https://doi.org/10.1099/jmm.0.000542
Alkasir R, Ma Y, Liu F, Li J, Lv N, Xue Y, Hu Y, Zhu B (2018) Characterization and transcriptome analysis of Acinetobacter baumannii persister cells. Microb Drug Resist 24:1466–1474. https://doi.org/10.1089/mdr.2017.0341
Kaur A, Sharma P, Capalash N (2018) Curcumin alleviates persistence of Acinetobacter baumannii against colistin. Sci Rep 8:11029. https://doi.org/10.1038/s41598-018-29291-z
CLSI (2018) Performance standards for antimicrobial susceptibility testing. CLSI supplement M100, 28th edn. CLSI, Malvern
Kashyap S, Kaur S, Sharma P, Capalash N (2021) Combination of colistin and tobramycin inhibits persistence of Acinetobacter baumannii by membrane hyperpolarization and down-regulation of efflux pumps. Microbes Infect 23:104795. https://doi.org/10.1016/j.micinf.2021.104795
Kim D, Paggi JM, Park C, Bennett C, Salzberg SL (2019) Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol 37:907–915. https://doi.org/10.1038/s41587-019-0201-4
Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L (2012) Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7:562–578. https://doi.org/10.1038/nprot.2012.016
Götz S, Garcia-Gomez JM, Terol J, Williams TD, Nagaraj SH, Nueda MJ, Robles M, Talon M, Dopazo J, Conesa A (2008) High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res 36:3420–3435. https://doi.org/10.1093/nar/gkn176
Hernández-González IL, Castillo-Ramírez S (2020) Antibiotic-resistant Acinetobacter baumannii is a One Health problem. Lancet Microbe 1:e279. https://doi.org/10.1016/S2666-5247(20)30167-1
Monem S, Furmanek-Blaszk B, Łupkowska A, Kuczyńska-Wiśnik D, Stojowska-Swędrzyńska K, Laskowska E (2020) Mechanisms protecting Acinetobacter baumannii against multiple stresses triggered by the host immune response, antibiotics and outside-host environment. Int J Mol Sci 21:5498. https://doi.org/10.3390/ijms21155498
Borovinskaya MA, Pai RD, Zhang W, Schuwirth BS, Holton JM, Hirokawa G, Kaji H, Kaji A, Cate JH (2007) Structural basis for aminoglycoside inhibition of bacterial ribosome recycling. Nat Struct Mol Biol 14:727–732. https://doi.org/10.1038/nsmb1271
Giuffrè A, Borisov VB, Arese M, Sarti P, Forte E (2014) Cytochrome bd oxidase and bacterial tolerance to oxidative and nitrosative stress. Biochim Biophys Acta 1837:1178–1187. https://doi.org/10.1016/j.bbabio.2014.01.016
Hammer ND, Reniere ML, Cassat JE, Zhang Y, Hirsch AO, Indriati Hood M, Skaar EP (2013) Two heme-dependent terminal oxidases power Staphylococcus aureus organ-specific colonization of the vertebrate host. MBio 4:e0024113. https://doi.org/10.1128/mbio.00241-13
Turner AK, Barber LZ, Wigley P, Muhammad S, Jones MA, Lovell MA, Hulme S, Barrow PA (2003) Contribution of proton-translocating proteins to the virulence of Salmonella enterica serovars Typhimurium, Gallinarum, and Dublin in chickens and mice. Infect Immun 71:3392–3401. https://doi.org/10.1128/iai.71.6.3392-3401.2003
Shepherd M, Achard ME, Idris A, Totsika M, Phan MD, Peters KM, Sarkar S, Ribeiro CA, Holyoake LV, Ladakis D, Ulett GC, Sweet MJ, Poole RK, McEwan AG, Schembri MA (2016) The cytochrome bd-I respiratory oxidase augments survival of multidrug-resistant Escherichia coli during infection. Sci Rep 6:35285. https://doi.org/10.1038/srep35285
Shi L, Salamon H, Eugenin EA, Pine R, Cooper A, Gennaro ML (2015) Infection with Mycobacterium tuberculosis induces the Warburg effect in mouse lungs. Sci Rep 5:18176. https://doi.org/10.1038/srep18176
Lu P, Heineke MH, Koul A, Andries K, Cook GM, Lill H, van Spanning R, Bald D (2015) The cytochrome bd-type quinol oxidase is important for survival of Mycobacterium smegmatis under peroxide and antibiotic-induced stress. Sci Rep 5:10333. https://doi.org/10.1038/srep10333
Berney M, Hartman TE, Jacobs WR Jr (2014) A Mycobacterium tuberculosis cytochrome bd oxidase mutant is hypersensitive to bedaquiline. MBio 5:e01275-14. https://doi.org/10.1128/mbio.01275-14
Brandt U (2006) Energy-converting NADH: quinone oxidoreductase (complex I). Annu Rev Biochem 75:69–92. https://doi.org/10.1146/annurev.biochem.75.103004.142539
Ma C, Sim S, Shi W, Du L, Xing D, Zhang Y (2010) Energy production genes sucB and ubiF are involved in persister survival and tolerance to multiple antibiotics and stresses in Escherichia coli. FEMS Microbiol Lett 303:33–40. https://doi.org/10.1111/j.1574-6968.2009.01857.x
Park C, Shin B, Park W (2020) Protective role of bacterial alkanesulfonate monooxygenase under oxidative stress. Appl Environ Microbiol 86:e00692–e00720. https://doi.org/10.1128/aem.00692-20
Hatzios SK, Bertozzi CR (2011) The regulation of sulfur metabolism in Mycobacterium tuberculosis. PLoS Pathog 7:e1002036. https://doi.org/10.1371/journal.ppat.1002036
Fu Y, Zhang L, Wang G, Lin Y, Ramanathan S, Yang G, Lin W, Lin X (2020) The LysR-type transcriptional regulator YeeY plays important roles in the regulatory of furazolidone resistance in Aeromonas hydrophila. Front Microbiol 11:577376. https://doi.org/10.3389/fmicb.2020.577376
Dwyer DJ, Belenky PA, Yang JH, MacDonald IC, Martell JD, Takahashi N, Chan CT, Lobritz MA, Braff D, Schwarz EG, Ye JD, Pati M, Vercruysse M, Ralifo PS, Allison KR, Khalil AS, Ting AY, Walker GC, Collins JJ (2014) Antibiotics induce redox-related physiological alterations as part of their lethality. Proc Natl Acad Sci USA 111:E2100–E2109. https://doi.org/10.1073/pnas.1401876111
Eijkelkamp BA, Stroeher UH, Hassan KA, Papadimitrious MS, Paulsen IT, Brown MH (2011) Adherence and motility characteristics of clinical Acinetobacter baumannii isolates. FEMS Microbiol Lett 323:44–51. https://doi.org/10.1111/j.1574-6968.2011.02362.x
French CE, Bell JM, Ward FB (2008) Diversity and distribution of hemerythrin-like proteins in prokaryotes. FEMS Microbiol Lett 279:131–145. https://doi.org/10.1111/j.1574-6968.2007.01011.x
Li X, Li J, Hu X, Huang L, Xiao J, Chan J, Mi K (2015) Differential roles of the hemerythrin-like proteins of Mycobacterium smegmatis in hydrogen peroxide and erythromycin susceptibility. Sci Rep 5:16130. https://doi.org/10.1038/srep16130
Hong H, Patel DR, Tamm LK, van den Berg B (2006) The outer membrane protein OmpW forms an eight-stranded β-barrel with a hydrophobic channel. J Biol Chem 281:7568–7577. https://doi.org/10.1074/jbc.m512365200
Touw DS, Patel DR, Van Den Berg B (2010) The crystal structure of OprG from Pseudomonas aeruginosa, a potential channel for transport of hydrophobic molecules across the outer membrane. PLoS ONE 5:e15016. https://doi.org/10.1371/journal.pone.0015016
Catel-Ferreira M, Marti S, Guillon L, Jara L, Coadou G, Molle V, Bouffartigues E, Bou G, Shalk I, Jouenne T, Vila-Farrés X, De E (2016) The outer membrane porin OmpW of Acinetobacter baumannii is involved in iron uptake and colistin binding. FEBS Lett 590:224–231. https://doi.org/10.1002/1873-3468.12050
Lei J, Feng D, Zhang Y, Dahanayaka S, Li X, Yao K, Wang J, Wu Z, Dai Z, Wu G (2012) Regulation of leucine catabolism by metabolic fuels in mammary epithelial cells. Amino Acids 43:2179–2189. https://doi.org/10.1007/s00726-012-1302-2
Arany Z, Neinast M (2018) Branched chain amino acids in metabolic disease. Curr Diab Rep 18:76. https://doi.org/10.1007/s11892-018-1048-7
Ma YP, Zhang SM, Meng LQ, Li WG (2019) Transcriptomic responses of Acinetobacter harbinensis HITLi 7 T at low temperatures. Biomed Environ Sci 32:371–375. https://doi.org/10.3967/bes2019.049
Maslowska KH, Makiela-Dzbenska K, Fijalkowska IJ (2019) The SOS system: a complex and tightly regulated response to DNA damage. Environ Mol Mutagen 60:368–384. https://doi.org/10.1002/em.22267
Baharoglu Z, Krin E, Mazel D (2013) RpoS plays a central role in the SOS induction by sub-lethal aminoglycoside concentrations in Vibrio cholerae. PLoS Genet 9:e1003421. https://doi.org/10.1371/journal.pgen.1003421
Kashyap S, Sharma P, Capalash N (2021) Potential genes associated with survival of Acinetobacter baumannii under ciprofloxacin stress. Microbes Infect. https://doi.org/10.1016/j.micinf.2021.104844
Butt A, Higman VA, Williams C, Crump MP, Hemsley CM, Harmer N, Titball RW (2014) The HicA toxin from Burkholderia pseudomallei has a role in persister cell formation. Biochem J 459:333–344. https://doi.org/10.1042/bj20140073
Funding
This study was financially supported by the Indian Council of Medical Research, New Delhi, India [OMI/17/2014-ECD-1].
Author information
Authors and Affiliations
Contributions
SK: Conceptualization, data curation, formal analysis, investigation, methodology, validation, visualization, writing—original draft, writing—review and editing. PS: Conceptualization, writing—review and editing. NC: Conceptualization, methodology, funding acquisition, resources, project administration, supervision, writing—review and editing. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Ethical Approval
Not applicable.
Informed Consent
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Kashyap, S., Sharma, P. & Capalash, N. Tobramycin Stress Induced Differential Gene Expression in Acinetobacter baumannii. Curr Microbiol 79, 88 (2022). https://doi.org/10.1007/s00284-022-02788-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00284-022-02788-7