
Abstract
Pleurotus strains are the most important fungi used in the agricultural industry. The exact characterization and identification of Pleurotus species is fundamental for correct identification of the individuals and exploiting their full potential in food industry. The amplified fragment length polymorphism (AFLP) method was applied for genomic fingerprinting of 21 Pleurotus isolates of Asian and European origin. Using one PstI restriction endonuclease and four selective primers in an AFLP assay, 371 DNA fragments were generated, including 308 polymorphic bands. The AFLP profiles were found to be highly specific for each strain and they unambiguously distinguished 21 Pleurotus sp. fungi. The coefficient of Jaccard’s genome profile similarity between the analyzed strains ranged from 0.0 (Pleurotus sp. I vs. P. sajor-caju 237 and P. eryngii 238) to 0.750 (P. ostreatus 246 vs. P. ostreatus 248), and the average was 0.378. The AFLP-based dendrogram generated by the UPGMA method grouped all the Pleurotus fungi studied into two major clusters and one independent lineage located on the outskirt of the tree occupied by naturally growing Pleurotus species strain I. The results of the present study suggest the possible applicability of the AFLP-PstI method in effective identification and molecular characterization of Pleurotus sp. strains.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The genus Pleurotus (Jacq.: Fr.) Kumm. (Pleurotaceae, higher Basidiomycetes) comprises a cosmopolitan group of mushrooms with high nutritional value, therapeutic properties, and various environmental and biotechnological applications [7]. These morphologically distinct fungi consist of microorganisms that share the common character, i.e., the ability to produce arthrospores from asexual fructifications on basidiomata and/or in mycelial cultures [38]. Several species of the genus Pleurotus have great commercial value in the global market of edible cultivated mushrooms, which reportedly grew from 2.13 million tons in 1996 to 3.43 million tons in 2007 [26]. In line with this, the production of oyster mushrooms is also increasing; they currently rank third behind Agaricus bisporus (J. Lge) Imbach and Lentinula edodes (Berk.) Pegler in annual production of 875,600 tons [6]. Beside unquestionable importance in the worldwide agriculture and food industry, all species belonging to the genus Pleurotus are producers of several enzymes including ligninolytic ones such as laccase and peroxidases [22, 27]. Traditionally, edible species of the genus Pleurotus were considered as medicinally important mushrooms [1, 9]. Nowadays, nearly every paper concerning these fungi brings to light the new bioactive substances exhibiting antibiotic, antiviral, antitumor, and anticholesterolic activities. These pharmaceutical substances have been purified and characterized, making them biotechnologically important agents that should be investigated clinically [7, 21, 28]. The genus Pleurotus is also one of the most diverse groups among cultivated fungi with many taxonomic problems [35]. According to Zervakis and Balis [37], the taxonomic disagreements in the genus Pleurotus have risen for the following reasons: initial misidentification, absence of type specimens, instability of morphological characters due to environmental changes, limited reports on physiological characteristics, and lack of mating compatibility studies. Thus, to clarify the taxonomic status of species in the genus Pleurotus (earlier determined mainly by morphological features), many researchers started to classify these fungi also by genomic criteria [2].
As all other cultivated fungi, the cultivated lines of Pleurotus sp. can undergo a drastic loss of diversity resulting from man’s selection [11, 14]. This genetic erosion increases genetic vulnerability and it may lead to dramatic effects on the production yield during cultivation [8]. The development of tools aimed at the clear-cut and safe identification, and assessment of genetic variability of the wild and cultivated strains is thus a fundamental goal of molecular genetics research [33]. The crucial challenge in developing new methods for tracking microorganisms and their identification is to acquire rapidly markers with a high level of discriminative power and interlaboratory reproducibility for specific and sensitive detection of a target organism in complex environmental or commercial samples. Markers based on amplified fragment length polymorphisms (AFLP) have potential in this respect [13, 36].
AFLP is a PCR-based technique for genotyping and fingerprinting DNA of any origin and complexity, i.e., human, plant, and microbial [3, 23, 29, 31, 34]. A simplified AFLP protocol was developed for rapid genomic characterization of genomic DNA. The major modifications to the standard AFLP procedure included: one-step digestion–ligation reaction with digestion with a single restriction endonuclease and amplification in one reaction [29, 32]. Reproducibility, reliability, and specificity are the main advantages of the AFLP technique, which has already been applied to establish differences among Pleurotus eryngii strains [17, 18, 24, 33].
In this paper, we describe the usefulness of a simplified AFLP technique for studying genomic diversity and for identification of Pleurotus sp. strains.
Materials and Methods
Fungal Strains and Their Cultivation
Pleurotus strains (Table 1) were obtained from different collections: the Laboratory for Anatomy and Physiology of Plants, the I.E. Purkyne University, Brno, Slovakia (LAPU); Agriculture University, Tokyo, Japan (FAT, FCTUA); The Fungal Collection of Lublin, the Department of Biochemistry, Maria Curie-Sklodowska University, Lublin, Poland (FCL); and the Botany Institute II, Regensburg University, Regensburg, Germany (BIUR). Environmental fungal isolate (Pleurotus sp. I) was collected in Lublin, Poland. Commercially available Pleurotus ostreatus II and III were derived from local mushrooms producers (L. W. Skrzypczyk and H. Kaczmarek mushroom farms, respectively), Poland. The stock culture of fungal strains was maintained on GPY slants (glucose 1 g/l, peptone 0.5 g/l, yeast extract 0.1 g/l, agar 20 g/l). The slants were inoculated with mycelia and incubated at 26 °C for 7 days, and then used for seed culture inoculation. The mycelia of Pleurotus strains were transferred into a 100 ml Erlenmeyer flask containing 40 ml stationary liquid Lindeberg–Holm (LH) medium [19] by punching out about 5 mm2 of the slants with a sterilized cutter. The seeds were cultivated for 14 days at 26 °C. Broth cultures were then harvested by centrifugation at 10,000×g for 10 min and used for DNA extraction.
Isolation of Total DNA
The fruit body or the mycelia from 40 ml liquid cultures were used for DNA extraction according to the method of Borges et al. [4]. To extract DNA, 0.5–2.0 g of fresh mycelium was ground in liquid nitrogen. The mycelial powder was transferred to a sterile test tube containing 15 ml of cold spermidine–SDS buffer (4 mM spermidine, 10 mM EDTA, 0.1 M NaCl, 0.5 % SDS, 10 mM β-mercaptoethanol, 40 mM Tris–HCl, pH 8.0) and thoroughly shaken for 20 min. The mixture was immediately extracted two times with one volume of double-distilled phenol. Subsequently, the aqueous phase was extracted with one volume of chloroform–isoamyl alcohol (24:1) and centrifuged (10,000×g, 10 min, 4 °C). 3 M sodium acetate (pH 5.5) was added to the aqueous phase at the proportion 9:1, respectively. DNA was then precipitated by addition of two volumes of ice-cold 96 % ethanol, and recovered by centrifugation at 10,000×g at 4 °C for 10 min. DNA was dried in a vacuum exsiccator (Sigma, USA), redissolved in 1 ml of sterile TE buffer, and stored at −20 °C.
Ribonuclease Treatment
Extracted nucleic acids were digested with the RNase A (Sigma, USA) according to the manufacturer’s protocol. The final RNase concentration was 10 μg/ml. The reaction mixture was incubated for 30 min at room temperature. The quality and quantity of genomic DNA were accurately measured with spectrophotometric absorbency at 260 and 280 nm, respectively.
AFLP Analysis
The AFLP reactions were performed as described by Vos et al. [36] with some modifications. Adapters and primers were synthesized by Genset Oligos, France and IBB PAN, Poland.
Restriction Ligation
The genomic DNA (1 μg) was digested in the final volume of 30 μl with 20 U of PstI restriction enzyme (Fermentas, Lithuania) for 18 h at 37 °C. The quality and quantity of the digested product were examined by gel electrophoresis, stained with ethidium bromide, and visualized under UV fluorescence as a smear across bromophenol blue.
The double-stranded PstI oligonucleotides adapters were formed in a total volume of 10 μl by incubating 10 μM PstIAF and 10 μM PstIAR adapters at 95 °C for 10 min, following 30 min at room temperature.
The ligation solution containing the double-stranded adapters (10 μl), DNA digested with PstI (850 ng), 5 U T4 DNA polymerase (Fermentas, Lithuania), and 1× T4 ligase buffer (40 mM Tris–HCl, 10 mM MgCl2, 10 mM DTT, 0.5 mM ATP, pH 7.8) was incubated for 4 h at 37 °C (25 μl final volume). Ligated DNA was then precipitated with a mixture of 3 M sodium acetate, pH 5.5 and ice-cold 96 % ethanol (1:25) at −18 °C for 30 min to remove unbound adapters. DNA was harvested by centrifugation (14,000 rpm, 4 °C, 20 min) and dried in a vacuum centrifuge. The debris of DNA was dissolved in 50 μl of sterile water and used as a template in the amplification reaction.
Nonselective PCR Amplification
Nonselective PCR was performed to check digestion and ligation reactions. PCR was carried out in 20 μl volume containing 5 μl of ligated with double-stranded adapters and purified DNA, 0.2 mM of each dNTP, 1.5 mM MgCl2, 0.4 U Taq DNA polymerase LC (recombinant) 1 U/μl (Fermentas, Lithuania), 1× PCR buffer (75 mM Tris–HCl, pH 8.8, 20 mM (NH4)2SO4, 0.01 % Tween 20), 750 nM PstIAF primer. Amplifications were carried out in a T-personal thermal cycler (Biometra, Germany) with the conditions as follows: 95 °C for 2 min 30 s followed by 45 cycles of 45 s at 94 °C, 45 s at 54 °C, and 45 s at 72 °C. The final cycle was followed by an additional 10 min at 72 °C.
Selective PCR Amplification
PCRs were performed in a 50 μl total volume which consisted of 1× PCR buffer (Fermentas, Lithuania), 2.5 mM MgCl2, 0.2 mM of each dNTP, 1 U of Taq DNA polymerase LC (recombinant) 1 U/μl (Fermentas, Lithuania), 10 pmol of each primer, 0.5 μl of targeted digested and ligated genomic DNA. All amplification reactions were performed in a T-personal thermal cycler (Biometra, Germany) with the conditions as follows: 94 °C for 2 min 30 s followed by seven cycles of amplification, with annealing temperature decreasing 1 °C/cycle: 94 °C for 30 s, first annealing for 30 s at 60–54 °C or 67–61 °C (annealing temperature depends on primers T m), 72 °C for 30 s, and next 33 amplification cycles of 94 °C for 45 s, 53 °C or 60 °C (annealing temperature depends on primers T m) for 45 s, and 72 °C for 45 s. The final cycle was followed by an additional 7 min at 72 °C. The PCR products were stored at 4 °C until further analysis. The adapters and primers employed for AFLP are shown in Table 2.
Electrophoresis and Imaging
A 25 μl aliquot of the PCR mixture was combined with 5 μl of loading buffer and the amplicons were separated by electrophoresis in 1.5 % agarose gel in 1× TBE buffer (89 mM Tris base, 89 mM boric acid, 2 mM EDTA, pH 8.0). The electrophoresis was run at 150 V in TBE buffer on a horizontal gel electrophoresis system (Agagel Mini, Biometra) for about 3 h. The gels were stained with ethidium bromide and photographed on a UV transilluminator (Vilber Lourmat, France).
Data Analysis
Electrophoretograms were analyzed using BIO 1D software (Vilber Lourmat, France). AFLP markers were manually scored as binary data for the presence or absence of fragments between 75 and 3,000 bp. This binary information was used to calculate Jaccard’s pairwise similarity coefficients as implemented in the program FreeTree version 0.9.1.50 [10]. The unweighted pair-group method with arithmetic averages (UPGMA) dendrograms were generated from DNA band patterns using Nei and Li [25] correlation coefficient. The phylogenetic tree was visualized and edited using NTSYSpc software version 2.01. (Exeter Software Co., New York).
Results
The rare cutting restriction endonuclease PstI and four primers were used in the AFLP analysis to fingerprint the genomes of 18 fungal strains belonging to six species of the genus Pleurotus: P. ostreatus, P. sajor-caju, P. eryngii, P. pulmonarius, P. florida, P. cystidiosus; two commercially available edible strains of P. ostreatus (II and III); and one environmental isolate determined as Pleurotus sp. I (Table 1).
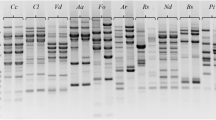
The smears obtained in the nonselective PCR amplification proved efficient degradation of DNA by PstI endonuclease. In the selective amplification reactions, all four primers successfully amplified AFLP bands in all the 21 fungi studied. Each of the four primers generated a fingerprint pattern markedly distinct from those of the other primers, even when the primers differed by only one selective nucleotide in the extension. A total of one to three selective bases were found to provide a sufficient complex pattern for the DNA polymorphism analysis (see Table 3). Although a variable number of amplified bands was obtained in the PCR with each primer, all of them generated polymorphic and unambiguously scored fragments, and numbers of scorable fragments.
The AFLP method applied has provided characteristic genomic markers to differentiate among the analyzed Pleurotus strains. The number of the scorable amplicons produced high variation and ranged from 1 to 12. A total of 371 AFLP markers were perceived using four primers. The large number of bands obtained in the PCR with PstG and PstGC primers demonstrate that the AFLP analysis is a robust and efficient method for detecting genomic differences between the strains in the genus Pleurotus. Primer PstGC amplified the highest number of fragments (192), while the fewest bands (18) were observed with the primer PstCAA. On average, 92.75 AFLP markers in the size range from 75 to 1,750 bp were amplified per primer. A total of 308 reliable polymorphic bands (90.4 % polymorphism) were observed across all 21 isolates from PCR with four primers, which corresponds to an average of 77 polymorphic bands per primer combination. The main AFLP characteristics for four primers are presented in Table 3.
Pairwise similarities between Pleurotus strains (Table 4) were calculated based on the number of polymorphic bands using Jaccard’s similarity coefficient [15]. The genomic similarity between Pleurotus species varied from 0.0 (Pleurotus sp. I vs. P. sajor-caju 237 and P. eryngii 238) to 0.750 (P. ostreatus 246 vs. P. ostreatus 248) and on average was 0.378. The average value of Jaccard’s pairwise genomic similarity within the group of P. ostreatus was 0.453. Lower variations were found in the groups of P. sajor-caju and P. eryngii: 0.320 and 0.313, respectively.
The genomic relationship between the studied Pleurotus strains is presented on the dendrogram constructed with an UPGMA cluster analysis (Fig. 1). Based on the combined data from the AFLP patterns obtained in the PCR with four primers, all the 21 Pleurotus species strains were classified into two main clusters at DNA profile similarity of 33 % and one separate lineage occupied by naturally growing Pleurotus sp. strain I. Cluster I combined 4 strains, i.e., 2 strains of P. eryngi, 1 strain of P. ostreatus, and 1 strain of P. sajor-caju. Cluster II grouped 17 strains classified as P. ostreatus (12 strains), P. sajor-caju (2 strains), P. cystidiosus (1 strain), P. florida (1 strain), and P. pulmonarius (1 strain). Pleurotus sp. strain I located on the outskirt of the tree and the other Pleurotus sp. strains included in the analysis displayed the AFLP profile similarity level in the range from 0.0 to 14.3 %.

UPGMA tree based on polymorphic AFLP markers showing genomic diversity of fungal strains examined in this study. All the 21 Pleurotus sp. strains were grouped into two main clusters (I and II) and one separate lineage. UPGMA cluster analysis was based on Nei and Li’s [25] genetic distance
Discussion
AFLP is capable of simultaneous screening of many different DNA regions distributed randomly throughout the genome after prior DNA digestion with the restriction enzyme [24].
AFLP can be applied to DNAs of many sources and complexity, and it has been reported to be suitable for identification and differentiation of microorganisms at the intraspecies level as well as for determining their genomic relationships [5, 12, 20]. To our knowledge, the AFLP technique has also been used for identification of fungi and analysis of their genomic diversity. Mueller et al. [23] used a simplified AFLP method with only PstI restriction enzyme to detect genomic differences among 14 symbiotic fungi of the fungus-growing ant, Cyphomyrmex minutus. Terashima and Matsumoto [30] demonstrated that AFLP analysis is suitable to strain typing with heat-dried fruiting bodies of shitake mushrooms. Urbanelli et al. [33] showed that AFLP distinguish ambiguously three studies by the genus Pleurotus ranks: P. ferulae, P. eryngii, and P. eryngii var. nebrodensis.
In this study, we used the AFLP method to assess genetic differences among 21 strains representing: P. ostreatus (12 strains), P. sajor-caju (3 strains), P. eryngi (2 strains), P. pulmunarius (1 strain), P. florida (1 strain), P. cystidiosus (1 strain), and 1 Pleurotus sp. isolated by us from natural environmental samples and classified into the genus Pleurotus by morphological features.
By using a combination of four AFLP primers, it was possible to distinguish clearly all the 21 strains of the genus Pleurotus at the species and intraspecies levels (Fig. 1). They exhibited very different banding patterns and their pairwise genomic similarities ranged from 0 between Pleurotus sp. I—a wild growing fungus and P. sajor-caju 237 as well as P. eryngii 238 (both of them are from culture collections) to 75 % between P. ostreatus 176 and P. sajor-caju 178. The significant genomic diversity of the studied Pleurotus sp. strains was demonstrated by cluster analysis of their AFLP profiles (Fig. 1). Each Pleurotus strain formed independent lineage on the AFLP tree and they were clustered into four groups and two separate branches at the AFLP genomic profile similarity of 33 %. Pleurotus sp. I from the natural environment was located on the outskirt of the AFLP tree (besides Pleurotus species from culture collection and two commercial P. ostreatus strains). This fact supports the thesis that wild populations of edible mushrooms (well adapted to changing environmental conditions) may be an important source of genomic variability for cultivated mushroom lineages that undergo loss of genomic diversity resulting from man’s selection [33].
Cluster analysis of Pleurotus sp. AFLP profiles also revealed that the geographically close P. ostreatus strain from Slovakian and Polish culture collections are genomically distant (their AFLP profile similarity is only 39 %). They fall into two different clusters similar to two commercial strains P. ostreatus II and III from Polish manufacturers (33 % AFLP profile similarity), which means that they are completely different lineages (Fig. 1).
It is known that in the AFLP analysis, the number of amplicons and the percentage of polymorphic DNA bands are determined by the number of selective nucleotides at the 3′ end of the restriction enzyme primers [16, 36]. This was supported in our studies with DNA of strains of Pleurotus genus. Primers with three selective nucleotides amplified fewer restriction fragments compared to those with one and two selective bases (Table 3).
Our results indicated that the AFLP method is very useful for fast, large scale, and reliable typing of strains of Pleurotus genus and practical identification of mushroom cultivars.
References
Ajith TA, Janardhanan KK (2007) Indian medicinal mushrooms as a source of antioxidant and antitumor agents. J Clin Biochem Nutr 40:157–162
Bao DP, Ishihara H, Mori N, Kitamoto Y (2004) Phylogenetic analysis of oyster mushrooms (Pleurotus spp.) based on restriction fragment length polymorphisms of the 5′ portion of 26S rDNA. J Wood Sci 50:169–176
Blears MJ, De Grandis SA, Lee H, Trevors JT (1998) Amplified fragment length polymorphism (AFLP): a review of the procedure and its applications. J Ind Microbiol Biotechnol 21:99–114
Borges MJ, Azevedo MO, Bonatelli JR, Felipe MSS, Astolfi-Filho S (1990) A practical method for the preparation of total DNA from filamentous fungi. Fungal Genetic Newslett 10:11
Brown JKM (1996) The choice of molecular marker methods for population genetic studies of plant pathogens. New Phytol 133:183–195
Chang ST (1999) World production of cultivated edible and medicinal mushrooms in 1997 with emphasis on Lentinus edodes (Berk.) Sing in China. Int J Med Mushrooms 1:291–300
Cohen R, Persky L, Hadar Y (2002) Biotechnological applications and potential of wood-degrading mushrooms of the genus Pleurotus. Appl Microbiol Biotechnol 58:582–594
Ellstrand NC, Elam DR (1993) Population genetic consequences of small population-size—implications for plant conservation. Annu Rev Ecol Systemat 24:217–242
Gunde-Cimerman N, Cimerman A (1995) Pleurotus fruiting bodies contain the inhibitor of 3-hydroxy-3-methylglutaryl-coenzyme A reductase-lovastatin. Exp Mycol 19:1–6
Hampl V, Pavlicek A, Flegr J (2001) Construction and bootstrap analysis of DNA fingerprinting-based phylogenetic trees with the freeware program FreeTree: application to trichomonad parasites. Int J Syst Evol Microbiol 51:731–735
Hamrick JL, Godt MJ (1989) Allozyme diversity in plant species. In: Brown AHD, Clegg MT, Weir BS, Kahler AL (eds) Plant population genetics, breeding and germplasm resources. Sinauer, Sunderland, pp 43–63
Hseu RS, Wang HH, Wang HF, Moncalvo JM (1996) Differentiation and grouping of isolates of the Ganoderma lucidum complex by random amplified polymorphic DNA-PCR compared with grouping on the basis of internal transcribed spacer sequences. Appl Environ Microbiol 62:1354–1363
Hynes SS, Chaudhry O, Providenti MA, Smith ML (2006) Development of AFLP-derived, functionally specific markers for environmental persistence studies of fungal strains. Can J Microbiol 52:451–461
Iracabal B, Zervakis G, Labarere J (1995) Molecular systematics of the genus Pleurotus—analysis of restriction polymorphisms in ribosomal DNA. Microbiology 141:1479–1490
Jaccard P (1912) The distribution of the flora in the alpine zone. New Phytol 11:37–50
Janssen P, Coopman R, Huys G, Swings J, Bleeker M, Vos P, Zabeau M, Kersters K (1996) Evaluation of the DNA fingerprinting method AFLP as an new tool in bacterial taxonomy. Microbiology 142:1881–1893
Karwasra SS, Mukherjee AK, Swain SC, Mohapatra T, Sharma RP (2002) Evaluation of RAPD, ISSR and AFLP markers for characterization of the loose smut fungus Ustilago tritici. J Plant Biochem Biotechnol 11:99–103
Kauserud H, Schmidt O, Elfstrand M, Hogberg N (2004) Extremely low AFLP variation in the European dry rot fungus (Serpula lacrymans): implications for self/nonself-recognition. Mycol Res 108:1264–1270
Lindeberg G, Holm G (1952) Occurrence of tyrosinase and laccase in fruit bodies and mycelia of some Hymenomycetes. Physiol Plant 5:100–114
Majer D, Mithen R, Lewis BG, Vos P, Oliver RP (1996) The use of AFLP fingerprinting for the detection of genetic variation in fungi. Mycol Res 100:1107–1111
Mizutani T, Inatomi S, Inazu A, Kawahara E (2010) Hypolipidemic effect of Pleurotus eryngii extract in fat-loaded mice. J Nutr Sci Vitaminol 56:48–53
Moussa TAA (2009) Molecular characterization of the phenol oxidase (pox2) gene from the ligninolytic fungus Pleurotus ostreatus. FEMS Microbiol Lett 298:131–142
Mueller UG, Lipari SE, Milgroom MG (1996) Amplified fragment length polymorphism (AFLP) fingerprinting of symbiotic fungi cultured by the fungus-growing ant Cyphomyrmex minutus. Mol Ecol 5:119–122
Mueller UG, Wolfenbarger LL (1999) AFLP genotyping and fingerprinting. Trends Ecol Evol 14:389–394
Nei M, Li WH (1979) Mathematical model for studying genetic variations in terms of restriction endonucleases. Proc Natl Acad Sci USA 76:5269–5273
Okuda Y, Murakami S, Matsumoto T (2009) A genetic linkage map of Pleurotus pulmonarius based on AFLP markers, and localization of the gene region for the sporeless mutation. Genome 52:438–446
Perez-Boada M, Ruiz-Duenas FJ, Pogni R, Basosi R, Choinowski T, Martinez MJ, Piontek K, Martinez AT (2005) Versatile peroxidase oxidation of high redox potential aromatic compounds: site-directed mutagenesis, spectroscopic and crystallographic investigation of three long-range electron transfer pathways. J Mol Biol 354:385–402
Selegean M, Putz MV, Rugea T (2009) Effect of the polysaccharide extract from the edible mushroom Pleurotus ostreatus against infectious bursal disease virus. Int J Mol Sci 10:3616–3634
Suazo A, Hall HG (1999) Modification of the AFLP protocol applied to honey bee (Apis mellifera L.) DNA. Biotechniques 26(704–705):708–709
Terashima K, Matsumoto T (2004) Strain typing of shiitake (Lentinula edodes) cultivars by AFLP analysis, focusing on a heat-dried fruiting body. Mycoscience 45:79–82
Terefework Z, Kaijalainen S, Lindstrom K (2001) AFLP fingerprinting as a tool to study the genetic diversity of Rhizobium galegae isolated from Galega orientalis and Galega officinalis. J Biotechnol 91:169–180
Tyrka M (2002) A simplified AFLP method for fingerprinting of common wheat (Triticum aestivum L.) cultivars. J Appl Genet 43:131–143
Urbanelli S, Della Rosa V, Punelli F, Porretta D, Reverberi M, Fabbri AA, Fanelli C (2007) DNA-fingerprinting (AFLP and RFLP) for genotypic identification in species of the Pleurotus eryngii complex. Appl Microbiol Biotechnol 74:592–600
Valsangiacomo C, Baggi F, Gaia V, Balmelli T, Peduzzi R, Piffaretti JC (1995) Use of amplified fragment length polymorphism in molecular typing of Legionella pneumophila and application to epidemiological studies. J Clin Microbiol 33:1716–1719
Vilgalys R, Moncalvo JM, Liou SR, Volovcek M (1996) Recent advances in molecular systematics of the genus Pleurotus. In: Royse DJ (ed) Mushroom biology and mushroom products. Pennsylvania State University Press, University Park, pp 91–101
Vos P, Hogers R, Bleeker M, Reijans M, Vandelee T, Hornes M, Frijters A, Pot J, Peleman J, Kuiper M, Zabeau M (1995) AFLP—a new technique for DNA-fingerprinting. Nucleic Acids Res 23:4407–4414
Zervakis G, Balis C (1996) A pluralistic approach in the study of Pleurotus species with emphasis on compatibility and physiology of the European morphotaxa. Mycol Res 100:717–731
Zervakis GI, Moncalvo JM, Vilgalys R (2004) Molecular phylogeny, biogeography and speciation of the mushroom species Pleurotus cystidiosus and allied taxa. Microbiology 150:715–726
Acknowledgments
This research was partially supported by the Polish Scientific Projects BS/ZBioch/Maria Curie-Sklodowska University.
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Pawlik, A., Janusz, G., Koszerny, J. et al. Genetic Diversity of the Edible Mushroom Pleurotus sp. by Amplified Fragment Length Polymorphism. Curr Microbiol 65, 438–445 (2012). https://doi.org/10.1007/s00284-012-0175-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00284-012-0175-7